- European Federation of Societies for Ultrasound in Medicine and Biology ~ Educating all for competence to practice ultrasound safely
Detection of a micrometastasis using elastography – a promising method [May 2023]
December 14, 2023
The use of ultrasound for cosmetic or aesthetic body shaping (2024)
January 29, 2024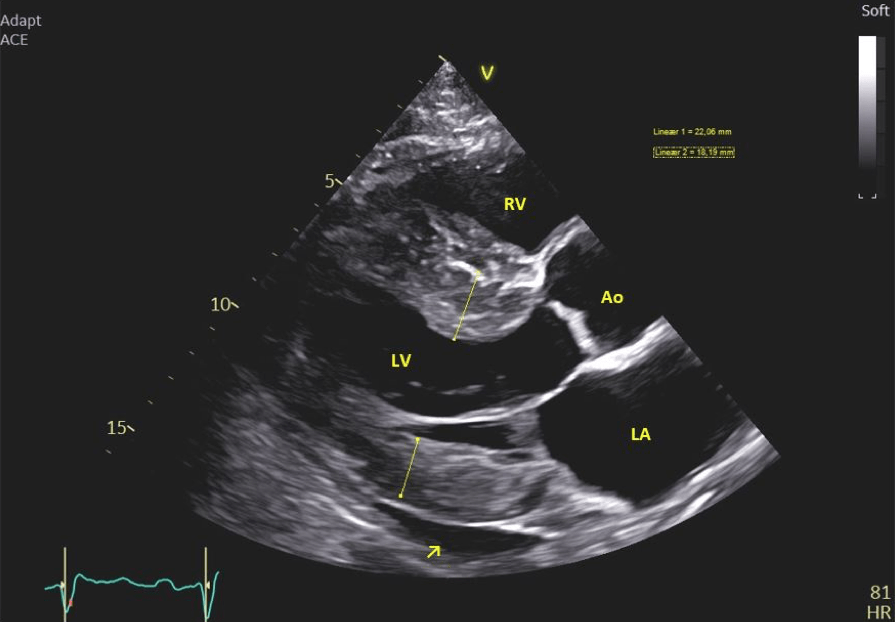
SUBMIT YOUR CASE
EFSUMB invites submission of interesting cases for the website section 'Case of the Month'. All CoM submissions are eligible for selection for free registration at the next Euroson congress. Two cases that receive the most 'likes' in a year will receive free registration for the next EUROSON congress and the third most liked liked case will receive a cash prize of 100 EUR.
Cardiac AL-amyloidosis
Authors: Jay Chotai, MD, Department of Emergency Medicine, Akershus University Hospital, Norway
Ai Phi Thuy Ho, MD, Department of Cardiology, Østfold Hospital Trust, Norway
1Clinical History
A 60 year old male patient with type 2 diabetes mellitus, asthma, and ischemic heart disease presented to the Emergency department (ER) with chest palpitations, and shortness of breath during physical activity since two weeks. Upon physical examination, a systolic cardiac murmur over the mitral valve was detected. He also had pitting edema in the distal parts of the lower limbs. Blood pressure was 114/71 mmHg.
ECG revealed a sinus rhythm with first-degree atrioventricular block, 1-2mm ST-T elevations in leads V2-V3 and minor ST-T wave depressions in leads V5-V6. His laboratory results returned with a normal complete blood count, liver enzymes, and creatinine. However, he had elevated levels of high-sensitivity troponin T (60 ng/L) and NT-proBNP (4014 ng/L). Consequently, the patient was admitted for further evaluation.
ECG revealed a sinus rhythm with first-degree atrioventricular block, 1-2mm ST-T elevations in leads V2-V3 and minor ST-T wave depressions in leads V5-V6. His laboratory results returned with a normal complete blood count, liver enzymes, and creatinine. However, he had elevated levels of high-sensitivity troponin T (60 ng/L) and NT-proBNP (4014 ng/L). Consequently, the patient was admitted for further evaluation.
2Image findings
ECHOCARDIOLOGY:
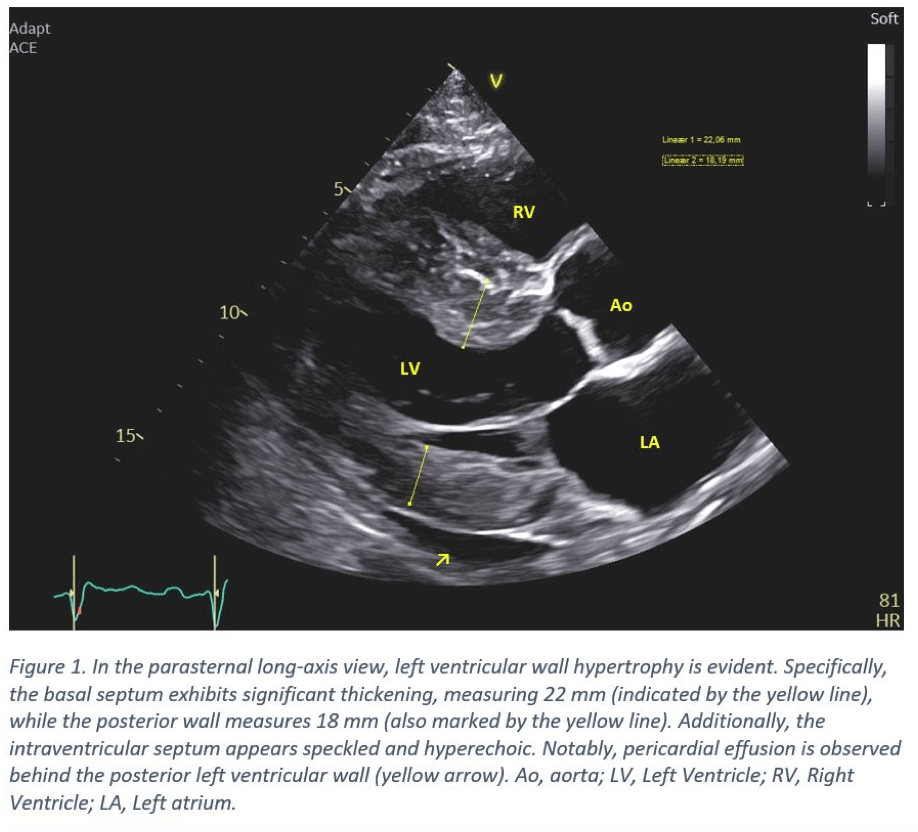
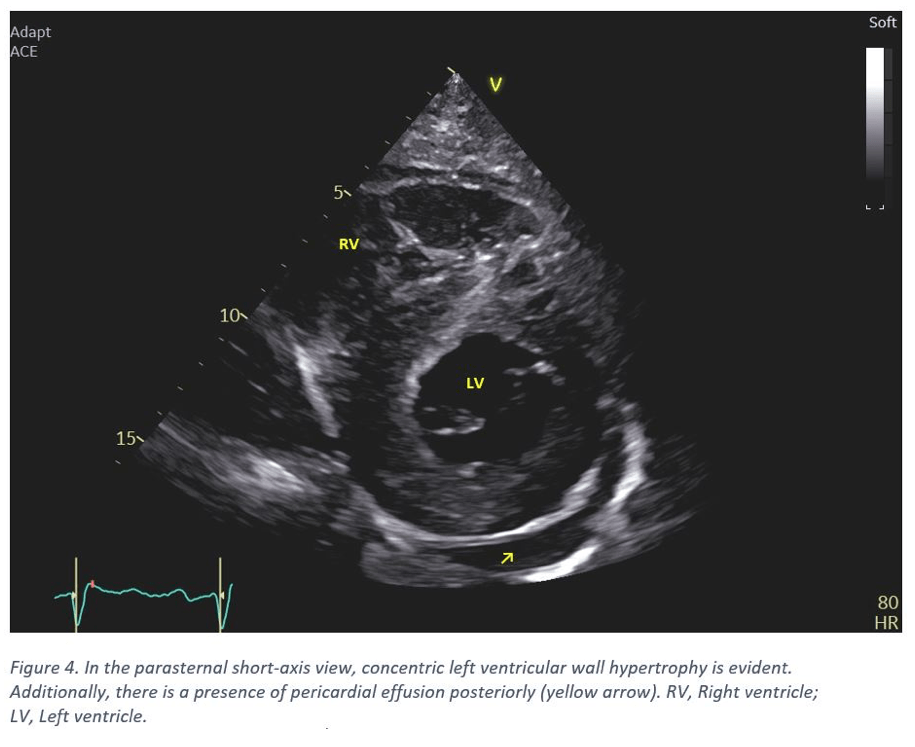
- The left ventricle displays concentric hypertrophy with the basal septum notably thickened (20-22 mm). Other septal regions also exhibiting thickening. The posterior wall measures 16-18 mm (Figure 1 & 4). The intraventricular septum appears speckled and hyperechoic (Figure 2).
- The interatrial septum is thickened, along with the mild thickening of the mitral valve leaflets (Figure 2).
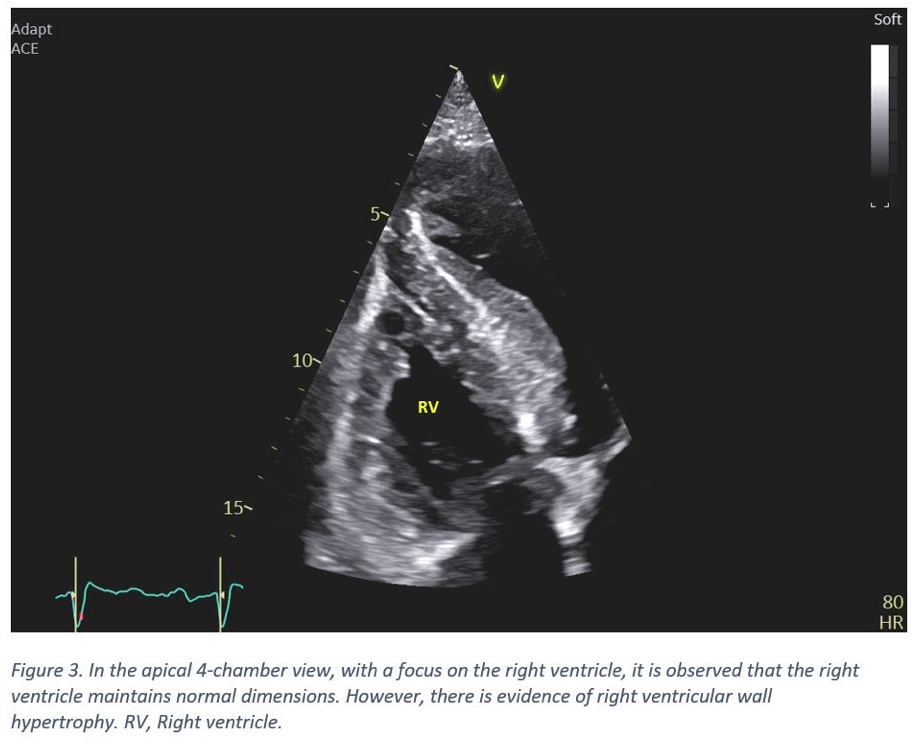
- The right ventricle demonstrates hypertrophy. Its dimensions remain within the normal range (Figure 3).
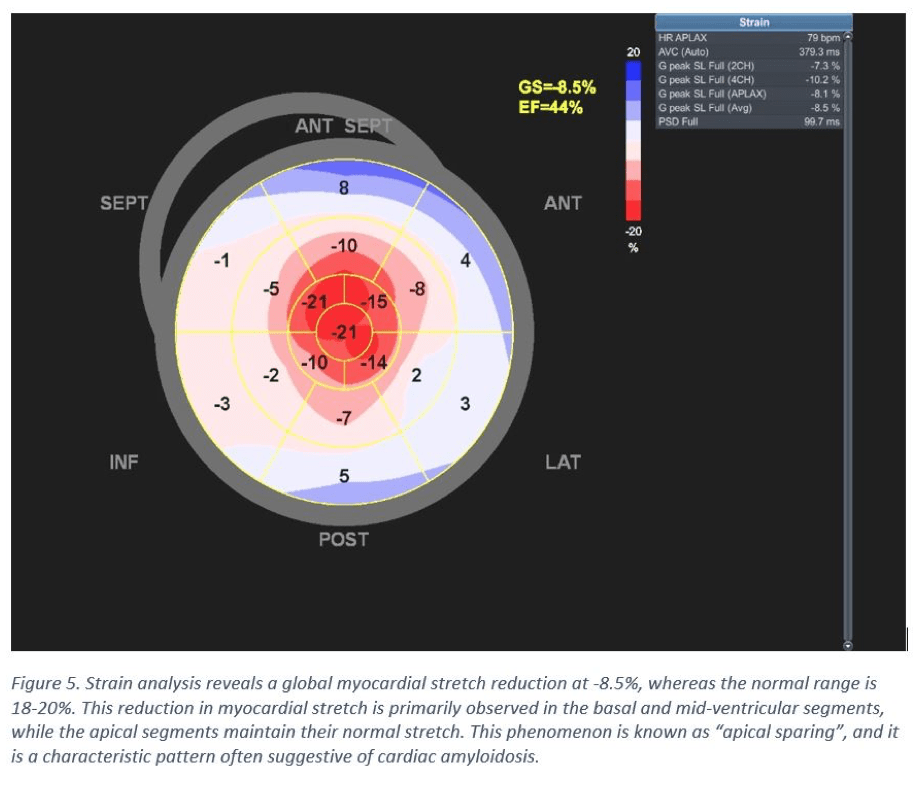
- Left ventricle global longitudinal strain (LV-GLS) is measured at -9%, indicating reduced systolic function (Figure 5). However, there are notable good apical contractions (Video clip). The biplanar ejection fraction (EF) was calculated to 50%, signifying severe longitudinal dysfunction.
MAGNETIC RESONANCE IMAGING (MRI):
MRI revealed a globally hypertrophic left ventricle with a normal ejection fraction. Significantly elevated T1 values were observed globally, and there was a notable increase in extracellular volume. These findings raise suspicion of cardiac amyloidosis.
CORONARY ANGIOGRAPHY:
No stenosis or occlusion were identified.
- The left ventricle displays concentric hypertrophy with the basal septum notably thickened (20-22 mm). Other septal regions also exhibiting thickening. The posterior wall measures 16-18 mm (Figure 1 & 4). The intraventricular septum appears speckled and hyperechoic (Figure 2).
- The interatrial septum is thickened, along with the mild thickening of the mitral valve leaflets (Figure 2).
- The right ventricle demonstrates hypertrophy. Its dimensions remain within the normal range (Figure 3).
- Left ventricle global longitudinal strain (LV-GLS) is measured at -9%, indicating reduced systolic function (Figure 5). However, there are notable good apical contractions (Video clip). The biplanar ejection fraction (EF) was calculated to 50%, signifying severe longitudinal dysfunction.
MAGNETIC RESONANCE IMAGING (MRI):
MRI revealed a globally hypertrophic left ventricle with a normal ejection fraction. Significantly elevated T1 values were observed globally, and there was a notable increase in extracellular volume. These findings raise suspicion of cardiac amyloidosis.
CORONARY ANGIOGRAPHY:
No stenosis or occlusion were identified.
3Diagnosis
Cardiac ultrasound and Cardiac MRI confirmed the presence of hypertrophic cardiomyopathy in our patient.
Subsequent blood workup showed elevated level of kappa free light chain (623 mg/L). Serum electrophoresis and immunoglobulin levels remained within the normal range. A bone marrow biopsy was performed and confirmed the presence of monoclonal plasma cells. Amyloid deposition was not detected in the bone marrow. However, an abdominal fat biopsy confirmed the diagnosis of systemic AL-amyloidosis.
Subsequent blood workup showed elevated level of kappa free light chain (623 mg/L). Serum electrophoresis and immunoglobulin levels remained within the normal range. A bone marrow biopsy was performed and confirmed the presence of monoclonal plasma cells. Amyloid deposition was not detected in the bone marrow. However, an abdominal fat biopsy confirmed the diagnosis of systemic AL-amyloidosis.
4Discussion
BACKGROUND:
In cardiac amyloidosis (CA), an amyloid protein accumulates in the interstitial space between cardiac myocytes, causing cellular injury. Patients with CA often also exhibit extracardiac manifestations affecting the kidney, nervous system, gastrointestinal tract, and musculoskeletal system (1).
There are primarily two subtypes of CA; transthyretin cardiac amyloidosis (ATTR-CA) and immunoglobulin light chain cardiac amyloidosis (AL-CA) (1).
Amyloid light chain (AL) amyloidosis is a rare disease with an annual incidence from 1 in 75,000 to 100,000 individuals. Approximately 50% of these patients have cardiac involvement (1). The AL-amyloid deposits consist of immunoglobulin light chains derived from clonal plasma cells in the bone marrow.
CLINICAL PERSPECTIVE:
Delays in diagnosing cardiac amyloidosis is common, hence it is important to be aware of the echocardiographic findings that may suggest the presence of cardiac amyloidosis. Typical echocardiographic findings include diastolic dysfunction, concentric biventricular thickening with involvement of the right ventricle, strain with relative apical sparing, and poor biventricular long axis with a normal/near-normal EF. Furthermore, the presence of biventricular, valvular, and interatrial septal thickening, accompanied by biatrial dilatation, is indicative of cardiac amyloidosis (2, 3).
Cardiac magnetic resonance (CMR) imaging offers improved myocardial imaging, making it valuable for distinguishing cardiac amyloidosis from other forms of cardiomyopathy.
Radionuclide imaging using 99mTc-DPD scintigraphy is highly sensitive and can effectively localize cardiac amyloid deposits, particularly in patients with ATTR-type amyloidosis.
While endomyocardial biopsy has traditionally been considered the gold standard for detecting and classifying amyloidosis (4), it is often adequate to diagnose AL-CA based on a combination of factors. These include the presence of AL-amyloid deposition in an extracardiac tissue biopsy, identification of a monoclonal light chain in blood or urine, evidence of clonal plasma cells via bone marrow biopsy, and typical findings on cardiac imaging (5).
THERAPY PLANNING:
The treatment for AL-CA involves anti-myeloma-based chemotherapy and immunotherapy. Autologous stem cell transplantation is considered in some cases. The treatment objective is to eliminate clonal plasma cells, thereby reducing the production of amyloid precursor protein.
Hematological remission is characterized by the clearance of the immunoglobulin light chains. This is typically observed within 3 to 6 months of initiating treatment, while organ-specific response usually becomes evident within 6 to 12 months (6).
OUTCOME & PROGNOSIS:
Various staging systems are employed to predict the prognosis of AL-amyloidosis. These staging systems primarily rely on the assessment of cardiac involvement severity, which is determined by the levels of Troponin T and NT-proBNP.
In advanced stages, AL amyloidosis has a significantly poor long-term prognosis, with a reported median survival as short as 5 months (6). However, recent advancements in more effective therapies have led to improved survival.
Our patient met the criteria for AL-CA, and received anti-myeloma-based immunochemotherapy. After completing six treatment cycles, hematological remission was achieved.
In cardiac amyloidosis (CA), an amyloid protein accumulates in the interstitial space between cardiac myocytes, causing cellular injury. Patients with CA often also exhibit extracardiac manifestations affecting the kidney, nervous system, gastrointestinal tract, and musculoskeletal system (1).
There are primarily two subtypes of CA; transthyretin cardiac amyloidosis (ATTR-CA) and immunoglobulin light chain cardiac amyloidosis (AL-CA) (1).
Amyloid light chain (AL) amyloidosis is a rare disease with an annual incidence from 1 in 75,000 to 100,000 individuals. Approximately 50% of these patients have cardiac involvement (1). The AL-amyloid deposits consist of immunoglobulin light chains derived from clonal plasma cells in the bone marrow.
CLINICAL PERSPECTIVE:
Delays in diagnosing cardiac amyloidosis is common, hence it is important to be aware of the echocardiographic findings that may suggest the presence of cardiac amyloidosis. Typical echocardiographic findings include diastolic dysfunction, concentric biventricular thickening with involvement of the right ventricle, strain with relative apical sparing, and poor biventricular long axis with a normal/near-normal EF. Furthermore, the presence of biventricular, valvular, and interatrial septal thickening, accompanied by biatrial dilatation, is indicative of cardiac amyloidosis (2, 3).
Cardiac magnetic resonance (CMR) imaging offers improved myocardial imaging, making it valuable for distinguishing cardiac amyloidosis from other forms of cardiomyopathy.
Radionuclide imaging using 99mTc-DPD scintigraphy is highly sensitive and can effectively localize cardiac amyloid deposits, particularly in patients with ATTR-type amyloidosis.
While endomyocardial biopsy has traditionally been considered the gold standard for detecting and classifying amyloidosis (4), it is often adequate to diagnose AL-CA based on a combination of factors. These include the presence of AL-amyloid deposition in an extracardiac tissue biopsy, identification of a monoclonal light chain in blood or urine, evidence of clonal plasma cells via bone marrow biopsy, and typical findings on cardiac imaging (5).
THERAPY PLANNING:
The treatment for AL-CA involves anti-myeloma-based chemotherapy and immunotherapy. Autologous stem cell transplantation is considered in some cases. The treatment objective is to eliminate clonal plasma cells, thereby reducing the production of amyloid precursor protein.
Hematological remission is characterized by the clearance of the immunoglobulin light chains. This is typically observed within 3 to 6 months of initiating treatment, while organ-specific response usually becomes evident within 6 to 12 months (6).
OUTCOME & PROGNOSIS:
Various staging systems are employed to predict the prognosis of AL-amyloidosis. These staging systems primarily rely on the assessment of cardiac involvement severity, which is determined by the levels of Troponin T and NT-proBNP.
In advanced stages, AL amyloidosis has a significantly poor long-term prognosis, with a reported median survival as short as 5 months (6). However, recent advancements in more effective therapies have led to improved survival.
Our patient met the criteria for AL-CA, and received anti-myeloma-based immunochemotherapy. After completing six treatment cycles, hematological remission was achieved.
5Teaching Points
Patients presenting with symptoms of heart failure and hypertrophic cardiomyopathy should undergo evaluation for amyloid cardiomyopathy.
Although a rare disease, CA have typical echocardiographic characteristics of which clinicians should be aware off. It is important to differentiate between the various subtypes of cardiac amyloidosis since the treatment options and prognosis vary depending on the underlying etiology.
Although a rare disease, CA have typical echocardiographic characteristics of which clinicians should be aware off. It is important to differentiate between the various subtypes of cardiac amyloidosis since the treatment options and prognosis vary depending on the underlying etiology.
6References
1.) Kittleson M, Ruberg F et al. 2023 ACC Expert consensus decision pathway on comprehensive multidisciplinary care for the patient with cardiac amyloidosis. J Am Coll Cardio. Mar 2023:8:1076–1126.
2.) Sanjay M. Banypersad, MRCP, et al. Updates in cardiac amyloidosis: a review. Journal of the American Heart Association. 2012, Volume 1, Issue 2.
3.) Melero Polo J, Roteta Unceta-Barrenechea A, Revilla Martí P, et al. Echocardiographic markers of cardiac amyloidosis in patients with heart failure and left ventricular hypertrophy. Cardiol J 2023; 30: 266-275.
4.) Prochorec-Sobieszek M, Bilińska ZT, Grzybowski J, et al. Cardiac amyloidosis diagnosed by endomyocardial biopsy. Clinical, histopathological, immunohistochemical and ultrastructural studies. Kardiol Pol 2005; 63: 20-35.
5.) Quarta CC, Gonzalez-Lopez E, Gilbertson JA, et al. Diagnostic sensitivity of abdominal fat aspiration in cardiac amyloidosis. . Eur Heart J 2017; 38: 1905-1908.
6.) Kyle RA, Gertz MA, Greipp PR, et al. A trial of three regimens for primary amyloidosis: colchicine alone, melphalan and prednisone, and melphalan, prednisone, and colchicine. N Engl J Med. 1997; 336: 1202-1207.
2.) Sanjay M. Banypersad, MRCP, et al. Updates in cardiac amyloidosis: a review. Journal of the American Heart Association. 2012, Volume 1, Issue 2.
3.) Melero Polo J, Roteta Unceta-Barrenechea A, Revilla Martí P, et al. Echocardiographic markers of cardiac amyloidosis in patients with heart failure and left ventricular hypertrophy. Cardiol J 2023; 30: 266-275.
4.) Prochorec-Sobieszek M, Bilińska ZT, Grzybowski J, et al. Cardiac amyloidosis diagnosed by endomyocardial biopsy. Clinical, histopathological, immunohistochemical and ultrastructural studies. Kardiol Pol 2005; 63: 20-35.
5.) Quarta CC, Gonzalez-Lopez E, Gilbertson JA, et al. Diagnostic sensitivity of abdominal fat aspiration in cardiac amyloidosis. . Eur Heart J 2017; 38: 1905-1908.
6.) Kyle RA, Gertz MA, Greipp PR, et al. A trial of three regimens for primary amyloidosis: colchicine alone, melphalan and prednisone, and melphalan, prednisone, and colchicine. N Engl J Med. 1997; 336: 1202-1207.
{kind=link}