Five-membered heteroaromatic rings, in particular, have gained prominence in medicinal chemistry as they offer enhanced metabolic stability, solubility and bioavailability, crucial factors in developing effective drugs. The unique physicochemical properties and biological effects of five-membered heterocycles have positioned them as key structural motifs in numerous clinically effective drugs.
1. Introduction
Heterocyclic compounds are a class of organic compounds that contain at least one atom other than carbon in the ring structure. The presence of heteroatoms such as nitrogen, oxygen and sulfur in the aromatic ring increases the polarity and offers the possibility of hydrogen bonds that can improve the binding affinity and selectivity of the drug at the target site. Heteroaromatic rings are considered preferred structures due to their ability to replace common motives in medicinal chemistry, enabling an improvement of metabolic stability, solubility and bioavailability, which are essential elements for the development of effective drugs
[1][2][3]. These properties can lead to the modulation of different biochemical metabolic pathways, resulting in therapeutic effects. Among active compounds with aromatic groups, five-membered heterocyclic compounds are of great importance in the field of pharmaceutical and medicinal chemistry. Due to their unique physicochemical properties and biological effects, these compounds are used as key structural motifs in many clinically effective drugs. Therefore, the development and synthesis of novel five-membered heterocycles, remains an important research area in medicinal chemistry, with the aim of finding new drugs for the treatment of various diseases. Recent advances in the development of histone deacetylase (HDAC) inhibitors incorporating five-membered heterocyclic scaffolds emphasized the importance of heterocycles as valuable scaffolds in this area of research
[4][5]. HDACs are encountered in many multiprotein complexes in the epigenetic apparatus, are involved in many cellular processes and are established targets for cancer, neurodegenerative diseases and other indication areas
[6][7] with five clinically approved drugs to date
[8][9][10][11][12]. The HDAC family is composed of 18 HDAC isozymes, which can be categorized as zinc or NAD+-dependent, whereas the latter are referred to as sirtuins (SIRT1-7)
[13][14]. The zinc-dependent HDAC isozymes can be grouped into four classes and vary in their size and localization as indicated in
Figure 1.
Figure 1. Schematic classification of HDAC isozymes into class I, IIa, IIb and IV. Indicated are catalytic domain size (blue box), protein length (size) and localization.
2. ZBG Scaffolds
2.1. 1,2,4-Oxadiazoles
1,2,4-oxadiazoles, also referred to as azoximes show diverse biological activities and are frequently employed in medicinal chemistry as hydrolysis resistant bioisosteric replacements for ester or amide functionalities
[3][15]. The scaffold is a weak base and can act only as hydrogen bond acceptor. X-ray measurements of azoximes show a planar ring and suggest a double bond character for both C-N distances, which is in agreement with a lower value of 39 for the aromatic character compared to furan with a value of 43, as determined by the bird index
[15][16]. The dipole moment for unsubstituted
1 (1.2 D) or substituted
2 (1.8 D) azoximes is lower compared to other oxadiazoles (
Figure 2)
[2][17].
Figure 2. Schematic illustration of bond lengths (Å), bond angles (°), dipole moments (D) and calculated hydrogen bond acceptor strengths in kcal/mol indicated in blue.
Azoximes are rather inert against electrophilic attacks as halogenation, nitration and Friedel Crafts acylation or alkylation do not occur
[17]. Formation of a diol moiety in
3 indicates the electron withdrawing nature of this scaffold
[17]. Reactivities observed for C3 and C5 substitutions in examples
4 and
5 can be rationalized by mesomeric structures of
6 and
7. Nucleophilic attack on C5 but not C3 can displace a leaving group (
Figure 3)
[17].
Figure 3. Properties and reactivities of 1,2,4-oxadiazole derivatives. The electron-withdrawing nature of azoximes is indicated by diol formation of 3. C3 and C5 reactivity is illustrated with 4 and 5 in the presence of electron withdrawing groups and can be rationalized with mesomeric structures of 6 and 7.
In 2013 Lobera et al. published a novel class IIa selective HDACi with a trifluoromethyl-1,2,4-oxadiazole (TFMO) motive as zinc binding group (ZBG) identified in a high-throughput screening (HTS) and used it to study gene regulation in phytohemagglutinin (PHA)-activated human peripheral blood mononuclear cells (PBMCs)
[4]. The obtained crystal structure (PDB-ID: 3ZNR, 2.35 Å) of
TMP269 bound to HDAC7 showed a U-shaped conformation of the inhibitor (
Figure 4).
Figure 4. Representation of
TMP269 bound to HDAC7 (PDB-ID: 3ZNR, 2.35 Å) illustrated using the UCSF ChimeraX visualization software (version ChimeraX-1.4)
[18][19]. The inhibitor and the amino acid side chains are displayed in the ball and stick or the stick style and are colored by element or heteroatom, respectively. (
A) Notable is the larger binding site of class IIa HDACs compared to other HDAC classes. Possible interactions between
TMP269 and the zinc ion are indicated as dotted red lines. (
B) Most prominent residues and interactions with
TMP269. The distal “cap”-phenyl moiety of
TMP269 occupies the foot pocket and displaces His843 with an edge-to-face orientation towards Phe679.
Compound TMP195 (Figure 5) was chosen as representative with an activity of 300 nM in cell-based class IIa HDAC assays and up- or downregulated only 16 and 60 genes by a factor of ≥1.4, respectively. Comparatively the pan-HDACi vorinostat up- or downregulated 1853 and 2703 genes, respectively.
Figure 5. SAR studies of the TFMO group are marked by a box. Inhibitory activity against class IIa HDACs is highlighted in red.
In 2020 Winter et al. explored the TFMO moiety as antifungal class II HDAC inhibitor. First results showed activity against
Phakopsora pachyrhizi, the major cause of soybean rust
[20]. Further SAR studies with the surrogate
Aspergillus nidulans led to potent inhibitors
8 and
9 of class II histone deacetylase HdaA and related HOS3-type histone deacetylase HosB (
Figure 5). The amide cap moiety was found to be variable, suggesting only a weak contribution. Interestingly no modifications of the TFMO group were acceptable, leading to a strong decrease in activity as seen with derivatives of
10 and
11 in
Figure 5.
Another antifungal application of TFMO containing compounds was reported by Yang et al.
[21]. The design of active compounds was guided by previous TFMO HDACi and diphenyl ether moieties, frequently used in pesticides. Unfortunately, only MD simulations were performed with class II HDAC and
12 containing a TFMO moiety (
Figure 5).
El-Awady et al. reported a TFMO containing HDACi with an imidazopydine moiety as a cap group
[22]. Compounds containing a TFMO group were selectively active against HDAC5 as frequently demonstrated by this class II selective motive. Compounds
13,
14 and
15 (
Figure 5) exhibited IC
50 values of 0.31 µM, 0.30 µM and 0.81 µM, respectively.
Compound
[18F]TMP195 is separatable from the precursor and could be produced with high radiochemical purity (>98%), acceptable radiochemical yield of 3–5% and higher molar activity of 0.33–0.49 GBq/µmol (8.9–13.4 mCi/µmol) when compared to previously reported PET tracers. Unfortunately,
[18F]TMP195 exhibited poor aqueous solubility and made in vivo application difficult. To circumvent the poor physiochemical properties of
TMP195, namely the low affinity of 800 nM in the hands of Turkman et al. and the aqueous solubility, a SAR study was conducted and
16 identified as potent inhibitor with sub nM affinity and improved aqueous solubility (
Figure 7)
[23].
Intrigued by a report showing to improve the phenotype of Huntington disease (HD) by a HDAC4 genetic suppression in the R6/2 mouse model
[24] Stott et al. took interest in targeting class IIa HDACs
[25]. Validation of a previously reported potent HDAC4 inhibitor
17 by Hebach et al.
[26] (
Figure 5) showed a 150-fold selectivity for class IIa in cells, high kinetic solubility, negligible P-gp efflux, high permeability, a short residence time of 2.5 min and poor metabolic stability in mouse liver microsomes. A SAR study of the cap moiety aimed to improve the metabolic stability led to a replacement of the basic amine with a pyrrolidine group and reduced the overall lipophilicity by a factor of 0.6 leading to
18 (
Figure 5). Further investigation showed the stereochemistry to be important and led to a D-alanine analogue
19, similar to previously undertaken studies.
To reduce the high toxicity of BTZ and omit the low aqueous solubility of TMP269 a novel class IIa selective HDACi was developed. An analysis via the Chou–Talalay method in four different leukemia cell lines (HL-60, K562, MONO-MAC-6 and THP-1) using a MTT assay showed strong synergistic effects for 20 and TMP269 with CI values below 0.5. In two of the four cell lines the synergistic effect of 20 was superior to that of TMP269 with concentrations that retained class IIa preference. Interestingly, 20 is 4–7.7-fold less cytotoxic in leukemia and non-cancer HEK293 cells but still increases caspase activation and apoptosis in a synergistic manner. Compound 20 showed synergistic effects at low concentrations of BTZ while synergistic effects for TMP269 were only observable at elevated concentrations. Another benefit is the induction of p21 in combination with BTZ compared to TMP269 plus BTZ and the stronger caspase-mediated apoptosis.
2.2. 1,3,4-Oxadiazoles
1,3,4-oxadiazole is a nearly flat and thermally stable heterocycle. It is not fully aromatic, with an index value of 50, compared to furan and thiophen with an index value of 43 and 66, respectively
[16]. Bond lengths, angles and dipole moments are depicted in
Figure 6 for
21 with a tilt of +3.3° and −3.3° for the pyridine rings. 1,3,4-oxadiazole exhibits bond orders of 1.3124, 1.9062 and 1.3348 for the O-C, C-N and N-N bond respectively
[15]. A systematic study conducted by Boström et al. on the interchangeability of 1,2,4- and 1,3,4-oxadiazoles, e.g.,
2 and
22, concluded that the 1,3,4-oxadiazole is superior to the 1,2,4-motive regarding the pharmaceutical properties
[2].
Figure 6. Bond lengths (Å), bond angles (°), dipole moments (D) and calculated hydrogen bond acceptor strengths in kcal/mol indicated in blue for 1,3,4-oxadiazole derivatives.
1,3,4-oxadiazoles like
23 are known to undergo hydrolysis in basic or acidic conditions and to fragment into carboxylic acids and hydrazine (
Figure 7)
[15].
Figure 7. Schematic illustration of the 1,3,4-oxadiazole reactivity.
Co-crystallization of 24 and zHDAC6-CD2 resulted in a structure with 1.6 Å resolution (PDB-ID: 8A8Z) and showed no major conformational changes compared to the ligand free structure of zHDAC6-CD2 (PDB-ID: 5EEM). Most interestingly, the electron density in the catalytic site did not match 24 and suggested ring opening of the DFMO moiety. This could be explained by a two-step hydrolytic conversion involving the catalytic activity of HDAC6 with 25 as intermediate, finally yielding a hydrazide as ZBG 26 (Figure 8).
Figure 8. Representation of
26 bound to the class IIb zHDAC6-CD2 (PDB-ID: 8A8Z, 1.60 Å) illustrated using the UCSF ChimeraX visualization software (version ChimeraX-1.4)
[18][19]. The inhibitor and the amino acid side chains are displayed in the ball and stick or the stick style and are colored by element or heteroatom, respectively. (
A) Notable is the smaller binding site compared to class IIa HDACs. Chelating interactions between
26 and the zinc ion are indicated as dotted red lines. (
B) Visualization of prominent active site residues and interactions with
26 indicated by dotted red lines. Similar to class I HDACs Tyr745 promotes stabilization by interacting with the carbonyl moiety via hydrogen bonding.
Additional synthesis of well-established HDACi and the replacement of their ZBGs with the DFMO moiety led to only moderate active HDAC6 inhibitors, indicating that other factors need to be considered when interchanging ZBGs. A co-crystallization of 27 with zHDAC6 CD2 led to a crystal structure with a resolution of 2.0 Å. An overlay of crystal structures containing 28 and ligand-free zHDAC6 CD2 (PDB-ID: 5EEM) showed a root-mean-square deviation of 0.177 Å indicating no major structural changes. Interestingly and supplementary to data of Cellupica et al., the found electron density matched acylhydrazine 28, the ring opening product perfectly. A comparison with the structure published by Cellupila et al. showed a shift in orientation of the ZBG by 0.9 Å and a coordination of the zinc ion by the primary amine of the hydrazide moiety. Further differences include a shorter hydrogen bond towards Y745 and the presence of water in the P571 pocket. To shed more light on the binding mechanism the acylhydrazine 28, the TFMO 29, and the methyl moiety 30 were synthesized and evaluated. Compounds 28 and 29 showed only weak inhibition of HDAC6 whereas 29 was more potent with an IC50 of 531 nM (Figure 9).
Figure 9. Summary of compounds and their inhibitory activity. Inhibitory activity against class IIb HDACs is highlighted in blue. Compounds showing no effects against HDAC isozymes were labeled as no effect (n.e).
Compounds 31 and 32 (Figure 9) were selected for biological evaluation. The compounds did not impact HDAC1 or HDAC4 levels and did not reduce cell viability.
In search of a therapeutic strategy for tauopathy, Onishi et al. synthesized and evaluated compound
33 (
Figure 9) via HTS and medicinal chemistry optimization
[27]. As expected on the basis of earlier findings
33 exhibited time dependent inhibitory activity (4.6 nM–60 min) but was highly potent even without preincubation (36 nM).
To evaluate HDAC6 inhibitors Ptacek et al. compared a DFMO containing HDACi side-by-side with common hydroxamate-based HDAC6 inhibitors frequently used in the field
[28]. In vitro evaluation showed HDAC10 as primary off-target isoform of hydroxamate-based HDACi while DFMO containing
34 (
Figure 9) had outstanding selectivity over all other isoforms. To address cellular potency nanoBRET assays were employed with HEK293T/T17 cells expressing the adequate HDAC isoforms.
2.3. Thiazolidine-2,4-Diones
Thiazolidine-2,4-diones (TZDs) are five-membered non-aromatic heterocycles which can structurally be derived from thiazole, its aromatic analogue. The TZD moiety can act as a hydrogen bond donor/acceptor and shows no significant deviation from planarity with bond angles and distances indicated in
Figure 10 [29].
Figure 10. Schematic illustration of bond lengths (Å), bond angles (°) and possible isomeric structures A to E.
In an effort to establish the TZD moiety as a ZBG group Tilekar et al.
[30] and Upadhyay et al.
[31] synthesized and evaluated TZD containing compounds based on prior studies
[32][33] as HDACis yielding compounds
35,
36 and
37 (
Figure 11).
Figure 11. Summary of compounds and their inhibitory activity.
Further optimization identified the benzothiazole moiety as the best cap group and 38 as the most potent compound (Figure 11) towards HDAC4 with an IC50 of 0.75 µM and a stabilization of 1 °C in thermal shift assays. Evaluation of 39 against several cancer cell lines showed inhibition of proliferation of hematological (CCRF-CEM) and solid tumor (MDA-MB-231) cells with low impact on non-cancerous HS-27 cells.
A few compounds were found to be dual inhibitors of HDAC4 and HDAC8 but most of the compounds were selective towards HDAC4 with a two-fold higher inhibitory activity. The most potent compound 40 (Figure 13) showed an inhibitory activity of 1.1 µM towards HDAC4 and an EC50 of 0.245 µM towards PPARγ. Additional evaluation of 41 in CCRF-CEM cells showed an CC50 of 2.8 µM and exhibited 14-fold selectivity against non-cancerous HS-27 cells. Further in vivo studies could show tumor regression in CCRF-CEM mice xenografts.
Compound 42 (Figure 11) was most potent with IC50 of 0.88 µM against HDAC4 and a remarkable selectivity against HDAC1-3, 7 and 8 but inhibited HDAC6 with an IC50 of 7.6 µM.
Compound 43 (Figure 11) was most potent with an IC50 of 18 µM, 45 µM, 16 µM, 0.36 µM, 15 µM, 6.3 µM and >50 µM against HDAC1-4 and HDAC6-8 but stabilized HDAC4 by only 0.6 °C. Docking into the open and closed conformation of HDAC4 (PDB-ID: 2VQJ and 4CBY, respectively) showed higher docking scores for the open conformation and higher binding scores for a (S)-enantiomer analogue.
First screening revealed 97 compounds with IC50 values under 50 µM which were used for further SAR analysis. A similarity mapping with the DataWarrior software yielded ten clusters (Figure 12).
Figure 12. Schematic representation of studied TZD clusters.
2.4. Miscellaneous Heterocycles
Interestingly Wang et al. synthesized natural substrate analogues and coupled the lysine ε-amine to different heterocycles yielding
44,
45,
46 and
47 (
Figure 13)
[34]. A screening with HeLa nuclear extracts at an inhibitor concentration of 100 µM revealed an inhibitory potential of 48.4%, 54.0%, 45.0% and 48.6% for the mentioned compounds, respectively.
Figure 13. Summary of compounds and their inhibitory activity mentioned. HDAC* inhibitory activity was measured with HeLa nuclear extract at an inhibitor concentration of 100 µM.
In an effort to develop class I HDAC inhibitors as latency-reversing agents for HIV treatment Lui et al. synthesized a series of compounds with different cap and protruding groups
[35]. An SAR study was performed using a ketone zinc binding group attached to an aryl moiety to optimize foot pocket interactions and yielded a series of compounds
48,
49a–
f and
50a–
f (
Figure 13).
3. Cap Group or Linker
3.1. 1,2,5-Oxadiazoles
The oxadiazole scaffold and its analogues are frequently used as cap groups or linking units as demonstrated in following studies. Prior to the presented studies, a short introduction on 1,2,5-oxadiazole and its oxo derivative will be given for a more complete understanding of the isoxazole isomers.
1,2,5-oxadiazole, also termed furazan is essentially a planar heterocycle which shows single bond character between the N2-O1 and O1-N5 with π-bond orders of 0.32–0.36 and exhibit significant π-electron delocalization for the bonds N2-C3 and C4-N5 with a π-bond order of 0.72–0.82 as well as a bond order of 0.45–0.52 for C3-C4
[15]. Furazans can formally be considered as π-excessive heterocycles with regard to the six electrons distributed over five atoms. Since the π-electron density is mostly located on the heteroatoms, the C-atoms exhibit a π-electron density value of lower than one. Furazans like
51 do not favor tautomerism and react only slowly with nucleophiles despite the low electron density on the C-atoms, but form α-oximinonitriles
52 as ring opening products when treated with strong bases (
Figure 14)
[15].
Figure 14. Schematic reactivity of furazans with strong bases.
The related 1,2,5-oxadiazole-2-oxide, also referred to as furoxane, is also nearly planar, with the exocyclic oxygen projecting by 0.05 Å out of the plane. It has an extended O1-N2 and a shortened N2-O
exo bond length. The symmetric distortion propagates extending the N2-C3 and shortening the C4-N5 bond. Consequently, the C3-C4 bond is shortened and exhibits about 30% double bond character. A distinctive feature of furoxanes is the ring-chain tautomerism from
53a to
53b, involving dinitroethene
54 as a transition state with an energy of about 120 kJmol
−1 above that of furoxan (
Figure 15)
[15].
Figure 15. Schematic illustration of the ring chain tautomerism of furoxanes.
Furazans
55a,
b and furoxanes
56a,
b (
Figure 16) have a comparably high dipole moment in the range of 4.04–5.01 D with a clear dipole vector shift towards the exocyclic oxygen atom present in furoxanes
[15].
Figure 16. Schematic illustration of differences in dipole moments and vectors between furazans and furoxans.
In an effort to design nitric oxide (NO)-donating scaffolds Tu et al.
[36] and Duan et al.
[37] synthesized and evaluated furoxane containing scaffolds. Compounds
57,
58 and
59 (
Figure 17) were most potent and raised NO levels as observed in the Griess assay. This was most desired since NO is a key signaling molecule involved in regulation of tumor cell proliferation, metastasis, angiogenesis and can modify proteins via S-nitrosylation or Tyr-nitration
[38]. Interestingly, the precursor
60 was also active against HDAC2 which was rationalized to originate from S-nitrosylation
[39][40].
Figure 17. Summary of compounds with their inhibitory activity. HDAC* inhibitory activity was measured using HeLa nuclear extract.
With the intent to replace the carbamate and amide functionalities with oxadiazole as isosteric moiety Cai et al.
[41][42] synthesized and evaluated analogues of
Entinostat [42] and
SAHA [41]. Selected compounds were evaluated against several cancer cell lines and docked into HDAC2 and HDAC8. Studies on
Entinostat [42] yielded
61 and
62 (
Figure 17) which exhibited better activity against HDAC1 and HDAC2 and complementary activity against HDAC8, respectively. Evaluation of
61 and
62 against several cancer cell lines yielded activities of HCT116 (2.33 µM and 29.3 µM), A549 (6.39 µM and >100 µM), NCI-H661 (4.73 µM and >100 µM), U937 (0.52 µM and 7.21 µM) and MDA-MB-231 (3.18 µM and 14.67 µM). Studies on
SAHA [41] yielded
63,
64 and
65 (
Figure 17), which showed better or comparable potency against HDAC1, HDAC2 and HDAC8. Again, evaluation in cancer cell lines yielded activity profiles of A549 (5.31 µM, 9.17 µM and 7.68 µM), NCI-H661 (3.09 µM, 0.41 µM and 0.52 µM) and U937 (0.29 µM, 0.46 µM and 0.41 µM) for
63,
64 and
65, respectively.
Several other approaches including substitution and optimization strategies yielded compounds
66,
67,
68,
69,
70 and
71 which are depicted in
Figure 17. Guant et al.
[43] evaluated
66 and
67 against MDA-MB-231 (4.69 µM and 1.21 µM), K562 (4.15 µM and 1.56 µM) and PC3 (7.75 µM and 3.60 µM) cancer cell lines, respectively. On the basis of previous results Yang et al.
[44] replaced a thiophen moiety with oxadiazole yielding
68 and
69, which performed better than
SAHA against HDAC1. Evaluation against cancer cell lines yielded following IC
50 values: HCCLM3 (5.19 µM and 6.56 µM) and HepG2 (1.07 µM and 1.03 µM) for
68 and
69, respectively. Pidugu et al.
[45] designed
70 and evaluated it in a follow up study
[46], showing it to inhibit growth of MDA-MB-231 and MCF7 cancer cell lines with IC
50 values of 0.23 µM and 1 µM, respectively. The compound
71, synthesized and evaluated by Yang et al.
[47], showed potencies ranging between 9.8 and 44.9 nM against 12 cancer cell lines. Application in a Daudi Burkitt’s lymphoma xenograft mice model showed tumor inhibition rates of 53.8% and 46.1% when administered orally with a dose of 20 mg/kg or 10 mg/kg, respectively.
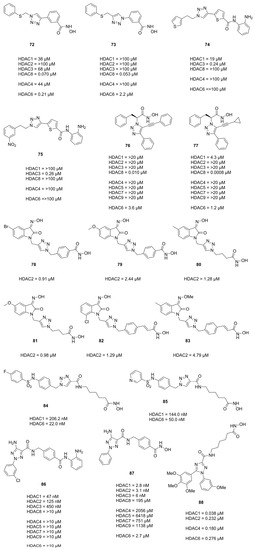
3.2. Triazoles
In 2010 Suzuki et al.
[48] identified the HDAC8 selective inhibitor
72 (
Figure 18) as part of a library prepared using the CuAAC reaction and followed up with further investigation and evaluation identifying the reversed triazole analogue
73 (
Figure 18) as a potent and selective HDAC8 inhibitor
[49]. Evaluation in four T-cell lymphoma cell lines (Jurkat, HH, MT4 and HUT78) revealed that compound
73, despite being HDAC8, selectively induced off-target α-tubulin acetylation at a concentration >10 µM and exhibited roughly two-fold higher GI
50 values compared to
72. Other studies evaluating a CuAAC synthesized triazole library yielded HDAC3 selective inhibitors
74 and
75 (
Figure 18)
[50] which exhibited GI
50 values for HCT116 (1.9 µM and 0.94 µM) and PC3 (1.4 µM and 1.0 µM) cell lines, respectively.
Figure 18. Summary of compounds mentioned with their inhibitory activities.
Studies by Ingham et al. identified compound
76 as potent and selective HDAC8 inhibitor and optimized the activity via the CuAAC reaction yielding
77 as best hit (
Figure 18)
[51].
In search for novel HDACi Huong et al.
[52] used the CuAAC reaction to synthesize compounds incorporating the 2-oxoindoline moiety as cap group. Compounds
78 and
79 (
Figure 18) were most potent and had comparable activities to
SAHA with regard to HDAC2 inhibition and potency against four cancer cell lines, respectively. Evaluation against four cancer cell lines showed IC
50 values of SW620 (8.73 µM and 2.06 µM), PC3 (7.98 µM and 2.62 µM), AsPC-1 (4.18 µM and 1.39 µM) and NCI-H23 (5.46 µM and 1.14 µM) for
78 and
79, respectively. Follow up studies by Dung et al.
[53] yielded compounds
80 and
81 (
Figure 18), with potencies better or comparable to
SAHA, exhibiting IC
50 values against cancer cell lines of SW620 (0.73 µM and 1.61 µM), PC3 (0.76 µM and 1.74 µM) and AsPC-1 (0.49 µM and 1.49 µM). Further screening and improvements by Dung et al.
[54] yielded representatives
82 and
83 (
Figure 18).
Mou et al. also used the click reaction to synthesize several triazole scaffolds as potential HDAC6 inhibitors. Evaluation yielded representatives 84 and 85 (Figure 18) as selective and potent HDAC6 inhibitors.
Profiling of pharmacokinetic properties in mice (20 mg·kg−1) of representative compounds 86 and 87 (Figure 18) yielded Kel (0.28·h−1 and·0.14·h−1), t1/2 (2.49 h and 5.03 h), tmax (0.42 h and 0.25 h), Cmax (9558 ng·mL−1 and 2715 ng·mL−1) and an AUC0–t (15278 h·ng·mL−1 and 2917 h·ng·mL−1), respectively with an oral bioavailability of 65.1% for 86.
In search of novel HDACi, Aboeldahab et al.
[55] synthesized two series of compounds, of which one was bearing a triazole moiety. Representative
88 of series II was not as potent compared to series I compounds but exhibited better antiproliferative activities presumably acting as tubulin inhibitor.
3.3. Thiazoles
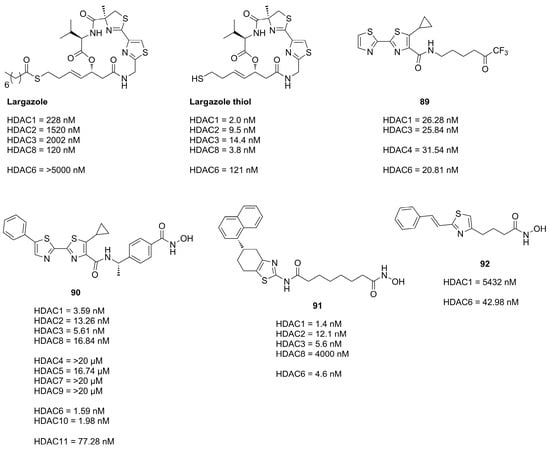
Thiazoles are frequently applied in HDACi scaffolds as cap groups as the bisthiazole motive is part of the well-known depsipeptide
Largazole (
Figure19), a class I selective HDACi, which interestingly shares the cap core motive with the approved
Romidepsin HDACi
[56][57].
Figure 19. Summary of compounds with their inhibitory activities.
The application of the bisthiazole cap in HDACi was studied by Gong et al.
[58] and Zhang et al.
[59] and yielded compounds
89 and
90 (
Figure 19). Compound
89, equipped with a trifluoromethyl ketone ZBG, exhibited the highest activity towards HDAC1-3 and HDAC6 and showed improved antiproliferative activity against cancer cell lines with IC
50 values of MM1S (0.39 µM), RPMI (0.085 µM), NCI-H929 (1.23 µM), LP1 (0.37 µM), Mino (0.12 µM) and JeKo-1 (0.064 µM)
[58]. Compound
90 (
Figure 19), equipped with a hydroxamic acid as ZBG showed potent activity towards HDAC isozymes and a high distribution in colon tissue, whereupon it was evaluated in human colorectal carcinoma HT-29 xenograft model, where it was more effective than
Panobinostat as control
[59].
Further studies incorporating the thiazole motive were conducted by Sun et al.
[60] yielding
91, which displayed high cytotoxicity in several cancer cell with IC
50 values better or comparable to
SAHA and exerted potent antitumor effects in the A-549 zebrafish xenograft model.
Another example is the HDAC6 selective compound
92 which was synthesized by Nam et al.
[61] based on previous studies and an SAR evaluation. The authors optimized linker lengths and cap group substituents and concluded based on docking results that cap rigidification was responsible for HDAC6 selectivity allowing the cap group to interact with the small hydrophobic groove of HDAC6.
3.4. Imidazoles and Oxazoles
Based on previous work and SAR studies aiming to target Plasmodium falciparum Bresciani et al. [62] identified potent and selective human HDAC class I inhibitors. Interestingly, an interdependence of used cap group and heterocycle was observed. Compound 93O (Figure 20), with an oxazole moiety, exhibited greater potency in combination with a 2-methoxyquinolin cap, whereas 94N with an imidazole moiety was more potent in combination with a naphthyl cap. Comparison of pharmacokinetic profiles of an imidazole and oxazole analogue showed improved properties for the oxazole analogue, likely originating from the increased lipophilicity contributing to a better absorption and membrane permeability [62]. Further SAR studies and improvements yielded compound 95, which had an improved pharmacokinetic profile and a comparable potency to 93O. Studies focusing on the HDAC3 selectivity yielded compound 96 (Figure 20), which was also synthesized as its oxazole analogue but failed to exhibit HDAC3 activity [62].
Figure 20. Summary of in chapter 3.4 mentioned compounds with their inhibitory activities. HDAC* inhibitory activity was measured using HeLa nuclear extract.
Similar scaffolds were utilized by Clausen et al.
[63], Yu et al.
[64] and by previously mentioned Liu et al.
[35] for HIV treatment studies via a “shock and kill” strategy. The shock is performed by HDAC inhibition reactivating latent HIV reservoirs via gene transcription in resting cells, making them susceptible for “kill” strategies. These studies yielded potent representatives
97,
98,
99 (
Figure 20) and
49 (
Figure 13). Crystal structures elucidated the interactions of the imidazole moiety with a water pocket (
Figure 20).
In an effort to optimize these interactions, Clausen et al. flipped the imidazole motive and installed additional groups pointing towards the water pocket
[63] (
Figure 21).
Figure 21. Shown are compounds
93N ((
A): PDB-ID: 6WBW, 1.46 Å) and
97 ((
B): PDB-ID: 6XEB, 1.50 Å) in complex with HDAC2 illustrated using the UCSF ChimeraX visualization software (version ChimeraX-1.4)
[18][19]. The inhibitor and the amino acid side chains are displayed in the ball and stick or the stick style and are colored by element or heteroatom, respectively. Ligands are colored dark grey, water is represented as red spheres, interactions are indicated with dotted red lines and amino acids are numbered according to crystal structure. (
A)
93N binds in canonical fashion to the active site and engages in interactions with surface bound water. (
B) The imidazole moiety was flipped and derivatized in
97 to optimize interactions with surface-bound water.
Bürli et al. synthesized several compounds in an effort to develop CNS-penetrant class IIa selective HDACi. The authors clearly demonstrated class IIa selectivity and high potency. Evaluation and optimization of ADME properties was challenging and yielded
100 (
Figure 20), which had suitable microsomal stability and limited P-gp efflux and could be used as tool compound for proof-of-concept studies in disease models. Senger et al. incorporated an oxazol moiety as linker yielding
101 and
102 (
Figure 20). Comparison to thiazole- and oxadiazole analogues validated oxazole as most potent and selective for HDAC6. Ahn et al. synthesized oxazole and thiazole hydroxamate inhibitors and evaluated them in three cancer cell lines. Representative compound
103 had comparable potencies to
SAHA when tested against HeLa nuclear extract and in three cancer cell lines.
3.5. Pyrazoles
The pyrazole scaffold was used in several publications as part of the cap group (Compounds
104,
105,
106,
107,
108,
109,
110,
111,
112,
113,
114,
115,
116 and
117,
Figure 22). Yao et al.
[65] identified the pyrazole moiety via a scaffold-hoping approach starting from
104. Several heterocyclic scaffolds were synthesized and evaluated. Out of the synthesized scaffolds the pyrazole isomer
105 was the most promising, exhibiting greater potencies than
106,
107 and
108 and better selectivity than
109 (
Figure 22). A SAR study identified compound
110 as most potent, which was evaluated with several other compounds against eight human cancer cell lines yielding GI
50 values for HeLa (1.23 µM), MCF-7 (1.81 µM), BGC823 (0.26 µM), A549 (0.26 µM), HT1080 (1.33 µM), K562 (0.46 µM), U973 (0.51 µM) and Molt-4 (0.17 µM).
[65]
Figure 22. Summary of compounds with their inhibitory activities. HDAC* inhibitory activity was measured using HeLa nuclear extract.
Studies by Wen et al.
[66] focused on thiol-based HDACis containing a pyrazole scaffold. Starting from previous work Wen et al. explored the chemical space via nitrogen modifications and regioisomer synthesis which yielded
111 as most potent compound (
Figure 22)
[66].
Subsequent studies by Wen et al.
[67] further optimized the scaffold and evaluated it against seven cancer cell lines. Gained insights of prior studies could be applied to enhance inhibitory activity by modifications at N1 but were omitted due to cell permeability concerns when evaluating the representative
112 (
Figure 22) against seven cancer cell lines.
Evaluation of
112 and the diol
113 yielded GI
50 values for HCT-116 (25.26 µM and 8.93 µM), HT-29 (10.20 µM and 6.92 µM), MCF-7 (12.23 µM and 7.15 µM), MDA-MB-231 (>50 µM and 27.31 µM), A549 (14.43 µM and 6.07 µM), PC-3 (11.03 µM and 4.47 µM) and AsPC-1 (38.85 µM and 25.31 µM), respectively
[67].
Further effort by Xu et al.
[68] aimed to improve HDAC selectivity by altering the cap motive. Most of the compounds displayed moderate inhibitory activity with
114 being the most selective for HDAC6 (
Figure 22)
[68].
More research was conducted by Zagni et al.
[69] focusing on pyrazole as surface-recognition motive leading to several compounds with inhibitory activity against HDACs and antiproliferative activity against SH-SY6Y tumor cells. It was observed that N1-aryl scaffolds performed significantly better than N1-H scaffolds, with compounds
115,
116 and
117 showing the most promising activity (
Figure 22)
[69].
3.6. Thiophenes
Thiophenes can be utilized as capping groups in HDAC inhibitors. Compounds with piperazine linker, 118 showed comparable values against HDAC6 (IC50 value of 13.5 μM). Yang et al. designed a series of thiophene-based N-bis-substituted aromatic amide hydroxamic acid derivatives. Thereby IC50 of 1.14 nM for HDAC1 was achieved for one compound, 119 (Figure 23). The hydrolysis of a specific trifluoroacetamide substrate, 120 was inhibited by the mutated HDAC4, whereby IC50 values in the nanomolar range were achieved.
Figure 23. Summary of compounds with their inhibitory activities.
4. Conclusions
Heterocyclic rings, particularly those with five members, are highly valued in the development of active pharmaceutical ingredients. These rings belong to a class of organic compounds that contain at least one non-carbon atom within their structure. The inclusion of heteroatoms such as nitrogen, oxygen and sulfur in the aromatic ring enhances the polarity of the compound and introduces the potential for multiple target-inhibitor interactions. This, in turn, improves the drug’s binding affinity and selectivity at the intended target site. Heteroaromatic rings are considered favorable structures in medicinal chemistry as they can replace common motifs, leading to enhanced metabolic stability, solubility and bioavailability. The five-ring heterocycles in HDACis occur in three different structural elements of typical HDACis. Five-ring heterocycles can act as ZBG, provide the correct geometry in the binding pocket in the linker part often forming additional hydrophobic or cation-Pi interactions or interact with the protein surface in the head group, which enables the modulation of isozyme selectivity. Of particular interest are five-ring heterocycles that can replace the most commonly used ZBGs in HDACis, such as hydroxamic acid or TFMK. This is significant because these groups potentially mediate non-specific metalloenzyme binding thus increasing the risk for undesired side effects. In addition, hydroxamic acids are suspected of having mutagenic effects and TFMKs are metabolized very rapidly. Five-ring heterocycles can not only replace ZBGs, but also add entirely new beneficial mechanistic properties. The special molecular properties of DFMKs, for example, allows specific cleavage of the oxadiazole ring, presumably by the catalytic machinery of HDAC6, which leads to highly selective HDAC6is with very long residence times.
 +1 point
+1 point