博文
Green Energy & Environment|识得根源,寻碳踪迹——果糖脱水反应初期胡敏素的生成网络构筑
|


研究背景
木质纤维素生物质是替代化石资源生产精细化学品和液体燃料的理想可再生资源,其主要成分是碳水化合物。近年来,将葡萄糖、果糖等碳水化合物转化为平台化学品的研究已取得了重大进展。其中,5-羟甲基糠醛(HMF)是葡萄糖和果糖脱水后的产物,由于其巨大的市场潜力,HMF被称为可持续化学领域“沉睡的巨人”。然而,HMF的产率低和分离纯化困难问题阻碍了该“巨人”的觉醒,这主要归因于HMF合成过程中易生成大量可溶性或不可溶性的胡敏素,造成10~50%的原料碳损失。为了提高HMF生产的经济可行性,应尽可能从源头上抑制胡敏素生成。然而,目前研究人员对胡敏素的生成路径尚缺乏系统的认识。 果糖脱水反应初期胡敏素的生成路径。 本文作者通过电喷雾电离质谱(ESI-MS)、衰减全反射红外光谱(ATR-IR)、拉曼光谱(Raman)、核磁共振谱(NMR)和量子化学计算,构筑了果糖在水溶液中脱水反应初期的详细反应网络,首次提出了生成可溶性胡敏素的三条双分子路径(即C12、C11和C11’路径),阐明了不同反应温度和时间条件下果糖互变异构体与可溶性胡敏素演化路径的密切关系,揭示了典型的极性非质子溶剂、Brønsted/Lewis酸催化剂和加热速率对可溶性胡敏素形成的影响规律,强调了控制果糖互变异构体分布和反应动力学对抑制胡敏素生成的重要性和有效性,为未来抑制胡敏素生成提供了重要的理论依据。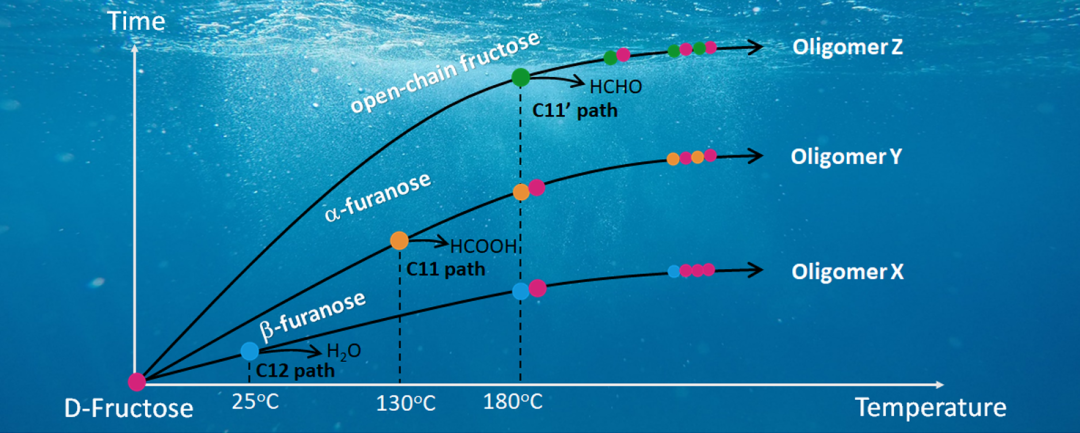
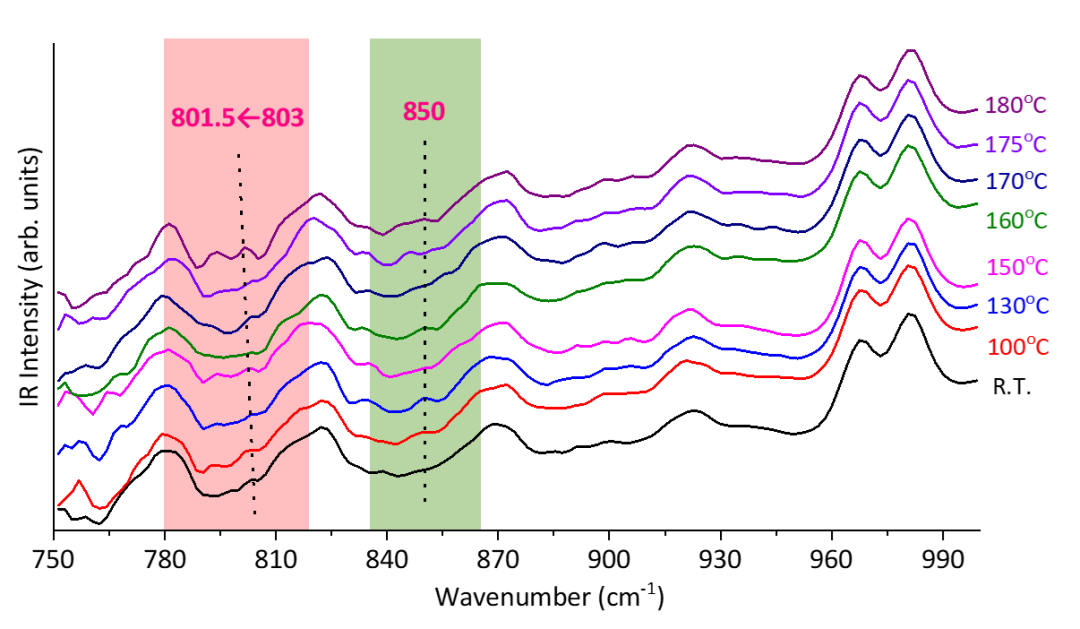
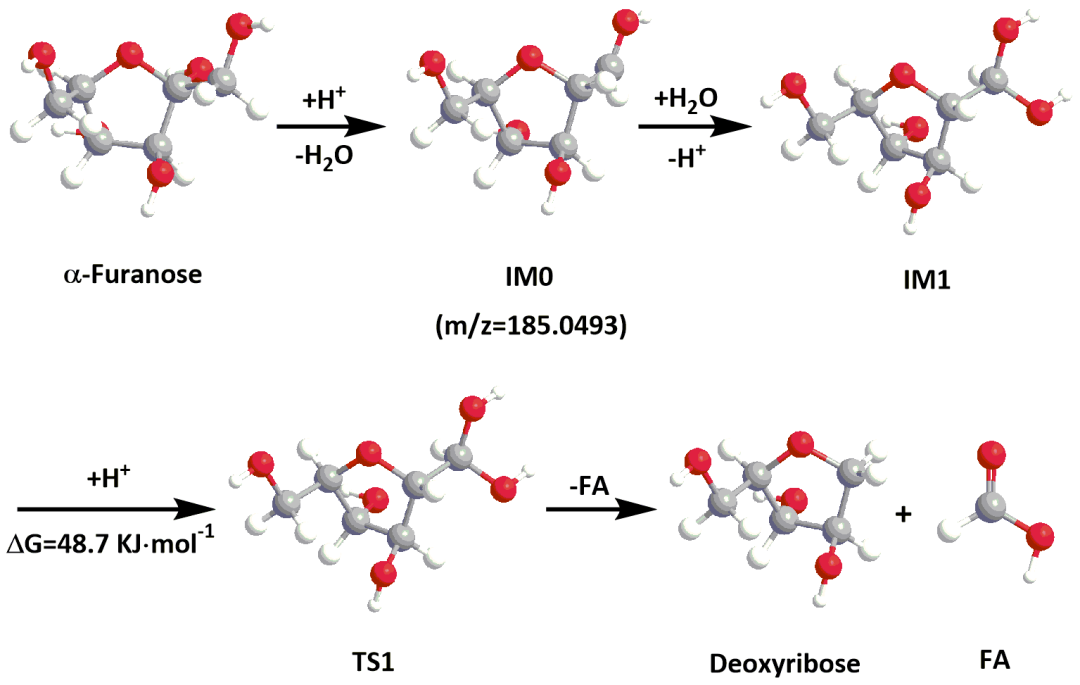
图文详解 通过淬灭反应液的ESI-MS谱图分析,揭示了果糖转化后的产物组成随反应温度和时间的变化规律。为了简化描述,定义C6为果糖(C6H12O6),C5为果糖脱除一分子甲酸后的中间体(C6-HCOOH),C5’为果糖脱除一分子甲醛后的中间体(C6-HCHO),C12为两分子果糖醚化脱水后的物种(C12=C6+C6-H2O),C11为果糖与C5缩合后的物种(C11=C5+C6-H2O),C11’为果糖与C5’缩合后的物种(C11'=C5'+C6-H2O)。 室温时,部分果糖发生分子间醚化形成稳定的二聚体C12物种。当反应温度升高至180oC并反应5分钟,依次生成C12脱除1~6分子水的物种;反应20分钟,发现[C12-6H2O]与果糖进一步缩合形成C18物种。如图1a所示,果糖通过分子间醚化、分子内脱水和缩合反应转化为可溶性胡敏素X,该路径被定义为C12路径。 此外,甲酸在150oC时生成,但未检测到C5中间体;180oC时,发现C5中间体与果糖通过分子间醚化或羟醛缩合反应生成C11物种[C5+C6-2H2O],延长时间,该C11物种发生逐级脱水;180oC反应2小时后,C11物种进一步与C5、C6或部分脱水的C6进行自缩合或交叉缩合,生成C16([C11+C5-2H2O])、C17([C11+C6-5H2O])、C22([C11+C11-3H2O])和C23([C11+2C6-6H2O])物种。如图1b所示,果糖通过降解缩合、分子内脱水和分子间进一步缩合反应后转化为可溶性胡敏素Y,该路径被定义为C11路径。 图1. 果糖在水中转化过程中寡聚物形成的双分子路径示意图. a) C12路径,b) C11路径,c) C11’路径。 180 °C时,还检测到C5’物种生成;反应10分钟后,发现C5’与果糖缩合并脱水生成[C5’+C6-3H2O]、[C5’+C6-4H2O]、[C5’+C6-5H2O]、[C6+C5′-7H2O]等C11’的脱水物种;反应2小时后,观察到C11’与果糖进一步缩合形成C17’和C23’物种,说明果糖通过脱除甲醛、分子内脱水和分子间缩合形成胡敏素Z1。此外,在180 °C分别反应20和25分钟后,还生成了[2HMF+HCHO]和[2HMF+HCHO-H2O]物种,说明脱除的甲醛可通过与HMF间的交叉缩合反应参与生成胡敏素Z2。如图1c所示,以上路径被定义为C11’路径。 图2. 不同反应温度和时间淬灭产物的ATR-IR光谱。 ATR-IR光谱中,850 cm-1处的特征IR峰归属为C12的C-C伸缩振动峰,从而证实了室温下C12物种的存在(图2)。果糖溶液的室温13C NMR谱图(102.7 ppm)也证实了b,b-呋喃果二糖的存在,该C12物种在100oC发生进一步转化。以b-呋喃果糖为底物的DFT计算表明,两个b-呋喃果糖分子在室温下醚化生成b,b-1,3-呋喃果二糖的表观活化能为43.5 KJ·mol-1,从理论上证实b-呋喃果糖室温下发生分子间醚化是可行的。随着反应温度升高,803 cm-1处的特征红外吸收峰向低波数移动,表明b-呋喃果糖的羟基氧被水中的水合氢离子所进攻,导致b-呋喃果糖发生连续脱水生成HMF,同时b,b-1,3-呋喃果二糖发生分子内脱水生成C12脱水物种。 通过比较果糖溶液在反应前和猝灭反应后的变旋异构体分布,以及果糖在重水中的变旋异构体分布和脱水反应,发现a-呋喃果糖与其脱甲酸反应相关,b-呋喃果糖与HMF生成相关。DFT计算结果表明,以a-呋喃果糖为底物时,中间体偕二醇呋喃糖(IM1)发生C-C断键生成脱氧核糖和甲酸的表观活化能为48.7 kJ·mol-1,从而从理论上证实a-呋喃果糖脱甲酸反应的可行性(图3)。 图3. 150 °C时a-呋喃果糖通过C1-C2键断裂生成FA和脱氧核糖的反应机理。 通过调节溶剂、催化剂和升温速率的系列对照实验,作者发现,通过构建水-有机共溶剂系统可调控胡敏素的生成路径。如,四氢呋喃的引入抑制了胡敏素X和Z生成,但促进胡敏素Y生成。加快升温速率,能通过改变互变异构体分布从而抑制胡敏素X和Z生成,但不影响胡敏素Y生成。Brønsted酸(HCl)抑制胡敏素X但促进胡敏素Y和Z生成;Lewis酸(AlCl3×6H2O)的作用规律与Brønsted酸一致,但能提高HMF的选择性。 总结与展望 本文结合实验和量子化学计算方法,系统地研究了纯水中果糖脱水转化初期的反应路径,提出了果糖经“醚化聚合”和“降解聚合”生成胡敏素的三条可能路径,完善了由果糖脱水转化制备HMF过程所涉及的反应网络。b-呋喃果糖在室温下通过分子间醚化形成C12中间体,a-呋喃果糖在130~150 °C通过脱羧和醚化形成C11中间体,链式果糖在180 °C通过脱羰和醚化形成C11’中间体,三条双分子路径形成的中间体经过后续的脱水和缩合反应而生成三种类型的可溶性胡敏素,合理地解释了HMF生成过程中碳损失的原因。以上对果糖脱水反应初期胡敏素生成网络的认识,可为提高生物质碳原子利用率提供必要的理论支持。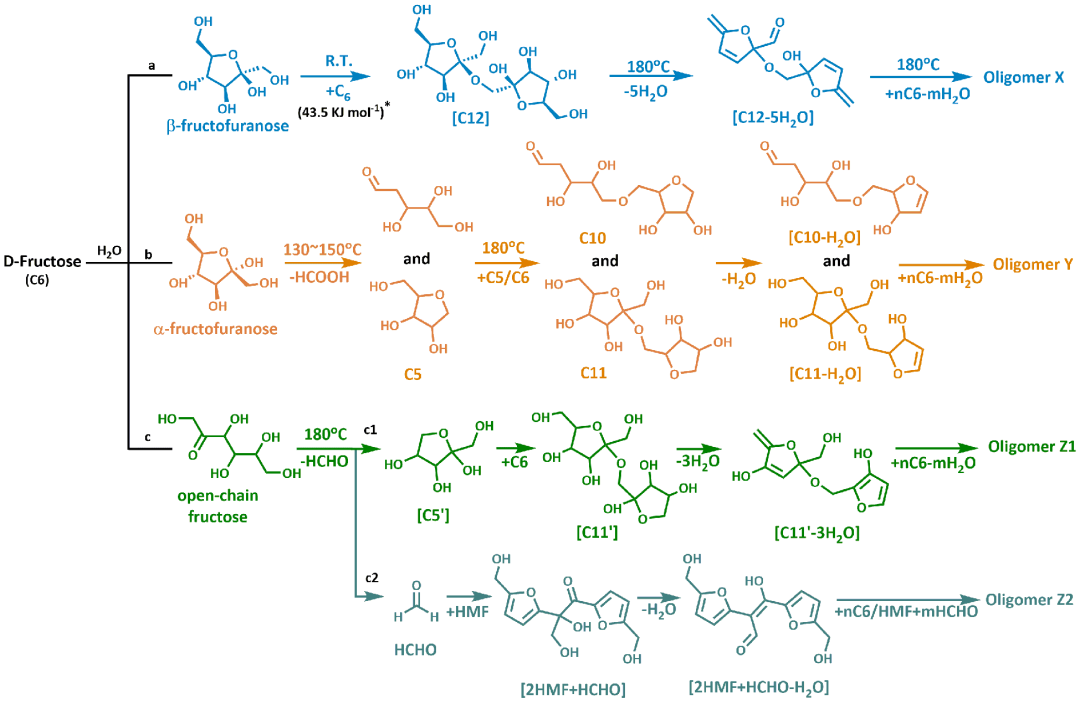
文章信息 本文以“Mapping out the reaction network of humin formation at the initial stage of fructose dehydration in water”为题发表在Green Energy & Environment期刊,第一作者为四川大学付兴博士(现为成都中医药大学讲师),通讯作者为四川大学祝良芳教授和胡常伟教授。本工作得到国家自然科学基金、高等学校学科创新引智计划、中央高校基本科研基金的支持。

https://blog.sciencenet.cn/blog-3393673-1369227.html
上一篇:Green Energy & Environment 荣获“中国最具国际影响力学术期刊”称号
下一篇:Green Energy & Environment|光驱动的低能耗CO2变温吸附