
Abstract
In stress conditions, as neoplastic transformation, amino acids serve not only as nutrients to maintain the cell survival but also as mediators of several regulatory pathways which are involved in apoptosis and autophagy. Especially, under glucose deprivation, in order to maintain the cell survival, proline and glutamine together with other glutamine-derived products such as glutamate, alpha-ketoglutarate, and ornithine serve as alternative sources of energy. They are substrates for production of pyrroline-5-carboxylate which is the product of conversion of proline by proline dehydrogenase/ proline oxidase (PRODH/POX) to produce ATP for protective autophagy or reactive oxygen species for apoptosis. Interconversion of proline, ornithine, and glutamate may therefore regulate PRODH/POX-dependent apoptosis/autophagy. The key amino acid is proline, circulating between mitochondria and cytoplasm in the proline cycle. This shuttle is known as proline cycle. It is coupled to pentose phosphate pathway producing nucleotides for DNA biosynthesis. PRODH/POX is also linked to p53 and AMP-activated protein kinase (AMPK)-dependent pathways. Proline availability for PRODH/POX-dependent apoptosis/autophagy is regulated at the level of collagen biosynthesis (proline utilizing process) and prolidase activity (proline supporting process). In this review, we suggest that amino acid metabolism linking TCA and Urea cycles affect PRODH/POX-dependent apoptosis/autophagy and the knowledge might be useful to targeted cancer therapy.
Similar content being viewed by others


Introduction
In stress conditions, cellular homeostasis is maintained by alteration of anabolic and catabolic processes. Anabolic processes are regulated by several factors affecting biosynthesis of cellular components. Major catabolic processes are mediated by the ubiquitin–proteasome system and autophagy [1]. In some cases, autophagy and apoptosis simultaneously occur in the same cell or autophagy precedes apoptosis via p53-dependent pathways or AMP-activated protein kinase (AMPK) [1]. Alternatively, autophagy can directly activate cell death pathway [1, 2]. Both p53 and AMPK are potent stimulators of proline dehydrogenase/proline oxidase (PRODH/POX) that has been implicated in the induction of autophagy and apoptosis [3,4,5,6,7,8,9,10]. Since PRODH/POX is linked to conversion of proline to pyrroline-5-carboxylate (P5C) [11], the availability of proline to this process is of critical importance. Proline and P5C are intermediates of interconversion of glutamine, glutamate, ornithine, and α-ketoglutarate suggesting the key role of these amino acids in the regulation of PRODH/POX-dependent apoptosis/autophagy. Therefore, this review aims to discuss the contribution of proline, glutamine, and its metabolites in regulation of PRODH/POX-dependent apoptosis/autophagy.
Regulatory mechanism of autophagy and apoptosis
Autophagy
Autophagy is a homeostatic, intracellular degradation process in which dispensable, long-lived, or aberrant proteins and damaged organelles are digested in lysosomes. The digestion products are recycled in cellular metabolism. It usually happens under stress conditions such as amino acid starvation [12,13,14]. Besides the removal of useless components retained in the cell, the other function of autophagy is to generate energy for synthesis of new building blocks in the process of homeostasis and cellular renovation [12, 13]. It suggests that autophagy has a profound impact on cancer cell survival [15]. Autophagy may also contribute to the suppression of cancer cell growth. The activation of autophagy explains a resistance mechanism in the course of cancer therapy. Therefore, the inhibition of autophagy was suggested as a potential pharmacotherapeutic approach for tumor growth suppression [13, 16].
A variety of proteins have been considered as autophagy markers for the assessment of presence or absence of autophagy in the cell. The first autophagy markers were found in yeast and identified more than 30 autophagy-related (ATG) genes, many of which have known orthologs in higher eukaryotes [17, 18]. Atg proteins have been classified into different groups based on their function in autophagy: (1) the Atg1/ULK complex (Atg1, Atg11, Atg13, Atg17, Atg29, and Atg31) regulates the induction of autophagosome formation; (2) the Atg9 complex (Atg2, Atg9, and Atg18), involved in membrane delivery to the expanding phagophore; (3) the PtdIns 3-kinase (PtdIns3K) complex (Vps34, Vps15, Vps30/Atg6, and Atg14) functions to recruit PtdIns3P-binding proteins; (4) two ubiquitin-like (Ubl) conjugation systems including the Atg12 complex (Atg5, Atg7, Atg10, Atg12, and Atg16) and a Atg8 complex (Atg3, Atg4, Atg7, and Atg8) that plays crucial role in vesicle expansion [19, 20] (Table 1). The mammalian ULK1/2 complex comprises ULK1/2 (mammalian homologs of Atg1), ATG13 (a homolog of yeast Atg13), RB1CC1/FIP200 (a putative Atg17 homolog), and C12orf44/ATG101 [21, 22]. The other study provided evidence that ULK1 kinase can be activated by AMP-activated protein kinase (AMPK) under glucose or amino acid starvation [23]. The ULK1/2 complex is inhibited by the phosphorylation of mTORC1 preventing interaction between ULK1 and AMPK. However, during induction of autophagy, the suppression of mTOR occurs and the protein complex of ULK1/2, ATG13, and RB1CC1 is formed to initiate the autophagy. Moreover, the autophagy process is mediated by Beclin-1 (autophagy-related gene, Atg 6) which codes for another autophagy protein [24, 25]. Some of these markers were linked to the PRODH/POX-dependent apoptosis/autophagy [3,4,5,6,7,8,9,10, 26, 27]. Since it has been proved that there is a cross-talk between autophagy and apoptosis [28], it cannot be excluded that the mechanism of this process may involve PRODH/POX.
Apoptosis
A concept of apoptosis was initially reported by Karl Vogt in 1872 then described by Walther Flemming who was the first to explain the mechanism of programmed cell death in 1885. Several studies suggested this mechanism as a program of cellular suicide where the cell destroys itself to maintain tissue homeostasis [29]. The machinery of apoptosis is mediated by a family of proteases, namely caspases which contain a cysteine at their active site and cleave the target proteins at a residue of aspartic acids [30]. Their precursors are called procaspases which are expressed as inactive forms in normal condition. These proteins, however, are cleaved to become active caspases triggering the apoptosis via energy-dependent cascade pathways [30]. The apoptosis is recruited through 3 different pathways: the extrinsic pathway, the intrinsic pathway, and Granzyme B-dependent pathway [31]. Among these pathways, the intrinsic and extrinsic pathways are the major mechanisms of apoptosis.
The intrinsic apoptosis pathway is activated by damages taking place within the cell. This mechanism involves the presence of pro-apoptotic proteins, BAX, and BID in the outer membrane of the mitochondria. They interact with the other protein, BAK to activate cytochrome c that binds to apoptotic protease activating factor-1 (Apaf-1) [32]. This binding activates active caspase 9 that triggers cascade downstream of effector caspases (such as caspase 3, caspase 7, and caspase 6), finally resulting in cell death [33]. The p53 protein is a key factor to activate the intrinsic pathway due to its contribution to activate BAX protein [34].
In contrast, the extrinsic pathway is initiated from extracellular events, triggered by ligand binding to plasma membrane death receptors, leading to activation of initiator caspase 8 [31]. Death receptors such as Fas/CD95 and tumor necrosis factor-related apoptosis inducing ligand (TRAIL) receptors DR-4 and DR-5 are transmembrane proteins that function to detect specific extracellular death signals [35, 36]. For instance, Adapter molecules like Fas Associated via Death Domain (FADD) contain death domain (DD) and a death effector domain (DED) which activate an active caspase-8 via a sequential action of a homotypic DED–DED interaction. Active caspase-8 generates a downstream of effector caspases contributing to cell death. However, they have the same execution pathway which is initiated by the activation of caspase-3 [31]. Typical biomarkers of apoptosis are listed in Table 2. Most of them were linked to PRODH/POX-dependent apoptosis [3,4,5,6,7,8,9,10].
PRODH/POX-dependent pathways relevant to apoptosis and autophagy
A variety of approaches to the inhibition of autophagy or activation of apoptosis have recently focused on proline dehydrogenase (PRODH), known also as proline oxidase (POX). PRODH/POX, a mitochondrial enzyme, converts proline to pyrroline-5-carboxylate (P5C) with the concomitant transfer of electrons to cytochrome c producing ATP or directly on oxygen generating reactive oxygen species (ROS) [5]. There are two human genes annotated as PRODH: PRODH1 (chromosome 22q11.21; NCBI Accession NM_016335) and PRODH2 (chromosome 19q13.12; NCBI Accession NM_021232). It has been suggested that the function of the enzyme may depend on substrate availability, proline. The main source of this amino acid is collagen which comprises 25% of total protein mass in animals [10, 30].
Briefly, these proteins are classified into major types which are type I in the skin, tendon, and bone, type II in cartilage, and type IV in basal laminae. Up to date, 28 types of collagen with 46 distinct polypeptide chains were found in vertebrates, as well as many other proteins containing collagenous domains [37, 38]. The predominant amino acids in collagen are proline and glycine, which enable triple-helical collagen structure. Extracellular degradation of collagens by tissue collagenases and further intracellular degradation of collagen degradation products in lysosomes release imidopeptides that are cleaved by cytoplasmic prolidase releasing a large amount of proline, the substrate for PRODH/POX.
After the conversion of proline to P5C, further proline metabolism is catalyzed by pyrroline-5-carboxylate dehydrogenase (P5CDH), transforming P5C into glutamate which is a precursor of α-ketoglutarate (α-KG) involved in the tricarboxylic acid (TCA) cycle. When the TCA cycle is overloaded by metabolites, the reversible reaction of conversion of P5C into proline by pyrroline-5-carboxylate reductase (P5CR) may occur, using NADPH or NADH as a cofactor. This interconversion of P5C-proline called proline cycle was first introduced in 1986 [39]. It has been demonstrated that the cellular proline, glutamine, and glutamate are linked to the proline pathway [40] regulating apoptosis/autophagy. The cycle is coupled to pentose phosphate shunt through NADPH from pentose pathway and NADP + from the proline cycle [4, 41]. Base on this mechanism, the role of PRODH/POX in the regulation of cellular metabolism has recently studied as an approach to cancer treatment. This cycle is responsible for the regulation of gene expression, purine biosynthesis, cellular redox state, apoptosis, and cell proliferation [3]. Moreover, PRODH/POX has a variety of regulatory functions, such as osmotic adjustment, protection against metabolic stress, and signaling in bacteria, plants, and mammals [10]. However, the most important function of PRODH/POX is donating electrons through flavin adenine dinucleotide (FAD) into the electron transport chain to generate ROS or ATP depending on environmental conditions [10].
PRODH/POX-induced apoptosis
Both intrinsic and extrinsic pathways of apoptosis may be induced by PRODH/POX [42]. Especially, in the extrinsic pathway (death receptor), PRODH/POX stimulates the expression of tumor necrosis factor-related apoptosis-activated ligand (TRAIL), DR5, and cleavage of caspase-8 [42, 43], and also activates caspase-9 and caspase-3 [44, 45]. In cancer cells, PRODH/POX is upregulated by a variety of factors, for example tumor suppressor p53 and inflammatory factor peroxisome proliferator-activated receptor gamma (PPARγ) [7, 10]. However, its level in cancer tissue is much lower than that in normal tissues from the patients [46, 47]. Regarding the overexpression of POX, the ROS generation is integrated with the p53-dependent mechanisms [5, 48], switching the apoptotic cell death in a variety of cancer cell types [5, 48,49,50,51]. The supporting evidence showed that the PRODH/POX coding gene induced the expression of p53 [52]. On the other hand, inactivation of proline oxidase reduced p53-induced upregulation of proline oxidase, a release of cytochrome c from mitochondria, and apoptosis in cancer cells [42, 49]. PRODH/POX acting as a driver of apoptosis was clearly evaluated in a model of PRODH/POX knockdown cancer cells [53].
PRODH/POX-induced autophagy
The recent study of Zareba et al., (2018) showed that in knocked down PRODH/POX MCF-7 breast cancer cells, cytoplasmic proline accumulation induced autophagy. However it was established that environmental conditions such as hypoxia or glucose deficiency may affect PRODH/POX-dependent autophagy/apoptosis [9]. It seems that proline availability may determine PRODH/POX-dependent apoptosis/autophagy. Although the mechanism of this process is not known, it has been suggested that hypoxia-inducible factor-1 alpha (HIF-1α) plays an important role in cancer cell metabolism. The availability of proline in the cell facilitates generation of α-KG that inhibits the transcriptional activity of HIF-1α. An increase in αKG concentration leads to an increase in the activity of a prolyl hydroxylase domain (PHD) of HIF-1α inducing proteasomal degradation of HIF-1α [43, 45, 54]. In contrast, proline through the same mechanism inhibits the activity of PHD, contributing to a decrease in HIF-1α proteasomal degradation and increase in its transcriptional activity.
It is well established that glutamine and proline metabolism, as well as other non-essential amino acids, are involved in oncometabolism of cells [9]. This process is called as “parametabolic pathway”. Particularly, the proline biosynthetic pathway was linked to glucose metabolism and POX-dependent apoptosis that is under the regulation of oncogene MYC.
Depending on the metabolic situation, proline can either be used for protein synthesis or oxidized in the mitochondria for energy production. Under nutrient deficiency and hypoxia, cancer cells may adopt to switch a survival mechanism which is the degradation of proline to produce the energy [26]. Therefore, hypoxia, glucose depletion, or treatment with rapamycin stimulated degradation of proline and POX-dependent autophagy.
The impact of amino acids on cell re-programming
Several amino acids have been linked to activation or inhibition of apoptosis/autophagy [55]. It is well recognized that they participate in the mTORC1 and GCN2/eIF2 pathways which function to regulate protein translation and control the cellular demand for amino acids by concomitantly regulating autophagy-dependent catabolism [56,57,58]. For instance, non-essential amino acids (NEA) as proline in condition of glucose deprivation activate anti-apoptotic pathways in cancer cells by inducing the expression of anti-apoptotic members of the Bcl-2 gene family and preventing the expression of pro-apoptotic proteins [59]. The study suggested that although under low glucose condition apoptosis could be induced in cancer cells, the non-essential amino acids may counteract the process. It was supported by the upregulation of amino acid transporter gene LAT1 in the membranes of cancer cells [27, 60, 61] under glucose stress [59].
Glutamine was proved to be a sustainable source of energy. Early findings indicated that tumor formation is significantly due to the mitochondrial vulnerability through the alteration of glycolysis [62]. The proliferation of cancer cells is mostly maintained by energy products derived from the TCA cycle [63, 64]. A larger majority of tumor suppressors and oncogenes have been linked to metabolic pathways [64,65,66,67]. Glutamine is an integral metabolite in the proliferation of mammalian cells. The consumption rate of glutamine in cancer cells is compared to that of other amino acids. However, the demand for glutamine was observed to be tenfold higher than that for other amino acids [68]. Glutamine has profound impact on the functional activity of mammalian target of rapamycin (mTOR) kinase, mitochondrial membrane potential, and NADPH production [69]. Glutamine is a nitrogen source both for purine and pyrimidine synthesis [70, 71]. In the non-essential amino acid synthetic pathways, glutamine-derived glutamic acid continues donating its amine group to accelerate the tricarboxylic acid (TCA) cycle metabolites for the production of α-ketoglutarate, serine, alanine, aspartate, and ornithine. Glutamine acts as a source of carbon and nitrogen for the synthesis of proline, ornithine, and arginine as well as a donor for the synthesis of asparagine from aspartic acid [69]. Lack of exogenous glutamine is one of the major causes for the death of cancer cells [72]. Several tumor cell lines, generated from pancreatic cancer, glioblastoma multiforme, acute myelogenous leukemia, and small cell lung cancer, are substantially vulnerable due to glutamine starvation [73]. The study suggested that derivatives of glutamine like glutamate, α-ketoglutarate, and glutathione are involved in the apoptotic pathway [74]. Similarly, proline interconvertibility with glutamate and arginine [3, 75] may play an important role in cell programming. However, recent data linked glutamine metabolism and apoptosis/autophagy through P5C to urea cycle.
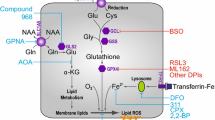
Ornithine and glutamate are important sources of P5C. Ornithine is converted into P5C in a reaction catalyzed by mitochondrial vitamin B6-dependent ornithine-δ-aminotransferase (OAT), while glutamate through a reduction reaction catalyzed by mitochondrial ATP- and NAD(P)H-dependent P5C synthase (P5CS) [76, 77]. This reaction can be reversed by mitochondrial P5C dehydrogenase (P5CDH) [76]. The role of this metabolic pathway in apoptosis/ autophagy was supported by data showing that degradation of ornithine by ornithine decarboxylase (ODC) play an important role in cell proliferation, differentiation, and cell death. It has been demonstrated that decreasing the activity of ODC by difluoromethylornithine (DFMO) causes accumulation of intracellular reactive oxygen species (ROS) and cell arrest, thus inducing cell death. These findings indicate that urea cycle contributes to the regulation of apoptosis and autophagy [78]. Since ornithine is easily convertible into P5C (products of catalytic activity of PRODH/POX), it may affect PRODH/POX-dependent apoptosis/autophagy. The results of these studies allow us to present a hypothesis on the regulation of PRODH/POX-dependent apoptosis/autophagy by key amino acids (Fig. 1). During conversion of PRO into P5C by PRODH/POX, ATP or ROS is generated inducing autophagy or apoptosis. PRO availability for this process is critical requirement for PRODH/POX-dependent function. PRO comes from collagen degradation products (last step of the degradation is catalyzed by prolidase) or proline convertible amino acids, mainly GLU and ORN. Conversion of PRO into P5C takes place in mitochondria, while P5C into PRO mainly in cytoplasm. This process is known as a “proline cycle” and is coupled to pentose phosphate pathway generating nucleotides for DNA biosynthesis. Interconversion of PRO, GLU, and ORN through intermediate GSA to P5C may represent an interface regulating PRODH/POX-dependent P5C generation and ATP/ROS for autophagy/apoptosis. The process links TCA and Urea cycles to proline cycle providing complex regulatory mechanism of PRODH/POX-dependent functions. Understanding the interplay between key amino acids and TCA/Urea metabolites and their role in the regulation of PRODH/POX-dependent apoptosis/autophagy might be a promising approach to targeted cancer therapy.

Regulation of PRODH/POX-dependent apoptosis/autophagy by key amino acids. PRO proline; GLU glutamate; ORN ornithine; GLN glutamine; GLYPRO glycyl-proline; PRODH/POX proline dehydrogenase (PRODH)/proline oxidase (POX); ROS reactive oxygen species; P5C pyrroline-5-carboxylate; P5CR pyrroline-5-carboxylate reductase; P5CDH pyrroline-5-carboxylate dehydrogenase; P5CS pyrroline-5-carboxylate synthase; OAT ornithine aminotransferase; GSA glutamic gamma-semialdehyde; αKG α-ketoglutarate; TCA tricarboxylic acid cycle; GS glutamine synthase; GLS glutaminase; GLUD glutamate dehydrogenase
Conclusion
Studies of last decade provided several lines of evidence for the regulatory role of proline availability in PRODH/POX-dependent apoptosis/autophagy in cancer cells. The enzyme expression is often downregulated in various tumors, limiting mitochondrial proline degradation and PRODH/POX-dependent apoptosis. Critical factor for the process is proline availability that depends on the activity of prolidase (enzyme supporting cytoplasmic proline level) and the rate of proline utilization in the process of collagen biosynthesis. However, proline also represents an energy-sensing molecule that reprograms cellular metabolism. Interconversion of proline, glutamate, and ornithine links TCA cycle, urea cycle, and amino acid metabolism to PRODH/POX-dependent apoptosis/autophagy. Deregulation of energetic metabolism in cancer cells due to Warburg’s effect facilitates protein degradation as an alternative source of energy. Therefore, when glucose supply is limited, cancer cells may select proline as an alternative energy source. Therefore, amino acid metabolism in specific environmental cellular conditions may represent interface of PRODH/POX-dependent apoptosis and autophagy. The hypothesis is outlined in Fig. 1.
References
Mariño G, Niso-Santano M, Baehrecke EH, Kroemer G (2014) Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol 15:81–94. https://doi.org/10.1038/nrm3735
Shen S, Kepp O, Kroemer G (2012) The end of autophagic cell death? Autophagy 8:1–3. https://doi.org/10.4161/auto.8.1.16618
Phang JM (1985) The regulatory functions of proline and pyrroline-5-carboxylic acid. Curr Top Cell Regul 25:91–132
Pandhare J, Donald SP, Cooper SK, Phang JM (2009) Regulation and function of proline oxidase under nutrient stress. J Cell Biochem 107:759–768. https://doi.org/10.1002/jcb.22174
Donald SP, Sun XY, Hu CA, Yu J, Mei JM, Valle D, Phang JM (2001) Proline oxidase, encoded by p53-induced gene-6, catalyzes the generation of proline-dependent reactive oxygen species. Cancer Res 61:1810–1815
Liu Y, Borchert GL, Surazynski A, Phang JM (2008) Proline oxidase, a p53-induced gene, targets COX-2/PGE2 signaling to induce apoptosis and inhibit tumor growth in colorectal cancers. Oncogene 27:6729–6737. https://doi.org/10.1038/onc.2008.322
Phang JM, Liu W, Zabirnyk O (2010) Proline metabolism and microenvironmental stress. Annu Rev Nutr 30:441–463. https://doi.org/10.1146/annurev.nutr.012809.104638
Phang JM, Liu W (2012) Proline metabolism and cancer. Front Biosci 17:1835–1845
Liu W, Phang JM (2012) Proline dehydrogenase (oxidase), a mitochondrial tumor suppressor, and autophagy under the hypoxia microenvironment. Autophagy 8:1407–1409. https://doi.org/10.4161/auto.21152
Liu W, Phang JM (2012) Proline dehydrogenase (oxidase) in cancer. BioFactors 38:398–406. https://doi.org/10.1002/biof.1036
Onodera J, Ohsumi Y (2005) Autophagy is required for maintenance of amino acid levels and protein synthesis under nitrogen starvation. J Biol Chem 280:31582–31586. https://doi.org/10.1074/jbc.M506736200
Mizushima N, Komatsu M (2011) Autophagy: renovation of cells and tissues. Cell 147:728–741. https://doi.org/10.1016/j.cell.2011.10.026
Yang ZJ, Chee CE, Huang S, Sinicrope FA (2011) The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther 10:1533–1541. https://doi.org/10.1158/1535-7163.MCT-11-0047
Vicencio JM, Galluzzi L, Tajeddine N, Ortiz C, Criollo A, Tasdemir E, Morselli E, Ben Younes A, Maiuri MC, Lavandero S, Kroemer G (2008) Senescence, apoptosis or autophagy? When a damaged cell must decide its path–a mini-review. Gerontology 54:92–99. https://doi.org/10.1159/000129697
Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, Cattoretti G, Levine B (2003) Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 112:1809–1820. https://doi.org/10.1172/JCI20039
Chude CI, Amaravadi RK (2017) Targeting autophagy in cancer: update on clinical trials and novel inhibitors. Int J Mol Sci 18: https://doi.org/10.3390/ijms18061279
Feng Y, He D, Yao Z, Klionsky DJ (2014) The machinery of macroautophagy. Cell Res 24:24–41. https://doi.org/10.1038/cr.2013.168
Mizushima N, Yoshimori T, Ohsumi Y (2011) The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 27:107–132. https://doi.org/10.1146/annurev-cellbio-092910-154005
Mizushima N (2007) Autophagy: process and function. Genes Dev 21:2861–2873. https://doi.org/10.1101/gad.1599207
Suzuki K, Kubota Y, Sekito T, Ohsumi Y (2007) Hierarchy of Atg proteins in pre-autophagosomal structure organization. Genes Cells 12:209–218. https://doi.org/10.1111/j.1365-2443.2007.01050.x
Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, Guan JL, Oshiro N, Mizushima N (2009) Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell 20:1981–1991. https://doi.org/10.1091/mbc.e08-12-1248
Hara T, Takamura A, Kishi C, Iemura S, Natsume T, Guan JL, Mizushima N (2008) FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol 181:497–510. https://doi.org/10.1083/jcb.200712064
Kim J, Kundu M, Viollet B, Guan KL (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13:132–141. https://doi.org/10.1038/ncb2152
Česen MH, Pegan K, Spes A, Turk B (2012) Lysosomal pathways to cell death and their therapeutic applications. Exp Cell Res 318:1245–1251. https://doi.org/10.1016/j.yexcr.2012.03.005
Lee JS, Kim YJ, Kim CL, Lee GM (2012) Differential induction of autophagy in caspase-3/7 down-regulating and Bcl-2 overexpressing recombinant CHO cells subjected to sodium butyrate treatment. J Biotechnol 161:34–41. https://doi.org/10.1016/j.jbiotec.2012.05.011
Phang JM, Pandhare J, Liu Y (2015S) The metabolism of proline as microenvironmental stress substrate. J Nutr 138:2008S–2015S. https://doi.org/10.1093/jn/138.10.2008S
Ichinoe M, Mikami T, Yoshida T, Igawa I, Tsuruta T, Nakada N, Anzai N, Suzuki Y, Endou H, Okayasu I (2011) High expression of L-type amino-acid transporter 1 (LAT1) in gastric carcinomas: comparison with non-cancerous lesions. Pathol Int 61:281–289. https://doi.org/10.1111/j.1440-1827.2011.02650.x
Maiuri MC, Zalckvar E, Kimchi A, Kroemer G (2007) Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 8:741–752. https://doi.org/10.1038/nrm2239
Goodman SR (2007) Medical cell biology. Elsevier/Academic Press, Amsterdam
Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P (2002) Molecular Biology of the Cell. Garland Science, New York
Logue SE, Martin SJ (2008) Caspase activation cascades in apoptosis. Biochem Soc Trans 36:1–9. https://doi.org/10.1042/BST0360001
Luo X, Budihardjo I, Zou H, Slaughter C, Wang X (1998) Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 94:481–490
Slee EA, Harte MT, Kluck RM, Wolf BB, Casiano CA, Newmeyer DD, Wang HG, Reed JC, Nicholson DW, Alnemri ES, Green DR, Martin SJ (1999) Ordering the cytochrome c-initiated caspase cascade: hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9-dependent manner. J Cell Biol 144:281–292. https://doi.org/10.1083/jcb.144.2.281
Maximov GK, Maximov KG (2008) The role of p53 tumor-supressor protein in apoptosis and cancerogenesis. Biotechnol Biotechnol Equipment 22:664–668
Fossati S, Ghiso J, Rostagno A (2012) TRAIL death receptors DR4 and DR5 mediate cerebral microvascular endothelial cell apoptosis induced by oligomeric Alzheimer's Aβ. Cell Death Dis 3:e321. https://doi.org/10.1038/cddis.2012.55
Ashkenazi A (2002) Targeting death and decoy receptors of the tumour-necrosis factor superfamily. Nat Rev Cancer 2:420–430. https://doi.org/10.1038/nrc821
Veit G, Kobbe B, Keene DR, Paulsson M, Koch M, Wagener R (2006) Collagen XXVIII, a novel von Willebrand factor A domain-containing protein with many imperfections in the collagenous domain. J Biol Chem 281:3494–3504. https://doi.org/10.1074/jbc.M509333200
Brinckmann J (2005) Collagens at a Glance. Springer, Berlin, Heidelberg
Hagedorn CH, Phang JM (1986) Catalytic transfer of hydride ions from NADPH to oxygen by the interconversions of proline and delta 1-pyrroline-5-carboxylate. Arch Biochem Biophys 248:166–174. https://doi.org/10.1016/0003-9861(86)90413-3
Cappelletti P, Tallarita E, Rabattoni V, Campomenosi P, Sacchi S, Pollegioni L (2018) Proline oxidase controls proline, glutamate, and glutamine cellular concentrations in a U87 glioblastoma cell line. PLoS ONE 13:e0196283. https://doi.org/10.1371/journal.pone.0196283
Phang JM, Liu W, Hancock C, Christian KJ (2012) The proline regulatory axis and cancer. Front Oncol 2:60. https://doi.org/10.3389/fonc.2012.00060
Liu Y, Borchert GL, Surazynski A, Hu CA, Phang JM (2006) Proline oxidase activates both intrinsic and extrinsic pathways for apoptosis: the role of ROS/superoxides, NFAT and MEK/ERK signaling. Oncogene 25:5640–5647. https://doi.org/10.1038/sj.onc.1209564
Kononczuk J, Czyzewska U, Moczydlowska J, Surażyński A, Palka J, Miltyk W (2015) Proline oxidase (POX) as a target for cancer therapy. Curr Drug Targets 16:1464–1469
Cooper SK, Pandhare J, Donald SP, Phang JM (2008) A novel function for hydroxyproline oxidase in apoptosis through generation of reactive oxygen species. J Biol Chem 283:10485–10492. https://doi.org/10.1074/jbc.M702181200
Zareba I, Celinska-Janowicz K, Surazynski A, Miltyk W, Palka J (2018) Proline oxidase silencing induces proline-dependent pro-survival pathways in MCF-7 cells. Oncotarget 9:13748–13757. https://doi.org/10.18632/oncotarget.24466
Liu Y, Borchert GL, Donald SP, Diwan BA, Anver M, Phang JM (2009) Proline oxidase functions as a mitochondrial tumor suppressor in human cancers. Cancer Res 69:6414–6422. https://doi.org/10.1158/0008-5472.CAN-09-1223
Liu W, Zabirnyk O, Wang H, Shiao YH, Nickerson ML, Khalil S, Anderson LM, Perantoni AO, Phang JM (2010) miR-23b targets proline oxidase, a novel tumor suppressor protein in renal cancer. Oncogene 29:4914–4924. https://doi.org/10.1038/onc.2010.237
Liu Y, Borchert GL, Donald SP, Surazynski A, Hu CA, Weydert CJ, Oberley LW, Phang JM (2005) MnSOD inhibits proline oxidase-induced apoptosis in colorectal cancer cells. Carcinogenesis 26:1335–1342. https://doi.org/10.1093/carcin/bgi083
Maxwell SA, Rivera A (2003) Proline oxidase induces apoptosis in tumor cells, and its expression is frequently absent or reduced in renal carcinomas. J Biol Chem 278:9784–9789. https://doi.org/10.1074/jbc.M210012200
Ferri KF, Kroemer G (2001) Organelle-specific initiation of cell death pathways. Nat Cell Biol 3:E255–E263. https://doi.org/10.1038/ncb1101-e255
Hu CA, Donald SP, Yu J, Lin WW, Liu Z, Steel G, Obie C, Valle D, Phang JM (2007) Overexpression of proline oxidase induces proline-dependent and mitochondria-mediated apoptosis. Mol Cell Biochem 295:85–92. https://doi.org/10.1007/s11010-006-9276-6
Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B (1997) A model for p53-induced apoptosis. Nature 389:300–305. https://doi.org/10.1038/38525
Zareba I, Surazynski A, Chrusciel M, Miltyk W, Doroszko M, Rahman N, Palka J (2017) Functional Consequences of Intracellular Proline Levels Manipulation Affecting PRODH/POX-Dependent Pro-Apoptotic Pathways in a Novel in Vitro Cell Culture Model. Cell Physiol Biochem 43:670–684. https://doi.org/10.1159/000480653
Myllyharju J (2013) Prolyl 4-hydroxylases, master regulators of the hypoxia response. Acta Physiol (Oxf) 208:148–165. https://doi.org/10.1111/apha.12096
Seglen PO, Gordon PB (1984) Amino acid control of autophagic sequestration and protein degradation in isolated rat hepatocytes. J Cell Biol 99:435–444. https://doi.org/10.1083/jcb.99.2.435
Jewell JL, Guan KL (2013) Nutrient signaling to mTOR and cell growth. Trends Biochem Sci 38:233–242. https://doi.org/10.1016/j.tibs.2013.01.004
Laplante M, Sabatini DM (2012) mTOR signaling in growth control and disease. Cell 149:274–293. https://doi.org/10.1016/j.cell.2012.03.017
Meijer AJ, Dubbelhuis PF (2004) Amino acid signalling and the integration of metabolism. Biochem Biophys Res Commun 313:397–403. https://doi.org/10.1016/j.bbrc.2003.07.012
Wang G, Dai L, Luo L, Xu W, Zhang C, Zhu Y, Chen Z, Hu W, Xu X, Pan W (2014) Non-essential amino acids attenuate apoptosis of gastric cancer cells induced by glucose starvation. Oncol Rep 32:332–340. https://doi.org/10.3892/or.2014.3205
Fukumoto S, Hanazono K, Komatsu T, Ueno H, Kadosawa T, Iwano H, Uchide T (2013) L-type amino acid transporter 1 (LAT1): a new therapeutic target for canine mammary gland tumour. Vet J 198:164–169. https://doi.org/10.1016/j.tvjl.2013.06.016
Kaira K, Oriuchi N, Takahashi T, Nakagawa K, Ohde Y, Okumura T, Murakami H, Shukuya T, Kenmotsu H, Naito T, Kanai Y, Endo M, Kondo H, Nakajima T, Yamamoto N (2011) L-type amino acid transporter 1 (LAT1) expression in malignant pleural mesothelioma. Anticancer Res 31:4075–4082
Warburg O (1956) On the origin of cancer cells. Science 123:309–314. https://doi.org/10.1126/science.123.3191.309
Boroughs LK, DeBerardinis RJ (2015) Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol 17:351–359. https://doi.org/10.1038/ncb3124
DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB (2008) The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 7:11–20. https://doi.org/10.1016/j.cmet.2007.10.002
Koppenol WH, Bounds PL, Dang CV (2011) Otto Warburg's contributions to current concepts of cancer metabolism. Nat Rev Cancer 11:325–337. https://doi.org/10.1038/nrc3038
Levine AJ, Puzio-Kuter AM (2010) The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 330:1340–1344. https://doi.org/10.1126/science.1193494
Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324:1029–1033. https://doi.org/10.1126/science.1160809
Eagle H, Oyama VI, Levy M, Horton CL, Fleischman R (1956) The growth response of mammalian cells in tissue culture to L-glutamine and L-glutamic acid. J Biol Chem 218:607–616
Wise DR, Thompson CB (2010) Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci 35:427–433. https://doi.org/10.1016/j.tibs.2010.05.003
Young VR, Ajami AM (2449S) Glutamine: the emperor or his clothes? J Nutr 131:2449S–S2459; discussion 2486S-7Shttps://doi.org/10.1093/jn/131.9.2449S
Ahluwalia GS, Grem JL, Hao Z, Cooney DA (1990) Metabolism and action of amino acid analog anti-cancer agents. Pharmacol Ther 46:243–271
Eagle H (1955) Nutrition needs of mammalian cells in tissue culture. Science 122:501–514. https://doi.org/10.1126/science.122.3168.501
Wu MC, Arimura GK, Yunis AA (1978) Mechanism of sensitivity of cultured pancreatic carcinoma to asparaginase. Int J Cancer 22:728–733
Matés JM, Segura JA, Alonso FJ, Márquez J (2009) Natural antioxidants: therapeutic prospects for cancer and neurological diseases. Mini Rev Med Chem 9:1202–1214
Adams E, Frank L (1980) Metabolism of proline and the hydroxyprolines. Annu Rev Biochem 49:1005–1061. https://doi.org/10.1146/annurev.bi.49.070180.005041
Hu CA, Lin WW, Valle D (1996) Cloning, characterization, and expression of cDNAs encoding human delta 1-pyrroline-5-carboxylate dehydrogenase. J Biol Chem 271:9795–9800. https://doi.org/10.1074/jbc.271.16.9795
Hu CA, Lin WW, Obie C, Valle D (1999) Molecular enzymology of mammalian Delta1-pyrroline-5-carboxylate synthase. Alternative splice donor utilization generates isoforms with different sensitivity to ornithine inhibition. J Biol Chem 274:6754–6762. https://doi.org/10.1074/jbc.274.10.6754
Liu GY, Hung YC, Hsu PC, Liao YF, Chang WH, Tsay GJ, Hung HC (2005) Ornithine decarboxylase prevents tumor necrosis factor alpha-induced apoptosis by decreasing intracellular reactive oxygen species. Apoptosis 10:569–581. https://doi.org/10.1007/s10495-005-1891-2
Papinski D, Schuschnig M, Reiter W, Wilhelm L, Barnes CA, Maiolica A, Hansmann I, Pfaffenwimmer T, Kijanska M, Stoffel I, Lee SS, Brezovich A, Lou JH, Turk BE, Aebersold R, Ammerer G, Peter M, Kraft C (2014) Early steps in autophagy depend on direct phosphorylation of Atg9 by the Atg1 kinase. Mol Cell 53:471–483. https://doi.org/10.1016/j.molcel.2013.12.011
Tucker KA, Reggiori F, Dunn WA, Klionsky DJ (2003) Atg23 is essential for the cytoplasm to vacuole targeting pathway and efficient autophagy but not pexophagy. J Biol Chem 278:48445–48452. https://doi.org/10.1074/jbc.M309238200
Shpilka T, Weidberg H, Pietrokovski S, Elazar Z (2011) Atg8: an autophagy-related ubiquitin-like protein family. Genome Biol 12:226. https://doi.org/10.1186/gb-2011-12-7-226
Ward TH, Cummings J, Dean E, Greystoke A, Hou JM, Backen A, Ranson M, Dive C (2008) Biomarkers of apoptosis. Br J Cancer 99:841–846. https://doi.org/10.1038/sj.bjc.6604519
Porter AG, Jänicke RU (1999) Emerging roles of caspase-3 in apoptosis. Cell Death Differ 6:99–104. https://doi.org/10.1038/sj.cdd.4400476
Butterick TA, Duffy CM, Lee RE, Billington CJ, Kotz CM, Nixon JP (2014) Use of a caspase multiplexing assay to determine apoptosis in a hypothalamic cell model. J Vis Exp. https://doi.org/10.3791/51305
Ehrnhoefer DE, Skotte NH, Savill J, Nguyen YT, Ladha S, Cao LP, Dullaghan E, Hayden MR (2011) A quantitative method for the specific assessment of caspase-6 activity in cell culture. PLoS ONE 6:e27680. https://doi.org/10.1371/journal.pone.0027680
Campos CB, Paim BA, Cosso RG, Castilho RF, Rottenberg H, Vercesi AE (2006) Method for monitoring of mitochondrial cytochrome c release during cell death: Immunodetection of cytochrome c by flow cytometry after selective permeabilization of the plasma membrane. Cytometry A 69:515–523. https://doi.org/10.1002/cyto.a.20273
Godin C, Louandre C, Bodeau S, Diouf M, Saidak Z, Conte MA, Chauffert B, Barbare JC, Barget N, Trinchet JC, Ganne N, Galmiche A (2015) Biomarkers of apoptosis and necrosis in patients with hepatocellular carcinoma treated with sorafenib. Anticancer Res 35:1803–1808
Weant AE, Michalek RD, Khan IU, Holbrook BC, Willingham MC, Grayson JM (2008) Apoptosis regulators Bim and Fas function concurrently to control autoimmunity and CD8+ T cell contraction. Immunity 28:218–230. https://doi.org/10.1016/j.immuni.2007.12.014
Montes-Berrueta D, Ramírez L, Salmen S, Berrueta L (2012) Fas and FasL expression in leukocytes from chronic granulomatous disease patients. Invest Clin 53:157–167
El-Karaksy SM, Kholoussi NM, Shahin RM, El-Ghar MM, Rl-S G (2013) TRAIL mRNA expression in peripheral blood mononuclear cells of Egyptian SLE patients. Gene 527:211–214. https://doi.org/10.1016/j.gene.2013.05.084
Zeestraten EC, Benard A, Reimers MS, Schouten PC, Liefers GJ, van de Velde CJ, Kuppen PJ (2013) The prognostic value of the apoptosis pathway in colorectal cancer: a review of the literature on biomarkers identified by immunohistochemistry. Biomark Cancer 5:13–29. https://doi.org/10.4137/BIC.S11475
Funding
This work was supported by the National Science Center [grant number 2017/25/B/NZ7/02183]; the European Union's Horizon 2020 research and innovation program under the Marie Sklodowska-Curie [grant agreement number 754432].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huynh, T.Y.L., Zareba, I., Baszanowska, W. et al. Understanding the role of key amino acids in regulation of proline dehydrogenase/proline oxidase (prodh/pox)-dependent apoptosis/autophagy as an approach to targeted cancer therapy. Mol Cell Biochem 466, 35–44 (2020). https://doi.org/10.1007/s11010-020-03685-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-020-03685-y