
Abstract
Systemic mastocytosis (SM) is a rare hematological neoplasm caused by the excessive proliferation of pathological mast cells that accumulate in the bone marrow (BM) and other extracutaneous organs leading to multi-organ damage and failure. Mast cell leukemia (MCL) is a rare form of systemic mastocytosis, accounting for < 1% of all cases of mastocytosis. MCL usually behaves aggressively with poor responses to current treatment options. Here, we report a diagnostic challenge with the leukemic subtype of MCL with a primary suspicion of pancreatic cancer. A cytomorphological, immunophenotypic, and histopathological examination of the bone marrow was performed. The diagnosis was based on the presence of ≥ 20% atypical and immature mast cells in the bone marrow and ≥ 10% mast cells among the peripheral white blood cells. The neoplastic cell population was identified as mast cell lineage by the expression of CD117 and tryptase. Only 3% of neoplastic cells displayed surface markers characteristic for clonal mast cells: CD25 and CD2. The D816V KIT mutation was not found. Neoplastic mast cells expressed CD30, a marker that is currently considered as a new minor criterion for SM. In the presented case, the primary suspicion of pancreatic cancer with osteosclerotic, lung, and pleural metastases was misleading, and a differential diagnosis based on hematological findings was performed. The patient’s severe symptoms were likely the result of organ damage from mast cell infiltration. Despite the use of intensive acute myeloid leukemia (AML)-like polychemotherapy, the patient died during the course of post-induction myelosuppression due to bleeding complications.
Similar content being viewed by others

Introduction
Mastocytosis is a clonal disease of the bone marrow stem cell leading to the expansion of morphologically and immunophenotypically abnormal mast cells (MCs) in one or more organ systems [1,2,3]. This is a rare and heterogeneous group of disorders. In the 2016 revision to the World Health Organization (WHO) classification, mastocytosis is no longer recognized as a myeloproliferative neoplasm (MPN) and is a distinct disease category delineated into cutaneous mastocytosis (CM), systemic mastocytosis (SM), and localized MC tumors. SM is defined by the major and minor criteria presented in Table 1 [4, 5]. Five subtypes of SM are recognized: indolent subtypes of SM include indolent systemic mastocytosis (ISM) and smoldering systemic mastocytosis (SSM), while advanced stages comprise systemic mastocytosis with an associated hematological neoplasm (SM-AHN), aggressive systemic mastocytosis (ASM), and mast cell leukemia (MCL). This division system is based on the mast cell burden and the presence/absence of secondary mast cell infiltration organ impairment [6].
Most cases of mastocytosis occur sporadically and are associated with somatic gain-of-function point mutations within the KIT. The KIT gene encodes a type III receptor tyrosine kinase known as mast cell growth factor/stem cell growth factor (SCFR/c-kit/CD117), whose activation leads to mast cell proliferation, maturation, adhesion, chemotaxis, and survival [1]. The oncogenic KIT mutation D816V is detected in > 80% of all patients with SM [4]. In advanced SM, KIT D816V is frequently accompanied by additional mutations in genes encoding signaling molecules (JAK2, KRAS, NRAS), transcription factors (RUNX1), epigenetic regulators (ASXL1, DNMT3A, EZH2, TET2), or splicing factors (SRSF2, SF3B1, U2AF1) [1, 9].
Neoplastic MCs are distinguished from normal MCs by the expression of CD25 and/or CD2 detected by flow cytometry or immunohistochemistry. Recent reports have demonstrated that CD25 expression may be a more reliable marker for neoplastic MCs. Immunostaining and immunophenotyping studies enhance the morphological distinction between normal (round and CD25-negative) and abnormal (spindle-shaped and CD25-positive) mast cells [2]. However, CD2 and CD25 markers were not found in 48% and 25% of MCL cases, respectively [2]. One-third of MCL cases reported in the literature had a double-negative CD2/CD25 immunophenotype [10]. It has been proposed that aggressive forms of mastocytosis, including MCL, exhibit an immature phenotype, in contrast to the more indolent forms characterized by an activated immunophenotype [2, 11]. Neoplastic MCs in MCL may express immaturity markers such as CD123, CD34, and HLA-DR [2, 11,12,13]. The immature phenotype is correlated with multilineage KIT mutations in the bone marrow and a poor prognosis [2].
Aggressive SM (ASM) is now divided into an untransformed variant and a variant in the transformation to MCL (ASM-t). In patients with ASM-t, the percentage of MCs in BM smears ranges from 5 to 20%. When the BM smear’s MCs percentage reaches 20%, the diagnosis changes from ASM-t to MCL [4]. Two subvariants of MCL can be distinguished: a (less frequent) classical variant in which MCs compose at least 10% of all blood cells irrespective of the WBC count and an aleukemic MCL (MCs < 10% in peripheral blood smears) [4, 7, 14, 15].
Patient categorization according to the 2016 WHO classification system is a crucial step in risk stratifying newly diagnosed SM patients. Secondly, assessing the molecular profile of SM patients is helpful in establishing a prognosis. The presence of poor-risk mutations (ASXL1, RUNX1, SRSF2, NRAS) is an independent risk factor of inferior survival [16]. In most MCL patients, the prognosis is poor, and the median survival time is < 1 year [8, 14, 16].
Treatment goals for patients with ASM are directed towards cytoreduction and ameliorating disease-related organ dysfunction. KIT D816V is an attractive treatment target because of its high frequency in SM. It confers primary resistance against the tyrosine kinase inhibitors: imatinib and masitinib [17, 18]. Nilotinib and dasatinib lack significant clinical activity [19, 20]. Midostaurin has in vitro activity against KIT D816V. In the phase 2 CPKC412D2201 trial, 16 patients with MCL were treated with midostaurin at 100 mg BID, and the response rate was 50% [21]. Responses occurred regardless of KIT D 816 V status. There are no head-to-head comparisons of midostaurin with other cytoreductive drugs. Allogeneic stem cell transplantation (allo-HSCT) is another treatment option that can prolong survival in MCL patients; however, primary refractoriness is often reported [22].
Case report
Clinical history
In March 2021, a 64-year-old woman was admitted to the Cancer Treatment Center with a suspicion of pancreatic cancer and/or myelofibrosis with extramedullary hematopoiesis. The patient complained of deteriorating health over the past few months and reported unintentional weight loss, flushing, progressive weakness, and increasing swelling of the lower extremities. Normocytic anemia in laboratory results and significant splenomegaly on physical examination were noted by the primary care physician who referred a patient to an oncology center. The liver was not enlarged. The patient’s chief concerns in the last month were the upper left abdominal pain, nausea, and vomiting. Pruritus and skin lesions were absent.
CT scan of the chest, abdomen, and pelvis revealed a tumor (30 mm) within the head of the pancreas, numerous osteosclerotic lesions, neoplastic cells infiltrating in the right lobe of the lung, fluid in both pleural cavities, enlarged spleen (200 mm) with limited tissue perfusion, and lumpy thickening of the paraspinal tissues at the Th2-Th11 level. As tumor marker levels (CA19-9 and CEA) were within normal ranges, pancreatic lymphoma or extramedullary hematopoiesis was suspected.
Complete blood count (CBC) showed normocytic anemia (hemoglobin level 7.6 g/dL; mean cell volume 92 fL), mild thrombocytopenia with a platelet (PLT) count of 142 G/L,and a normal white blood cell (WBC) count (4.26 G/L). The peripheral blood (PB) smear revealed 11% of undifferentiated cells. Undifferentiated cells were not recognized as circulating mast cells (MCs) in microscopic examinations. Elevated fibrin degradation products (D-dimer 2.86 pg/mL and fibrinogen 377 mg/dL) and slightly prolonged APTT (41 s) were detected. Other laboratory test results at admission were unremarkable.
Morphological, immunophenotypic, and histopathological examination of bone marrow (BM) was performed due to undifferentiated cells in the peripheral blood and a suspicion of acute leukemia. Bone marrow flow cytometry (FC) analysis showed the presence of 45% immature cells. Even though only 3% of blasts displayed CD2 and CD25 antigens, these findings, together with tryptase levels above 200 mg/L, prompted the diagnosis of mast cell leukemia.
After the initial MCL diagnosis, the patient was transferred to the Department of Hematology, Institute of Hematology and Transfusion Medicine, Warsaw, for further treatment.
The patient received remission-induction chemotherapy according to the DA scheme “3 + 7” (DA, daunorubicin at a dose of 60 mg/m2/day on days 1–3, cytosine arabinoside at a dose of 100 mg/m2/day on days 1–7). Chemotherapy caused myelosuppression with thrombocytopenia refractory to platelet transfusions and complicated by recurrent upper gastrointestinal bleeding and death. The patient died within 30 days of hospital admission.
Materials and methods
May-Grunwald-Giemsa–stained bone marrow aspirate smears and squashes were examined under optical microscopy with 100 × and 1000 × magnification.
Multidimensional flow cytometry was used to characterize the immunophenotype of malignant cells in BM with stain-lyse-wash standard sample preparation protocol. Twelve-color combinations of the monoclonal antibodies (MoAbs) were used, and the panel of MoAbs consisted of CD22 FITC, CD36 FITC, CD66c FITC, CD7 PE, CD13 PE, CD16 PE, CD64 PE, CD30 PE, CD11c PerCP-Cy5.5, CD33 PerCP-Cy5.5, CD34 PerCP-Cy5.5, CD25 PE-Cy7, CD117 PE-Cy7, CD19 APC, CD123 APC, CD14 APC-H7, CD38 APC-H7, CD56 APC-R700, HLA-DR V450, CD45 V500, CD11b BV605, CD71 BV711, and CD15 BV786. All MoAbs were obtained from BD Biosciences (San Jose, CA, USA). Data acquisition was performed in a FACSLyric flow cytometer (BD Biosciences) with the FACSuite software. For data analysis, the Flow Jo v7.6.5 software (Tree Star) was used.
The immunohistochemical examination was performed using routine diagnostic procedures with standardized IVD antibodies (Dako Autostainer and Dako Omnis).
Genomic DNA was extracted from peripheral blood leukocytes using the QIAamp DNA Blood Mini Kit (Qiagen). KIT (exon 17), IDH1 (exon 4), IDH2 (exon 4), TP53 (exons 2–11), ASXL1 (exon 12), SRSF2 (exon 1 fragment), and RUNX1 (exons 2–9) were each amplified by polymerase chain reaction (PCR) using suitably positioned primers. Primer sequences were checked for the presence of single-nucleotide polymorphism (SNPs) on their complementary DNA strands. PCR products were sequenced using internal primers and analyzed using an ABI 310 Genetic Analyzer (Applied Biosystems). The presence of JAK2 V617F was excluded by allele-specific PCR.
Results
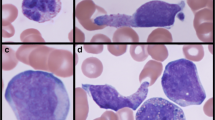
Bone marrow aspirate smears were hypercellular, displaying indisputable morphologic features of erythroid and myeloid cell dysplasia. Atypical cells accounted for 26% of all bone marrow aspirate cells and additionally formed numerous compact aggregates. Morphologically atypical cells did not resemble MCs but did appear similar to atypical monocytes (Fig. 1). Their cytoplasm contained small, faintly visible granules. The chromatin pattern appeared mature. In some microscopic fields of view, cells with elongated or bilobed nuclei were found. Metachromatic blasts and spindle-shaped MCs were not present in bone marrow smears. Additional bone marrow studies allowed the diagnosis to be established.

Bone marrow aspirate smears in MGG staining. Cytomorphologic features of hypogranulated mast cells (a–e); asterisks indicate dysplastic erythroid precursors and dysplastic granulopoiesis; arrows indicate bilobed nuclei and finer, immature chromatin pattern (promastocytes), magnification 1000 × . Aggregate of mast cells (f), magnification 100 ×
Immunophenotypic examination of bone marrow showed the presence of 45% immature cells SSC low, CD45 + dim (blast gate) expressing myeloid associated markers: CD117 + (bright expression), CD13 + , CD33 + , CD64 + and lymphoid markers CD22 + (low expression), and CD30 + . Cells were negative for other immaturity markers, such as CD34, HLA-DR, and CD123, and did not express CD7, CD11b, CD11c, CD14, CD15, CD16, CD19, CD36, CD38, CD56, or CD71. Only 3% of the blasts displayed surface markers of clonal MCs: CD25 and CD2. Representative flow cytometry dot plots are shown in Fig. 2.

Representative flow cytometry dot plots. Blast population (SSC low, CD45 + dim) shown in pink, lymphocytes (SSC low, CD45 + bright) shown in blue
At a later stage of treatment, the results of the trephine biopsy were obtained, which demonstrated infiltration by atypical cells occupying > 95% of the intertrabecular marrow space and at first indicated low-differentiated acute myeloid leukemia. Further histopathological image analysis and additional immunohistochemical staining confirmed the diffuse infiltration of atypical mast cells presenting the immunophenotype: MCT( +), CD117( +), CD30( ±), CD25( ±), CD2( −), CD1a( −), MPO( −), and Ki-67( +) in 5% of nuclei (Fig. 3).

Bone marrow histopathological image of MCL, including the immunohistochemical staining with mast cell tryptase (MCT), CD117, and CD30
Cytogenetic BM examination revealed a normal female karyotype. Molecular tests for the KIT D816V mutation, JAK2 V617F mutation, and SRS2 codon P95 mutation were negative. Sanger sequencing of KIT (exon 17), IDH1/IDH2, TP53 (exons 2–11), ASXL1 (exon 12), and RUNX1 (exons 2–9) identified no pathogenic or likely pathogenic variants.
Discussion
MCL is one of the rarest types of leukemia, accounting for approximately 1% of all systemic mastocytosis cases [2]. MCL is diagnosed when criteria for systemic mastocytosis are fulfilled and bone marrow biopsy shows ≥ 20% leukemic infiltration by atypical/immature MCs. Leukemic mast cells may exhibit immature forms rarely seen in other types of SM: promastocytes, metachromatic blasts, and even ungranulated blasts, which pose a diagnostic challenge to identify due to the diverse cytomorphological characteristics [2, 23]. Research shows that spindle-shaped MCs are not found or are very rare in patients with acute MCL [2]. In the presented case, the bone marrow examination demonstrated a multifocal infiltration by atypical mast cells/blasts which were not spindle-shaped and had a hypogranulated cytoplasm. The morphology of some of the cells resembled that of promastocytes (bilobed or intended bilobed with a finer chromatin pattern). Neoplastic MCs type II (promastocytes), with indented, bilobed nuclei, and an abundant cytoplasm, are repeatedly reported in MCL cases [24,25,26,27]. Cells with identical cytomorphology were found in May-Grünwald-Giemsa-stained peripheral blood smears. These atypical MCs represented 11% of nucleated cells, indicating a “leukemic” subvariant of MCL.
In the presented case, the activating driver KIT mutation was not detected. In contrast to other SM types, where KIT D816V is present in 80–90% of cases, only 40–67% of MCL cases carry this hallmark mutation [2, 28, 29]. In the study of Jahwar et al., KIT mutations were detected in 25 (89%) of the 28 MCL patients: D816V (n = 19), D816H/Y (n = 5), and F522C (n = 1) [30]. In our case, Sanger sequencing was only performed for exon 17 of the KIT gene, where the most common somatic mutation in SM, KIT D816V, occurs [29]. Nevertheless, KIT mutations other than D816V in the extracellular domain or juxtamembrane domain may occur in MCL, but the remaining exons were not tested in our case [31, 32]. The Sanger sequencing VAF (variant allele frequency) detection limit was estimated at 20%. It is possible that sequencing PB samples in which mast cells accounted for only 11% of nucleated cells failed to detect mutations in the KIT gene. However, according to Hoerman et al., KIT D816V allele burden is not correlated with MC infiltration but rather with serum tryptase levels [33]. Recent data have shown that the KIT D816V allele burden measured using digital droplet PCR in formalin-fixed, paraffin-embedded BM sections reflects the burden of neoplastic cells in SM better than established mutant allele burden measurements in PB or BM aspirates [34]. In our case, a thorough examination of the BM for the presence of KIT D816V or NGS sequencing of the entire KIT gene to exclude mutations in exons outside of exon 17 was not possible. Efforts have been made to improve the sensitivity of KIT mutation detection using different approaches, including the enrichment of MCs from BM samples via laser micro-dissection, fluorescence-activated cell sorting (FACS), or a sensitive PCR assay; however, these techniques are not routinely used for diagnostic algorithms [31, 34].
The immunophenotypic identification of MCs was based on their strong reactivity for CD117 and low CD45 expression in the absence of expression for CD34 and CD38 to discriminate MC from CD34 progenitors and CD117 + plasma cells. The panel of antibodies used did not allow a clear classification of the MS type, as proposed by Teodosio et al. [13], and the final diagnosis was based on histopathological studies and the clinical picture. In this case, only a small number of all atypical mast cells met the minor immunophenotypic SM criterion and displayed the aberrant expression of CD2 and CD25. In turn, all of those cells expressed CD30. The co-expression of CD2 and CD25 on MCs is more frequent among KIT D816V-positive MCL patients (66%) compared to KIT D816V-negative MCL patients (25%) [2, 23]. Recent data suggest that aberrant CD30 expression on mast cells is strongly associated with SM [23, 35]. CD30-positive MCs are found even in patients lacking CD2 and CD25 expression. Therefore, CD30 was proposed as a new addition to the existing minor SM criterion [35]. Well-differentiated systemic mastocytosis (WDSM) patients do not typically carry the D816V KIT mutation without CD2 and CD25 expression. MCs in WDSM are also typically CD30-positive, although they are characterized by mature, hypergranulated morphology FSChigh/SSChigh without atypical features, in contrast to our case [13]. CD64 (immunoglobulin receptor FCγRI) is not typically expressed on normal/reactive bone marrow MCs. In turn, as an activation-associated molecule, the increased expression of CD64 was observed in approximately 84% of SM cases, and its expression on MCs can be significantly upregulated by IFN-γ, leading to degranulation and enhanced cytokine expression [36]. According to Teodosio et al., an increased reactivity for CD64 and a lack of CD16 (FcγRIIIa) expression may characterize MCL cases [12].
Severe dysplasia found in more than 10% of patient’s BM cells suggests that myelodysplastic syndrome criteria are met and a diagnosis of MCL with an associated hematologic neoplasm (MCL-AHN) can be made. Although additional somatic mutations have been found in 90% of advanced SM patients (most with an SM-AHN) [37], neither mutations within SRS2, IDH1/IDH2, TP53, ASXL1, or the RUNX1 genes nor cytogenetic abnormalities were detected in our case.
Because neoplastic cells in our case lack a KIT D816V mutation, most of them did not express CD25, so CD2 differential diagnoses of tryptase-positive acute myeloid leukemia (T + AML) and myelomastocytic leukemia (MML) were taken into consideration. T + AML blasts co-express CD34 and tryptase but not CD117 [15]. In our case, blasts were strongly positive for CD117, the hallmark marker for MC lineage. In MML, mast cells express CD117 and tryptase but lack CD2 and CD25 and usually comprise more than 10% of nucleated cells in bone marrow; however, in contrast to our case, they never form compact focal infiltrates [15, 27]. Additionally, serum tryptase levels in MML rarely exceed 100 ng/mL [27].
Clinical, radiological, and laboratory results in the reported case showed the presence of multiple “cytoreduction-requiring” findings, suggesting the diagnosis of the acute form of MCL and a progressive disease course. Dysplasia was found in erythroid and myeloid lineages in BM smears. Serum tryptase was highly elevated, and the patient presented splenomegaly. The patient suffered from abdominal pain and gastrointestinal distress. All of these symptoms may indicate organ impairment due to MC infiltrates. The patient was therefore classified into the poorest-risk group whose overall survival (OS) was historically short, with a median of 6 to 18 months [29, 37]. Durable responses and survival of 3–5 years regardless of KIT mutation status can be seen in some MCL patients, when treated with kinase inhibitors, including midostaurin [2, 37]. In the presented case, firstly, remission-induction treatment was administered, and midostaurin treatment was planned at a later stage. Midostaurin was not reimbursed in Poland but was available in the Novartis Managed Access Program.
The vigilance of clinical laboratory scientists, along with a proper differential diagnosis, a histopathologist, and a medical doctor, are all necessary to make an exact MCL diagnosis. Then, individualized MCL treatment should be selected. The outcomes in patients with MCL are not satisfactory, and new drugs with the potential to improve the treatment efficacy and prolong survival are needed. Understanding the complex genetic and molecular pathogenesis of MCL may support the development of targeted therapy.
Data availability
Not applicable.
References
Lewandowski K (2020) Mastocytosis. Oncology in Clinical. Practice 6:86–98
Georgin-Lavialle S, Lhermitte L, Dubreuil P et al (2013) Mast cell leukemia. Blood 121:1285–1295. https://doi.org/10.1182/blood-2012-07-442400
Valent P, Oude Elberink JNG, Gorska A et al (2019) The Data Registry of the European Competence Network on Mastocytosis (ECNM): set up, projects, and perspectives. J Allergy Clin Immunol Pract 7:81–87. https://doi.org/10.1016/j.jaip.2018.09.024
Valent P, Akin C, Metcalfe DD (2017) Mastocytosis: 2016 updated WHO classification and novel emerging treatment concepts. Blood 129:1420–1427. https://doi.org/10.1182/blood-2016-09-731893
Horny HP, Akin C, Arber DA, Peterson LC,Tefferi A (2016) Mastocytosis. In: Swerdlow E et al, (ed) World Health Organization (WHO) Classification of Tumours. Pathology & Genetics. Tumours of Haematopoietic and Lymphoid Tissues, 4th edn. IARC Press, Lyon, France.
Valent P, Akin C, Hartmann K et al (2017) Advances in the classification and treatment of mastocytosis: current status and outlook toward the future. Cancer Res 77:1261–1270. https://doi.org/10.1158/0008-5472.CAN-16-2234
Hans-Peter H, Sotlar K, Metzgeroth G, Reiter A,Valent P (2018) Mastocytosis and the updated WHO classification (2016, 2017): what is really new? Ann Hematol Oncol. 5(2):1194.
Pardanani A (2016) Systemic mastocytosis in adults: 2017 update on diagnosis, risk stratification and management. Am J Hematol 91:1146–1159. https://doi.org/10.1002/ajh.24553
Jawhar M, Schwaab J, Schnittger S et al (2015) Molecular profiling of myeloid progenitor cells in multi-mutated advanced systemic mastocytosis identifies KIT D816V as a distinct and late event. Leukemia 29:1115–1122. https://doi.org/10.1038/leu.2015.4
Costopoulos M, Uzunov M, Bories D et al (2018) Acute mast cell leukemia: a rare but highly aggressive hematopoietic neoplasm. Diagn Cytopathol 46(7):639–641
Horny HP, Sotlar K, Valent P (2014) Mastocytosis: immunophenotypical features of the transformed mast cells are unique among hematopoietic cells. Immunol Allergy Clin North Am 34:315–321. https://doi.org/10.1016/j.iac.2014.01.005
Teodosio C, Garcia-Montero AC, Jara-Acevedo M et al (2010) Mast cells from different molecular and prognostic subtypes of systemic mastocytosis display distinct immunophenotypes. J Allergy Clin Immunol 125:719–726. https://doi.org/10.1016/j.jaci.2009.10.020
Teodosio C, Mayado A, Sanchez-Munoz L et al (2015) The immunophenotype of mast cells and its utility in the diagnostic work-up of systemic mastocytosis. J Leukoc Biol 97:49–59. https://doi.org/10.1189/jlb.5RU0614-296R
Bosch-Vilaseca A, Monter-Rovira A, Cisa-Wieczorek S et al (2019) Ultrastructural, cytogenetic, and molecular findings in mast cell leukemia: case report. Clin Case Rep 7:1395–1398. https://doi.org/10.1002/ccr3.2208
Zheng Y, Nong L, Liang L, Wang W, Li T (2018) De novo mast cell leukemia without CD25 expression and KIT mutations: a rare case report in a 13-year-old child. Diagn Pathol 13:14. https://doi.org/10.1186/s13000-018-0691-2
Pardanani A (2021) Systemic mastocytosis in adults: 2021 update on diagnosis, risk stratification and management. Am J Hematol 96:508–525. https://doi.org/10.1002/ajh.26118
Growney JD, Clark JJ, Adelsperger J et al (2005) Activation mutations of human c-KIT resistant to imatinib mesylate are sensitive to the tyrosine kinase inhibitor PKC412. Blood 106:721–724. https://doi.org/10.1182/blood-2004-12-4617
Dubreuil P, Letard S, Ciufolini M et al (2009) Masitinib (AB1010), a potent and selective tyrosine kinase inhibitor targeting KIT. PLoS ONE 4:e7258. https://doi.org/10.1371/journal.pone.0007258
Hochhaus A, Baccarani M, Giles FJ et al (2015) Nilotinib in patients with systemic mastocytosis: analysis of the phase 2, open-label, single-arm nilotinib registration study. J Cancer Res Clin Oncol 141:2047–2060. https://doi.org/10.1007/s00432-015-1988-0
Verstovsek S, Tefferi A, Cortes J et al (2008) Phase II study of dasatinib in Philadelphia chromosome-negative acute and chronic myeloid diseases, including systemic mastocytosis. Clin Cancer Res 14:3906–3915. https://doi.org/10.1158/1078-0432.CCR-08-0366
Gotlib J, Kluin-Nelemans HC, George TI et al (2016) Efficacy and safety of midostaurin in advanced systemic mastocytosis. N Engl J Med 374:2530–2541. https://doi.org/10.1056/NEJMoa1513098
Ustun C, Reiter A, Scott BL et al (2014) Hematopoietic stem-cell transplantation for advanced systemic mastocytosis. J Clin Oncol 32:3264–3274. https://doi.org/10.1200/JCO.2014.55.2018
Valent P, Sotlar K, Sperr WR et al (2015) Chronic mast cell leukemia: a novel leukemia-variant with distinct morphological and clinical features. Leuk Res 39:1–5. https://doi.org/10.1016/j.leukres.2014.09.010
Soliman DS, Al-Sabbagh A, Ibrahim F et al (2019) Highly aggressive CD4-positive mast cell leukaemia (leukaemic variant) associated with isolated trisomy 19 and hemophagocytosis by neoplastic mast cells: a case report with challenging experience and review. Case Rep Hematol 2019:1805270. https://doi.org/10.1155/2019/1805270
Bae MH, Kim HK, Park CJ et al (2013) A case of systemic mastocytosis associated with acute myeloid leukemia terminating as aleukemic mast cell leukemia after allogeneic hematopoietic stem cell transplantation. Ann Lab Med 33(2):125–129
Lopes M, Teixeira MDA, Casais C et al (2018) KIT D816V positive acute mast cell leukemia associated with normal karyotype acute myeloid leukemia. Case Rep Hematol 2018:3890361. https://doi.org/10.1155/2018/3890361
Wang RC, Ward D, Dunn P, Chang CC (2017) Acute mast cell leukemia associated with t(4;5)(q21;q33). Hum Pathol 67:198–204. https://doi.org/10.1016/j.humpath.2017.03.014
Galura GM, Cherukuri SV, Hakim N, Gaur S, Orazi A (2020) Acute aleukemic mast cell leukemia: report of a case and review of the literature. Leuk Res Rep 14:100230. https://doi.org/10.1016/j.lrr.2020.100230
Gilreath JA, Tchertanov L, Deininger MW (2019) Novel approaches to treating advanced systemic mastocytosis. Clin Pharmacol 11:77–92. https://doi.org/10.2147/CPAA.S206615
Jawhar M, Schwaab J, Meggendorfer M et al (2017) The clinical and molecular diversity of mast cell leukemia with or without associated hematologic neoplasm. Haematologica 102(6):1035–1043
Arock M, Sotlar K, Akin C et al (2015) KIT mutation analysis in mast cell neoplasms: recommendations of the European Competence Network on Mastocytosis. Leukemia 29:1223–1232. https://doi.org/10.1038/leu.2015.24
Chatterjee A, Ghosh J, Kapur R (2015) Mastocytosis: a mutated KIT receptor induced myeloproliferative disorder. Oncotarget 6(21):18250–18264
Hoermann G, Gleixner KV, Dinu GE et al (2014) The KIT D816V allele burden predicts survival in patients with mastocytosis and correlates with the WHO type of the disease. Allergy 69:810–813. https://doi.org/10.1111/all.12409
Greiner G, Gurbisz M, Ratzinger F et al (2020) Molecular quantification of tissue disease burden is a new biomarker and independent predictor of survival in mastocytosis. Haematologica 105:366–374. https://doi.org/10.3324/haematol.2019.217950
Valent P, Akin C, Hartmann K et al (2021) Updated diagnostic criteria and classification of mast cell disorders: a consensus proposal. Hemasphere 5:e646. https://doi.org/10.1097/HS9.0000000000000646
Woolhiser MR, Okayama Y, Gilfillan AM, Metcalfe DD (2001) IgG-dependent activation of human mast cells following up-regulation of FcgammaRI by IFN-gamma. Eur J Immunol 31:3298–3307. https://doi.org/10.1002/1521-4141(200111)31:11%3c3298::aid-immu3298%3e3.0.co;2-u
Reiter A, George TI, Gotlib J (2020) New developments in diagnosis, prognostication, and treatment of advanced systemic mastocytosis. Blood 135:1365–1376. https://doi.org/10.1182/blood.2019000932
Acknowledgements
We thank Dr. Hubert Heleniak and Dr. Aleksandra Gołos for providing additional clinical information.
Funding
No funding was received.
Author information
Authors and Affiliations
Contributions
EZP and JCT wrote the manuscript. JCT and AK performed the flow cytometry. EZP performed the cytomorphological staining and analysis. EP analyzed the clinical data. ASC performed the histopathology and IHC staining and analysis. All authors edited and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical approval
Not applicable.
Consent for publication
All authors gave consent for the manuscript publication.
Informed consent
Not applicable.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zaremba-Pataj, E., Patkowska, E., Krzywdzińska, A. et al. Acute mast cell leukemia without KIT D816V mutation and lack of CD2 and CD25—a case report of rare entity. J Hematopathol 16, 39–47 (2023). https://doi.org/10.1007/s12308-022-00526-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12308-022-00526-3