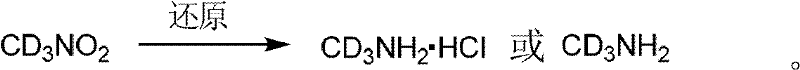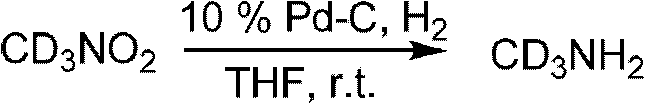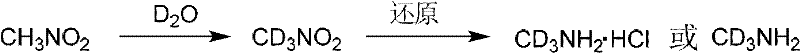发明内容
本发明的目的就是提供一种简便、高效和/或价廉的制备氘代甲胺及其盐的方法。
在本发明的第一方面,提供了一种氘代甲胺或其盐的制备方法,包括步骤:
(i)在碱和相转移催化剂下,将硝基甲烷,和氘水反应得到氘代硝基甲烷
(ii-a)在惰性溶剂中,还原氘代硝基甲烷,形成氘代甲胺;然后任选地,将氘代甲胺与酸反应,形成氘代甲胺的盐;或者,
(ii-b)在惰性溶剂中,在酸存在下,还原氘代硝基甲烷,直接形成氘代甲胺的盐
在另一优选例中,所述的碱选自下组:氢化钠、氢化钾、氘代氢氧化钠、氘代氢氧化钾、碳酸钾或其组合。
在另一优选例中,在步骤(ii-a)或(ii-b)中,使用锌粉、镁粉、铁、镍作为催化剂。
在另一优选例中,所述的酸选自下组:盐酸、硫酸、甲酸、乙酸、或其组合。
在另一优选例中,在步骤(ii-a)或(ii-b)中,所述的惰性溶剂选自下组:甲醇、乙醇、水、四氢呋喃、异丙醇、或其组合。
在本发明的第二方面,提供了一种氘代甲胺或其盐的制备方法,所述方法包括步骤:
(a1)在惰性溶剂中,在催化剂存在下,将邻苯二甲酰亚胺与氘代甲醇反应,形成N-(1,1,1-三氘代甲基)苯并丁二酰亚胺;
或者(a2)在惰性溶剂中,将邻苯二甲酰亚胺的碱金属盐与式A化合物进行反应,
式中,Z为CH3,O-CD3或其中R为甲基、硝基或卤素(F、Cl或Br),从而形成N-(1,1,1-三氘代甲基)苯并丁二酰亚胺;
步骤(b):将N-(1,1,1-三氘代甲基)苯并丁二酰亚胺与酸反应,形成氘代甲胺盐;以及
任选的步骤(c):将氘代甲胺盐与碱反应,形成氘代甲胺。
在另一优选例中,在步骤(a1)中,所述的惰性溶剂选自:四氢呋喃。
在另一优选例中,所述的酸选自下组:盐酸、硫酸、甲酸、乙酸、或其组合。
在另一优选例中,在步骤(a1)中,所述的催化剂选自:偶氮二甲酸二乙酯(DEAD)、偶氮二甲酸二异丙酯(DIAD)、三苯基膦、三丁基膦、或其组合。
在另一优选例中,在步骤(a2)中,所述的惰性溶剂包括:N,N-二甲基甲酰胺(DMF)、N,N-二甲基乙酰胺(DMA)、二甲基亚砜(DMSO)、N-甲基吡咯烷酮(NMP)、或其组合。
在另一优选例中,步骤(a2)中,反应温度为-10℃至回流温度,较佳地为-4℃至100℃,更佳地为20-80℃。
在另一优选例中,反应时间为0.1-24小时,较佳地为0.3-5小时,更佳地为0.5-2小时。
在另一优选例中,步骤(a2)中,所述的邻苯二甲酰亚胺碱金属盐包括:邻苯二甲酰亚胺钾盐、邻苯二甲酰亚胺钠盐、邻苯二甲酰亚胺锂盐或其组合。
在另一优选例中,步骤(a2)中,所述的式A化合物包括4-甲基苯磺酸-(1,1,1-三氘代甲基)酯、3-硝基苯磺酸-(1,1,1-三氘代甲基)酯,或4-硝基苯磺酸-(1,1,1-三氘代甲基)酯。
在另一优选例中,所述方法在步骤(a2)之前还包括步骤:在碱性条件下和惰性溶剂中,将氘代甲醇与对甲苯磺酰氯进行反应,从而形成4-甲基苯磺酸-(1,1,1-三氘代甲基)酯。较佳地,该步骤中所述的惰性溶剂包括水、四氢呋喃、或其混合溶剂。
在本发明的第三方面,提供了一种制备1,1,1-三氘代甲胺盐的方法,包括步骤:
在水性溶剂中,将N-(1,1,1-三氘代甲基)苯并丁二酰亚胺与酸进行反应,从而形成1,1,1-三氘代甲胺盐,其中所述的酸包括:盐酸、硫酸、氢溴酸、三氟乙酸、或其组合。
在另一优选例中,所述的反应温度为30至回流温度(如120℃),较佳地为40-110℃。
在另一优选例中,反应时间为0.5-48小时,较佳地为1-36小时,更佳地为2-24小时。
在另一优选例中,所述方法包括步骤:
在本发明的第四方面,提供了用本发明制备的氘代甲胺或其盐制备化合物N-(4-氯-3-(三氟甲基)苯基)-N’-(4-(2-(N-1’,1’,1’-三氘甲基氨基甲酰基)-4-吡啶基氧)苯基)脲的方法,
应理解,在本发明范围内中,本发明的上述各技术特征和在下文(如实施例)中具体描述的各技术特征可以互相组合,从而构成新的或优选的技术方案。限于篇幅,在此不再一一累述。
具体实施方式
本发明人经过广泛而深入的研究,开发了简便、高效而经济地生产氘代甲胺及其盐酸盐的的方法和工艺。在此基础上完成了本发明。
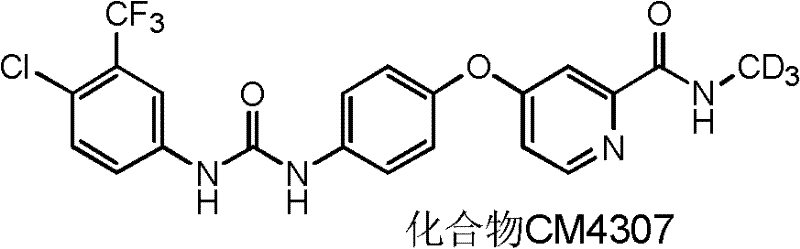
此外,本发明人还合成了可更有效抑制磷酸激酶的氘代的ω-二苯基脲化合物,以最优选的氘代化合物N-(4-氯-3-(三氟甲基)苯基)-N’-(4-(2-(N-1’,1’,1’-三氘甲基氨基甲酰基)-4-吡啶基氧)苯基)脲(化合物CM4307)和未氘代的N-(4-氯-3-(三氟甲基)苯基)-N’-(4-(2-(N-甲基氨基甲酰基)-4-吡啶基氧)苯基)脲(化合物CM4306)为例,
药代动力学实验结果显示,CM4307比CM4306的半衰期T1/2延长,曲线下面积AUC0-∞CM4307比CM4306显著增加,CM4307比CM4306表观清除率减少。
在人肝细胞癌SMMC-7721裸鼠移植模型进行的药效学实验结果显示,在100mg/kg每日的剂量下每日灌胃给药连续2周,CM4306的抗肿瘤活性的评价指标相对肿瘤增值率T/C(%)为32.2%;而CM4307的抗肿瘤活性的评价指标相对肿瘤增值率T/C(%)为19.6%,故抗肿瘤活性的绝对值提高10%以上,相对值提高约60%(32.2%/19.6%-1=64%),表现出更为显著的抑制肿瘤生长的作用。
术语
如本文所用,“卤素”指F、Cl、Br、和I。更佳地,卤原子选自F、Cl和Br。
如本文所用,“烷基”包括直链或支链的烷基。优选的烷基是C1-C4烷基,例如甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基等。
如本文所用,“氘代”指化合物或基团中的一个或多个氢被氘所取代。氘代可以是一取代、二取代、多取代或全取代。术语“一个或多个氘代的”与“一次或多次氘代”可互换使用。
在另一优选例中,氘在氘取代位置的氘同位素含量是大于天然氘同位素含量(0.015%),更佳地大于50%,更佳地大于75%,更佳地大于95%,更佳地大于97%,更佳地大于99%,更佳地大于99.5%。
如本文所用,术语“化合物CM4306”指化合物4-(4-(3-(4-氯-3-(三氟甲基)苯基]酰脲)-苯氧基)-N-甲基吡啶酰胺。
如本文所用,术语“化合物CM4307”指化合物4-(4-(3-(4-氯-3-(三氟甲基)苯基]酰脲)-苯氧基)-2-(N-1’,1’,1’-三氘代甲基)吡啶酰胺。
如本文所用,术语“TsOH”表示对甲苯磺酸。因此,CM4307·TsOH表示化合物CM4307的对甲苯磺酸盐。CM4309·TsOH表示化合物CM4309的对甲苯磺酸盐。
本发明的一种关键中间体是N-(1,1,1-三氘代甲基)苯并丁二酰亚胺:
该中间体也可称为“氘代甲基邻苯二甲酰亚胺”。上式化合物中的除H之外的其他元素(如N、C、O等)全部或基本上(>99wt%)为丰度最高的天然存在的元素,例如14N、12C和16O。
制备方法
下面更具体地描述本发明化合物的制备方法,但这些具体方法不对本发明构成任何限制。本发明化合物还可以任选将在本说明书中描述的或本领域已知的各种合成方法组合起来而方便的制得,这样的组合可由本发明所属领域的技术人员容易的进行。
通常,在制备流程中,各反应通常在惰性溶剂中,在室温至回流温度(如0℃~80℃,优选0℃~50℃)下进行。反应时间通常为0.1小时-60小时,较佳地为0.5-48小时。
以化合物CM4307为例,一种优选的制备流程如下:
合成路线一
如合成路线一所示,对羟基苯胺(化合物I)和3-三氟甲基-4-氯-苯胺(化合物II)在N,N’-羰基二咪唑、光气或三光气作用下,反应得到1-(4-氯-3-(三氟甲基)苯基)-3-(4-羟基苯基)脲(化合物III)。吡啶甲酸甲酯(化合物IV)和氘代甲胺或氘代甲胺盐酸盐在碱(例如碳酸钠、碳酸钾、氢氧化钠、三乙胺、吡啶等)的作用下、或直接混合反应,得到吡啶-2-(N-1’,1’,1’-三氘代甲基)甲酰胺(化合物V)。化合物III和化合物V在碱(如叔丁醇钾、氢化钠、氢化钾,碳酸钾、碳酸铯、磷酸钾、氢氧化钾、氢氧化钠)和任选的催化剂(如碘化亚铜和脯氨酸、或碘化亚铜和吡啶甲酸)的作用下,得到化合物CM-4307。上述反应在惰性溶剂,如二氯甲烷、二氯乙烷、乙腈、正己烷、甲苯、四氢呋喃、N,N-二甲基甲酰胺、二甲基亚砜等中,温度0~200℃下进行。
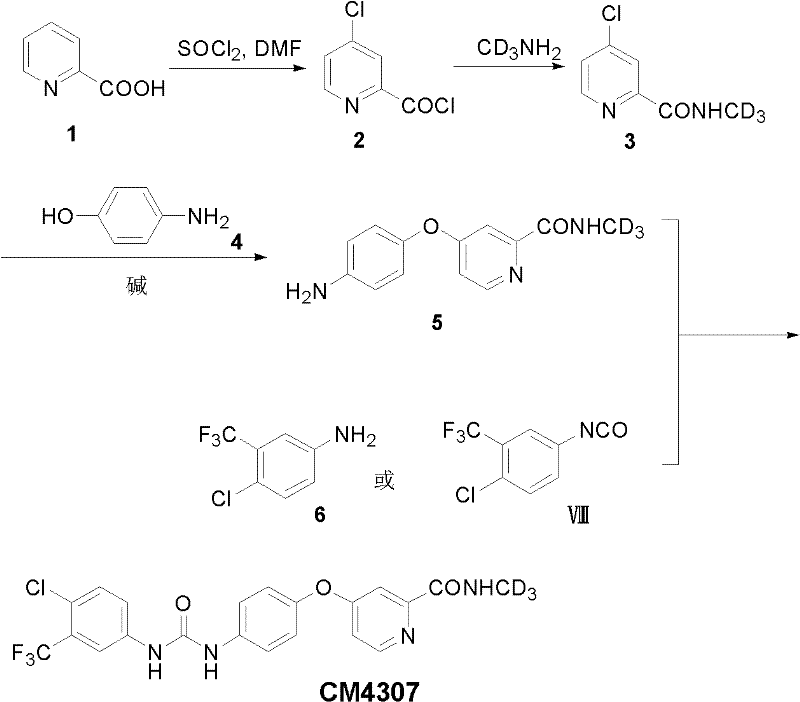
以化合物CM4307为例,另一种特别优选的制备流程如下:
其中,氘代可以通过氘代甲胺引入的。氘代甲胺也可以通过已知的文献方法如下方法制备,如氘代硝基甲烷的氢化加氢反应。
式中,r.t.表示室温。
或者,可通过以下反应得到氘代甲胺或其盐酸盐。硝基甲烷在碱(氢化钠、氢化钾、氘代氢氧化钠、氘代氢氧化钾、碳酸钾等),或在相转移催化剂下,和氘水反应得到氘代硝基甲烷,如有必要,重复上述实验,以得到高纯度的氘代硝基甲烷。氘代硝基甲烷还原,如锌粉、镁粉、铁或镍等作用下,得到氘代甲胺或其盐酸盐。
再者可以通过以下反应得到氘代甲胺或其盐酸盐。
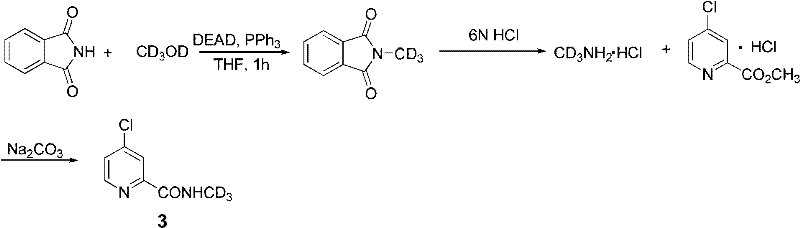
关键中间体3也可以通过如下方法从氘代甲醇合成。
其具体合成方法在实施例1中有详细的说明。
本发明的氘代甲胺或其盐的制法的主要优点包括:
(1)制法简便、高效且成本较低。
(2)产品纯度高。
(3)通用性高。
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。除非另外说明,否则份数和百分比为重量份和重量百分比。
实施例1:N-(4-氯-3-(三氟甲基)苯基)-N’-(4-(2-(N-1’,1’,1’-三氘甲基氨基甲酰基)-4-吡啶基氧)苯基)脲(化合物CM4307)
1、4-氯吡啶-2-(N-1’,1’,1’-三氘代甲基)甲酰胺(3)的制备
在配有尾气处理装置的250mL单颈圆底烧瓶中,加入氯化亚砜(60mL),维持温度在40~50℃之间,向其中缓慢的滴加无水DMF(2mL),滴加完毕后,继续搅拌10分钟,在20分钟内向其中分批加入烟酸(20g,162.6mmol),溶液的颜色逐渐由绿色转变为浅紫色。将温度升到72℃,搅拌回流16小时,产生大量的固体沉淀物。冷却到室温,用甲苯(100mL)稀释,浓缩至近干,然后再用甲苯稀释,浓缩至干。过滤,用甲苯洗涤,得到淡黄色的3-氯-吡啶-2-甲酰氯固体。冰浴下将此固体慢慢的加入到氘代甲胺的四氢呋喃饱和溶液中,维持温度低于5℃,继续搅拌5小时。浓缩,加乙酸乙酯,析出白色固体,滤除,滤液用饱和食盐水洗涤,无水硫酸钠干燥,浓缩至干,得到淡黄色的4-氯吡啶-2-(N-1’,1’,1’-三氘代甲基)甲酰胺(3)(20.68g),收率73%。
1H NMR(CDCl3,300MHz):8.37(d,1H),8.13(s,1H),7.96(br,1H),7.37(d,1H).
2、4-(4-氨苯氧基)-2-吡啶-(N-1’,1’,1’-三氘代甲基)甲酰胺(5)的制备
向100mL干燥的无水DMF中依次加入对氨基苯酚(9.54g,0.087mol),叔丁醇钾(10.3g,0.092mol),溶液变成深褐色,室温下搅拌2小时后,向其中加入4-氯吡啶-2-(N-1’,1’,1’-三氘代甲基)甲酰胺(3)(13.68g,0.079mol),无水碳酸钾(6.5g,0.0467mol),将反应液温度升到80℃后继续搅拌过夜。TLC检测反应完毕,冷却到室温,将反应液倒入乙酸乙酯(150mL)和饱和食盐水(150mL)的混和溶液中,搅拌分层,静置后分液,水层用乙酸乙酯萃取(100mL×3),合并萃取液,用饱和水洗涤(100mL×3),无水硫酸钠干燥,浓缩,得到淡黄色的4-(4-氨苯氧基)-2-吡啶-(N-1’,1’,1’-三氘代甲基)甲酰胺(18.00g),收率92%。
1H NMR(CDCl3,300MHz):8.32(d,1H),7.99(br,1H),7.66(s,1H),6.91~6.85(m,3H),6.69(m,2H),3.70(br,s,2H).
3、N-(4-氯-3-(三氟甲基)苯基)-N’-(4-(2-(N-1’,1’,1’-三氘甲基氨基甲酰基)-4-吡啶基氧)苯基)脲(CM4307)的制备
向120mL二氯甲烷中加入5-氨基-2-氯-三氟甲基苯(15.39g,78.69mol),N,N’-羰基二咪唑(CDI)(13.55g,83.6mmol),室温搅拌16小时后,向其中缓慢的滴加4-(4-氨苯氧基)-2-吡啶-(N-1’,1’,1’-三氘代甲基)甲酰胺(18g,73mmol)的二氯甲烷(180mL)溶液,室温下继续搅拌18小时。TLC检测反应完毕,旋去部分二氯甲烷溶剂至100mL左右,室温放置数小时,有大量白色固体析出,抽滤,固体用大量二氯甲烷洗涤。滤液浓缩去除部分溶剂后,又析出部分固体,合并两次固体,用大量二氯甲烷再次洗涤,得到白色粉状的N-(4-氯-3-(三氟甲基)苯基)-N’-(4-(2-(N-1’,1’,1’-三氘甲基氨基甲酰基)-4-吡啶基氧)苯基)脲CM4307纯品(20.04g),收率58%。
1H NMR(CD3OD,300MHz):8.48(d,1H),8.00(d,1H),7.55(m,5H),7.12(d,1H),7.08(s,2H),ESI-HRMS m/z:C21H13D3ClF3N4O3,Calcd.467.11,Found490.07(M+Na)+.
另外,可将化合物CM4307溶于二氯甲烷中,与过氧苯甲酸进行反应,制得相应的氧化产物:4-(4-(3-(4-氯-3-(三氟甲基)苯基)脲基)苯氧基)-2-(N-1’,1’,1’-三氘甲基氨基甲酰基)吡啶-1-氧化物。
实施例2:4-氯吡啶-2-(N-1’,1’,1’-三氘代甲基)甲酰胺(3)的制备
a)将邻苯二甲酰亚胺(14.7g,0.1mol),氘代甲醇(3.78g,0.105mol,1.05eq),三苯基膦(28.8g,0.11mol,1.1eq)溶于无水四氢呋喃中,冰浴下滴加DEAD(1.1eq)的四氢呋喃溶液,滴加完毕后室温搅拌一小时。过柱提纯,或者溶剂旋干后,加适量DCM于冰箱冷冻析出固体后过滤,滤液旋干,再快速过柱,得纯品氘代甲基邻苯二甲酰亚胺14.8g。收率90%。
b)氘代甲基邻苯二甲酰亚胺(12.5g,0.077mol)溶于适量盐酸(6N,50ml)中,于封管中回流24-30小时,反应液冷却至室温后,置于冰箱中冷却到零度以下,过滤析出的固体,用冷的去离子水洗涤,收集滤液,旋蒸除水并干燥得到氘代甲胺盐酸盐。加入无水DCM(100ml)于氘代甲胺盐酸盐中,并加入4-氯烟酸甲酯盐酸盐(6.52g,0.038mol,0.5eq),碳酸钠(12.2g,0.12mol,1.5eq),反应瓶密封,置于冰箱中反应一天。TLC检测反应,完毕后水洗,干燥,浓缩,过柱提纯。得化合物4-氯吡啶-2-(N-1’,1’,1’-三氘代甲基)甲酰胺(3),5.67g,收率86%。其结构特征与实施例1一致。
实施例3
氘代甲胺盐酸盐的合成
1.氘代硝基甲烷
将硝基甲烷(0.61g,10mmol,1.0eq)溶于重水(5.0g,250mmol,25.0eq)中,氮气置换3次,回流反应16小时。冷却至室温,加入无水乙醚(20mL×2)萃取。无水硫酸钠干燥,过滤减压除去溶剂,得到标题化合物,为黄色液体0.1g,核磁的结果表明有部分氘代或全氘代的硝基甲烷生成。
2.氘代甲胺盐酸盐
将氘代硝基甲烷(0.64g,10.0mmol)溶于甲醇(25.0mL)中,加入钯炭(10%,0.1g),通上气球氢气置换三次后,室温搅拌16小时,滴加盐酸酸化反应液,过滤,滤液减压除去溶剂,得0.60g标题化合物,为淡黄色产品,核磁的结果表明有氘代甲胺盐酸盐。
1H NMR(DMSO-d6,400MHz):δ8.05(br,2H)。
实施例4
氘代甲胺盐酸盐的合成
1:4-甲基苯磺酸-(1,1,1-三氘代甲基)酯的制备
将氢氧化钠(180g,4.5mol,5.0eq)加入到水(288mL)中,在0℃下,加入氘代甲醇(32.4g,900mmol,1.0eq),并缓慢滴加对甲苯磺酰氯(206g,1.1mmol,1.2eq)的四氢呋喃(288mL)溶液。升至室温搅拌过夜。在25℃以下滴加醋酸(206g)中和至中性,过滤,分液,水层用乙酸乙酯(100mL)萃取,滤饼用水(300mL)溶解,并用乙酸乙酯萃取(200mL),合并有机相,用饱和碳酸钠(100mL)洗涤,用饱和食盐水(100mL)洗涤,有机相无水硫酸钠干燥,过滤,减压除去溶剂,得到标题化合物,为浅黄色液体160.5g,纯度99%,收率94%。
1H NMR(CDCl3-d,400MHz):δ3.20(s,3H),7.71-7.75(m,2H),7.84-7.88(m,2H).
2:N-(1,1,1-三氘代甲基)苯并丁二酰亚胺的制备
将邻苯二甲酰亚胺钾盐(166.7g,0.9mol,2.0eq)加入至N,N-二甲基甲酰胺(DMF,225mL)中,室温下滴加4-甲基苯磺酸-(1,1,1-三氘代甲基)酯(85.2g,0.45mmol,1.0eq),在60℃下搅拌0.5小时。趁热过滤,滤饼用DMF(250mL)洗涤,过滤取出滤饼,再次用DMF(100mL)洗涤,合并DMF溶液,在0℃下,滴加水(1150mL),析出白色固体,过滤,并用水(100mL×2)洗涤,真空干燥得到标题化合物,为白色固体64g,纯度99.6%,收率85%。
1H NMR(CDCl3-d,400MHz):δ7.71-7.77(m,2H),7.84-7.88(m,2H).
3:1,1,1-三氘代甲胺盐酸盐的制备
在室温下,将N-(1,1,1-三氘代甲基)苯并丁二酰亚胺(82g,0.5mol,1eq)加入至蒸馏水(625mL)与浓盐酸(625mL,7.5mol,15eq)的混合液中,升温至105度回流过夜。冷却至室温,过滤,并用蒸馏水洗涤(50mL×2),减压除去盐酸,得到淡黄色固体,加入无水乙醇(140mL),回流1小时,冷却至室温,过滤,并用乙醇(30mL)洗涤,滤饼真空干燥得到标题化合物,为白色固体28g,收率80%。
1H NMR(DMSO-d6,400MHz):δ8.05(br,2H)。
实施例4:大鼠中的药代动力学评价
8只雄性Sprague-Dawley大鼠,7-8周龄,体重约210g,分成2组,每组4只(大鼠编号:对照组为13-16;实验组为9-12),单次口服给予3mg/kg剂量的(a)对组合物:未氘代的N-(4-氯-3-(三氟甲基)苯基)-N’-(4-(2-(N-甲基氨基甲酰基)-4-吡啶基氧)苯基)脲(对照化合物CM4306)或(b)实施例1制备的N-(4-氯-3-(三氟甲基)苯基)-N’-(4-(2-(N-1’,1’,1’-三氘甲基氨基甲酰基)-4-吡啶基氧)苯基)脲(本发明化合物CM4307),比较其药代动力学差异。
大鼠采用标准饲料饲养,给予水和利眠宁。实验的前一天晚上停止给予利眠宁,给药后2小时重新给予利眠宁。试验前16小时开始禁食。药物用30%PEG400溶解。眼眶采血,采血的时间点为给药后0.083小时,0.25小时,0.5小时,1小时,2小时,4小时,6小时,8小时和24小时。
令大鼠吸入乙醚后短暂麻醉,眼眶采集300uL血样于试管。试管内有30ul1%肝素盐溶液。使用前,试管于60℃烘干过夜。在随后一个时间点血样采集完成之后,大鼠乙醚麻醉后处死。
血样采集后,立即温和地颠倒试管至少5次,保证混合充分后放置于冰上。血样在4℃5000rpm离心5分钟,将血清与红细胞分离。用移液器吸出100uL血清到干净的塑料离心管中,表明化合物的名称和时间点。血清在进行LC-MS分析前保存在-80℃。
结果显示,CM4307比CM4306的半衰期T1/2延长[分别为11.3±2.1小时和8.6±1.4小时],曲线下面积AUC0-∞CM4307比CM4306显著增加[分别为11255±2472ng·h/mL和7328±336ng·h/mL],CM4307比CM4306表观清除率减少[分别为275±52mL/h/kg和410±18.7mL/h/kg]。
从上面结果看出,本发明化合物在动物体内具有更好的药物动力学,因而具有更好的药效学和治理效果。
实施例5:CM4307对人肝细胞癌SMMC-7721裸小鼠移植瘤生长抑制的
药效学评价
Balb/c nu/nu裸小鼠,6周龄,雌性,70只,购自上海试验动物资源中心(上海西普尔-必凯实验动物有限公司)。
SMMC-7721细胞购自中科院上海生命科学院(上海,中国)。
肿瘤裸鼠移植模型的建立:收获对数生长期的SMMC-7721细胞,计数后将细胞悬于1×PBS,调整细胞悬液浓度至1.5×107/ml。用1ml注射器在裸鼠右侧腋下皮下接种肿瘤细胞,3×106/0.2ml/鼠。共接种70只裸鼠。
在肿瘤体积达到30-130mm3时,将动物进行随机分组,共获得58只动物,使各组肿瘤差异小于均值的10%,并开始给药。
试验剂量分组设置见下表:
| 组别 |
动物 |
化合物 |
给药方式 |
剂量(mg/kg) |
方案 |
| 1 |
10 |
空白对照(溶剂) |
po |
0.1ml/10gBW |
qdx2周 |
| 2 |
8 |
CM4306 |
po |
10mg/kg |
qdx2周 |
| 3 |
8 |
CM4306 |
po |
30mg/kg |
qdx2周 |
| 4 |
8 |
CM4306 |
po |
100mg/kg |
qdx2周 |
| 5 |
8 |
CM4307 |
po |
10mg/kg |
qdx2周 |
| 6 |
8 |
CM4307 |
po |
30mg/kg |
qdx2周 |
| 7 |
8 |
CM4307 |
po |
100mg/kg |
qdx2周 |
试验期间每周测定两次动物体重和肿瘤大小。每日观察记录临床症状。给药结束时,拍照记录肿瘤大小。每组处死一只小鼠取肿瘤组织,固定于4%多聚甲醛。给药结束后,继续观察,当肿瘤均值大于2000mm3,或动物出现濒死状态时,处死动物,做大体解剖,取肿瘤组织固定于4%多聚甲醛。
肿瘤体积(Tumor volume,TV)的计算公式为:TV=a×b2/2。其中a、b分别代表肿瘤测量长和宽。相对肿瘤体积(relative tumor volume,RTV)计算公式为:RTV=Vt/V0。其中V0为分组给药时的肿瘤体积,Vt为测量时的肿瘤体重。抗肿瘤活性的评价指标为相对肿瘤增值率T/C(%),计算公式为:T/C(%)=(TRTV/CRTV)×100%。TRTV为治疗组RTV,CRTV为阴性对照组RTV。
疗效评价标准:相对肿瘤增值率T/C(%)≤40%并经统计学分析p<0.05为有效。
结果表明,CM4306和CM4307单剂量10、30、100mg/kg每日灌胃给药连续2周,两个化合物均表现剂量依赖性的抑制肿瘤生长作用。给药结束时,CM4306的T/C%分别为56.9%、40.6%和32.2%。CM4307的T/C(%)分别为53.6%、40.8%和19.6%。其中100mg/kg剂量组的T/C%均<40%,肿瘤体积与对照组比较有显著差异(p<0.01),表现显著的抑制肿瘤生长的作用。
CM4307的高剂量100mg/kg组比CM4306高剂量组肿瘤抑制作用强(最佳T/C%分别为19.6%和32.2%,d15),瘤体积组间比较有显著差异(p<0.01)。与CM4306相比,CM4307的抑瘤率的绝对值提高10%以上,相对幅度提高约60%(32.2%/19.6%-1=64%),表现出更为显著的抑制肿瘤生长的作用。
此外,试验过程中未见其他药物相关毒性反应。
在本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。