Practice Essentials
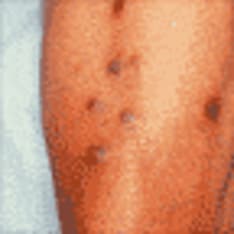
Porphyria is the common term for a group of syndromes, largely hereditary, that result from defects in porphyrins (the enzymes involved in heme synthesis). Depending on the specific enzyme affected, porphyria may manifest clinically in an acute or non-acute manner, and the signs and symptoms may be predominantly neurovisceral, psychiatric, cutaneous, or some combination of those. See the image below.
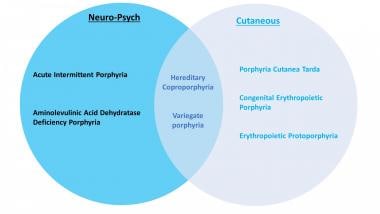
Types of porphyria
Porphyrias can be inherited or (rarely) acquired. [1] There are at least 8 different types of porphyrias. Acute intermittent porphyria (AIP), hereditary coproporphyria (HCP), variegate porphyria (VP), and the familial form of porphyria cutanea tarda (PCT) follow an autosomal dominant inheritance pattern with low penetration. Aminolevulinic acid dehydratase deficiency porphyria (ADP), congenital erythropoietic porphyria (CEP), erythropoietic protoporphyria (EPP), and hepatoerythropoietic porphyria (HEP) are autosomal recessive. [2] There is also an X-linked dominant inherited porphyria called X-linked protoporphyria (XLP).
The mutations that underlie porphyria result in accumulation and increased excretion of porphyrins and their precursors. Clinical manifestations depend on the step in the heme production pathway in which the enzymatic defect occurs. [3] There are many ways to classify porphyrias; the most common are by pathophysiology, depending on where pathway intermediates accumulate (hepatic vs erythropoietic) or by clinical manifestation (acute vs cutaneous).
The acute porphyrias include the following:
The cutaneous porphyrias include the following:
-
Erythropoietic protoporphyria
-
Hepatoerythropoietic porphyria
-
X-linked protoporphyria
Signs and symptoms
In acute porphyrias, presenting features of attacks may include the following:
-
Abdominal pain - The most common presenting symptom, occurring in 90% of attacks
-
Muscle weakness
-
Focal neurologic deficits (eg, quadriparesis, hyponatremia)
-
Chest, back, or limb pain
-
Psychiatric symptoms (eg, psychosis, confusion, hallucinations, anxiety)
-
Discolored urine (turns red or dark on exposure to light)
-
HCP and VP can also present as blistering cutaneous lesions
The cutaneous porphyrias are dermatologic diseases that may or may not involve the liver and nervous system. Cutaneous signs often result from photosensitivity (eg, skin fragility and blistering in porphyria cutanea tarda).
Workup
Urine, blood, and stool studies are used in the diagnosis of porphyrias, as follows:
-
Urine porphyrin studies are the mainstay in the diagnosis of acute porphyria attacks. Test for increased 5-aminolevulinic acid and porphobilinogen (PBG) in a single-void urine collected during an attack.
-
Significantly increased ALA and PBG in urine have 100% specificity (ie, rules in) for AIP, ADP, HCP, and VP. A normal urine PBG result has a sensitivity of almost 100% (ie, rules out) in the diagnosis of porphyria in acutely symptomatic patients.
-
To assess for cutaneous porphyria, the plasma porphyrin level should be measured, using fluorescence emission spectroscopy. Whole blood for porphyrin analysis is used to identify protoporphyria plasma porphyrins.
-
Stool studies: The ratio of fecal coproporphyrin to fecal protoporphyrin varies among the porphyrias. For example, fecal protoporphyrin always exceeds coproporphyrin in VP, whereas the reverse is true in HCP.
-
Erythrocyte uroporphyrinogen decarboxylase activity is a reliable diagnostic test for PCT.
-
Erythrocyte total protoporphyrin is the initial screening exam to diagnose EPP and XLP, if the total is elevated with metal-free protoporphyrin it is suggestive of EPP or XLP.
-
Genetic testing is helpful in confirming the diagnosis of porphyrias
Treatment
Treatment of acute episodes addresses symptoms and includes parenteral narcotics for pain. Hemin (plasma-derived intravenous heme) is the definitive treatment and mainstay of management.
Avoidance of sunlight is the key in preventing attacks of cutaneous porphyrias. Treatment of PCT involves reduction of hepatic iron, such as with phlebotomy or low-dose hydroxychloroquine. Afamelanotide, an α-melanocyte–stimulating hormone analogue, may permit increased duration of sun exposure in patients with EPP or XLP.
Background
Porphyria is named from the ancient Greek word porphura, meaning purple. The name refers to the color of the urine that may occur during an attack of one form of the disease, porphyria cutanea tarda. [4]
King George III of England reportedly had abdominal pain, rashes, reddish urine, and psychotic episodes that are consistent with porphyria, although the account is disputed by many. [5] During the period 1955-1959, approximately 4000 people in southeast Anatolia (Turkey) developed porphyria due to the ingestion of hexachlorobenzene (HCB), a fungicide that was added to wheat seedlings. [6]
Pathophysiology
Heme synthesis is summarized in the image below. In some porphyria patients and families, however, diagnostic evaluation can reveal simultaneous findings that are compatible with two different forms of porphyria, a phenomenon referred to as dual porphyria. [7]
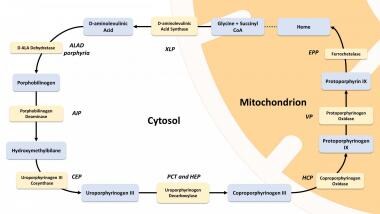
Acute porphyrias
The acute porphyrias are characterized by periodic acute attacks of abdominal pain, neurologic deficits, psychiatric symptoms, and discolored (red) urine. These disorders may stay occult for a long time. Overproduction of the heme precursor delta-ALA is the cause of pain in these disorders. [8] Four major disorders in this group are as follows:
Cutaneous porphyrias
The chronic porphyrias are dermatologic diseases that may or may not involve the liver and nervous system and do not present as acute attacks, as described for the acute porphyrias above. These include the following:
-
Erythropoietic porphyria
-
Hepatoerythropoietic porphyria
-
X-linked protoporphyria
Specific porphyria syndromes
Acute intermittent porphyria
AIP is an autosomal dominant disease that results from defects in the enzyme porphobilinogen-deaminase. [9, 10, 11, 12] Episodes of AIP are marked by abdominal pain, psychiatric symptoms, and neuropathies, but not cutaneous involvement. Attacks can be precipitated by certain lipophilic drugs (see the Drugs to Avoid section) or fasting; mild attacks may respond to high doses of glucose. [13]
Aminolevulinic acid dehydratase (ALAD) deficiency porphyria/Doss porphyria
Aminolevulinic acid dehydratase (ALAD) deficiency porphyria, also known as Doss porphyria, is extremely rare. [14] Abdominal pain and polyneuropathy are typical of this syndrome. Urinary ALA and coproporphyrin levels are markedly increased. Molecular genetic studies of the ALAD gene reveal the mutated nucleotides. In some reported cases, the acute porphyria syndrome developed while the patient received pharmacologic doses of erythropoietin and then resolved when the drug was stopped, which suggests that by stimulating heme synthesis, erythropoietin may unmask an enzyme deficiency resulting in the clinical expression of ALAD porphyria. [15]
Lead is a potent inhibitor of ALAD. Lead poisoning may produce a clinical picture that mimics ALAD deficiency porphyria, or may unmask occult ALAD deficiency porphyria; this condition is termed plumboporphyria. [16]
Hereditary coproporphyria
Hereditary coproporphyria results in most cases from half-normal activity (50%) of coproporphyrin oxidase. [17] The disease is an acute hepatic porphyria that is characterized by abdominal pain, neuropsychiatric symptoms, and cutaneous photosensitivity. [18] In rare homozygous cases, enzyme activity decreases to < 10% and the term harderoporphyria is used. [19]
Variegate porphyria
Variegate porphyria is an autosomal dominant inherited trait that results in decreased activity of protoporphyrinogen oxidase. It is characterized clinically by photosensitive skin disease and a propensity to acute neurovisceral crises. It presents as chronic blistering skin lesions; these are less prevalent in colder regions and dark-skinned individuals. [20] Variegate porphyria is found worldwide but has an exceptionally high frequency in South Africa. [21]
Erythropoietic protoporphyria
Erythropoietic protoporphyria (EPP) is caused by loss-of-function mutations in the ferrochelatase (FECH) gene.22 In EPP, the protoporphyrin molecule accumulates and can be excited by absorbing light energy. This causes the generation of free radicals and, thereby, photosensitivity of all tissues exposed to light. Mechanisms of toxicity in tissues not exposed to light include the following:
-
Deposition of protoporphyrin crystals in hepatocytes and bile canaliculi
-
Interference with redox systems
-
Formation of cytotoxic bile
Clinical manifestations of EPP are photosensitivity, insignificant hematologic abnormalities, and liver disease. [22] The hepatic manifestations of the disease are diverse: mildly disturbed liver enzymes in 20% to fatal hepatic failure in less than 5%. [23]
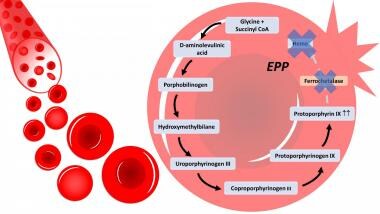 In erythropoietic protoporphyria (EPP), there is a deficiency in the enzyme ferrochetalase, causing a building up of protoporphyrin III in the RBC and leading to defective RBCs, hemolysis and porphyrias in the bloodstream.
In erythropoietic protoporphyria (EPP), there is a deficiency in the enzyme ferrochetalase, causing a building up of protoporphyrin III in the RBC and leading to defective RBCs, hemolysis and porphyrias in the bloodstream.
Congenital erythropoietic porphyria (Gunther disease)
Congenital erythropoietic porphyria, or Gunther disease, is one of the least common porphyrias. [24] It is a rare autosomal recessive disease that results from a deficient activity of uroporphyrinogen III synthase (UROS). Disease severity depends on the level of residual UROS activity. Clinical characteristics include cutaneous photosensitivity, erythodontia (brown discoloration of teeth), and hemolytic anemia. [25]
X-linked dominant protoporphyria
X-linked dominant protoporphyria results from a gain of mutation function in the ALAS2 gene. [23] First described in 2008, this subtype is similar in presentation to erythropoietic protoporphyria but has a higher incidence of liver disease and higher levels of erythrocyte protoporphyrin, with an X-linked dominant inheritance pattern. [26]
Porphyria cutanea tarda
Porphyria cutanea tarda (PCT) results from deficiency of the heme synthetic enzyme uroporphyrinogen decarboxylase (UROD). The majority of cases are acquired (type I). Familial cases most often involve autosomal dominant inheritance of a single mutation of the UROD gene (type II). Patients present with skin fragility, erosions, vesicles, bullae, and milia in sun-exposed skin. Less common dermatologic manifestations are periorbital mottled hyperpigmentation and hypertrichosis, sclerodermoid changes, and ulceration. [3]
In both type I and type II PCT, clinical expression most often follows exposure to environmental hepatotoxins, such as polyhalogenated aromatic hydrocarbons or ethanol, or to infectious agents such as hepatitis C virus (HCV). [27, 28] Risk may also be increased by coexisting conditions that adversely affect hepatocytes, such as carriage of the hemochromatosis gene (HFE) mutation. Up to two thirds of Saxon patients with PCT carry the classic HFE mutations (C282Y and/or H63D). In a French cohort of PCT patients, both C282Y and H63D were more frequent in PCT-positive patients than in controls. [27]
A Swedish cohort identified the following as possible risk factors for PCT [29] :
-
Alcohol abuse (38% of male patients)
-
Estrogen treatment (55% of female patients)
-
Diabetes (17%) or impaired glucose tolerance (45% of tested patients)
-
Hepatitis C, HIV
-
Smoking
-
Hemachromatosis
Hepatoerythropoietic porphyria
Hepatoerythropoietic porphyria is a rare autosomal recessive disease that results from severely deficient uroporphyrinogen decarboxylase (UROD) activity due to the mutation of both UROD alleles (homozygosity or compound heterozygosity). [30] It is a homozygous form of type II PCT. The clinical features of HEP are identical to PCT but HEP usually manifests in infancy or childhood. Clinical features include blistering skin lesions, hypertrichosis, and red urine. [31] See the image below.
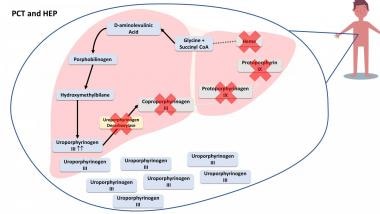 In porphyria cutanea tarda (PCT) and hepatoerythropoietic porphyria (HEP), the enzyme uroporphyrinogen decarboxylase is deficient and it leads to a buildup of porphyrias and precursors that spill out of the liver and accumulate in other parts of the body, such as skin, urine, and stool.
In porphyria cutanea tarda (PCT) and hepatoerythropoietic porphyria (HEP), the enzyme uroporphyrinogen decarboxylase is deficient and it leads to a buildup of porphyrias and precursors that spill out of the liver and accumulate in other parts of the body, such as skin, urine, and stool.
Prevalence
The combined prevalence of the acute porphyrias is approximately 5 cases per 100,000 persons. [32] Porphyria cutanea tarda is the most common porphyria, with a prevalence of 1 in 10,000. The most common acute porphyria, acute intermittent porphyria, has a prevalence of approximately 1 in 20,000, and the prevalence of the most common erythropoietic porphyria, erythropoietic protoporphyria, is estimated at 1 in 50,000 to 75,000. [2] Congenital erythropoietic porphyria is extremely rare, with an estimated prevalence of 1 in 1,000,000 or less. [2] Fewer than 200 cases have been reported in the literature. [33]
A specialist porphyria center network (European Porphyria Network [EPnet]) estimated the risk of recurrent attacks at 4% of porphyria patients in Europe. The incidence of symptomatic porphyria was around 0.13 case per million per year in most European countries, except Sweden, where it was 0.51 case per million per year. [34, 35]
History
Abdominal pain
The most common presenting symptom (90%) of an acute porphyria is abdominal pain. [36] This is usually colicky in nature, located in the left lower abdomen, and lasts hours to days. The abdominal pain is rarely accompanied by fever, leukocytosis, or peritoneal signs. Accompanying nausea and vomiting appear common. [37]
There is a very characteristic discrepancy between the severity of the patient's complaints and the objective clinical findings. [38] When a patient has repetitive visits to the emergency department because of severe abdominal pain without reasonable causes and needs narcotics for pain control, acute porphyria should be taken into consideration. [39]
Muscle weakness and neurologic deficits
Muscle weakness and focal neurologic deficits such as tetraparesis [40] may be the presenting feature, [41] especially in women of reproductive age. Limb pain is common. A motor, axon-predominant neuropathy is a strong clue when accompanied by abdominal and psychiatric symptoms. [42] A minority of patients present with paresis only, and no abdominal pain. [43]
The lifetime prevalence of acute intermittent porphyria–associated seizures has been reported as 2.2% of all those with known acute intermittent porphyria and 5.1% of all those with manifest acute intermittent porphyria. Epileptic seizures among persons with acute intermittent porphyria are less common than has been previously described. [44]
Psychiatric symptoms
Some patients develop psychiatric symptoms such as psychosis similar to schizophrenia. Diagnostic difficulty may lead to underdiagnosis of porphyria in patients who present with strictly psychiatric symptoms. This assumption is supported by a high prevalence of acute intermittent porphyria in psychiatric hospitals. [45]
Anxiety is raised in patients with acute intermittent porphyria and with variegate porphyria, in males and females, compared with the normal population. [46]
Other
Acute intermittent porphyria should be suspected in individuals presenting with unexplained acute abdominal pain following international air travel. [47]
The relative risk of an acute attack in acute intermittent porphyria compared with that in variegate porphyria was 14 in a series of 112 patients from South Africa with porphyria attacks. [48]
A thorough family history for porphyria and the patient's occupational history must be obtained.
Physical Examination
In acute porphyrias presenting as abdominal pain, attention should be paid to peritoneal signs, which are typically absent in porphyria. The presence of localized tenderness, rebound tenderness, vaginal discharge, cervical motion tenderness, and/or genitourinary bleeding should raise "red flags" even in patients known to carry a diagnosis of porphyria, and alternative diagnoses should be sought.
Jaundice may or may not be present. [49]
A focused neurologic exam should be performed to identify motor and sensory deficits and peripheral neuropathy.
In some but not all acute porphyrias, a skin rash may be seen. Variegate porphyria and, much less commonly, hereditary coproporphyria can also cause chronic, blistering lesions on sun-exposed skin that are identical to those in porphyria cutanea tarda, a much more common condition.
Gross, biochemical, and microscopic examination of the patient's urine is paramount if porphyria is in the differential diagnosis. In acute intermittent porphyria, for example, urine may be red to brown urine and may darken when exposed to air, light, and warmth. [50] The urine of patients with porphyria cutanea tarda is red to brown in natural light (red-wine urine) and pink to red in fluorescent light. [51]
Differential Diagnosis
The differential diagnosis in porphyria includes the following:
-
Acute abdomen
-
Hereditary tyrosinemia type I [52]
-
Pseudoporphyria is a bullous photosensitivity that most commonly arises as an adverse effect of a variety of medications, such as the antifungal agent voriconazole [53]
Diagnostic considerations
The clinical criteria of an acute attack of intermittent porphyria include the paroxysmal nature of the episode, the absence of other obvious causes, and the combination of various signs and symptoms, such as the following [54] :
-
Abdominal pain
-
Autonomic dysfunction
-
Hyponatremia
-
Muscle weakness
-
Psychiatric symptoms
The key manifestations that indicate possible acute porphyria are intensive abdominal pain without peritoneal signs; acute peripheral neuropathy; and encephalopathy, usually with seizures or psychosis. The presence of typical signs and symptoms, along with a more than fivefold elevation of urinary porphobilinogen excretion, justify starting treatment. [54]
Laboratory
Urine, stool, and blood studies in various types of porphyria are summarized in the image below.
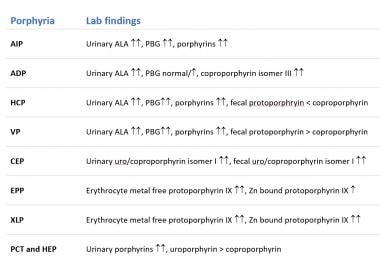
Urine
Urine porphyrin studies are the mainstay in the diagnosis of acute porphyria attacks. Establish the diagnosis promptly by testing for increased porphobilinogen (PBG) in a single-void urine collected during an attack. An expert guidelines panel recommended the Trace PBG Kit (Thermo Trace/DMA, Arlington, Texas). [36]
Urine samples for PBG testing should be protected from light (eg, by wrapping the specimen container in aluminum foil or placing it in a brown paper bag). However, brief exposure to light will not compromise the test results. Urine study results are unreliable if the urine creatinine level is below 2 mmol/L. [55]
Patients with acute exacerbations of porphyria have logarithmic increases (5-100 times) in metabolic precursors (eg, aminolevulinic acid [ALA], PBG). Minor elevations of these precursors are nondiagnostic and nonspecific. [56]
Significantly increased ALA and PBG in urine have 100% specificity (ie, rules in) for acute intermittent (hepatic) porphyria, variegate porphyria, and coproporphyria. A normal urine PBG result has a sensitivity of almost 100% (ie, rules out) in the diagnosis of porphyria in acutely symptomatic patients. [57]
Blood
To assess for cutaneous porphyria, the plasma porphyrin level should be measured, using fluorescence emission spectroscopy. Whole blood for porphyrin analysis is used to identify protoporphyria plasma porphyrins. [55]
Stool
Stool coproporphyrin and protoporphyrin are the most commonly measured porphyrins in feces. The ratio of fecal coproporphyrin to fecal protoporphyrin varies among the porphyrias. For example, fecal protoporphyrin always exceeds coproporphyrin (P > C = V) in variegate porphyria, whereas the reverse is true in hereditary coproprophyria. [58]
Erythrocyte uroporphyrinogen decarboxylase
Erythrocyte uroporphyrinogen decarboxylase activity is a specific and intrinsic defect in porphyria cutanea tarda. Measurement of this enzyme is a reliable diagnostic test for this disease. [59]
Electrolytes
Hyponatremia is typical. [60] In 1966, lesions of the median eminence of the hypothalamus and both hypothalamic–hypophyseal tracts were described in a patient with acute intermittent porphyria and syndrome of inappropriate antidiuretic hormone (SIADH). It was suggested that SIADH occurred because of damage to these areas of the brain from excessive exposure to porphyrins. [61]
Genetic mutations
Testing for genetic mutations is increasingly available for the diagnosis of porphyrias. Prenatal diagnosis is possible in some types of porphyria.
For example, in congenital erythropoietic porphyria, pink fluorescence of the amniotic fluid examined fortuitously in sunlight is suggestive. DNA analysis may show the mutation C73R in the gene for URO-synthase. [62] Another detectable mutation is in PPOX, encoding the mitochondrial enzyme protoporphyrinogen oxidase in variegate porphyria. [20] Heterozygous mutation in CPOX (encoding coproporphyrinogen-III oxidase) confirms the diagnosis of hereditary coproporphyria. [63] However, the utility of prenatal testing is questionable as many heterozygous carriers live healthy lives. The prenatal testing thus should not be done unless pregnancy termination is being considered.
A mutation screening for family members should be conducted to identify symptom-free carriers, especially in cases with a positive family history. [40]
Imaging
Computed tomography (CT)
CT scanning of the abdomen and pelvis helps clinicians to rule out other causes of excruciating abdominal pain, such as a ruptured viscus or vessel, and may help to pick up concomitant pathology, such as intussusception or infarction. [64] Focal, fatty nodularity of the liver may be noted in some patients. [65]
Magnetic resonance imaging (MRI)
MRI of the brain in acute intermittent porphyria demonstrates multiple large, contrast-enhancing, subcortical white matter lesions, which regress with glucose and hematin infusions. Diffusion-weighted MRI is normal, and MR spectroscopy excludes acute demyelination or tissue necrosis. MRI findings of acute intermittent porphyria can differ from those in posterior reversible encephalopathy syndrome by virtue of intense contrast enhancement. Because diffusion-weighted MRI and MR spectroscopy findings are normal, the lesions are likely caused by reversible vasogenic edema and transient breakdown of the blood-brain barrier. [66]
T2-weighted MRI sequences demonstrated multiple white-matter, high-signal lesions in 4 of 16 acute intermittent porphyria gene carriers (25%). [67] Kupferschmidt and colleagues [68] described 2 patients with acute intermittent porphyria who presented with cortical blindness. MRI showed bilateral occipital lesions, and the investigators speculated that these lesions were caused by vasospasm-induced ischemia due to unopposed cerebral vasoconstriction that resulted from a deficiency of nitrous oxide synthase, a major vascular dilator.
The striking feature of the MRI findings in these cases and in those of acute intermittent porphyria described in the literature is that the lesions are bioccipital and partially or totally reversible. These characteristics are typical of MRI findings seen in patients with hypertensive encephalopathy, and many patients presenting with acute intermittent porphyria attacks have high blood pressure on presentation. [69]
In cases of porphyria cutanea tarda, MRI of the liver shows poorly defined areas, which, on T2-weighted sequences, exhibit a hypersignal with fat saturation. Treatment of porphyria cutanea tarda may lead to clinical remission and resolution of radiologic abnormalities. [70]
Treatment of Porphyria
Therapeutic measures include the following:
-
Withdrawal of any culprit medications, alcohol, drugs, toxins, and chemicals is the key to therapy; smokers should be extensively counseled about cessation
-
Supportive care such as fluid, electrolytes, and nutrition is paramount
-
Monitor for hyponatremia or hypomagnesemia, and treat vigorously if found
-
Aggressively treat respiratory failure, which may ensue once muscle weakness involves the diaphragm, and support ventilation in an intensive care unit as appropriate
-
Treat pain with parenteral narcotics; complicated and debilitating chronic cases may require celiac plexus injection [71]
-
Administer phenothiazines for manifestations such as nausea, vomiting, agitation; ondansetron or related drugs can also be used for nausea [20]
-
Treat tachycardia and/or hypertension with propranolol or nadolol, which can be safely used for beta blockade once dehydration is ruled out
-
Promptly start glucose infusion in the form of 10% dextrose. At least 300-400 g should be given in 24 hours. The infusion is a time-buying measure to bridge the patient to more definitive treatment with hemin; by itself, glucose infusion is only effective for mild symptoms. [72] However, British and Irish Porphyria Network guidelines warn that administering intravenous glucose-in-water solutions may worsen hyponatremia, and instead recommend infusing glucose with normal saline. [13]
-
Treat acute porphyria attacks with hemin (plasma-derived intravenous heme); 1-4 mg/kg/d for up to 14 days is the definitive treatment and mainstay of management. Both heme arginate and heme hydroxide have been used. [20] Thrombophlebitis is the major adverse effect. At least two thirds of patients have a good response, with resolution of pain and neurologic deficits. [73, 74, 75] Tin protoporphyrin appears to have a synergistic effect with hemin. [76]
Other therapies include the following:
-
Hemodialysis has been used in dire circumstances with some benefit when hemin was not available. [77]
-
Termination of pregnancy may have to be considered as a last resort in acute fulminant attacks presenting during pregnancy. [78]
-
Perioperative care should involve intravenous glucose and nonbarbiturate induction of anesthesia, as well as appropriate protection from intraoperative surgical lights with filters. [63]
-
Magnesium sulfate has been used to control seizures, as many regular antiseizure medications are contraindicated. [80]
-
Neither red blood cell exchange transfusion nor plasmapheresis prevented progressive hepatic deterioration in 2 cases of advanced hepatic erythropoietic protoporphyria despite a significant decrease in protoporphyrin levels. [81]
-
Givosiran (Givlaari) is approved by the FDA for adults with acute hepatic porphyrias (ie, acute intermittent porphyria, variegate porphyria, hereditary coproporphyria, ALA dehydratase deficiency porphyria), in which attacks are caused induction of the enzyme 5-aminolevulinic acid synthase 1 (ALAS1). Givosiran is a small-interfering RNA that causes degradation of aminolevulinate synthase 1 (ALAS1) mRNA in hepatocytes through RNA interference, reducing the elevated levels of liver ALAS1 mRNA. [84] The ENVISION study demonstrated that long-term givosiran has an acceptable safety profile and significantly reduces the frequency of attacks, hemin use, and pain severity [85] .
-
Recombinant human porphobilinogen deaminase was tested in phase I studies of acute hepatic porphyria and proved safe and effective for removing porphobilinogen from plasma and urine. [86] However, a phase IIb study found insufficient correlation between reduction in porphobilinogen levels and the desired clinical effect, and the parent company discontinued clinical development of the product. [87]
Cutaneous porphyrias
Treatment measures include the following:
-
Avoidance of sunlight is the key in treating cutaneous porphyrias. [88]
-
The use of beta-carotene has shown some benefit for cutaneous porphyrias.
-
High-dose chloroquine therapy for porphyria cutanea tarda is rarely used now because of its hepatic effects. [91] Low-dose chloroquine or hydroxychloroquine appears at least as effective as phlebotomy. [92] A meta-analysis found that relapse rates in the year following treatment were 20% with phlebotomy and 35-36% with chloroquine. [93]
-
Iron chelation therapy (eg, with deferasirox, deferiprone, or desferrioxamine) may be considered for patients with porphyria cutanea tarda if phlebotomy is contraindicated. In a pilot study of 10 patients with porphyria cutanea tarda, iron chelation with deferasirox reduced new blisters and ferritin levels in most patients. [94]
-
Afamelanotide, an α-melanocyte–stimulating hormone analogue, has been shown to permit increased duration of sun exposure without pain and improve quality of life in patients with erythropoietic protoporphyria. [95, 96] It is administered in a subcutaneous implant containing a 60-day supply of the medication. The US Food and Drug Administration approved afamelanotide (Scenesse) in October 2019. [97]
Congenital erythropoietic porphyria
Treatment measures include the following:
-
In congenital erythropoietic porphyria mice, the proteasome inhibitor bortezomib has been used with some success to reverse the premature mutant protein degradation caused by deficiency of uroporphyrinogen III synthase. [103]
-
Pharmacological chaperone molecules and gene therapy are being investigated in congenital erythropoietic porphyria. [104]
Prognosis
Of 206 adult Finnish patients with acute intermittent porphyria or variegate porphyria, 47 patients had a total of 117 acute attacks during the period of 1967-1989. [105] Six of these patients died during an attack, and 21 attacks were associated with paresis; the frequency of severe attacks was significantly lower than before 1967. Most pareses and deaths occurred because of a delay in diagnosis and inappropriate treatment of porphyria.
In cases of acute intermittent porphyria, the risk of attacks correlated with the excretion of PBG in the urine during remission among adults; a low rate of excretion predicted freedom from acute attacks. Two percent of the surgical operations and 4% of the pregnancies were associated with acute attacks. Nearly one third of the women had symptoms of porphyria associated with the menstrual cycle, but these seldom progressed to an acute attack. Forty-six percent of the women had used sex-hormone preparations regularly; 2 (4.5%) of the women experienced associated acute attacks.
Long-term complications in patients with acute intermittent porphyria include chronic hypertension, chronic kidney insufficiency, chronic pain syndrome, and hepatocellular carcinoma (HCC). [54] A meta-analysis by Baravelli et al suggested an association between acute hepatic porphyria and increased risks of kidney cancer and endometrial cancer. [106] A systematic review by Ramai et al found that patients with acute hepatic porphyria have a substantially higher risk for primary liver cancer—principally HCC but also cholangiocarcinoma—which may occur even in the absence of cirrhosis. These authors recommend that patients with acute hepatic porphyria who are older than 50 years undergo screening for HCC as frequently as every 6-12 months, using alfa-fetoprotein measurement in conjunction with imaging modalities. [105]
Erythropoietic porphyria is a persistent, severely painful, socially disabling disease with a marked impact on quality of life. [107]
Prevention of Porphyria
Educate patients about their form of porphyria, its inheritance, precipitating drugs and events, and the importance of seeking treatment in early stages of an attack. [108] Encourage patients to wear medical alert bracelets. [36]
Patients who smoke should be strongly encouraged to quit. Smoking, which increases hepatic cytochrome P450 enzymes and presumably heme synthesis, is associated with more frequent porphyria attacks. [109]
Perioperative management includes the use of filters on operative lights to prevent skin burns and intestinal perforation. [110] To prevent burn injuries, astral lamps in the operating room are covered with yellow film filters. [111]
A high risk of primary hepatocellular carcinoma has been demonstrated in acute hepatic porphyrias, and periodic surveillance is recommended. [112]
Expertise, Testing, and Centers of Excellence
A listing of experts, testing, and centers of excellence in the porphyrias is available at the American Porphyria Foundation Website (www.porphyriafoundation.com).
The European Porphyia Initiative (EPI) (www.porphyria-europe.org) network was formed in 2001 to compare the experience among countries in an attempt to develop a common approach to the management of the porphyrias. [113]
Drugs to Avoid
The list of drugs to avoid continues to grow. Major culprits include the following:
-
Barbiturates
-
Anticonvulsants
-
Progestins
-
Rifampin
Individual medications can be checked against a safe and unsafe drug database that is maintained by the American Porphyria Foundation.
Acute intermittent porphyrias are rare complications of ovulation induction with clomiphene citrate, but these syndromes should be considered in patients who develop unexplained hyponatremia or neurovisceral symptoms. [114]
Different drugs in the same class may have different effects in the porphyrias. For example, although lidocaine should be avoided, bupivacaine or levobupivacaine provided successful and safe local anesthesia for dental procedures in 5 children with a diagnosis of latent acute intermittent porphyria or who had a family history of acute intermittent porphyria. [115]
Questions & Answers
Overview
What are the signs and symptoms of porphyria?
How are acute episodes of porphyria treated?
What is the role of heme biosynthesis in the pathophysiology of porphyria?
What are specific porphyria syndromes?
What is the pathophysiology of variegate porphyria?
What is the pathophysiology of erythropoietic protoporphyria (EPP)?
What is the pathophysiology of porphyria cutanea tarda (PCT)?
What is the pathophysiology of congenital erythropoietic porphyria (Gunther disease)?"
What is the pathophysiology of x-linked dominant protoporphyria?
What is the prevalence of porphyria?
How is abdominal pain characterized in porphyria?
What are the neurologic signs and symptoms of porphyria?
What are the psychiatric signs and symptoms of porphyria?
Which clinical history findings are characteristic of acute intermittent porphyria?
Which physical findings are characteristic of porphyria?
Which conditions are included in the differential diagnoses of porphyria?
What is the role of urine and stool studies in the workup of porphyria?
What is the role of blood tests in the workup of porphyria?
How is porphyria cutanea tarda diagnosed?
What is the role of electrolyte measurement in the workup of porphyria?
What is the role of genetic testing in the workup of porphyria?
What is the role of CT scanning in the workup of porphyria?
What is the role of MRI in the workup of porphyria?
How is cutaneous porphyria treated?
How is congenital erythropoietic porphyria treated?
What is the prognosis of porphyria?
What is included in patient education about porphyria?
How are burn injuries prevented in patients with porphyria?
What is included in long-term monitoring of patients with porphyria?
Where are professional resources found for the diagnosis and management of porphyria?
Which medications should be avoided by patients with porphyria?
-
Clinical classification of the different porphyrias.
-
Heme biosynthesis pathway.
-
In erythropoietic protoporphyria (EPP), there is a deficiency in the enzyme ferrochetalase, causing a building up of protoporphyrin III in the RBC and leading to defective RBCs, hemolysis and porphyrias in the bloodstream.
-
In porphyria cutanea tarda (PCT) and hepatoerythropoietic porphyria (HEP), the enzyme uroporphyrinogen decarboxylase is deficient and it leads to a buildup of porphyrias and precursors that spill out of the liver and accumulate in other parts of the body, such as skin, urine, and stool.
-
Urine, stool and blood findings for the different porphyrias.
Tables

What would you like to print?
- Practice Essentials
- Background
- Pathophysiology
- Prevalence
- History
- Physical Examination
- Differential Diagnosis
- Laboratory
- Imaging
- Treatment of Porphyria
- Prognosis
- Prevention of Porphyria
- Expertise, Testing, and Centers of Excellence
- Drugs to Avoid
- Questions & Answers
- Show All
- Media Gallery
- References