Leonardo Cuda, Federico Tielli, Daniela Orellana, Juan Pedro Díaz Vélez, Guadalupe Toledo, Humberto Mimura, Carlos Falbo
Servicio de Gastroenterología y Endoscopía Digestiva, HIGA P. Fiorito, Avellaneda. Buenos Aires, Argentina.
Acta Gastroenterol Latinoam 2020;50(4):470-473
Recibido: 14/04/2020 / Aprobado: 13/11/2020 / Publicado en www.actagastro.org el 14/12/2020

Resumen
La fiebre mediterránea familiar es una enfermedad hereditaria autosómica recesiva, que se caracteriza por episodios recurrentes y breves de fiebre con serositis (peritoneo, pleural, pericárdico) y dolor abdominal. El MEFV es el gen responsable de la enfermedad. La amiloidosis es la complicación más importante de esta enfermedad y es la principal causa de muerte en este caso. La fiebre mediterránea familiar se presenta en especial en las etnias mediterráneas y se manifiesta principalmente en las dos primeras décadas de vida. A continuación, se presenta el caso de un paciente masculino de 32 años de edad, que consulta por dolor abdominal crónico que cede parcialmente en la posición de plegaria mahometana y con AINEs, asociado a fiebre, diarrea, náuseas y vómitos. Luego de la evaluación semiológica e interpretación de los estudios complementarios, se arriba al diagnóstico de Fiebre mediterránea familiar, y se indica tratamiento con colchicina, con franca mejoría clínica.
Palabras claves. Fiebre mediterránea familiar, dolor abdominal recurrente, gen MEFV.
Familial Mediterranean Fever: a Rare Cause of Abdominal Pain
Summary
Familial Mediterranean Fever is an inherited autosomal recessive disease characterized by recurrent and brief episodes of fever with serositis, (peritoneum, pleural, pericardial) and abdominal pain. The MEFV is the responsible gene for the disease. Amyloidosis is the most important complication of this disease leading to the main cause of death in this case. Familial mediterranean fever occurs especially within Mediterranean ethnic groups and itis mainly manifested within the first two decades of life. The following is the case of a 32-year-old male patient who consults for chronic abdominal pain, that is partially controlled with mohammedan prayer and with NSAIDs, associated with fever, diarrhea, nausea, and vomiting. After semiological evaluation and interpretation of complementary studies, the Familial mediterranean fever diagnosis was reached, indicating Colchicine treatment with clinical improvement.
Key words. Familial mediterranean fever, recurrent abdominal pain, MEFV gen.
Abreviaturas
FMF: Fiebre mediterránea familiar.
AINEs: Antiinflamatorios no esteroideos.
PCR: Proteína C reactiva.
ERS: Eritrosedimentación.
HP: Helicobacter pylori.
SII: Síndrome de intestino irritable.
IBP: Inhibidores de la bomba de protones.
ATB: Antibiótico.
Introducción
La fiebre mediterránea familiar (FMF) es la forma más frecuente de fiebre periódica hereditaria. Es una enfermedad autosómica recesiva, que se caracteriza por episodios cortos y recurrentes de fiebre y dolor secundario a serositis (peritoneo, pleura, pericardio, sinovial y túnica vaginal testicular).1
El MEFV es el gen de la FMF, cuya detección confirma el diagnóstico.1 La amiloidosis es la segunda complicación más importante y suele ser la principal causa de muerte en los casos en que se presenta.1 Su prevalencia es variable y depende de factores étnicos (es mayor en judíos sefarditas y turcos).2
Objetivo
Presentar un caso atípico de dolor abdominal.
Caso clínico
Paciente masculino de 32 años de edad, que consulta por cuadro de dolor abdominal de quince años de evolución en fosa ilíaca derecha y flanco derecho, que se generaliza y cede parcialmente en la posición de plegaria mahometana y con AINEs, asociado a fiebre mayor a 38 ºC, diarrea, náuseas y vómitos. Los episodios se repiten cada quince días, con una duración promedio de 48 horas, con períodos intercrisis asintomáticos.
En la primera consulta se interpretó el cuadro como abdomen agudo secundario a apendicitis aguda, y no se realizó intervención quirúrgica por negativa de los familiares.
Durante esos años continuó con crisis, con un impacto negativo en su calidad de vida.
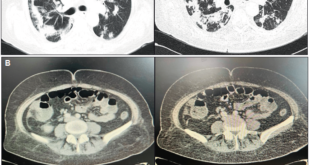
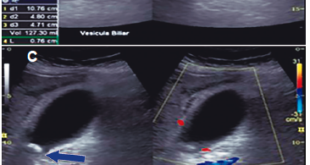
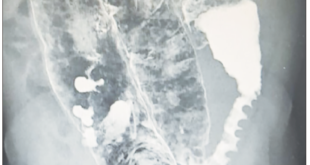
Consultó a numerosos especialistas de distintos centros, sin obtener un diagnóstico preciso. Se realizaron múltiples estudios analíticos, serológicos, autoinmunes, imagenológicos y endoscópicos, que arrojaroncomo único dato de relevancia un aumento de la amilasa tres puntos sobre el valor normal (el cual inicialmente fue relacionado con patología pancreática), PCR Y ERS elevadas, esteatosis hepática, esplenomegalia leve, hernia inguinal izquierda y gastritis superficial leve.
Diagnósticos previos: en los años anteriores el paciente había recibido múltiples diagnósticos por el cuadro: gastritis por HP, SII, dolor abdominal funcional, porfiria aguda intermitente, pancreatitis recurrente, realizando múltiples tratamientos con IBP, ATB, espasmolíticos, hernioplastia, sin respuesta. A mediados del año 2015, consulta al servicio de gastroenterología de nuestro nosocomio. Considerando la clínica, los antecedentes personales, el origen armenio y los estudios complementarios, se sospecha FMF, y ante la imposibilidad de realizar una prueba genética, se inicia una prueba terapéutica con colchicina (1 mg/día), con mejoría clínica. En mayo de 2017 se realizan los estudios genéticos, y se detecta mutación p. Val726Ala y p. Met694Val en heterocigosis en el exón 10 del gen MEFV, confirmando de esta manera el diagnóstico.
Discusión
La FMF es una enfermedad de herencia autosómica recesiva que afecta principalmente a descendientes de poblaciones mediterráneas, tales como las comunidades judías sefardíes, asquenazíes, armenios, árabes y turcos. La frecuencia de portadores se ha calculado de 1:5 a 1:7; se estima que hay 100.000 afectados en todo el mundo3 y se presenta en el 90% de los casos antes de los 20 años de edad,4 siendo más frecuente en hombres.
Clínicamente se manifiesta por episodios recurrentes de fiebre, acompañados de dolor abdominal agudo, que muchas veces es difícil de diferenciar de un abdomen agudo quirúrgico. La peritonitis puede ser periódica y formar parte de un síndrome de poliserositis con compromiso de pleura, pericardio y articulaciones. Otra manifestación característica es la formación de un eritema erisiopeloide en la cara anterior de pierna y el dorso del pie.5
Entre los episodios febriles los pacientes cursan asintomáticos y recurren en forma irregular, la mayoría de las veces sin un pródromo identificable. Los exámenes de laboratorio durante el ataque revelan elevación de los reactantes de fase aguda.
La FMF se produce por mutaciones en el gen MEFV 16p13.6 Se han identificado hasta la fecha más de 150 mutaciones del gen,7, 8 las más frecuentes son M694V, V726A y el E148Q. La mutación M694V muestra una asociación significativa con el desarrollo de amiloidosis renal, al igual que la mutación compuesta V726A-E148Q, por lo que es un determinante de peor pronóstico.
Este gen codifica una proteína denominada Pirina, compuesta por 781 aminoácidos y que se encuentra en el núcleo de granulocitos, células dendríticas y fibroblastos sinoviales.9 Además, participa en la vía del factor nuclear Κb (NFκB), regulador central de genes de inflamación y apoptosis.10 Esta proteína tendría un efecto inhibitorio sobre la activación de la IL-1β, por lo que las mutaciones de pérdida de función de esta causarían una regulación de la inflamación.10
Nuestro caso se trata de un paciente con dolor abdominal y fiebre recurrentes, en quien la búsqueda etiológica orientada a elementos infecciosos, inmunes y tumorales resultó negativa.
El primer elemento diagnóstico es la presentación clínica, clásica de la FMF, con episodios febriles de inicio en la infancia, recurrentes, autolimitados, que duran pocos días, con compromiso de serosas.
El segundo elemento diagnóstico es el origen poblacional del paciente; se trata de un descendiente de armenios tanto por línea paterna como materna. Determinadas mutaciones en el gen MEVF tienen una elevada frecuencia en ciertas poblaciones de la cuenca mediterránea, particularmente los armenios.11
El diagnóstico se establece sobre la base de los criterios de Tel-Hashomer (véase la Tabla 1).19 Nuestro paciente reúne dos criterios mayores (episodios de fiebre recurrente con serositis y respuesta favorable al tratamiento con colchicina), por lo que la sospecha de FMF se confirma.
Tabla 1. Criterios diagnósticos de Tel-Hashomer

Dentro del diagnóstico diferencial debemos plantear otras causas de fiebre de origen desconocido.
En nuestro medio, es habitual que se presenten dificultades en el diagnóstico, ya que no es considerada dentro de los diagnósticos diferenciales de fiebre de origen desconocido debido a la falta de notificación de casos en nuestro país.
Ante la sospecha de FMF (criterios de Tel-Hashomer), está indicado el estudio molecular, tal como fue realizado en este caso, el cual confirma el diagnóstico, habiéndose hallado homocigosis o heterocigosis compuesta en el gen MEVF. Habitualmente, ante la presencia de síntomas similares a los descriptos, se buscan las siguientes mutaciones: M694V, V726A, V680I, E148Q, V694I, las cuales representan entre el 70 y el 80% de los casos. Se han identificado portadores asintomáticos con mutaciones en ambos alelos, lo que demuestra una gran variabilidad en la penetrancia.12 El estudio genético molecular también permite el mapeo de otros miembros de la familia asintomáticos o paucisintomáticos, la detección de heterocigotos y el diagnóstico prenatal.
En cuanto al tratamiento, se basa en: control de las crisis, evitar su aparición y disminuir el riesgo de desarrollar amiloidosis. Para el tratamiento sintomático de las crisis se emplean los AINES. El fármaco de primera línea es la colchicina, que inhibe la quimiotaxis de los neutrófilos y ha demostrado mejorar el pronóstico de la enfermedad, consiguiendo una remisión completa de las crisis en un 60% de los pacientes y una disminución de la frecuencia en el 30%.13 Esta respuesta terapéutica es el último criterio diagnóstico mayor fundamental.
En este caso el paciente padece de mutaciones en ambos genes, lo que implica un peor pronóstico, siendo la amiloidosis renal su principal complicación, por lo que debe realizar controles periódicos de su función renal.
Un 5 a 10% de los pacientes no responden a la terapia convencional; en ellos las terapias propuestas son: interferón alfa y la adición de colchicina endovenosa al tratamiento oral. Se ha reportado también el uso de talidomida,14 infliximab,15 inhibidores de la recaptación de serotonina,16 etarnecept17 y anakinra.18
Conclusión
De lo analizado surge la necesidad de realizar una correcta evaluación de los pacientes portadores de patología crónica. En nuestro paciente, la reexaminación integral del caso, en conjunto con su presentación clínica y etnia, contribuyeron a realizar un correcto diagnóstico, que, sumado a un tratamiento adecuado, mejoró considerablemente su calidad de vida. Consideramos de vital importancia difundir la información sobre esta enfermedad poco frecuente, para que sea contemplada como diagnóstico diferencial en pacientes que padecen de dolor abdominal crónico intermitente.
Referencias
- Romero-Santana F, García-Cruz L, Jaén-Sánchez N, Hervás-García M, Santana-Rodríguez A, Pérez-Arellano JL. Fiebre mediterránea familiar. Nueva mutación MEFV con presentación clínica inusual Rev Esp Casos Clin Med Intern (RECCMI) 2018; 3 (3): 127-129.
- Sohar E, Gafni J, Pras M, Heller H. Familial Mediterranean Fever. A survey of 470 cases and review of the literature. Am J Med 1967; 43: 227-253.
- Simon A, Van der Meer J, Drenth J, Firestein. Familial Autoinflammatory Síndromes. En: Kelley’s Textbook of Rheumatology. Philadelphia: WB Saunders Company 2008; 8: capítulo 113.
- Inocencio J. Síndromes febriles recurrentes en pediatría [en línea]. En: 34º Congreso Argentino de Pediatría, Córdoba 2006. Disponible en: http://www.sap.org.ar/staticfiles/actividades/congresos/congre2006/conarpe34/material/reu_inocencio.pdf [acceso el 20 de mayo de 2013].
- Drenth JPH, Van der Meer JWM. Hereditary periodic fever. N Engl J Med 2001; 345: 1748-1757.
- Dodé C, Pécheux C, Cazeneuve C, Cattan D, Dervichian M, Goossens M, et al. Mutations in the MEFV gene in a large series of patients with a clinical diagnosis of familial Mediterranean fever. Am J Med Genet 2000; 92: 241-246.
- Babior BM, Matzner Y. The familial Mediterranean fever gene – cloned at last. N Engl J Med 1997; 337 (21): 1548-1549.
- Infevers Database (The registry of MEFV sequence variants). [Base de datos en línea]. 2001. Disponible en: http://fmf.igh.cnrs.fr/ISSAID/infevers/disease_menu. php?n=1 [acceso en julio de 2009].
- Chae JJ, Wood G, Masters SL, Richard K, Park G, Smith BJ, et al. The B30.2 domain of pyrin, the familial Mediterranean fever protein, interacts directly with caspase-1 to modulate IL-1 production. Proc Natl Acad Sci USA 2006; 103 (26): 9982-9987.
- Simon A, Van der Meer JW. Pathogenesis of familial periodic fever syndromes or hereditary autoinflammatory syndromes. Am J Physiol Regul Integr Comp Physiol 2006; 292: R86-R98.
- Yepiskoposyan L, Harutyunyan A. Population genetics of familial Mediterranean fever: a review. Eur J Hum Genet 2007; 15 (9): 911-916.
- Shohat M, Halpern GJ. Familial Mediterranean Fever. En: Pagon RA, Bird TD, Dolan CR, Stephens K, Adam, M, editors. GeneReviews [En línea]. Seattle (WA): University of Washington, Seattle; 1993 Disponible en: http://www.ncbi.nlm.nih.gov/books/NBK1227/ [acceso en julio 2009].
- Kallinich T, Haffner D, Niehues T, Huss K, Lainka E, Neudorf U, et al. Colchicine use in children and adolescents with familial Mediterranean fever: literature review and consensus statement. Pediatrics. 2007; 119 (2): e474-e783.
- Seyahi E, Ozdogan H, Masatlioglu S, Yazici H. Successful treatment of familial Mediterranean fever attacks with thalidomide in a colchicine resistant patient. Clin Exp Rheumatol. 2002; 20 (4, Suppl 26): S43-S44.
- Metyas S, Arkfeld DG, Forrester DM, Ehresmann GR. Infliximab Treatment of Familial Mediterranean Fever and Its Effect on Secondary AA Amyloidosis. J Clin Rheumatol. 2004; 10 (3): 134-137.
- Onat AM, Ozturk MA, Ozcazar L, Ureten K, Kaymak SU, Kiraz S, et al. Selective serotonin reuptake inhibitors reduce the attack frequency in Familial Mediterranean Fever. Tohoku J Exp Med 2007; 211: 9-14.
- Mor A, Pillinger MH, Kishimoto M, Abeles AM, Livneh A. Familial Mediterranean fever successfully treated with etanercept. J Clin Rheumatol 2007; 13 (1): 38-40.
- Roldan R, Ruiz AM, Miranda MD, Collantes E. Anakinra: new therapeutic approach in children with Familial Mediterranean Fever resistant to colchicines. Joint Bone Spine 2008; 75 (4): 504-505.
- Pras M, Kastner DL. Familial Mediterranean fever. In: Klippel JH, Dieppe PA, editors. Rheumatology. London: Mosby 1997; 2: 23.1-23.4.
Correspondencia: Leonardo Cuda
Correo electrónico: leonardocuda1983@gmail.com
Acta Gastroenterol Latinoam 2020;50(4):470-473