
Abstract
The complex of beech taxa in western Eurasia (Fagus sylvatica L. s.l.) represents one of the most widespread broad-leaved trees in the temperate forests of the northern hemisphere. Two subspecies are generally recognized, F. sylvatica ssp. sylvatica in Europe and F. sylvatica ssp. orientalis in southwestern Asia, but several strongly differentiated regional populations were identified within this complex. To reveal geographical and genomic patterns of hybridization and introgression, genetic variation at 6789 single-nucleotide polymorphisms (SNP) resulting from ddRAD sequencing was studied in 288 specimens representing 207 populations distributed across the whole range. Two clusters resulting from a Bayesian analysis of population Structure well coincided with the subspecies, while increasing the number of clusters allowed to distinguish divergent lineages within subspecies. Two zones of potential hybridization between such lineages were analyzed: western Balkans at the contact of both subspecies and the Alpine area at the contact of populations coming from different glacial refugia. Geographical distribution of hybrid indices in the Balkan hybrid zone indicates asymmetric gene flow preferentially directed from F. orientalis to F. sylvatica. Eight SNPs showed a significant shift of genomic cline centers, seven of them toward excess F. orientalis ancestry, suggesting that introgression irregularly affects different regions of the beech genome. On the other hand, no genomic cline outliers were found in the Alpine hybrid zone. Implications for assisted gene flow are shortly discussed.
Similar content being viewed by others


Introduction
In the past, hybridization has often been viewed as a lapsus of nature. Under the biological species concept (Mayr 1942), reproductive isolation has been regarded as a phenomenon defining species boundaries and a basic prerequisite for the persistence of species. With the advent of molecular methods in systematics and evolutionary studies, it became obvious that hybridization is rather a common process; one quarter of plant species and one-tenth of animal species are supposed to be involved in interspecific crossing (Arnold and Hodges 1995; Mallet 2005, 2007).
Viability and fertility of F1 hybrids and backcrosses in comparison with parental species may vary. In wild outcrossing and mixed-mating plants, average fitness is typically lower in hybrids than in parental taxa, except in specific habitats (Rieseberg et al. 1999), but this is not always the case. Hybrids may both profit from somatic or adaptive heterosis (Miller et al. 2015) and suffer from outbreeding depression (Ellison and Burton 2008; Rosas et al. 2010; Sambatti et al. 2008), or the hybrid vigor is just transient and hybrid breakdown occurs in later generations (Johansen-Morris and Latta 2006; Johnson et al. 2010; Latta et al. 2007). In general, hybrid fitness can vary between environments (Leinonen et al. 2011), and hybrids and backcrosses may eventually outperform pure species (Arnold and Martin 2010; Arnold et al. 2012; Dagilis et al. 2019). Hybrid zones appearing at the secondary contact of divergent genetic lineages represent natural laboratories allowing to evaluate how gene flow and selection regulate the spread of genetic material across species boundaries (Lexer et al. 2007; Rieseberg et al. 1999). Genes can be exchanged between lineages at different rates ranging from zero (or almost zero) for the genomic regions involved in reproductive isolation to free (or almost free) exchange for the selectively neutral segments or parts of the genome under selective pressures driven by factors common for both lineages (Barton 1979; Capblanq et al. 2020; Hamilton et al. 2013; Harrison 1993).
Hybridization is commonly understood as gene exchange between different species, or at least recognized subspecific taxa. However, mating between divergent lineages within a species may lead to similar consequences as mating between closely related species (Burke and Arnold 2001; Keller and Taylor 2010; Lucek et al. 2010).
The complex of beech taxa in western Eurasia (Fagus sylvatica L. sensu lato) is a good model for the study of hybridization and introgression at the subspecific level. Native old-growth forests distributed across the whole range from the Carpathians through Dinaric mountains, Pontic mountains to the Alborz, where beech trees reach heights over 45 m, document ecological stability and production potential of beech-dominated forest communities (Holeksa et al 2009; Leibundgut 1982; Tabari et al. 2005). Although in some regions beech suffers from the ongoing climate changes (Czúcz et al. 2011; Leuschner 2020), it continues to expand in mixed montane forests on the expense of coniferous species (Keren et al. 2014). Currently, Flora Europaea (Akeroyd 1993) recognized beech populations in western Eurasia as a single species, Fagus sylvatica L. However, as a taxon with very large and partly discontinuous range, it has been object of many botanical, phytosociological and taxonomical studies, resulting in the description of several subspecific taxa, although the opinions about their number and rank are divergent, sometimes even controversial. Recent systematic revisions (Denk 1999a, b) confirmed the view of Greuter and Burdet (1981), who classified western Eurasian beeches in two subspecies: F. sylvatica subsp. sylvatica and F. sylvatica subsp. orientalis. Nevertheless, the latter subspecies is regarded as a separate species F. orientalis Lipsky not only by local botanists and foresters (Gulisashvili et al. 1975; Mayer and Aksoy 1986; Mobayen and Tregubov 1970) but also authoritative sources such as the Plants of the World Online database (https://powo.science.kew.org/taxon/urn:lsid:ipni.org:names:295615-1) or World Flora Online (WFO 2023). For brevity, the subspecies are hereafter labeled as “F. sylvatica” and “F. orientalis,” respectively. Moreover, a plenty of local or regional populations were described as separate taxa, mostly on the subspecies or variety level (for a historical overview, see Denk et al. 2002); among them, the names “F. moesiaca (K. Malý) Czeczott” and “F. taurica Popl.” are widely used by local botanists for the populations distributed in the southern Balkans and the Crimean Peninsula, respectively. Also, beech in the Caucasus and the Alborz Mountains was described as a separate species F. hohenackeriana Palib., but this name is rarely used.
Such a plethora of names is by far not exceptional in botanical taxonomy and is partly justified. The range of beech is discontinuous, especially in the eastern part, and the hitherto genetic studies have documented that individual regional populations are highly differentiated. The range of F. orientalis is divided into three fragments: the Alborz, the Caucasus and Asia Minor (the last reaching into the southeastern Balkans), which are strongly differentiated from each other, both genetically and morphologically (Denk 1999a; Denk et al. 2002; Gömöry et al. 2007). Phylogeny reconstruction based on allozyme genes even indicated that the split between the Iranian population and the remaining two regional populations of F. orientalis occurred earlier than the divergence of F. sylvatica from F. orientalis (Gömöry et al. 2018). The range of F. sylvatica is restricted to Europe. However, Pleistocene glaciations led to a disruption of the distribution range and a subsequent differentiation of populations surviving in glacial refugia also in this species. Genetic studies documented at least four refugial areas serving as sources for the Holocene recolonization: Slovenia/Istria, southern Balkans, Apennine Peninsula and Massif Central, along with several other refugia which have not expanded at the onset of the Holocene (Magri et al. 2006). While most sutures between expanding refugial populations were obliterated, and a nearly continuous distribution has been restored in some regions (western Balkans, southeastern France), big rivers such as the Danube or the Po still represent barriers for migration by seeds. Contact of populations with different gene pools offers a potential for the formation of hybrid zones, where gene flow by pollen leads to hybridization between genetic lineages and introgression. Western Balkans is an example of such a hybrid zone, where gene frequencies change clinally from Slovenia down to western Turkey (Gömöry et al. 1999).
This study focused on the identification of the extent and direction of gene exchange among different parental lineages within putative hybrid zones. At the same time, we tried to identify SNP loci with patterns of introgression deviating from most of the genome as a consequence of adaptation or reproductive isolation.
Materials and methods
Experimental material
Material for this study was partly collected from natural populations (especially F. orientalis) and partly in international provenance experiments with European beech. Provenance trials included two Slovak plots of the experiment coordinated by the Institute of Forest Genetics of the J. H. von Thünen Institute in Grosshansdorf, Germany, which was established in two series in 1995 and 1998 with 2-year-old seedlings (100 and 32 provenances covering a major part of the distribution range of beech in Europe were planted at the localities Vrchdobroč and Tále, central Slovakia, respectively). At these plots, material from 84 and 15 provenances, respectively, was collected for this study. Another provenance trial serving as a source of material was a plot of the international experiment with southeast European provenances at the locality Medvednica, Croatia, where 19 provenances were sampled. The remaining material was collected in natural populations (Fig. 1; detailed information on populations is provided at https://doi.org/10.5281/zenodo.7794256).

Distribution of the analyzed specimens: natural populations (yellow), provenance experiment Vrchdobroč (red), provenance experiment Tále (black), provenance experiment Medvednica (light blue). (Color figure online)
DNA extraction and sequencing
Twigs with dormant buds and leaves were collected from trees growing in provenance trials and natural populations, respectively. Total genomic DNA was extracted from the silica-dried leaves or buds using a modified CTAB protocol following Doyle and Doyle (1987). DNA concentration and quality were assessed with a Qubit 4 Fluorometer (Thermo Fisher, Waltham MA, USA) and NanoDrop (Thermo Fisher, Waltham MA, USA), and samples were sent for sequencing to IGA Technology Services, Udine, Italy.
Double-digest restriction-site-associated DNA sequencing (ddRAD) was applied to reveal single-nucleotide polymorphisms (SNP) in the nuclear genome. ddRAD libraries were produced by IGA Technology Services using a custom protocol, with minor modifications with respect to Peterson’s double-digest restriction-site-associated DNA preparation (Peterson et al. 2012). 300 ng of genomic DNA was double-digested with 2.4 U of both SphI and MboI endonucleases (New England Biolabs) in 30 µl reaction supplemented with CutSmart Buffer and incubated at 37 °C for 90 min and then at 65 °C for 20 min. Fragmented DNA was ligated with 180 U of T4 DNA ligase (New England BioLabs, Ipswisch MA, USA) to 2.5 pmol of overhang barcoded adapter for rare cut sites and to 5 pmol of overhang barcoded adapter for frequent cut sites in 50 µl reaction incubated at 23 °C for 60 min and at 20 °C for 60 min followed by 20 min at 65 °C. Samples were pooled and beads-purified. Targeted fragments distribution was collected on BluePippin instrument (Sage Science, Beverly MA, USA) setting the range of 450–700 bp. Gel-eluted fraction was amplified with indexed primers using Phusion High-Fidelity PCR Master Mix (New England BioLabs) in final volume of 50 µl and subjected to the following thermal protocol: [95 °C, 3 min] – [95 °C, 30 s – 60 °C, 30 s – 72 °C, 45 s] × 10 cycles – [72 °C, 2 min]. After cleanup, libraries were checked with both Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, CA, USA) and Bioanalyzer DNA assay (Agilent Technologies, Santa Clara, CA, USA). Libraries were sequenced with 150 cycles in paired-end mode on NovaSeq 6000 instrument following the manufacturer’s instructions (Illumina, San Diego, CA, USA).
Data analysis
The initial bioinformatic analyses performed by IGA Technology Services consisted of demultiplexing of raw Illumina reads using the process_radtags utility included in Stacks v2.4 (Catchen et al. 2013). Alignment to the reference F. sylvatica genome (version 2 scaffold level assembly; Mishra et al. 2018; http://thines-lab.senckenberg.de/beechgenome/) using BWA-MEM (Li and Durbin 2009) with default parameters and selection of uniquely aligned reads (with a mapping quality > 4). Detection of the covered loci from the aligned reads using the gstacks program in Stacks v2.61. Filtering of detected loci using the populations program in Stacks v2.61, with option -R = 0.75 in order to retain only loci that are represented in at least the 75% of the whole metapopulation. To remove loci with possible technical errors, a cut-off max-obs-het = 0.8 was used in order to process a nucleotide site at a locus with observed heterozygosity at maximum of 80%. After the initial bioinformatic analyses in IGA Technology Services, we additionally filtered the SNPs based on several criteria: quality value Q > 13 (Q = 26 was the actual lowest value); markers with missing data in > 20% samples of any of the two species; samples with > 20% missing data; samples with read depth deviating ± 3 SD from average; < 5% minor allele frequency (MAF); and read depth further than ± 2 standard deviations from the mean. One randomly chosen SNP per contig was retained for further analysis. In total, 6789 SNP loci passed filtering; they are quite regularly distributed across the genome, maybe with the exception of chromosome 1 with a generally lower density of SNPs (Fig. 2a).

Density of SNPs per 1 Mbp distributed over 12 reference chromosomes: a all 6789 SNPs, b the 220 SNPs exhibiting allele frequency difference Δp > 0.85 between F. sylvatica and F. orientalis (b). Green points denote the positions of outlier SNPs. (Color figure online)
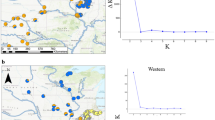
To reveal potential substructure, the Bayesian clustering algorithm implemented in STRUCTURE version 2.3.4 (Pritchard et al. 2000) was applied to cluster individuals into K groups, employing the admixture model with correlated allele frequencies. K values were tested from 1 to 10. A burn-in period of 50,000 iterations was followed by 100,000 iterations for estimation of the membership coefficients. Fifteen independent Markov chains were run for each K. The most likely number of clusters K was inferred from plotting log posterior probability against K, as well as the ΔK measure of Evanno et al. (2005) (Fig. 3a). Clusters were aligned using CLUMPP 1.1.2 (Jakobsson and Rosenberg 2007) with the Greedy algorithm testing 1000 random input orders.

Results of the Bayesian Structure analysis superimposed over the map of Europe and the eastern Mediterranean: a Assessment of the number of groups in the Bayesian analysis (Evanno et al. 2005), b inferred cluster membership proportions at K = 2, c cluster membership proportions at K = 3 (c), d cluster membership proportions at K = 5
Bayesian genomic clines (bgc; Gompert and Buerkle 2011) were used to analyze locus-specific ancestry using the bgc software (Gompert and Buerkle 2012). To define parental reference populations, we relied on earlier experience from allozyme and chloroplast DNA studies (Gömöry et al. 1999, 2007; Magri et al. 2006). For the putative hybrid zone in the Balkans, populations in Asia Minor were considered to be the F. orientalis parental population (12 individuals), while populations located close to the putative glacial refugium in Slovenia as well as those located farther to the North and East were considered pure F. sylvatica parental population (63 individuals). To be on the safe side, we checked whether the summary proportion of the clusters typical for either side of the hybrid zone under K = 5 exceeded 0.95; all samples in the parental zones fulfilled this criterion. In accordance with Denk (1999b) and Gömöry et al. (1999), beech populations in between (including Strandzha Mts. in eastern Bulgaria, usually classified as F. orientalis; c.f. Denk 1999a; Kandemir and Kaya 2009; Tzonev et al. 2006) were regarded as a broad introgression zone with increasing proportion of F. orientalis genes toward the southeast (admixed population; 39 individuals). For the putative hybrid zone between populations originated from glacial refugia in the Apennine peninsula and those originated from refugia in the foothills of Eastern Alps, the Po valley was considered a migration barrier (Magri et al. 2006) and gene flow (possibly bidirectional) was expected to have been accomplished through beechwoods in Western Alps. Therefore, the whole region comprising the Alps, their southern, western and northern foothills and northern Apennines (north of 43°N) were considered the admixed population (21 individuals), while the rest of the Apennine peninsula as well as samples from western Germany, the Netherlands and Luxembourg were taken as reference populations (10 and 27 specimens, respectively). Samples included in each of the hybrid zones and samples removed from the analysis because of missing data are listed in supplementary materials (Table S1).
Since each of these two hybrid zones consisted a subset of individuals from the total dataset, we again removed markers with < 5% minor allele frequency for each of these two sets of individuals. In the case of the Balkans hybrid zone 5038 SNPs were retained, while in the case of Apennine peninsula hybrid zone 4206 markers were left. Hybrid index was calculated using the bgc software (Gompert and Buerkle 2012). Analysis was run with 400,000 burn-in MCMC steps followed by 100,000 subsequent iterations and a thinning interval of 100. Genomic clines were assessed for SNPs with an allele frequency differential between parental populations Δp > 0.85; the distribution of these SNPs over the reference chromosomes was displayed using the R-package RIdeogram (Hao et al. 2020). Two parameters were of interest, namely genomic cline center (α) and cline rate (β) (Gompert and Buerkle 2011). Cline center indicates the direction of introgression, where outliers indicate excess ancestry from one or the other reference population. Cline rate is an indicator of the progression of SNP frequency change; a steeper cline indicated by positive values means a sharp transition of ancestry from one reference population to the other. Point estimates and credible intervals for parameters were generated using the software estpost (Gompert and Buerkle 2012). Outliers were considered significant if the 95% credible intervals for α or β did not overlap zero (Gompert and Buerkle 2011). To assess the sensitivity of the analysis of genomic clines, we performed resampling with replacement of individuals within each group (two reference populations and hybrid zone); 100 bootstrap replicates were created using an ad hoc R-script and the proportions of significant values were used as a support of the calculated genomic cline parameters. Inter-lineage heterozygosity (He, the proportion of loci where alleles originate from different parental populations) was also calculated using bgc. However, as the authors recommend calculation of He only if most loci are fixed or nearly fixed in the parental populations, the dataset was reduced to 220 SNPs, for which the allele frequency differential between parental populations exceeded 0.85. As this criterion was not fulfilled for any SNP in the Alpine hybrid zone, He was calculated only for the Balkans.
Basic diversity and differentiation statistics (nucleotide diversity Pi, Nei 1987; coefficient of differentiation FST, Hudson et al. 1992; pairwise net number of nucleotide substitutions per site between populations Da, Nei 1987) were calculated using DnaSP v. 6.12 (Rozas et al. 2017) for the subspecies, the Balkan hybrid zone and the Alpine hybrid zone.
Results
Population Structure
Basic summary statistics showed that nucleotide diversity is higher in F. orientalis compared to F. sylvatica (Table 1). The subspecies are also strongly differentiated (FST = 0.4722). However, the differentiation among regional populations of F. orientalis (Asia Minor, Caucasus, Alborz) is almost equally strong (FST = 0.4256). This pattern was partly confirmed also in the Balkan hybrid zone: nucleotide diversity was the highest in the reference F. orientalis population, but differentiation between parental populations belonging to different subspecies was substantially weaker (FST = 0.2677); it must be reminded that the reference F. orientalis population comprised only Turkish samples, excluding more divergent Caucasian and Iranian populations. Expectedly, the differentiation between the Apennine and Central European lineages in the Alpine hybrid zone was the weakest (FST = 0.0276). The pairwise net numbers of nucleotide substitutions per site exhibited a similar pattern. The population in Asia Minor is strongly divergent, while the distance between the Alborz and the Caucasus is not so pronounced. In both hybrid zones, parental populations showed the highest divergence, while admixture zones were expectedly less differentiated.
The ΔK measure after Evanno et al. (2005) clearly peaked at K = 2. However, a secondary peak at K = 5 and the course of the log posterior probability showing a decrease after K = 6 indicate that a substructure with higher numbers of clusters is also possible (Fig. 3a). The distribution of cluster proportions at K = 2 corresponded partly with the current taxonomical view of two subspecies: one cluster clearly dominated in Europe (including western Balkans), the other one in the Caucasus and the Alborz Mts, while populations classified as F. orientalis in Asia Minor and eastern Bulgaria showed a mixture of both gene pools (Fig. 3b). At K = 3, F. sylvatica remained a separate group, while F. orientalis was split into western Anatolian and east Bulgarian specimens on one side, Caucasian and Iranian specimens on the other side, and populations from the eastern part of the Pontic range and Amanus Mts. in southern Turkey containing a mixture of both clusters (Fig. 3c). With higher numbers of clusters, this Structure was retained, and additional gene pools appeared within the range of F. sylvatica (Fig. 3d), namely a cluster (black pies) with proportions of 8–27% in the specimens from the Apennines (but also dispersed across the southern and western Balkans, southern Alps and the Carpathians), and another cluster present in western Europe (Spain, France, Switzerland, Germany), but again with proportions not exceeding 15% (green pies). Increasing the number of clusters K to 6 or more did not bring any interpretable pattern (data not shown). In contrast to expectations relying on earlier studies (Gömöry et al. 2007; Kurz et al. 2023), the population from the Alborz Mts. (Iran) was not distinguished from the remaining F. orientalis regional populations at any K. In one specimen from Bosnia (Tomislavgrad), the admixture proportion (Q-value) of the cluster predominating in Turkey and eastern Bulgaria (F. orientalis) was unusually high, in spite of being located within the range of F. sylvatica. As Bosnia had been a part of the Ottoman Empire until 1905, we suspect the parental population of this specimen to be artificially introduced; therefore, the sample was excluded from the assessment of genomic clines.
Hybrid zones
In the putative Balkan hybrid zone, hybrid index changes very abruptly at the boundary between F. sylvatica and F. orientalis in eastern Bulgaria. The four specimens classified as F. orientalis showed hybrid indices close to one, i.e., absolute dominance of F. orientalis genomes (hi > 0.85). On the other hand, in the remaining Bulgarian and western Balkan beech populations classified as F. sylvatica (or F. moesiaca) hybrid indices range between 0.002 and 0.412, with a clear trend of increase toward the boundary with F. orientalis (Fig. 4a). Interspecific heterozygosities showed a partly similar pattern (Fig. 5): High heterozygosities were observed in the Bulgarian F. sylvatica populations (especially those geographically proximate to the range of F. orientalis), and rapidly decrease northwestwards, being close to zero in the Croatian specimens. However, low interspecific heterozygosity was also observed in the Bulgarian F. orientalis.

Distribution of hybrid indices across the hybrid zones: a Balkan hybrid zone, b Alpine hybrid zone. White and black diamonds: parental reference populations

Distribution of inter-lineage heterozygosity across the Balkan hybrid zone
Out of 5038 SNPs studied in the Balkan hybrid zone, 220 showed an allele frequency differential between reference populations of more than 0.85. Their distribution over the reference chromosomes was quite irregular (Fig. 2b); none was found on the longest chromosome 1 and just 4 on chromosome 5, while most of them were concentrated on chromosomes 9, 11 and 12 (33, 30 and 24, respectively). Seven SNPs exhibited genomic cline center α significantly differing from zero. Excess F. orientalis ancestry (α < 0) was identified for six SNPs; bootstrap support for α was over 80% in all cases. In addition, another SNP showed significantly outlying cline center with the highest bootstrap support (95%), but allele frequency differential in this case was 0.73 only. Excess F. sylvatica ancestry (α > 0) was found for one SNP, but the bootstrap support was quite small in this case (56%). Genomic cline rate β was non-significant for all loci, indicating a uniform genomic cline steepness across the genome (Table 2). Outlier SNPs were distributed across five reference chromosomes; three of them are localized in exons, two in introns, and three outside a gene—in these cases, Table 2 gives the localization and description of the most proximate genes.
In the hybrid zone crossing the Alps, the pattern is quite different. Even in the northernmost specimens from the Apennines hybrid index does not exceed 0.5 (Fig. 4b). Relatively high values (hi > 0.25) were also observed in the southern and southwestern foothills of the Alps and rapidly decreased toward southern Germany. Allelic frequency differential between reference populations does not exceed 0.65 for any locus; therefore, interspecific heterozygosity (in this case ‘inter-lineage heterozygosity) was not calculated. Even when we gave up the Δp > 0.85 criterion for this hybrid zone, none of genome cline centers (α) or rates (β) differed significantly from zero.
Discussion
Population Structure
Whatever number of clusters (K) was chosen for the Bayesian analysis of population Structure, the outcomes diverged from earlier studies relying on chloroplast, allozyme or microsatellite markers. The most striking difference is a lack of differentiation between the Iranian and the Caucasian populations. All studies covering the whole range of F. orientalis, both allozyme-based (Gömöry et al. 2007) and nuclear microsatellites-based (Bijarpasi et al. 2020; Budde et al. 2023; Kurz et al. 2023) clearly separated populations from the Alborz Mts. from all remaining F. orientalis. The same applies to morphometry based on leaf and fruit traits (Denk 1999a, b; Denk et al. 2002). A phylogeny simulation relying on approximate Bayesian computations and using allozyme data even suggested the Iranian beech to be the earliest diverged regional subpopulation within the whole complex of western Eurasian beeches (Gömöry et al. 2018). Another contrast to earlier studies is a lack of clear differentiation between east-Anatolian and Caucasian beech populations, observed both using allozymes (Gömöry et al. 2007) and microsatellite markers (Kurz et al. 2023). Differentiation statistics conformed well with the outcomes of the Structure analysis: A strong differentiation was observed among F. orientalis regional populations (comparable to differentiation between subspecies), suggesting that the more distant beech populations in the Caucasus and the Alborz did not contribute to the recent gene exchange with F. sylvatica; at the same time, these two regions do not strongly differ from each other. In any case, such a strong differentiation within F. orientalis implies that the population Structure best supported by ΔK (Evanno et al. 2005) is misleading, while the Structures at higher numbers of clusters than K = 2 better reflect biological reality.
In Europe, the results of the Structure analysis conformed better with what is currently known about the Quaternary history of beech (Giesecke et al. 2007; Magri et al. 2006; Magri 2008; Vettori et al. 2004). At K > 3, a substructure within F. sylvatica was revealed. Even though one main cluster remained predominant across the distribution range, two other clusters appeared at low frequencies, one shared by the Balkan and Italian populations and another one represented north and west of the Alps. Sharing a substantial portion of the genome is not surprising, as all European beech populations belong to the same subspecies, which has been distributed across a major part of the continent during most of the Pleistocene (Denk and Grimm 2009), and the current differentiation is mainly due to range fragmentation during the last glacial (Magri et al. 2006). Most of the range was colonized from a single refugium localized in eastern foothills of the Alps (Magri et al. 2006). The presence of a specific gene pool in the Apennine Peninsula shared with southern Balkans may reflect both adaptation to the Mediterranean climate and gene exchange during earlier phases of the Pleistocene. Sharing of gene pools between the southern European peninsulas was found also in other plants (Musacchio et al. 2006). Similarly, the admixture of another gene pool in the western part of the F. sylvatica range is probably related to the existence of glacial refugia in the Massif Central and the Rhône valley (Magri 2008).
Gene exchange between divergent genetic lineages
Even when the outcomes of the Bayesian population Structure analysis did not properly show it, there is enough paleobotanical and genetic evidence for the existence of a number of highly divergent genetic lineages within F. sylvatica s.l., potentially connected by gene flow. As shown by Postolache et al. (2021), the distribution of geographical barriers to gene flow (mountain ranges, seas) does not fully overlap with potential boundaries between refugial areas; in spite of this, hybrid zones may be much more numerous than just the two addressed by this study. However, a limited number of available samples and irregular sampling density (especially within the range of F. orientalis) forced us to focus on just the Balkans and the Alpine area. The results of the Structure analysis were partly disappointing from the point of view of the definition of reference parental populations and delineation of hybrid zones for the study of introgression. For this purpose, we finally decided to combine the patterns shown by Structure with earlier knowledge about the distribution of chloroplast haplotypes or lineages defined by nuclear markers (Vettori et al. 2004; Magri et al. 2006; Gömöry et al. 2007).
The information about the preferential direction of inter-taxon gene flow is not without ambiguity. A study in Greek and Turkish beech populations (Müller et al. 2019) suggested a very narrow hybrid zone. The study identified only one population with a noteworthy level of admixture, namely a Greek F. orientalis population, while in all F. sylvatica populations admixture was negligible; this implies gene flow directed primarily into F. orientalis. The comparison of the distributions of hybrid index and interspecific heterozygosity in our study also indicates that in the southeastern Balkans, interspecific gene flow seems to be quite limited and asymmetric, but with much more intense gene flow from F. orientalis to F. sylvatica than vice versa. This confirms our earlier observations with allozyme markers, when frequencies of several alleles changed abruptly between F. orientalis and F. sylvatica at very short distances and subsequently changed clinally toward central Europe (Gömöry et al. 1999, 2007). Hypothetically, a phenological gap may hamper gene exchange in this area. In fact, we have no exact data about differentiation in floral phenology among beech populations resulting from direct observations. However, in beech, the development of vegetative and generative Structures within the annual cycle is synchronized: Male and female flowers appear concurrently with bud flushing (Packham et al. 2012). Spring phenology observations are thus relevant for assessing the floral phenology overlap between beech subspecies. Several provenance experiments contain a mixture of F. sylvatica and F. orientalis provenances. In general, they show that F. orientalis provenances belong to early flushers (Robson et al. 2011; Gömöry and Paule 2011); however, delay in flowering of eastern F. sylvatica provenances is generally small. Recently, Kurz et al. (2023) assessed spring phenology of the introduced F. orientalis individuals admixed in German and Swiss beechwoods and found a phenological gap of more than 4 days. Such temporal difference is too small to hamper gene flow from F. orientalis to F. sylvatica in mixed stands, where both subspecies grow under identical climate. However, it remains questionable to what extent these observations are relevant to the potential of gene exchange between subspecies in natural populations. Even though in the contact zone (eastern Bulgaria and Greece), the stands of both subspecies grow close to each other, F. orientalis occurs typically at low to medium altitudes in warm and dry habitats related to deciduous Quercus forests, while F. sylvatica populations grow at higher elevations in moderate to cool climates (Bergmeier and Dimopoulos 2001). A contrasting floristic composition is also a strong indicator of a difference in climate requirements (Tzonev et al. 2006). Temporal shift of flowering between subspecies caused by climatic contrast linked along with a lack of stepping stones for gene flow may thus be much larger in situ than in artificially established stands. Given a slight protogyny of beech (Nielsen and Schaffalitzky de Muckadell 1954), earlier male flowering of F. orientalis induced by warmer microclimate combined with protogynous mating in the proximate F. sylvatica populations may explain the observed unidirectional gene flow. Excess F. orientalis ancestry was found in six cases, also confirming that gene exchange between subspecies is not equal in both directions. Asymmetry of hybridization rates is not exceptional in the Fagaceae, it was also observed in mixed populations of Quercus robur and Q. petraea. Different fertility rates were also observed in controlled crosses; unequal success of artificial crossing in different directions suggests genetic incompatibilities (Aas 1990; Steinhoff 1993), as factors relevant in natural populations such as phenological gap, differences in pollen cloud density, spatial setup, etc., are eliminated in this case. For beech, no such direct evidence coming from crossing experiments is available. Experience with the occurrence of hybrids in the plantations of F. orientalis in central Europe gives just partial information about the frequency and direction of hybridization. Kurz et al (2023) identified a higher proportion of hybrids in the vicinity of F. orientalis maternal trees than under F. sylvatica trees, but they admit that this may be due to a dominance of European beech in the pollen cloud. Budde et al. (2023) considered interspecific gene flow to be bidirectional, but most hybrids identified based on nuclear nSSRs in their study had chloroplast haplotypes of F. sylvatica, indicating the same asymmetry as that observed in our study.
Nevertheless, genetic isolation remains an alternative explanation of the observed introgression patterns. The outliers in genomic clines imply the existence of some type of genetic reproductive barriers. We found eight loci with genomic cline centers significantly shifted compared to neutral expectations, out of which three SNPs are localized in exons, thus potentially affecting functionality of gene products. The functional annotation of loci is known, but detecting the mechanisms underlying their contribution to reproductive isolation between subspecies is hardly possible. Even if we knew the exact function of the expressed proteins, it would be difficult to assess how exactly they contribute to physiological traits affecting fitness of inter-lineage hybrids. Irregular distribution of the outlier SNPs and their concentration on chromosomes showing high numbers of highly differentiated loci suggest that certain regions of the beech genome may resist introgression more than others (Mallet 2005; Gompert and Buerkle 2011).
In the case of the Alpine area, it is difficult to talk about a true hybrid zone. The populations on both sides of the Po valley are classified as the same subspecies. Even when both nuclear and chloroplast markers showed that the genetic lineage (more exactly, the mixture of genetic lineages) spreading across the Apennine Peninsula is differentiated from the rest of the distribution range (Magri et al. 2006; Vettori et al. 2004), there is no indication that they are reproductively isolated, even partly.
Implications for forestry
Under the progressing climate change, European beech has become a species with uncertain future because of a limited drought tolerance (Leuschner 2020; Martinez del Castillo et al. 2022; Rasztovits et al. 2014). Transfer of putatively pre-adapted forest reproductive materials from southern sites (assisted migration) and future introgression of established populations with the local ones (assisted gene flow) are often suggested as mitigation measures (Aitken and Whitlock 2013). In the case of beech, the southern European peninsulas (Iberian, Apennine, Balkan) or even F. orientalis are suggested as donor regional populations (Mellert and Šeho 2022; Kurz et al. 2023; Petrík et al. 2023). The hitherto experience from provenance experiments or plantations of F. orientalis in central Europe justifies this concept at least partly; on most sites, the performance of southern provenances or F. orientalis in large-scale international experiments or in the plantations is similar or at least not substantially worse compared to the local ones (Alía et al. 2011; Bogunović et al. 2020). Nevertheless, assisted gene flow requires that the introduced provenances hybridize with the local beech. Along with the first information about the extent of hybridization and survival of hybrids in juvenile stages provided recently by Kurz et al. (2023) and Budde et al. (2023), our results suggest that hybridization and introgression between divergent lineages within F. sylvatica s.l. have been ongoing since the contact between them was established, without detrimental effects on fitness.
Availability of data and material
Geographical coordinates of specimens, SNPs produced by ddRAD sequencing and annotation of SNP loci are available as open-access data in the Zenodo repository at https://doi.org/10.5281/zenodo.8388693.
References
Aas G (1990) Kreuzbarkeit und Unterscheidung von Stiel- und Traubeneiche. AFZ/der Wald 45:219–221
Aitken SN, Whitlock MC (2013) Assisted gene flow to facilitate local adaptation to climate change. Annu Rev Ecol Evol S 44:367–388. https://doi.org/10.1146/annurev-ecolsys-110512-135747
Akeroyd JR (1993) Fagus L. In: Tutin TG, Heywood VH, Burges NA, Valentine DH, Moore DM (eds) Flora Europaea, Volume 1 Psilotaceae to Platanaceae. Cambridge University Press, Cambridge, p 72
Alía R, Božič G, Gömöry D, Huber G, Rasztovits E, von Wühlisch G (2011) The survival and performance of beech provenances over a Europe-wide gradient of climate. In: Aranda I (eds.) Monografias INIA: Serie Forestale No. 22. Genetic Resources of European Beech (Fagus sylvatica L.). Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria, Madrid, pp 115–126.
Arnold ML, Hodges SA (1995) Are natural hybrids fit or unfit relative to their parents? Trends Ecol Evol 10:67–71. https://doi.org/10.1016/S0169-5347(00)88979-X
Arnold ML, Martin NH (2010) Hybrid fitness across time and habitats. Trends Ecol Evol 25:530–536. https://doi.org/10.1016/j.tree.2010.06.005
Arnold M, Ballerini E, Brothers A (2012) Hybrid fitness, adaptation and evolutionary diversification: lessons learned from Louisiana Irises. Heredity 108:159–166. https://doi.org/10.1038/hdy.2011.65
Barton NH (1979) The dynamics of hybrid zones. Heredity 43:341–359. https://doi.org/10.1038/hdy.1979.87
Bergmeier E, Dimopoulos P (2001) Fagus sylvatica forest vegetation in Greece: syntaxonomy and gradient analysis. J Veg Sci 12:109–126. https://doi.org/10.1111/j.1654-1103.2001.tb02622.x
Bijarpasi MM, Müller M, Gailing O (2020) Genetic diversity and structure of oriental and European beech populations from Iran and Europe. Silvae Genet 69:55–62. https://doi.org/10.2478/sg-2020-0008
Bogunović S, Bogdan S, Lanšćak M, Ćelepirović N, Ivanković M (2020) Use of a common garden experiment in selecting adapted beech provenances for artificial stand restoration. SE Eur for 11:1–10. https://doi.org/10.15177/seefor.20-07
Budde KB, Hötzel S, Müller M, Samsonidze N, Papageorgiou AC, Gailing O (2023) Bidirectional gene flow between Fagus sylvatica L. and F. orientalis Lipsky despite strong genetic divergence. For Ecol Manage 537:120947. https://doi.org/10.1016/j.foreco.2023.120947
Burke JM, Arnold ML (2001) Genetics and the fitness of hybrids. Annu Rev Genet 35:31–52. https://doi.org/10.1146/annurev.genet.35.102401.085719
Capblancq T, Després L, Mavárez J (2020) Genetic, morphological and ecological variation across a sharp hybrid zone between two alpine butterfly species. Evol Appl 13:1435–1450. https://doi.org/10.1111/eva.12925
Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA (2013) Stacks: an analysis tool set for population genomics. Mol Ecol 22:3124–3140. https://doi.org/10.1111/mec.12354
Czúcz B, Gálhidy L, Mátyás C (2011) Present and forecasted xeric climatic limits of beech and sessile oak distribution at low altitudes in Central Europe. Ann for Sci 68:99–108. https://doi.org/10.1007/s13595-011-0011-4
Dagilis AJ, Kirkpatrick M, Bolnick DI (2019) The evolution of hybrid fitness during speciation. PLoS Genet 15:e1008125. https://doi.org/10.1371/journal.pgen.1008125
Denk T (1999a) The taxonomy of Fagus in western Eurasia, 1: Fagus sylvatica subsp. orientalis (= F. orientalis). Feddes Repert 110:177–200. https://doi.org/10.1002/fedr.19991100305
Denk T (1999b) The taxonomy of Fagus in western Eurasia, 2: Fagus sylvatica subsp. sylvatica. Feddes Repert 110:381–412. https://doi.org/10.1002/fedr.19991100510
Denk T, Grimm GW (2009) The biogeographic history of beech trees. Rev Palaeobot Palynol 158:83–100. https://doi.org/10.1016/j.revpalbo.2009.08.007
Denk T, Grimm G, Stögerer K, Langer M, Hembelen V (2002) The evolutionary history of Fagus in western Eurasia: the evidence from genes, morphology and the fossil record. Plant Syst Evol 232:213–236. https://doi.org/10.1007/s006060200044
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19:11–15
Ellison CK, Burton RS (2008) Interpopulation hybrid breakdown maps to the mitochondrial genome. Evolution 62:631–663. https://doi.org/10.1111/j.1558-5646.2007.00305.x
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software Structure: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Giesecke T, Hickler T, Kunkel T, Sykes MT, Bradshaw RHW (2007) Towards an understanding of the Holocene distribution of Fagus sylvatica L. J Biogeogr 34:118–131. https://doi.org/10.1111/j.1365-2699.2006.01580.x
Gömöry D, Paule L (2011) Trade-off between height growth and spring flushing in common beech (Fagus sylvatica L.). Ann For Sci 68:975–984. https://doi.org/10.1007/s13595-011-0103-1
Gömöry D, Paule L, Brus R, Zhelev P, Tomović Z, Gračan J (1999) Genetic differentiation and phylogeny of beech on the Balkan Peninsula. J Evol Biol 12:746–754. https://doi.org/10.1046/j.1420-9101.1999.00076.x
Gömöry D, Paule L, Vyšný J (2007) Patterns of allozyme variation in western-Eurasian beeches. Bot J Linn Soc 154:165–174. https://doi.org/10.1111/j.1095-8339.2007.00666.x
Gömöry D, Paule L, Mačejovský V (2018) Phylogeny of beech in western Eurasia as inferred by approximate Bayesian computation. Acta Soc Bot Pol 87:3582. https://doi.org/10.5586/asbp.3582
Gompert Z, Buerkle CA (2011) Bayesian estimation of genomic clines. Mol Ecol 20:2111–2127. https://doi.org/10.1111/j.1365-294X.2011.05074.x
Gompert Z, Buerkle CA (2012) BGC: software for Bayesian estimation of genomic clines. Mol Ecol Resour 12:1168–1176
Greuter W, Burdet HM (1981) Fagus sylvatica ssp. orientalis. Willdenowia 11:271–280
Gulisashvili VZ, Makhatadze LB, Prilipko LI (1975) Rastitel’nost’ Kavkaza. Nauka, Moskva
Hamilton JA, Lexer L, Aitken SN (2013) Differential introgression reveals candidate genes for selection across a spruce (Picea sitchensis × P. glauca) hybrid zone. New Phytol 197:927–938. https://doi.org/10.1111/nph.12055
Hao Z, Lv D, Ge Y, Shi J, Weijers D, Yu G, Chen J (2020) RIdeogram: drawing SVG graphics to visualize and map genome-wide data on the idiograms. PeerJ Comput Sc 6:e251. https://doi.org/10.7717/peerj-cs.251
Harrison RG (1993) Hybrids and hybrid zones: historical perspective. In: Harrison RG (ed) Hybrid zones and the evolutionary process. Oxford University Press, Oxford, pp 3–12
Holeksa J, Saniga M, Szwagrzyk J, Czerniak M, Staszynska K, Kapusta P (2009) A giant tree stand in the West Carpathians – An exception or a relic of formerly widespread mountain European forests? For Ecol Manage 257:1577–1585. https://doi.org/10.1016/j.foreco.2009.01.008
Hudson RR, Slatkin M, Maddison WP (1992) Estimation of levels of gene flow from DNA sequence data. Genetics 132:583–589. https://doi.org/10.1093/genetics/132.2.583
Jakobsson M, Rosenberg NA (2007) CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806. https://doi.org/10.1093/bioinformatics/btm233
Johansen-Morris AD, Latta RG (2006) Fitness consequences of hybridization between ecotypes of Avena barbata: Hybrid breakdown, hybrid vigor, and transgressive segregation. Evolution 60:1585–1595. https://doi.org/10.1111/j.0014-3820.2006.tb00503.x
Johnson JR, Fitzpatrick BM, Shaffer HB (2010) Retention of low-fitness genotypes over six decades of admixture between native and introduced tiger salamanders. BMC Evol Biol 10:147. https://doi.org/10.1186/1471-2148-10-147
Kandemir G, Kaya Z (2009) EUFORGEN Technical guidelines for genetic conservation and use of oriental beech (Fagus orientalis). Bioversity International, Rome, Italy
Keller SR, Taylor DR (2010) Genomic admixture increases fitness during a biological invasion. J Evol Biol 23:1720–1731. https://doi.org/10.1111/j.1420-9101.2010.02037.x
Keren S, Motta R, Govedar Z, Lučić R, Medarević M, Diaci J (2014) Comparative structural dynamics of the Janj mixed old-growth mountain forest in Bosnia and Herzegovina: are conifers in a long-term decline? Forests 5:1243–1266. https://doi.org/10.3390/f5061243
Kurz M, Kölz A, Gorges J, Pablo Carmona B, Brang P, Vitasse Y, Kohler M, Rezzonico F et al (2023) Tracing the origin of Oriental beech stands across Western Europe and reporting hybridization with European beech – Implications for assisted gene flow. For Ecol Manag 531:120801. https://doi.org/10.1016/j.foreco.2023.120801
Latta RG, Gardner KM, Johansen-Morris AD (2007) Hybridization, recombination, and the genetic basis of fitness variation across environments in Avena barbata. Genetica 129:167–177. https://doi.org/10.1007/s10709-006-9012-x
Leibundgut H (1982) Europäische Urwälder der Bergstufe. Haupt Verlag AG, Bern, Switzerland
Leinonen PH, Remington DL, Savolainen O (2011) Local adaptation, phenotypic differentiation, and hybrid fitness in diverged natural populations of Arabidopsis lyrata. Evolution 65:90–107. https://doi.org/10.1111/j.1558-5646.2010.01119.x
Leuschner C (2020) Drought response of European beech (Fagus sylvatica L.) - a review. Perspect Plant Ecol 47:125576. https://doi.org/10.1016/j.ppees.2020.125576
Lexer C, Buerkle CA, Joseph JA, Heinze B, Fay MF (2007) Admixture in European Populus hybrid zones makes feasible the mapping of loci that contribute to reproductive isolation and trait differences. Heredity 98:74–84. https://doi.org/10.1038/sj.hdy.6800898
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. https://doi.org/10.1093/bioinformatics/btp324
Lucek K, Roy D, Bezault E, Sivasundar A, Seehausen O (2010) Hybridization between distant lineages increases adaptive variation during a biological invasion: stickleback in Switzerland. Mol Ecol 19:3995–4011. https://doi.org/10.1111/j.1365-294X.2010.04781.x
Magri D (2008) Patterns of post-glacial spread and the extent of glacial refugia of European beech (Fagus sylvatica). J Biogeogr 35:450–463. https://doi.org/10.1111/j.1365-2699.2007.01803.x
Magri D, Vendramin GG, Comps B, Dupanloup I, Geburek T, Gömöry D, Latalowa M, Litt T, Paule L, Roure JM, Tantau I, van der Knaap WO, Petit RJ, de Beaulieu J-L (2006) A new scenario for the Quaternary history of European beech populations: palaeobotanical evidence and genetic consequences. New Phytol 171:199–222. https://doi.org/10.1111/j.1469-8137.2006.01740.x
Mallet J (2005) Hybridization as an invasion of the genome. Trends Ecol Evol 20:229–237. https://doi.org/10.1016/j.tree.2005.02.010
Mallet J (2007) Hybrid speciation. Nature 446(7133):279–283. https://doi.org/10.1038/nature05706
Martinez del Castillo E, Zang CS, Buras A, Hacket-Pain A, Esper J, Serrano-Notivoli R, Hartl C et al (2022) Climate-change-driven growth decline of European beech forests. Commun Biol 5:163. https://doi.org/10.1038/s42003-022-03107-3
Mayer H, Aksoy H (1986) Wälder der Türkei. Gustav Fischer, Stuttgart
Mayr E (1942) Systematics and the origin of species. Columbia University Press, New York
Mellert KH, Šeho M (2022) Suitability of Fagus orientalis Lipsky at marginal Fagus sylvatica L. forest sites in Southern Germany. iForest 15:417–423. https://doi.org/10.3832/ifor4077-015
Miller M, Song Q, Shi X, Juenger TE, Chen ZJ (2015) Natural variation in timing of stress-responsive gene expression predicts heterosis in intraspecific hybrids of Arabidopsis. Nat Commun 6:7453. https://doi.org/10.1038/ncomms8453
Mishra B, Gupta DK, Pfenninger M, Hickler T, Langer E, Nam B, Paule J, Sharma R, Ulaszewski B, Warmbier J, Burczyk J, Thines M (2018) A reference genome of the European beech (Fagus sylvatica L.). GigaScience 7:063. https://doi.org/10.1093/gigascience/giy063
Mobayen S, Tregubov V (1970) La carte de la végétation naturelle de l’Iran. Université de Tehran, Tehran
Müller M, Lopez PA, Papageorgiou AC, Tsiripidis I, Gailing O (2019) Indications of genetic admixture in the transition zone between Fagus sylvatica L. and Fagus sylvatica ssp. orientalis Greut. & Burd. Diversity 11:90. https://doi.org/10.3390/d11060090
Musacchio A, Pellegrino G, Cafasso D, Widmer A, Cozzolino S (2006) A unique A. palustris lineage across tine Otranto strait: botanical evidence for a past land-bridge? Plant Syst Evol 262:103–111. https://doi.org/10.1007/s00606-006-0469-y
Nei M (1987) Molecular Evolutionary Genetics. Columbia University Press, New York
Nielsen PC, Schaffalitzky de Muckadell M (1954) Flower observations and controlled pollinations in Fagus. Silvae Genet 3:6–17
Packham JR, Thomas PA, Atkinson MD, Degen T (2012) Biological flora of the British Isles: Fagus sylvatica. J Ecol 100:1557–1608. https://doi.org/10.1111/j.1365-2745.2012.02017.x
Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE (2012) Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE 7:e37135. https://doi.org/10.1371/journal.pone.0037135
Petrík P, Grote R, Gömöry D, Kurjak D, Petek-Petrik A, Lamarque LJ, Sliacka Konôpková A, Mukarram M, Debta H, Fleischer P Jr (2023) The role of provenance for the projected growth of juvenile European beech under climate change. Forests 14:26. https://doi.org/10.3390/f14010026
Postolache D, Oddou-Muratorio S, Vajana E, Bagnoli F, Guichoux E, Hampe A, Le Provost G, Lesur I, Popescu F, Scotti I, Piotti A, G.G. Vendramin GG, (2021) Genetic signatures of divergent selection in European beech (Fagus sylvatica L.) are associated with the variation in temperature and precipitation across its distribution range. Mol Ecol 30:5029–5047. https://doi.org/10.1111/mec.16115
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure from multilocus genotype data. Genetics 155:945–959. https://doi.org/10.1093/genetics/155.2.945
Rasztovits E, Berki I, Mátyás C, Czimber K, Pötzelsberger E, Móricz N (2014) The incorporation of extreme drought events improves models for beech persistence at its distribution limit. Ann for Sci 71:201–210. https://doi.org/10.1007/s13595-013-0346-0
Rieseberg LH, Whitton J, Gardner K (1999) Hybrid zones and the genetic architecture of a barrier to gene flow between two sunflower species. Genetics 152:713–727. https://doi.org/10.1093/genetics/152.2.713
Robson TM, Alía R, Božič G, Clark J, Forsteuter M, Gömöry D, Liesebach M, Mertens P, Rasztovits E, Zitová M, von Wühlisch G (2011) The timing of leaf flush in European beech (Fagus sylvatica L.) saplings. . In: Aranda I (ed) Monografias INIA: Serie Forestal No. 22. Genetic Resources of European Beech (Fagus sylvatica L.). Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria, Madrid, pp. 61–80
Rosas U, Barton NH, Copsey L, de Reuille PB, Coen E (2010) Cryptic variation between species and the basis of hybrid performance. PLoS Biol 8:e1000429. https://doi.org/10.1371/journal.pbio.1000429
Rozas J, Ferrer-Mata A, Sánchez-Del Barrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, Sánchez-Gracia A (2017) DnaSP 6: DNA sequence polymorphism analysis of large datasets. Mol Biol Evol 34:3299–3302. https://doi.org/10.1093/molbev/msx248
Sambatti JBM, Ortiz-Barrientos D, Baack EJ, Rieseberg LH (2008) Ecological selection maintains cytonuclear incompatibilities in hybridizing sunflowers. Ecology Lett 11:1082–1091. https://doi.org/10.1111/j.1461-0248.2008.01224.x
Steinhoff S (1993) Results of species hybridization with Quercus robur L. and Quercus petraea (Matt) Liebl. Ann Sci for 50:137–143. https://doi.org/10.1051/forest:19930713
Tabari M, Fayaz P, Espahbodi K, Staelens J, Nachtergale L (2005) Response of oriental beech (Fagus orientalis Lipsky) seedlings to canopy gap size. Forestry 78:443–450. https://doi.org/10.1093/forestry/cpi032
Tzonev R, Dimitrov M, Chytrý M, Roussakova V, Dimova D, Gussev C, Pavlov D, Vulchev V, Vitkova A, Gogoushev G, Nikolov I, Borisova D, Ganeva A (2006) Beech forest communities in Bulgaria. Phytocenologia 36:247–279. https://doi.org/10.1127/0340-269X/2006/0036-0247
Vettori C, Vendramin GG, Anzidei M, Pastorelli R, Paffetti D, Giannini R (2004) Geographic distribution of chloroplast variation in Italian populations of beech (Fagus sylvatica L.). Theor Appl Genet 109:1–9. https://doi.org/10.1007/s00122-004-1609-9
WFO (2023) Fagus orientalis Lipsky. The World Flora Online. http://www.worldfloraonline.org/taxon/wfo-0000966486. Accessed 02 Apr 2023
Acknowledgements
A substantial part of the material was collected in an international provenance experiment with European beech established through the realization of the project European Network for the Evaluation of the Genetic Resources of Beech for Appropriate Use in Sustainable Forestry Management (AIR3-CT94-2091) under the coordination of H.-J. Muhs and G. von Wühlisch. The authors thank P. Salehi-Shanjani, D. Ballian, G.S. Dzhamirzoev, Z. Manvelidze, S. Litvinskaya, S.N. Sannikov, A.C. Papageorgiou and V. Otte for providing samples from natural populations and A. Gavranović for help with sampling in the provenance trial Medvednica. The study was supported by a research grant of the Slovak Grant Agency for Science VEGA 1/0029/20 and the Shota Rustaveli National Science Foundation (project #FR17_621). Technical assistance of G. Arendášová is greatly appreciated. We also thank K. Willingham for linguistic revision.
Funding
Open access funding provided by The Ministry of Education, Science, Research and Sport of the Slovak Republic in cooperation with Centre for Scientific and Technical Information of the Slovak Republic. This study was funded by Slovak Grant Agency for Science, grant no. VEGA 1/0029/20, and Shota Rustaveli National Science Foundation, project #FR17_621.
Author information
Authors and Affiliations
Contributions
MH was involved in methodology, data analysis, data curation and visualization. DK was involved in methodology, investigation and data curation. LP was involved in conceptualization and resources. PZ, HS, MI, NG and JP were involved in resources. DG was involved in conceptualization, funding acquisition, writing—original draft and project administration. All authors commented and revised the original draft.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interests.
Additional information
Communicated by Oliver Gailing.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hrivnák, M., Krajmerová, D., Paule, L. et al. Are there hybrid zones in Fagus sylvatica L. sensu lato?. Eur J Forest Res 143, 451–464 (2024). https://doi.org/10.1007/s10342-023-01634-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10342-023-01634-0