Laboratory Studies
GSD type I
Hypoglycemia (serum glucose < 60mg/dL), lactic acidosis (blood lactate >2.5 mmol/L), hyperuricemia (uric acid >5.0 mg/dL), hypertriglyceridemia (triglyceride level >250 mg/dL), and hypercholesterolemia (cholesterol level >200 mg/dL) are present in patients with GSD type 1. Urea and creatinine levels might be elevated when renal function is impaired. [35] The following laboratory values should be obtained:
-
Serum glucose and electrolyte levels (Higher anion gap [also see the anion gap calculator] may suggest lactic acidosis.)
-
Serum lactate level
-
Blood pH
-
Serum uric acid level
-
Serum triglyceride and cholesterol levels (see HDL cholesterol and LDL cholesterol)
-
CBC and differential (eg, anemia, leukopenia, neutropenia)
-
Coagulation
-
Urinalysis for aminoaciduria, proteinuria, and microalbuminuria in older patients
-
Urinary excretion levels of uric acid and calcium
-
Serum alkaline phosphatase, calcium, phosphorus, urea, and creatinine levels
GSD type II
Findings on laboratory analyses are usually normal. Serum creatinine kinase (CK), lactic dehydrogenase and aspartate aminotransferase can be elevated.
Molecular diagnosis:
Definitive diagnosis requires measurement of the activity of acid alpha-1,4-glucosidase in the whole blood or dried blood spots, This assay should be use as initial test. [41, 42] Gene sequencing is used to confirm diagnosis, two pathogenic mutations in the GAA gene is considered confirmatory. [42] In case of indeterminate results, enzyme activity can be measure in skin fibroblast or muscle samples.
Urine glucose tetrasaccharide (Glc(4)) can be use adjunctive diagnostic test, moreover for monitoring of effect of enzyme replacement therapy.
Newborn screening is still under investigation. Studies have tried to demonstrate advantages of screening, even though the incidence of GSD II in screened and unscreened population was similar. Those screened were diagnosed and treated earlier. [43, 44, 45]
GSD type III
Fasting hypoglycemia and ketonuria may be noted.
Hyperlipidemia may be present.
Serum aminotransferase and CK levels may be elevated. Normal CK do not exclude GSD type III. In GSD type IIIb, serum aminotransferase levels are elevated during childhood but usually normalize at puberty.
Usually, serum lactate and uric acid levels are in the reference range.
GSD type IV
Liver function test: Serum aminotransferase levels are elevated. Synthetic function of the liver is also affected: elevated PTT, PT and hypoalbuminemia can be seen as liver failure progress. Fasting hypoglycemia is present in some patients. In neuromuscular variants CK is elevated.
In skin fibroblast cultures, muscle and liver tissues, glycogen branching enzyme activity (GBE assay) can be measured.
Molecular testing and GBE Assay after histologic work-up is indicated: identification of biallelic pathogenic variants in GBE1 gene, makes the diagnosis of GSD Type IV. If biopsy cannot be performed genetic testing or GBE assay can be performed in the setting of high suspicion of disease.
In families with prior history of GSD type IV, carried testing should be performed. Prenatal diagnosis can be done by culture of amniocytes. [40]
GSD type V
The main laboratory sign of disease is elevated levels of serum CK at rest. After intensive exercise, CK levels increase further.
At the same time, the blood ammonia, inosine, hypoxanthine, and uric acid concentrations are above the reference range. Activities of muscle phosphorylase may be extremely low.
Molecular gene testing of PYGM is to confirm diagnosis of GSD type V. If genetic diagnosis is unclear, myophosphorylase enzyme activity assay can confirm the diagnosis as well. [46]
Differentiate patients with McArdle disease from patients with other inflammatory myopathies. In addition, GSD type VII has the same clinical manifestations and can be differentiated on the basis of enzymatic study only. The forearm ischemic test, a useful diagnostic test, can produce abnormal results in patients with GSD type VII and in patients with debranching enzyme deficiency (GSD type III) when it is performed after fasting.
GSD type VI
Serum aminotransferase levels are elevated. Hypoglycemia, ketosis, and hyperlipidemia are rare and usually mild. CK is normal. Uric acid and lactic acid are normal, however post prandial lactic acidosis has also been described. Glucose does not increase after glucagon administration. Biotinidase activity is elevated.
Molecular testing is the preferred method for diagnosis, testing of PYGL, is diagnostic. Enzyme assay are available but low specificity.
Molecular testing is recommended in families with known mutations, to provide early treatment.
GSD type VII
CK levels are elevated.
Erythrocyte, hemoglobin, and reticulocyte counts, and serum unconjugated bilirubin concentration are important diagnostic measurements in patients with hemolysis.
Imaging Studies
In GSD type I, liver and kidney ultrasonography should be performed for follow-up of organomegaly and detection of hepatic adenomas and nephrocalcinosis.
Because of the risk of long-term complications, liver ultrasonography should be performed every 12-24 months in patients less than 16 years of age. In adults, imaging of the liver, in the form of CT or MRI should be performed at least every 12 months. Current guidelines recommend abdominal ultrasonography with tumor marker levels (eg, alpha-fetoprotein [AFP], carcinoembryonic antigen [CEA]) every 3 months if the patient develops hepatic lesions. [47] Abdominal CT scanning or MRI is advised whenever the lesions are large, poorly defined, or are growing rapidly. See the image below.
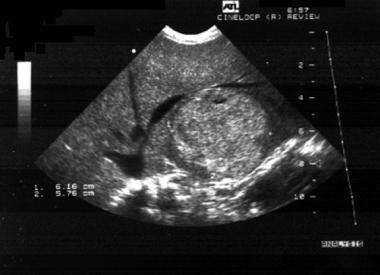 Glycogen storage disease type I. Abdominal sonogram showing large nodules in the liver.
Glycogen storage disease type I. Abdominal sonogram showing large nodules in the liver.
In GSD type II, echocardiography may be performed. It is noninvasive and useful for detection of cardiac muscle involvement. Occasionally, only the left ventricle may be affected. In advanced disease, evaluating the functional reserve of the heart may be helpful.
In GSD type III, echosonography may be performed. It is a noninvasive method that can provide useful information about the size of the liver, spleen, and heart.
In GSD type IV abdominal ultrasound reveal enlarged, fibrotic or cirrhotic liver.
In GSD types V and VII, MRI with phosphate-31 is a noninvasive method for the investigation of muscle metabolism.
In GSD type VI, ultrasound is performed for liver measurement and to rule out hepatic adenoma. Bone scan should be performed once the patient stops growing,
Other Tests
GSD type I
Glucagon or epinephrine challenge test causes little to no increase in glucose levels, but plasma levels of lactic acid are significantly raised. [35]
Orally administered galactose and fructose (1.75 g/kg) do not increase glucose levels, but plasma lactic acid levels do increase.
Glucose tolerance test (1.75 g/kg PO) progressively lowers lactic acid levels over several hours after the administration of glucose.
GSD type II
ECG is characteristic with shortening of the PR interval and large QRS complex.
Electromyography (EMG) reveals a myopathic pattern in all patients with pseudomyotonic discharge. Many patients have fibrillation potentials.
Nerve conduction velocities are in the reference range.
In pulmonary function test, the force vital capacity is usually reduced.
Genetic counseling and carrier testing is advised to affected families.
GSD type III
In the glucose tolerance test, serum lactate levels increase from the basal levels during the test, gradually returning to baseline values thereafter.
Orally administered galactose and fructose are converted into glucose because gluconeogenesis is unaffected.
Ingested amino acids and proteins induce a moderate but prolonged increase in blood glucose levels.
The response of blood glucose levels to the administration of glucagon and epinephrine varies. Glucagon administered after a fasting period does not induce a rise in glycemia; however, if glucagon is administered 2 hours after a meal, it produces an increase in blood glucose levels.
EMG findings are compatible with skeletal myopathy, and peripheral nerve conduction velocities may be abnormal.
ECG changes suggest ventricular hypertrophy, but signs of significant cardiac dysfunction are rarely observed.
GSD type IV
Glucose tolerance test results are in the reference range.
Glucagon and epinephrine test results vary.
GSD type V
The forearm ischemic test is a useful diagnostic test. Lack of an increase in blood lactate concentration and exaggerated increase in ammonia concentration simultaneously are reliable signs of disturbed glycogen metabolism in the skeletal muscle.
Occasionally, EMG changes may be similar to those of some nonspecific inflammatory myopathies.
GSD type VI
Diagnosis rests with histologic analysis of liver tissue or determination of the activity of the enzymes hepatic phosphorylase in the liver and phosphorylase b kinase in the liver, skeletal muscle, and heart.
GSD type VII
The forearm ischemic exercise test is a useful diagnostic test.
EMG should be performed.
Procedures
GSD type I
For diagnostic purposes, 13C nuclear magnetic resonance spectroscopy may be used for enzyme function assessment.
Definitive diagnosis requires determination of G6Pase activity in fresh and frozen liver tissue specimens and/or DNA-based analysis. When assaying for translocases, an open surgical liver biopsy is needed for sampling an adequate tissue specimen.
GSD type II
Skin biopsy should be performed to determine the activity of the enzyme in fibroblast culture.
If the presence of the mutations is known in the family, amniocentesis is necessary for amniotic fluid or chorion biopsy with the aim of prenatal diagnosis.
GSD type III
Biopsy of the liver and skeletal muscle should be performed for enzyme activity measurements.
GSD type IV
Liver and skeletal muscle biopsies are needed for enzyme activity and microscopic analysis.
Glycogen content in tissues is usually in the reference range, but its structure is abnormal.
GSD type V
Muscle biopsy should be performed.
Molecular DNA analysis or analysis of the functional activities of myophosphorylase is necessary for definitive diagnosis of McArdle disease.
Prenatal diagnosis is unnecessary.
GSD type VI
Skeletal muscle and liver biopsy should be performed for microscopic and enzymatic analysis.
GSD type VII
Muscle biopsy should be performed for microscopic and enzymatic analysis.
Histologic Findings
A liver biopsy may reveal chronic histologic changes of varying severity despite mild disease. All GSDs may exhibit these findings on liver biopsy. Hence, definitive classification requires a mutational analysis. [48]
In GSD type I, no specific findings occur in the liver, but higher amounts of normal glycogen, as well as fatty infiltration, are found. There may be distention of hepatocytes due to glycogen and lipid deposition. In GSD type I, liver fibrosis and cirrhosis do not occur. [35] Histologic findings in the kidneys comprise focal glomerular sclerosis, interstitial fibrosis, tubule atrophy or vacuolization, and significant atherosclerosis. A conspicuous glomerular hypertrophy occurs, and less commonly, numerous lipid deposits occur in the glomerular mesangium, tubular epithelial cells, and interstitium. Electron microscopy may reveal diffuse thickening of the glomerular basement membrane and lipid droplets in the mesangium.
In GSD type II, ultrastructural analysis of a large number of different tissue samples reveals large amounts of normal glycogen. In a muscle biopsy under a light microscope and hematoxylin and eosin staining, a “lace-work pattern can be identified. On periodic acid-Schiff staining, muscle fibers stain strong in infantile form. Under the electron microscope, muscle biopsies show vacuolar myopathy with glycogen storage in lysosomes and free glycogen in the cytoplasm, however, failure to identify this finding cannot rule out the diagnosis.
See the images below.
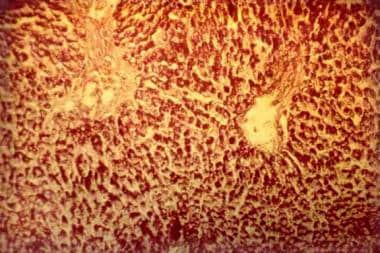 Glycogen storage disease type II. Photomicrograph of the liver. Note the intensively stained vacuoles in the hepatocytes (periodic acid-Schiff, original magnification X 27).
Glycogen storage disease type II. Photomicrograph of the liver. Note the intensively stained vacuoles in the hepatocytes (periodic acid-Schiff, original magnification X 27).
 Glycogen storage disease type II. Photomicrograph of the liver. Note the regular reticular net and hepatocytes vacuolization (Gordon-Sweet stain, original magnification X 25).
Glycogen storage disease type II. Photomicrograph of the liver. Note the regular reticular net and hepatocytes vacuolization (Gordon-Sweet stain, original magnification X 25).
The histologic picture of the liver in patients with GSD type III is characterized by generalized distension of the hepatic cells by glycogen and fibrous tissue. The fibrotic process may be characterized by minimal periportal disease or micronodular cirrhosis. This is usually nonprogressive.
In GSD type IV, biopsy of the liver and muscles shows enlarged, PAS- positive hepatocytes, and diastase -resistance inclusions. In the liver, foamy histiocytes in the reticuloendothelial system can also be seen. Interstitial fibrosis is also present, Fibrillar aggregates of amylopectin-like material are seen under a electron-microscope.
In GSD type V, histologic findings are nonspecific.
In GSD type VI, hepatocytes distended by the accumulated frayed or burst glycogen (ie, alpha particles, rosette form) may be observed in the liver and are less compact than in classic glycogenoses types I and III.
In GSD type VII, the abnormal polysaccharide accumulates, with fibrillar morphology, in the skeletal muscle.
-
An infant with glycogen storage disease type Ia. Note the typical facial aspect resembling a doll's face.
-
Glycogen storage disease type I. Abdominal sonogram showing large nodules in the liver.
-
A child with glycogen storage disease type Ia.
-
Glycogen storage disease type II. Photomicrograph of the liver. Note the intensively stained vacuoles in the hepatocytes (periodic acid-Schiff, original magnification X 27).
-
Glycogen storage disease type II. Photomicrograph of the liver. Note the regular reticular net and hepatocytes vacuolization (Gordon-Sweet stain, original magnification X 25).








