
Abstract
Antarctic notothenioid fishes are recognised as one of the rare examples of adaptive radiation in the marine system. Withstanding the freezing temperatures of Antarctic waters, these fishes have diversified into over 100 species within no more than 10–20 million years. However, the exact species richness of the radiation remains contested. In the genus Channichthys, between one and nine species are recognised by different authors. To resolve the number of Channichthys species, genetic information would be highly valuable; however, so far, only sequences of a single species, C. rhinoceratus, are available. Here, we present the nearly complete sequence of the mitochondrial genome of C. rugosus, obtained from a formalin-fixed museum specimen sampled in 1974. This sequence differs from the mitochondrial genome of C. rhinoceratus in no more than 27 positions, suggesting that the two species may be synonymous.
Similar content being viewed by others


Introduction
The diversification of fishes of the perciform suborder Notothenioidei in Antarctic and Sub-Antarctic waters is a rare example of adaptive radiation in the marine environment (Clarke and Johnston 1996; Rüber and Zardoya 2005; Eastman 2005; Near et al. 2012; Matschiner et al. 2015). The radiating group of notothenioid fishes is composed of five families [Nototheniidae, Harpagiferidae, Artedidraconidae, Bathydraconidae, and Channichthyidae, jointly called “Cryonotothenioidea” (Near et al. 2015)] that together include over 100 species, distributed primarily on the shelf areas surrounding Antarctica and sub-Antarctic islands (Gon and Heemstra 1990; Eastman and Eakin 2021). The validity of most species within this radiation is well established and in many cases corroborated by genetic data. However, in other cases, species are known only from few specimens and distinguished from congeners based on minute morphological differences alone. One such example is the genus Pogonophryne (Artedidraconidae), for which close to twenty species have been described within the last four decades (Eastman and Eakin 2021), primarily on the basis of variation in the morphology of the mental barbel (e.g. Spodareva and Balushkin 2014). This species richness within Pogonophryne could not be confirmed in a recent genetic analysis, which instead led to the synonymization of 24 out of 29 valid species (Parker et al. 2022).
Outside Pogonophryne, the validity of species described in the genus Channichthys (Channichthyidae) remains particularly questionable. As many as nine species have been recognised, including the unicorn icefish (C. rhinoceratus Richardson, 1844), the red icefish (C. rugosus Regan, 1913), the sailfin pike (C. velifer Meisner, 1974), and six species described by Shandikov between 1995 and 2011 (Aelita icefish, C. aelitae; big-eyed icefish, C. bospori; pygmy icefish, C. irinae; charcoal icefish, C. panticapaei; green icefish, C. mithridatis; and robust icefish, C. richardsoni) (Shandikov 1995a, b, 2008, 2011) (Table 1). All species of the genus are endemic to the Kerguelen–Heard plateau and appear to share largely overlapping distributions (Shandikov 2011), implying that they either represent a small radiation on their own or that at least some of the described taxa are de facto morphs of one and the same species. The members of the genus are morphologically similar, with total lengths around 30–50 cm, a wide and spatulated snout, a tall first dorsal fin, and the rostral spine that gave its name to the first described species, C. rhinoceratus (Richardson 1844).
As a second species in the genus Channichthys, C. rugosus was described on the basis of two specimens that were found to differ from the known C. rhinoceratus specimens in eye diameter, roughness of the head, the position of supraorbital edges, and the length of the maxillary (Regan 1913). As additional specimens became available, the diagnosis of the two species became refined (Norman 1937), but the lack of clearly species-defining traits led some authors to question their separation even before further members of the genus were described (Hureau 1964). The third species to be described was C. velifer (Meisner 1974), which was claimed to differ from C. rhinoceratus in the number of spines of the first dorsal fin and the presence of a single median series of bony plates on the posterior part of the body (Meisner 1974). However, most of the specimens assigned to this new taxon were females, suggesting that sexual dimorphism could have explained the observed differences (Gon and Heemstra 1990). The six remaining described species were added to the list to accommodate previously unseen combinations of ray numbers, interorbital width, fin membrane height, and a duplicated row of gill rakers, amongst others (Shandikov 1995a, b, 2008, 2011). In most cases, these species were described on the basis of few specimens only.
To address a felt demand for a “complete overhaul” (Duhamel et al. 2005; Eastman and Eakin 2021) of the systematics of the genus Channichthys, Nikolaeva and Balushkin began a series of investigations based on comprehensive comparisons of specimens in the collections of the Zoological Institute of the Russian Academy of Sciences in Saint Petersburg, the Ukrainian National Museum of Natural History in Kyiv, and the British Natural History Museum in London. Their analyses indicated that the duplicate gill rakers observed in C. rhinoceratus, C. panticapaei, C. bospori, and C. irinae, but also in more distantly related icefishes, has evolved repeatedly (Balushkin and Nikolaeva 2015), leading them to suggest the synonymization of the latter three Channichthys species (Nikolaeva 2019). Their work further resulted in redescriptions of C. velifer (Nikolaeva and Balushkin 2019), C. rhinoceratus (Nikolaeva 2020), and C. rugosus (Nikolaeva 2021), as well as the suggested synonymization of C. aelitae, C. mithridatis, and C. richardsoni with C. rhinoceratus (Nikolaeva 2020).
Thus, according to Nikolaeva and Balushkin, the following four species are currently recognised in the genus Channichthys: C. rhinoceratus, C. rugosus, C. velifer, and C. panticapaei. The redescribed C. rugosus differs from C. rhinoceratus in four characters: greater height of the anterior dorsal fin, a fin membrane extending to the apexes of the longest rays, a narrower and concave interorbital space, and a more uniformly reddish body colour (Nikolaeva 2021). Channichthys rugosus can be further distinguished from C. velifer by numbers of fin rays in the first dorsal and the pectoral fin, bone plaques on the lateral line, and its colouration. Additionally, C. panticapaei was said to differ from C. rugosus in having a duplicated row of gill rakers and a more brownish black colouration (Nikolaeva 2021).
To complement the morphological analyses of Channichthys species and further test their validity, genetic data would be essential. Unfortunately, however, molecular information is so far only available for a single species, C. rhinoceratus. The genetic data available for this species include a set of ten nuclear markers commonly used in phylogenetic studies, cytochrome c oxidase I (CO1) barcodes (Smith et al. 2012), and the recently published complete sequence of a mitochondrial genome with a length of 17,408 bp (Andriyono et al. 2019), as well as restriction site-associated DNA (RAD) markers (Near et al. 2018). These sequences are available from the National Centre for Biotechnology Information (NCBI). For the other three potentially valid species of the genus, the unavailability of sequence information is at least in part due to the rarity of suitable tissue samples. To the best of our knowledge no more than 3–83 specimens are present for these species in museum collections (Table 1). Moreover, most specimens of these species were caught decades ago and fixed in formalin, which leads to degradation and chemical modification of DNA, making the recovery of genetic information challenging. To overcome this limitation, protocols for DNA extraction and sequencing library preparation tailored for formalin-fixed specimens have recently become available and proved to be remarkably successful (Gansauge et al. 2017, 2020; Gould et al. 2021).
Here, we apply recently developed methods to retrieve DNA sequences from a specimen of C. rugosus that was collected and formalin fixed in 1974 and then stored in 70 percent ethanol since. Modifications to a standard DNA extraction protocol from animal tissue maximise the yield of short DNA fragments, whilst application of a single-stranded DNA library preparation method (Gansauge et al. 2017, 2020) allows to convert even minute amounts of degraded DNA into sequencing libraries. We reconstruct the complete sequence of the specimen’s mitochondrial genome based on Illumina read data and compare this sequence to the mitochondrial genome of C. rhinoceratus to assess the genetic divergence between these two species. We find that the two mitochondrial genomes are nearly identical, with only 27 nucleotide differences between them. Such close similarity lends support to the hypothesis that C. rugosus is a synonym of C. rhinoceratus and suggests that like Pogonophryne, Channichthys comprises fewer differentiated species than previously thought.
Materials and methods
Sampling
The C. rugosus specimen used for sequencing was collected on 28.06.1974 during voyage 7 of the Soviet scientific trawler ‘Skif’ (‘Скиф’). It was obtained by bottom-trawling (trawl 188) on the Kerguelen shelf, at a depth of 115–120 m to the North-East of the Kerguelen Islands (48\(^{\circ } 34^{\prime }\) 1 S, 70\(^{\circ } 37^{\prime }\) 1 E). The specimen (ZIN 56294) has a standard length of 252 mm and is located in the collection of the Zoological Institute of the Russian Academy of Sciences in Saint Petersburg, Russia. It was formalin fixed upon arrival in Saint Petersburg and remained in 40% formalin for several years before being transferred to 70% ethanol. The specimen had been identified as C. rugosus based on the height of its first dorsal fin, the shape of its interorbital space, its body colouration, numbers of fin rays, and the absence of a second row of gill rakers on the first gill arch (Nikolaeva 2021) (Fig. 1).

Channichthys rugosus specimen ZIN 56294. Photograph (a) and X-ray image (b) of the C. rugosus specimen used for DNA extraction. The specimen has a standard length of 252 mm
DNA extraction and sequencing
To reduce contamination, DNA extraction, sequencing library preparation, and set-up of qPCR and PCR were conducted in a dedicated laboratory for low-copy DNA analysis, featuring intermittent UV-C irradiation, rigorous decontamination procedures, and positive pressure HEPA-filtrated ventilation. DNA was extracted from a small piece of muscle tissue (5.8 mg dry weight) that was dried in a vacuum centrifuge and then immersed in lysis buffer (260 µL ATL buffer (Qiagen) and 40 µL Proteinase K [20 mg/mL]). Two extraction negative control reactions received the same lysis buffer, but did not contain sample. After 24-h incubation at 56 °C, the lysates were centrifuged at 17,000×g for 5 min and 300 µL supernatant mixed with 3000 µL Buffer PB (Qiagen). The mixtures were loaded onto Minelute silica columns (Qiagen) in steps of 600 µL and then washed twice with 600 µL Buffer PE (Qiagen). Centrifugation was carried out for 1 minute each at 8000×g, which is lower than recommended by the manufacturer, in order to increase the retention of short DNA fragments. The columns were dry spun for 1 min at 16,000×g to remove residual wash buffer. To elute the DNA, 50 µL Buffer AE (Qiagen) were placed directly onto the silica membrane, incubated for 10 min, and centrifuged at 16,000×g for 2 min. DNA concentration was determined using 5 µL of the extract in a Qubit dsDNA High-Sensitivity assay. The sample yielded 6.6 ng/µL, whilst the extraction negatives contained 0.0244 ng/µL or were below the detection threshold of 0.02 ng/µL, respectively. Eight µL of the extract (= 52.8 ng DNA) and 30 µL each of the negative controls were then used to build Illumina sequencing libraries by single-stranded DNA library preparation as described by Gansauge et al. (2017, 2020), using the same reagents as listed in Gansauge et al. (2020). Briefly, the DNA extract and two extraction negative controls, along with one library negative control (water) and one library positive control (0.1 pmol of oligonucleotide CL104) were used for five separate library build reactions. Furthermore, all reactions received 10 amol CL104 as internal control. Samples were dephosphorylated and the \(3^{\prime }\)-adapter (TL181/TL110) attached by splinted ligation. Adapter molecules were bound to streptavidin-coated magnetic beads which were carried through the reactions and washed after each enzymatic reaction. An adapter-complementary primer (CL128) was used to prime the fill-in reaction, creating 5′-blunt-ended double-stranded molecules. Another ligation attached the second adapter (CL53/TL178). Libraries were eluted into 50 µL Tween-20-supplemented Tris–EDTA buffer (i.e. TET buffer) by heat denaturation. 1 µL each of a 1:50 dilution of libraries was used in two qPCR assays to determine control molecule numbers and required PCR-cycle numbers for amplification. The remaining 49 µL of libraries, with the exception of the positive library control, were then uniquely dual indexed (7-bp index length) (Kircher et al. 2012) and amplified until the end of the exponential phase and then purified using the Minelute PCR purification kit (Qiagen). Libraries were pooled and size selected to 160–250 bp on a Blue Pippin instrument using a 3% cassette with internal markers. The selected size fraction was measured with both, the Qubit dsDNA High-Sensitivity assay and the Tapestation HS1000 assay to adjust the input molarity for the sequencing run. The pool was then sequenced on an Illumina NextSeq500 instrument using a high-output single-end 75-bp read length kit with custom primers for read 1 (CL72) and index 2 (Gesaffelstein) (Paijmans et al. 2017).
Raw read files were demultiplexed using bcl2fastq v.2.19.1 (https://support.illumina.com/sequencing/sequencing_software/bcl2fastq-conversion-software.html), allowing for a maximum combined distance of 1 between barcodes and saved to fastq format. In the same step, prevalent adapter sequences were trimmed from the ends of reads and reads under 20 bp of length were omitted.
To ensure the presence of endogenous reads, we matched each read to the NCBI non-redundant (NR) sequence database (downloaded on 12 February 2022) with the BLASTX algorithm as implemented in Diamond v.2.0.4 (Altschul et al. 1990; Buchfink et al. 2015). The resulting taxon assignments were plotted with MEGAN v.6.21.4 (Huson et al. 2016).
Reference-based sequence analysis
We mapped reads to the C. rhinoceratus mitochondrial genome (Genbank accession number NC_057120) using BWA v.0.7.17 (Li and Durbin 2010), with its “aln” algorithm and the maximum fraction of missing alignments set to 0.05. The C. rhinoceratus mitochondrial genome was obtained from an individual that was collected in 2018 from Antarctic Subarea 58.5.2 (Heard Island and McDonald Island; Sapto Andriyono and Hyun-Woo Kim, priv. comm.). Resulting alignments were filtered to a Phred-scaled mapping quality of 25 or higher with Samtools v.1.9 (Li et al. 2009). Duplicated reads, e.g. from PCR duplicates, were flagged with Picard’s v.2.21.3 (http://broadinstitute.github.io/picard/) MarkDuplicate function and filtered with Samtools. Sites with a minimum read depth of 3 were consensus called using ANGSD v.0.933 (Korneliussen et al. 2014). To recover the terminal positions of the sequence, the first 200 bp of the reference were cut and appended and the mapping repeated. Regions of low-complexity and larger repeats exceeding the read length were edited manually or replaced with “N” for the length of the reference, as described in the Results section. The degradation state of DNA was assessed by inspection of the distribution of read lengths and analysis of substitution frequencies per base position within reads, using mapDamage v.2.0.8 (Jónsson et al. 2013).
Reference-independent sequence analysis
To exclude potential reference bias, we also performed analyses of the mitochondrial genome of C. rugosus based on de novo assembly. We performed local assembly of individual mitochondrial markers with aTRAM v.2.0 (Allen et al. 2018). As queries, we used nucleotide and protein sequences from all notothenioid mitochondrial genomes available on NCBI. To identify these mitochondrial genomes on NCBI, we used the search string ““Notothenioidei”[Organism] AND (“mitochondrial”[Title] OR “mitochondrion”[Title]) AND “complete genome”[Title]” on 4 December 2021. This set of mitochondrial genomes included the one for Channichthys rhinoceratus and 40 other unique mitochondrial genomes. From each of these 41 mitochondrial genomes, we extracted each gene (rRNA, tRNA, or protein coding) in nucleotide format, and protein-coding features in amino acid format, and used all of these in aTRAM analyses. All aTRAM analyses were performed separately with seven different e-value thresholds (1e-2, 1e-3, 1e-4, 1e-5, 1e-6, 1e-8, 1e-10) for the BLASTN and TBLASTN v.2.10.1 (Altschul et al. 1990) searches that aTRAM runs internally. As the assembler tool internally employed by aTRAM, we selected Trinity v.2.10.0 (Grabherr et al. 2011). Each aTRAM analysis was continued for 20 iterations.
All contigs produced by aTRAM were jointly used as input for a second assembler tool, MIRA v.4.9.6 (Chevreux et al. 1999). We set MIRA’s “nasty repeat ratio” to 25 (“-KS:nrr=25”), the “maximum megahub ratio” to 40 (“-SK:mmhr=40”), specified the lack of quality information (“-AS:epoq=no”), and turned off the checks for average coverage (“-NW:cac=no”) and maximum read name length (“-NW:cmrnl=warn”), according to the format of the input data. The contigs produced by MIRA (or their reverse complements) were then individually aligned to the mitochondrial genome for C. rhinoceratus with MAFFT v.7.470, using a gap opening penalty of 2, a gap extension penalty of 1, and the program’s “6merpair” and “addfragments” options.
Comparative analyses
The reference-based and reference-independent mitochondrial genome sequences for C. rugosus were compared visually using AliView v.1.2.6 (Larsson 2014). Nucleotide sequences of the 13 protein-coding genes were extracted from the mitochondrial genome of C. rugosus and all other notothenioid mitochondrial genomes and aligned per gene with MAFFT. The 13 alignments were concatenated and a distance matrix was calculated from the concatenated alignment with the Python script “convert.py” (available from GitHub: https://github.com/mmatschiner/supergenes/blob/main/gadidae_phylogenomics/src/convert.py), ignoring all sites with missing data. The concatenated alignment was further used for maximum-likelihood phylogenetic inference with IQ-TREE v.2.1.2 (Minh et al. 2020), applying the programme’s automated substitution model selection and 1000 ultrafast bootstrap (Minh et al. 2013) iterations.
To gain a more complete view of sequence variation in Channichthys, we took advantage of the CO1 barcode sequences available on NCBI (Smith et al. 2012). We downloaded these sequences and aligned them together with the homologous sequences extracted from the mitochondrial genomes of C. rhinoceratus and C. rugosus, using MAFFT. To illustrate the CO1 sequence variation in Channichthys, we first applied maximum-likelihood phylogenetic inference with IQ-TREE and then used the estimated phylogeny jointly with the sequence alignment to draw a haplotype genealogy graph with the program Fitchi v.1.1.4 (Matschiner 2016).
Results
Sequencing
For the C. rugosus specimen, a total of 63,158,791 reads passed initial trimming and length filtering. For extraction negative controls 1 and 2 (exneg 1, exneg 2) and library negative control (libneg), the number of reads were 22,393,754, 21,343,774 and 3,080,057, respectively. Analysis with Diamond and MEGAN assigned a substantial proportion of the reads from the sample to Notothenioidei, confirming that these were likely endogenous (Fig. 4).
Reference-based sequence analysis
Mapping reads to the C. rhinoceratus mitochondrial genome generated 48,313 hits for the sample, corresponding to 0.0765% of total reads from the sample. After filtering duplicates, 18,687 unique reads were used for further analysis. These reads had an average length of 32.98 bp, with only 0.1% of reads being 70 bp or longer. Figure 2 shows the distribution of read lengths of hits. Negative controls only produced 5 (exneg 1), 5 (exneg 2), and 1 unique hit (libneg) to the C. rhinoceratus mitochondrial genome sequence, which were not analysed further. The resulting coverage had an average depth of 35.4 and extended over 98.1% of the reference. The alignment showed two apparent gaps in the D-loop and spurious alignments directly adjacent to them, therefore positions 15,192–15,294 and 16,856–17,262, respectively, were called as “N” for the length of the reference. A 14-mer C repeat at reference positions 15,986–15,999 was spanned by two reads only, both indicating an indel with one fewer repeat in C. rugosus, which was called manually. Mapped reads showed an elevated rate of C to T changes in both their 3′- and 5′-ends, a pattern of cytosine deamination that is typical for degraded DNA (Fig. 3).

Distribution of read lengths of hits to the C. rhinoceratus mitochondrial genome. The solid line and bars indicate the number of hits for a given read length, and the dotted line indicates the amount of data in nucleotides gathered from reads of a given length

Frequency of post-mortem C to T changes by nucleotide position within reads. Deaminated cytosines are being read as thymine and are especially prevalent at molecule ends. Red line: frequency of C (reference)/T (sequencing data) mismatches by position in read from 5′-end (left) and 3′-end (right); blue line: frequency of G/A mismatches. Other types of mismatches are indicated with thin grey lines but are largely concealed. Here, the deamination signal manifests only as C/T and not G/A, because single-stranded, rather than double-stranded DNA library preparation was used

Taxonomic assignment of individual reads. Reads were mapped to the NCBI non-redundant (NR) database. The sizes of circles on internal and terminal nodes are proportional to the numbers of reads mapping to the corresponding taxon; a single read mapped to taxa without visible circles
Reference-independent sequence analysis
Assembly with MIRA produced 14 contigs with lengths between 317 and 1471 basepairs (bp) (mean length: 677.4 bp). One of these contigs overlapped fully with another one and two other contigs overlapped by 13 bp. The total length of the mitochondrial genome covered by these contigs was 9849 bp. No nucleotide differences were observed between overlapping contigs.
Comparative analyses
Comparison of the reference-based and the reference-independent mitochondrial genome sequences showed that these were completely identical across the 9849 bp covered by both. Compared to the mitochondrial genome of C. rhinoceratus, the reference-based sequence differed at 27 sites (Table 2). Seventeen of these nucleotide differences were within regions also covered by the reference-independent sequence for C. rugosus, and all of these were confirmed by that sequence. The differences between the C. rhinoceratus and C. rugosus mitochondrial genomes included five transversions (two A/C, one G/C, one G/T, and one T/G substitution) and 21 transitions (seven A/G, two C/T, six G/A, and six T/C substitutions), as well as one C/-indel. The nucleotide differences were distributed unevenly across the mitochondrial genome and mostly found in the ND6 gene and the D-loop region (Fig. 5). Compared to the mitochondrial genome-wide background of 0.0011 substitutions per bp, the ND6/D-loop region had an elevated divergence of 0.0065 substitutions per bp.

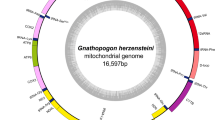
Mitochondrial genome of C. rugosus. Coordinates on the outside of the circle are given in units of basepairs. The grey inner circle indicates the nearly full coverage achieved in the reference-based analysis. Two gaps with a total length of 510 bp remain in this sequence between positions 15,192 and 15,294, and between positions 16,856 and 17,262. The black fragments inside the grey circle show the positions of contigs from the reference-independent approach. Substitutions compared to the mitochondrial genome of C. rhinoceratus (NCBI accession NC_057120) are marked in orange, and non-synonymous substitutions are labelled with the resulting amino acid change
Phylogenetic inference with IQ-TREE based on protein-coding mitochondrial sequences grouped the two Channichthys genomes with full bootstrap support. The two taxa were separated only by very short branches that had a combined length of 0.0012 substitutions per bp. The Channichthys mitochondrial genomes were most similar to those of Chionobathyscus dewitti and Cryodraco antarcticus, with which they formed a clade that also received full bootstrap support. This clade appeared as the sister group to a clade formed by the mitochondrial genomes of Chaenodraco wilsoni and three members of the genus Chionodraco, albeit with low bootstrap support of 70% (Fig. 6).

Mitochondrial phylogeny of Channichthyidae. Maximum-likelihood phylogeny of all available mitochondrial genomes for Channichthyidae. Short branches connect Channichthys rhinoceratus and C. rugosus. The order of coding genes is identical in all mitochondrial genomes
The haplotype genealogy graph for 14 available Channichthys CO1 sequences showed that eleven of them shared the same haplotype. Three other haplotypes differed by a single substitution from the majority haplotype and were each represented by a single individual. One of these three private haplotypes was found in the published mitochondrial genome sequence for C. rhinoceratus, whilst the C. rugosus mitochondrial genome had the majority haplotype (Fig. 7).

Haplotype genealogy graph for CO1. Circles represent four distinct Channichthys CO1 haplotypes, found in 13 C. rhinoceratus and one C. rugosus individuals. Radii are drawn according to the number of individuals with that haplotype (indicated with labels on circles). Each of the three edges represents one single substitution. mt., mitochondrial
Discussion
Our comparison of the mitochondrial genomes of C. rhinoceratus and C. rugosus showed that these two mitochondrial genomes are highly similar, with only one indel, 26 nucleotide substitutions, and six amino acid substitutions between them. The sequence divergence is 0.16%, and most of this divergence is concentrated in the ND6/D-loop region where the divergence reaches 0.65%. Despite very short read lengths, reconstruction of C. rugosus’ mitochondrial genome was possible, except for two loci in the ND6/D-loop region. Those are most likely repeats that were too large to be spanned by single reads and could not be mapped correctly. Whilst larger rearrangements of notothenioid mitochondrial genomes have been reported by Papetti et al. (2021), gene order was shown to be identical for eight of the species in Fig. 6 (all except Chionobathyscus dewitti, Cryodraco antarcticus, and Channichthys sp.). Hence, whilst possible, the gaps in the alignments are unlikely to be caused by rearrangements or larger indels and are probably artefacts due to short read length. This highlights a shortcoming of the use of degraded DNA, where read lengths are usually limited by DNA molecule lengths rather than by sequencing technology, making the detection of structural variants challenging.
Analysis of post-mortem damage shows a robust pattern of cytosine deamination in single-stranded overhangs, manifesting as an elevated number of reads with apparent C to T changes towards either end of their sequence. Given the appreciable depth of coverage, however, and the fact that intact cytosines vastly outnumber deaminated ones overall, such erroneous base calls in individual reads are unlikely to cause wrong calls of alignment positions. Here, only C to T changes are seen as we used single-stranded DNA library preparation. In double-stranded DNA library preparation, deaminated cytosines would also be apparent as G to A changes in the sequencing data. The presence of this deamination signal can be interpreted as evidence for the authenticity of the sequences, as modern contamination would not show it. However, this is more relevant for ancient samples, for which the age of endogenous DNA and ex situ contamination would be very different and for cases where contamination is more likely to be confused with endogenous sequences, such as ancient human samples. Here, contamination is unlikely to affect our results, as negative controls did not produce concerning numbers of reads mapping to the reference, and no other samples or DNA were handled in the laboratory environment which could not be readily distinguished from our sample.
The available molecular data for the genus Channichthys does not allow us to perform a formal genetic species delimitation analysis as was recently done for Pogonophryne (Parker et al. 2022). Nevertheless, a comparison of the divergence between the two Channichthys mitochondrial genomes with the levels of between- and within-species sequence divergence in other notothenioid fishes can inform about the existence of one or several species within the genus. As shown in Fig. 6, the divergence within Channichthys is far smaller than that between any other pairs of Channichthyidae species. Besides Channichthys rhinoceratus and C. rugosus, the most closely related species (judged on the basis of their mitochondrial genomes) appear to be Chionodraco hamatus and Chionodraco rastrospinosus. However, despite their recent divergence around a million years ago (Colombo et al. 2015), the mitochondrial genomes of these two species are connected by a total branch length of 0.019 substitutions per bp, around 16 times as long as the branches connecting the two Channichthys mitochondrial genomes. Of note, the two Chionodraco species appear not to be fully separated, given that hybrids and signals of introgression have been observed (Schiavon et al. 2021).
On the other hand, the divergence between the two Channichthys mitochondrial genomes is comparable to the within-species divergence in other notothenioid species. Mitochondrial genomes of two individuals are available for five notothenioid species: Trematomus borchgrevinki, Notothenia coriiceps, Notothenia rossi, Chaenodraco wilsoni, and Chionodraco hamatus. These two genomes per species differ by 16–92 substitutions and 3–22 indels across the mitochondrial genome or by 11–69 substitutions and 2–10 indels when the ND6 / D-loop region is excluded (Table 3). The divergence of the two Channichthys mitochondrial genomes is close to or below the lower ends of these ranges, considering that we found these two genomes to differ in 26 nucleotide substitutions (17 outside the ND6/D-loop region) and one 1-bp indel only. In contrast, the two most closely related pairs of sister species within Channichthyidae (Chionodraco hamatus, Chionodraco rastrospinosus, and Chionobathyscus dewitti, Cryodraco antarcticus; Fig. 2) differ by 421–540 substitutions and 25–34 indels (253–272 substitutions and 6–11 indels when the ND6/D-loop region is excluded; Table 3). Thus, the divergence of the mitochondrial genomes of Channichthys rhinoceratus and C. rugosus is consistent with the existence of a single species within the genus Channichthys (Eastman and Eakin 2021). If this result should be confirmed by further molecular data, Channichthys rugosus may need to be synonymized with C. rhinoceratus.
Low divergence in mitochondrial genome sequence can be indicative of a close—e.g. intraspecific—phylogenetic relationship, but could also be due to alternative scenarios. Mitochondrial capture, where one lineage fixes the mitochondrial genome received from another by introgression, can result in two valid, reproductively largely isolated species with appreciable nuclear genome divergence having no or little mitochondrial genome divergence. This phenomenon can also apply to large, rapidly diversifying clades where many species have arisen from nuclear genomic diversity created through hybridisation of divergent ancestral lineages, for example, in Lake Victoria’s haplochromine cichlids (Meier et al. 2017). In that case low mitochondrial diversity and rampant haplotype sharing would conceal a large species diversity. It would need to be tested using genome-wide nuclear data if such a scenario applies to Channichthys, rather than a previous overestimation of species diversity in the genus.
Genetic data can be vital to corroborate or reject taxonomic assessments based on morphology and to more accurately estimate organismal diversity. The fixation of specimens with formalin, which severely hampers genetic analyses, has long been recognised and indeed lamented, as a major obstacle for tapping the theoretically vast potential of museum collections for addressing long-standing questions in systematics, taxonomy, evolutionary, and conservation biology (Card et al. 2021; Raxworthy and Smith 2021). Time-structured samples, including extinct species, can reveal details about the recent decline of biodiversity. Specimens from places that are difficult to access could fill gaps in studies otherwise relying on a smaller geographical sampling. Given this potential it is unsurprising that the research community continues to undertake great efforts in refining methodology to maximise the data yield from wet-collection specimens (Schander and Kenneth 2003; Campos and Gilbert 2012; Hykin et al. 2015; Straube et al. 2021; Hahn et al. 2021). Here, we demonstrated that recently proposed methods that build on advances in the study of ancient DNA and high-throughput shotgun sequencing can recover usable genetic data from a formalin-fixed fish specimen. The short molecule lengths and very low DNA amounts recovered from such samples mandate the use of specialised, sensitive methods, such as lower centrifugation speeds and greater excess of binding buffer in DNA extraction (Dabney et al. 2013). The chemical cross-linking of DNA with DNA and proteins requires a harsher treatment of the tissue sample during lysis, for example, using high amounts of proteinase, as done here. Subsequently, the use of very efficient single-stranded DNA library preparation can convert appreciable numbers of molecules even if chemical damage and cross-linking are prevalent. Caution should be taken when analysing such low-yield samples, as levels of contamination regarded minor in other circumstances can obscure signal from the target or lead to incorrect interpretations. Working in a clean laboratory that is dedicated to the analysis of low biomass, degraded samples is therefore recommended, as well as including appropriate negative controls to monitor contamination. This extra effort, however, is greatly rewarded when open questions can be addressed with otherwise unobtainable data.
Code availability
Analysis code is available from GitHub (https://github.com/mmatschiner/unsequenced).
References
Allen JM, LaFrance R, Folk RA et al (2018) aTRAM 2.0: an improved, flexible locus assembler for NGS data. Evol Bioinf 14:1176934318774546. https://doi.org/10.1177/1176934318774546
Altschul SF, Gish W, Miller W et al (1990) Basic local alignment search tool. J Mol Biol 215(3):403–410. https://doi.org/10.1016/s0022-2836(05)80360-2
Andriyono S, Alam MJ, Lee SR et al (2019) Characterization of the complete mitochondrial genome of Chionobathyscus dewitti (Perciformes, Channichthyidae). Mitochondrial DNA Part B 4(2):3914–3915. https://doi.org/10.1080/23802359.2019.1688112
Balushkin AV, Nikolaeva EA (2015) “Dolichobranchiata’’ mutation in the Antarctic representatives from the families of plunderfishes (Artedidraconidae)and white-blooded (Channichthyidae) fish (Notothenioidei). J Ichthyol 55(1):9–15. https://doi.org/10.1134/S0032945215010014
Buchfink B, Xie C, Huson DH (2015) Fast and sensitive protein alignment using DIAMOND. Nat Methods 12(1):59–60. https://doi.org/10.1038/nmeth.3176
Campos PF, Gilbert TMP (2012) DNA extraction from formalin-fixed material. Methods Mol Biol (Clifton, NJ) 840:81–5. https://doi.org/10.1007/978-1-61779-516-9_11
Card DC, Shapiro B, Giribet G et al (2021) Museum genomics. Annu Rev Genet 55(1):1–27. https://doi.org/10.1146/annurev-genet-071719-020506
Chevreux B, Wetter T, Suhai S (1999) Genome sequence assembly using trace signals and additional sequence information. Comput Sci Biol 18:1–12
Clarke A, Johnston IA (1996) Evolution and adaptive radiation of Antarctic fishes. Trends Ecol Evol 11(5):212–218. https://doi.org/10.1016/0169-5347(96)10029-X
Colombo M, Damerau M, Hanel R et al (2015) Diversity and disparity through time in the adaptive radiation of Antarctic notothenioid fishes. J Evol Biol 28(2):376–394. https://doi.org/10.1111/jeb.12570
Dabney J, Knapp M, Glocke I et al (2013) Complete mitochondrial genome sequence of a Middle Pleistocene cave bear reconstructed from ultrashort DNA fragments. Proc Natl Acad Sci USA 110(39):15,758-15,763. https://doi.org/10.1073/pnas.1314445110
Duhamel G, Gasco N, Davaine P (eds) (2005) Poissons des Iles Kerguelen et Crozet. Guide Régional de l’Océan Austral. Patrimoines Naturels, vol 63. Muséum National d’Histoire Naturelle, Paris
Eastman JT (2005) The nature of the diversity of Antarctic fishes. Polar Biol 28(2):93–107. https://doi.org/10.1007/s00300-004-0667-4
Eastman JT, Eakin RR (2021) Checklist of the species of notothenioid fishes. Antarct Sci 33(3):273–280. https://doi.org/10.1017/S0954102020000632
Gansauge MT, Gerber T, Glocke I, et al (2017) Single-stranded DNA library preparation from highly degraded DNA using T4 DNA ligase. Nucleic Acids Res. https://doi.org/10.1093/nar/gkx033
Gansauge MT, Aximu-Petri A, Nagel S et al (2020) Manual and automated preparation of single-stranded DNA libraries for the sequencing of DNA from ancient biological remains and other sources of highly degraded DNA. Nat Protoc 15(8):2279–2300. https://doi.org/10.1038/s41596-020-0338-0
Gon O, Heemstra PC (1990) Fishes of the Southern Ocean. J.L.B. Smith Institute of Ichthyology, Grahamstown, South Africa, p 196
Gould AL, Fritts-Penniman A, Gaisner A (2021) Museum genomics illuminate the high specificity of a bioluminescent symbiosis for a genus of reef fish. Front Ecol Evol 9(630):207. https://doi.org/10.3389/fevo.2021.630207
Grabherr MG, Haas BJ, Yassour M et al (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29(7):644–652. https://doi.org/10.1101/gr.229202
Hahn EE, Alexander MR, Grealy A et al (2021) Unlocking inaccessible historical genomes preserved in formalin. Mol Ecol Resour. https://doi.org/10.1111/1755-0998.13505
Hureau JC (1964) Sur la probable identité des deux espèces du genre Chaenichthys, de la familie des Channichthyidae (poissons à “sang blanc’’). Bulletin du Muséum National d’Histoire Naturelle 36(4):450–456
Huson DH, Beier S, Flade I et al (2016) MEGAN Community edition-interactive exploration and analysis of large-scale microbiome sequencing data. PLOS Comput Biol 12(6):e1004957. https://doi.org/10.1371/journal.pcbi.1004957
Hykin SM, Bi K, Mcguire JA (2015) Fixing formalin: a method to recover genomic-scale DNA sequence data from formalin-fixed museum specimens using high-throughput sequencing. PLoS ONE 10(10):e0141579-16. https://doi.org/10.1371/journal.pone.0141579
Jónsson H, Ginolhac A, Schubert M et al (2013) mapDamage2.0: fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 29(13):1682–1684. https://doi.org/10.1093/bioinformatics/btt193
Kircher M, Sawyer S, Meyer M (2012) Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Res 40(1):e3–e3. https://doi.org/10.1093/nar/gkr771
Korneliussen TS, Albrechtsen A, Nielsen R (2014) ANGSD: analysis of next generation sequencing data. BMC Bioinform 15(1):356. https://doi.org/10.1186/s12859-014-0356-4
Larsson A (2014) AliView: a fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 30(22):3276–3278. https://doi.org/10.1093/bioinformatics/btu531
Li H, Durbin R (2010) Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26(5):589–595. https://doi.org/10.1093/bioinformatics/btp698
Li H, Handsaker B, Wysoker A et al (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25(16):2078–2079. https://doi.org/10.1093/bioinformatics/btp352
Matschiner M (2016) Fitchi: haplotype genealogy graphs based on the Fitch algorithm. Bioinformatics 32(8):1250–1252. https://doi.org/10.1093/bioinformatics/btv717
Matschiner M, Colombo M, Damerau M, et al (2015) The adaptive radiation of notothenioid fishes in the waters of Antarctica. In: Extremophile Fishes: Ecology, Evolution, and Physiology of Teleosts in Extreme Environments, vol 36. Springer, Cham. https://doi.org/10.1111/j.1365-294X.2006.03105.x
Meier JI, Marques DA, Mwaiko S et al (2017) Ancient hybridization fuels rapid cichlid fish adaptive radiations. Nat Commun 8(14):363. https://doi.org/10.1038/ncomms14363
Meisner EE (1974) New species of the icefishes from the Southern Ocean. Vestnik Zoologii 6:50–55
Minh BQ, Nguyen MAT, von Haeseler A (2013) Ultrafast approximation for phylogenetic bootstrap. Mol Biol Evol 30(5):1188–1195. https://doi.org/10.1093/oxfordjournals.molbev.a025811
Minh BQ, Schmidt HA, Chernomor O et al (2020) IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol 37(5):1530–1534. https://doi.org/10.1093/molbev/msaa015
Near TJ, Dornburg A, Kuhn KL et al (2012) Ancient climate change, antifreeze, and the evolutionary diversification of Antarctic fishes. Proc Natl Acad Sci USA 109(9):3434–3439. https://doi.org/10.1073/pnas.1115169109
Near TJ, Dornburg A, Harrington RC et al (2015) Identification of the notothenioid sister lineage illuminates the biogeographic history of an Antarctic adaptive radiation. BMC Evol Biol 15:109. https://doi.org/10.1186/s12862-015-0362-9
Near TJ, MacGuigan DJ, Parker E et al (2018) Phylogenetic analysis of Antarctic notothenioids illuminates the utility of RADseq for resolving Cenozoic adaptive radiations. Mol Phylogenet Evol 129:268–279. https://doi.org/10.1016/j.ympev.2018.09.001
Nikolaeva EA (2019) A review of the icefish species from the genus Channichthys Richardson, 1844 (Channichthyidae) with double-rowed gill rakers. Proc Zool Inst RAS 323(4):558–567. https://doi.org/10.31610/trudyzin/2019.323.4.558
Nikolaeva EA (2020) Redescription of the unicorn icefish Channichthys rhinoceratus Richardson (Notothenioidei: Channichthyidae) with synonymization of three similar species. Proc Zool Inst RAS 324(4):485–496. https://doi.org/10.31610/trudyzin/2020.324.4.485
Nikolaeva EA (2021) On the taxonomic status of the red icefish Channichthys rugosus (Notothenioidei: Channichthyidae) from the Kerguelen Islands (South Ocean). Proc Zool Inst RAS 325:485–494. https://doi.org/10.31610/trudyzin/2021.325.4.485
Nikolaeva EA, Balushkin AV (2019) Morphological characteristics of sailfish pike Channichthys velifer (Channichthyidae) from the Kerguelen Islands (Southern Ocean). J Ichthyol 59(6):834–842. https://doi.org/10.1134/S0032945219060079
Norman JR (1937) Fishes. B.A.N.Z. Antarctic research expedition 1929–31. Repts Ser B Zool Bot 1(2):50–88
Paijmans JLA, Baleka S, Henneberger K, et al (2017) Sequencing single-stranded libraries on the Illumina NextSeq 500 platform. arXiv:1711.11004
Papetti C, Babbucci M, Dettai A et al (2021) Not frozen in the ice: large and dynamic rearrangements in the mitochondrial genomes of the Antarctic fish. Genome Biol Evol 13(3):evab017. https://doi.org/10.1093/gbe/evab017
Parker E, Dornburg A, Struthers CD et al (2022) Phylogenomic species delimitation dramatically reduces species diversity in an Antarctic adaptive radiation. Syst Biol 71(1):58–77. https://doi.org/10.1093/sysbio/syab057
Raxworthy CJ, Smith BT (2021) Mining museums for historical DNA: advances and challenges in museomics. Trends Ecol Evol 36(11):1049–1060. https://doi.org/10.1016/j.tree.2021.07.009
Regan CT (1913) II—The Antarctic fishes of the Scottish National Antarctic expedition. Trans R Soc Edinburgh 49(2):229–292
Richardson J (1844) LII—Descriptions of a new Genus of Gobioid Fish. Ann Mag Nat Hist 13(86):461–462. https://doi.org/10.1080/03745484409442631
Rüber L, Zardoya R (2005) Rapid cladogenesis in marine fishes revisited. Evolution 59(5):1119–1127. https://doi.org/10.1111/j.0014-3820.2005.tb01048.x
Schander C, Kenneth HM (2003) DNA, PCR and formalinized animal tissue—a short review and protocols. Organ Divers Evol 3(3):195–205. https://doi.org/10.1078/1439-6092-00071
Schiavon L, Dulière V, La Mesa M et al (2021) Species distribution, hybridization and connectivity in the genus Chionodraco: unveiling unknown icefish diversity in antarctica. Divers Distrib 27(5):766–783. https://doi.org/10.1111/ddi.13249
Shandikov GA (1995a) A new species of icefish, Channichthys panticapei sp. n. (Channichthyidae, Notothenioidei) from the Kerguelen Island (Antarctica). Proceedings of South Research Institute of Marine Fishery and Oceanography (YugNIRO) Special Issue, 1:1–10
Shandikov GA (1995b) To the question about the composition of icefish species of the genus Channichthys in the Kerguelen Islands area with description of three new species. Proceedings of South Research Institute of Marine Fishery and Oceanography (YugNIRO) Special Issue, 2:1–18
Shandikov GA (2008) Channichthys mithridatis, a new species of icefishes (Perciformes: Notothenioidei: Channichthyidae) from the Kerguelen Islands (East Antarctica), with comments on the taxonomic status of Channichthys normani. Visnyk Charkivs’koho Universytetu Imeni V N Karazina, Ser Biologija, Charkiv 14(917):123–131
Shandikov GA (2011) Channichthys richardsoni sp. n., a new Antarctic icefish (Perciformes: Notothenioidei: Channichthyidae) from the Kerguelen Islands area, Indian sector of the Southern Ocean. Journal of V N Karazin Kharkiv National University, Series: Biology 14:125–134
Smith PJ, Steinke D, Dettai A et al (2012) DNA barcodes and species identifications in Ross Sea and Southern Ocean fishes. Polar Biol 35(9):1297–1310. https://doi.org/10.1007/s00300-012-1173-8
Spodareva VV, Balushkin AV (2014) Description of a new species of plunderfish of genus Pogonophryne (Perciformes: Artedidraconidae) from the Bransfield Strait (Antarctica) with a key for the identification of species of the group “marmorata’’. J Ichthyol 54(1):1–6. https://doi.org/10.1134/S0032945214010135
Straube N, Lyra ML, Paijmans JLA et al (2021) Successful application of ancient DNA extraction and library construction protocols to museum wet collection specimens. Mol Ecol Resour 21(7):2299–2315. https://doi.org/10.1111/1755-0998.13433
Acknowledgements
We thank Arcady V. Balushkin for providing the C. rugosus specimen and Marcelo Sánchez-Villagra for financially supporting the sequencing of its mitochondrial genome. Sapto Andriyono and Hyun-Woo Kim provided helpful information about the C. rhinoceratus mitochondrial genome. Mark Lever (ETH Zurich) kindly provided lab space. The Genetic Diversity Centre (ETH Zurich) provided access to laboratory and HPC facilities. The Functional Genomics Centre Zürich (ETH and University of Zurich) provided assistance with sequencing. We further thank the reviewers Chiara Papetti and Joe Eastman for comments that helped improve the manuscript.
Funding
Open access funding provided by University of Oslo (incl Oslo University Hospital). M. Muschick was supported by the SNSF Sinergia grant CRSII5_183566. M. Matschiner was supported by the Norwegian Research Council with FRIPRO grant 275869. E. Nikolaeva was supported by a research programme of the Zoological Institute of the Russian Academy of Sciences (Project Number 122031100285-3).
Author information
Authors and Affiliations
Contributions
MMu performed molecular lab work, bioinformatic analyses, and contributed to the manuscript. EN initiated the study and contributed the tissue sample of C. rugosus. LR established the collaboration. MMa performed bioinformatic analyses and wrote most of the manuscript. All authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that no competing interest exists, as well as that there is no financial support or relationships that may pose any kind of conflict. Likewise, the authors declare that contributed to the text, agreed with its content and approved it for submission.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Muschick, M., Nikolaeva, E., Rüber, L. et al. The mitochondrial genome of the red icefish (Channichthys rugosus) casts doubt on its species status. Polar Biol 45, 1541–1552 (2022). https://doi.org/10.1007/s00300-022-03083-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00300-022-03083-8