
Abstract
The increasing number of new fungal species described from all over the world along with the use of genetics to define taxa, has dramatically changed the classification system of early-diverging fungi over the past several decades. The number of phyla established for non-Dikarya fungi has increased from 2 to 17. However, to date, both the classification and phylogeny of the basal fungi are still unresolved. In this article, we review the recent taxonomy of the basal fungi and re-evaluate the relationships among early-diverging lineages of fungal phyla. We also provide information on the ecology and distribution in Mucoromycota and highlight the impact of chytrids on amphibian populations. Species concepts in Chytridiomycota, Aphelidiomycota, Rozellomycota, Neocallimastigomycota are discussed in this paper. To preserve the current application of the genus Nephridiophaga (Chytridiomycota: Nephridiophagales), a new type species, Nephridiophaga blattellae, is proposed.
Similar content being viewed by others


Introduction
Fungi are primarily heterotrophic, nutrition-absorptive (osmotrophic) eukaryotes that exist in every ecological niche. They grow within their food, digesting it externally and absorbing nutrients across a semi-rigid chitinous cell wall during key phases of their vegetative life cycle (Voigt 2012a; James and Berbee 2012; Stajich et al. 2009). A total of 150,246 species of fungi—which match the above-mentioned definition—are currently recognized (Species Fungorum at www.speciesfungorum.org, accessed 19th April, 2021). This is a significant increase on the estimated number (ca. 100,000) reported in 2008 (Kirk et al. 2008). Concurrently, the number of new species annually described in the twenty-first century is rising. Estimations of the extant fungal species range between 1.5 and 3.8 million (Hawksworth 1991, 2001a, b; Hawksworth and Lücking 2017). Assuming no redundancy or error, only 3.8–8.8% of the estimated fungal species were described. This makes a twofold up to 11-fold increase. Molecular operational taxonomic units (MOTUs), in particular, represent a hidden treasure of likely undescribed taxa. Expanding databases to enable the molecular identification of these undescribed taxa should be a priority for fungal taxonomists (Kõljalg et al. 2005). For instance, a study of the basal genus Mortierella revealed a large contribution of reference collections to the identification of fungal environmental samples (Nagy et al. 2011).
Compared to higher fungi (Dikarya), taxonomic and evolutionary studies on the basal fungal lineages are few. There are more than 200 orders of fungi that are classified into a total of 19 fungal phyla (Wijayawardene et al. 2020). Phylogenomic studies are increasingly used to reveal the evolution and phylogeny of fungal taxa. Recently, Galindo et al. (2021) proposed a new phylum, Sanchytriomycota, a sister clade to Blastocladiomycota, which would increase the number of fungal phyla to 20 of which 17 are basal. Members of basal fungal groups produce both motile and non-motile sporangiospores (Fig. 1). Some species of basal fungi are important in biotechnological areas, such as production of enzymes, lipids and antifungal proteins, and also known as opportunistic pathogens (Walther et al. 2019, 2020). The chytrid fungus Batrachochytrium dendrobatidis has been linked to regional and global declines of amphibian populations (Fisher et al. 2009, 2018; Hyde et al. 2018), resulting from disease outbreaks of amphibian chytridiomycosis (Farrer et al. 2011; James et al. 2009; Schloegel et al. 2009). Anaerobic fungi colonize the digestive tracts of herbivorous vertebrates and play a significant role in the breakdown of lignocellulosic feed, providing a source of fermentable sugars for other microbes and the host (Flad et al. 2020). They possess a range of cell wall-degrading enzymes, making them efficient degraders of plant biomass and inexpensive carbohydrates materials (Haitjema et al. 2014). These potent enzymes have received much attention in recent years for their biotechnological and industrial applications.

Diversity of basal lineage fungi. a Gongronella koreana EML-TS2Bp (sporangiophore with sporangium). b Absidia koreana EML-IFS45-1 (sporangiophore with a sporangium). c Mucor cheongyangensis CNUFC ICL1 (sporangiophore and mature sporangium). d Cunninghamella elegans EML-RUS1-1 (vesicle bearing sporangiola). e Syncephalastrum monosporum EML-BT5-1 (vesicle bearing merosporangiola). f Backusella locustae CNUFC-SFB2 (multispored sporangiola). g Pilobolus crystallinus CNUFC-EGF1-4 (yellow and black sporangia at the tips of the sporangiophores on water deer dung). h Syzygites megalocarpus CNUFC SM01 (zygosporangium with suspensors). i Mucor orantomantidis CNUFC MID1-1 (zygosporangium with suspensors). j Chytriomyces hyalinus CNUFC HRW1-3 (mature zoosporangium with branched rhizoids). k isolate CNUFC AMS2 (mature thallus with two prominent discharge papillae). l isolate CNUFC CHS1-1 (zoospores). m isolate CNUFC 19JW3 (mature thallus). n isolate CNUFC PS10 (multi-spored sporangiolum). o isolate CNUFC IS1 (multi-spored sporangiolum borne on circinate branches). Scale bars = 20 μm, except h, i = 50 μm
In this review, we discuss current species concepts, the ecology and distribution of basal lineages of fungi, and propose novel insights into their phylogeny.
Phylogeny and evolution of early-diverging fungi
Traditional taxonomy splits the fungi into two derived lineages, the Ascomycota and the Basidiomycota, and two basal lineages, the chytrids or zoosporic (flagellate) fungi (Chytridiomycota) and the zygosporic fungi (Zygomycota), of which the former phylum as defined by Barr (2001) is aquatic and ancestral to the terrestrial fungi and the latter one among the first terrestrial fungi appearing on Earth (Berbee and Taylor 2001; Liu and Voigt 2010; Mendoza et al. 2014). The morphological diversity of species, which are representative for basal lineage fungi, is illustrated in Fig. 1. The use of gene trees based on DNA sequence data to define taxa has largely accelerated the research on fungal phylogenetics influencing and revolutionizing the systematics of the fungi, especially the classification of the Zygomycota, which has changed significantly over the past four decades (Hawksworth et al. 1983, 1995; Kirk et al. 2001, 2008; Voigt and Kirk 2014).
Analysis of large-scale multigene phylogenies and morphological data suggested paraphyletic origins of both basal phyla (James et al. 2006a). The chytrids form zoospores terminally equipped with a single posterior flagellum of the whiplash type (opisthokont) during the vegetative and generative phases of their life cycle. They split into five clades, four of which were given the rank of classes (or even phyla); these are: Blastocladiomycetes Doweld (Blastocladiomycota T. Y. James: James et al. 2006b); Chytridiomycetes Caval.-Sm. (Cavalier-Smith 1998; Chytridiomycota Doweld); Neocallimastigomycetes M. J. Powell (Hibbett et al. 2007); Monoblepharidomycetes J. H. Schaffn. (Schaffner 1909; Monoblepharidomycota Doweld). Whilst the Blastocladiomycetes, the Chytridiomycetes and the Monoblepharidomycetes are aerobic-flagellate fungi occurring mainly as saprobes or parasites of plants, animals, protists or algae primarily inhabiting aquatic environments, the Neocallimastigomycetes are anaerobic-flagellate fungi that inhabit the digestive systems of herbivorous mammals and reptiles, with the more specialized forms populating the rumen of ruminants (Fliegerova et al. 2012, 2015). Synapomorphic characters are the presence and absence of a prominent basal cell adjacent to the reproductive structure, sexual conjugation via anisogamy, isogamy, oogamy and anaerobic growth for the Blastocladiomycetes, the Chytridiomycetes, the Monoblepharidomycetes and the Neocallimastigomycetes, respectively (Voigt 2012b; Shelest and Voigt 2014). The fifth clade of the chytrids consists of the genus Olpidium, which comprises unicellular, obligate parasites of plants that reproduce with flagellate, swimming zoospores (Voigt et al. 2013). This genus remains a distinct clade at present, though Olpidium was postulated to form a monophyletic group with taxa traditionally classified in the Zygomycota (Sekimoto et al. 2011; Chang et al. 2021). Topology tests rejected all alternative trees that constrained the species of Olpidium to cluster with other groups of flagellate fungi. The Zygomycota is considered to depict the most basal terrestrial phylum to have evolved from flagellate, aquatic ancestors. Molecular phylogenetic analyses revealed dispersal into five subphyla containing one to four orders (Hibbett et al. 2007; Hoffmann et al. 2011).
Based on the potential of all five subphyla to produce zygospores during sexual conjugation of two yoke-shaped gametangia, they are referred to a morphological group named the Zygomycota for zygosporic fungi as a whole, which share morphological features but consist of subphyla whose phylogenetic relationships are not completely resolved. Revisions using large-scale multigene phylogenies resulted in conflicting phylogenetic relationships among the basal fungal clades (James et al. 2006a; Liu et al. 2006). The Zygomycota group (including Mucorales, Glomerales, Entomophthorales and Harpellales) appears monophyletic in a phylogenetic analysis based on the deduced amino acid sequences of the genes encoding RNA polymerase II subunits 1 and 2 (RPB1 and RPB2; Liu et al. 2006), but polyphyletic in a six gene phylogeny using RPB1 and RPB2 in addition to the genes encoding translation elongation factor 1α (TEF), β-tubulin (BTUB) as well as the nuclear small (18S) and the large (28S) subunit ribosomal RNA (rRNA) encoded by the 18S (SSU) and the 28S (LSU) rDNA, respectively (James et al. 2006a). RPB1 and RPB2 were shown to be highly informative and discriminate elegantly at species level, whilst the other marker genes are homoplasious due to convergent or parallel evolution (Schoch et al. 2012).
Nevertheless, the deep-level phylogenetic relationships among the basal lineage fungi remained unsatisfactorily resolved when an oligogenic approach was used for phylogenetic reconstruction. In an endeavour to increase the phylogenetic signal by augmentation of the number of informative characters phylogenomic studies arose, which applied a multitude of orthologous genes and proteins to resolve the deep branches in the fungal tree (Spatafora et al. 2016; Chang et al. 2021).
Members of the phylum Cryptomycota M.D.M. Jones & T.A. Richards (Jones et al. 2011a) were proposed to represent evolutionary intermediates between animals and fungi, which cluster at the base of the fungal tree. Contemporarily the new phylum Rozellomycota was proposed by James and Berbee (2012) and was validly published by Doweld (2013). The name Rozellomycota is used in this review.
Members of the Rozellomycota most strikingly lack a chitinous cell wall during food absorption (James and Berbee 2012) and are almost exclusively known from ubiquitous environmental samples by sequencing genes encoding ribosomal RNA (rRNA) (Lara et al. 2010; Jones et al. 2011b). It has been proposed that the Rozellomycota may be divergent fungi that evolved from an ancestor with a nearly complete suite of classical fungal-specific characters (James and Berbee 2012). The nuclear genome of Rozella allomycis encodes four chitin synthases, including one with a myosin domain, and lacks a large number of genes for primary metabolism (James et al. 2013). Rozella is a genus of endoparasites of a range of primarily water mold hosts and a key to a total of 27 species of the genus was published by Letcher and Powell (2018). Molecular phylogenies based on SSU rRNA revealed the existence of a large and widespread group of eukaryotes, branching at the base of the fungal tree (Corsaro et al. 2014). This group, comprising almost exclusively environmental clones, includes the endoparasitic Rozella as the unique known representative and two endonuclear rozellids, which have microsporidia-like ultrastructural features and parasitize free-living naked amoebae. Similar to microsporidia, these endoparasites form unflagellated walled spores and grow inside the host cells as unwalled non-phagotrophic meronts and were named Paramicrosporidium, appearing to be the morphological missing link between fungi and Microsporidia. Classification system of basal fungi is still controversy. In this paper, we reviewed the current classification of basal fungi in Wijayawardene et al. (2020), and Galindo et al. (2021). Seventeen phyla of basal fungi are shown in Table 1.
The basal lineages of the fungi appear in a multitude of paraphyletic relationships to the Dikarya, encompassing Ascomycota, Basidiomycota, and Entorrhizomycota (Fig. 2). Based on current tree inferences from a concatenated alignment of SSU and LSU rRNA genes of nine nephridiophagid species, the order Nephridiophagales has clearly been shown to be affiliated to the Chytridiomycota (Strassert et al. 2021). This relationship is confirmed in our phylogenetic analysis (Fig. 2). The analysis performed on the basis of the LSU and SSU combined sequence data (Fig. 2) showed that CNUFC CHS1-1, CNUFC AMS2, CNUFC CPW7-3, CNUFC GFW2, CNUFC 19JW3, and CNUFC AFW4-2 appear to be more related to the phylum Chytridiomycota; whereas CNUFC PS10 and CNUFC IS1 grouped within the Mucorales (Mucoromycotina, Mucoromycota). However, they form distinct separate lineages, which may present them as interesting new taxa.

Phylogram generated from Maximum Likelihood analysis based on LSU and SSU combined sequence data showing evolutionary relationships of basal fungal phyla and relatives. The tree was inferred using 89 taxa and 3,790 aligned nucleotide sites. Support in nodes is indicated above or below branches and is represented by bootstrap values (ML analysis) of 70% and higher. Full-supported branches (100% BS) are highlighted by thickened branches. Analysis performed using RAxML-HPC2 on the CIPRES Science Gateway server with 1,000 bootstrap replicates and the GTRGAMMA model of nucleotide substitution. The tree includes six outgroup taxa including Acanthoeca spectabilis, Capsaspora owczarzaki, Creolimax fragrantissima, Monosiga brevicollis, Paramonosiga thecata and Salpingoeca rosetta. Strains in bold blue refer to taxa not designated yet to a specific group. The corresponding sequences were generated during this study
Species concepts
Species concepts in Chytridiomycota
Overview of the problems and methods
In the last 20 years our understanding of the phylogenetic relationships and the taxonomy of the zoosporic true fungi has been completely revised. Phylogenetic studies upended the taxonomy of Chytridiomycota by showing it was not a natural group, but rather a paraphyletic grade of lineages retaining motility and diverging long before terrestrial fungi diversified (Letcher et al. 2006; James et al. 2006b; Sekimoto et al. 2011). Robust inference of monophyletic groups was made possible by the use of phylogenies based on DNA sequence data. These data helped to explain the incredible diversity of zoospore ultrastructure and showed that ultrastructural characters of the zoospore could be used to revise the systematics of the group (James et al. 2000; Letcher et al. 2005, 2008a, b; Mozley-Standridge et al. 2009; Simmons et al. 2009). Currently a dual approach combining both DNA sequence and ultrastructural data is used for the taxonomy of zoosporic fungi, and the majority of new taxa, from orders down to species are accompanied with both forms of data (Simmons et al. 2009; Karpov et al. 2014a; Longcore et al. 2020; Seto et al. 2020).
In this period of increased appreciation of zoosporic fungal diversity, the number of phyla has increased to 5 or 7 depending on whether Rozellomycota and Aphelidiomycota are included within fungi (Karpov et al. 2014b; Tedersoo et al. 2018; James et al. 2020), and the number of orders has blossomed to 22. Many of the new orders reflect taxa that are unculturable, the majority of them phycoparasites or mycoparasites (Karpov et al. 2014a, 2016; Seto et al. 2020). The uncultured and unnamed diversity, the so called “dark matter fungi” (Grossart et al. 2016) apparently comprises a large amount of taxonomic diversity of chytrids. Although the deep relationships among the chytrid phyla and orders have yet to be resolved, orders are now used to describe clearly monophyletic groups, whose ultrastructure is relatively consistent. Revisionary work has been conducted on several of the major orders. During these monographic treatments a number of taxonomic questions have arisen. Firstly, how best should families and genera be defined, and specifically what combination of characters should be used? Gross morphological characters of the thallus have been shown to be nearly useless due to homoplasy (James et al. 2000). Ultrastructural characters are less likely to be homoplastic, but a careful analysis of which characters are more consistent has not been accomplished. Moreover, expertise in interpretation of electron microscopy data is waning and the equipment is not fully accessible to all researchers.
A second set of questions arising from the monographic work is how we should define species, and specifically what species concepts should be employed. In general, the mycological community is focused on using an evolutionary species concept that relies on phylogenetic data to delimit species that are not undergoing genetic exchange (Taylor et al. 2000). Most studies of zoosporic fungal diversity use conserved rRNA (nuclear large and small subunits) genes to generate phylogenies. This tool may not be appropriate to split species, and the use of the non-coding rRNA loci, such as the internal transcribed spacer (ITS) could help (Schoch et al. 2012). Does this move to the use of DNA sequences and ultrastructure in systematic taxonomy leave any room for morphology? Specifically, will morphological or even ecological species concepts have any utility when applied to zoosporic fungi at the species level? Systematists struggle with the fact that most of the named species are recorded in papers published before both widespread culturing of taxa and molecular data existed. In the absence of host information, it is often hard to ascribe isolated chytrid taxa with previous names, particularly given the evidence that suggests most morphological characters are poor indicators of phylogenetic relatedness. Some taxa, however, such as Lobulomyces poculatus and Podochytrium dentatum are highly distinctive, suggesting that with the right lens or mindset the proper characters could be found for morphological species definitions. For pathogenic species, how stable and useful is host range for delimiting taxa? Could recommendations be provided for future research on zoosporic fungal diversity with respect to defining species concepts across labs? In the following sections we summarize the current state of the taxonomy of three groups of chytrids discussing these taxonomic and evolutionary issues in turn. We include a discussion on how to apply names to observations, cultures, and environmental DNA sequences.
Species concepts in Cladochytriales
The current members of Cladochytriales (seven genera) were previously classified within the Chytridiales based on morphological characters of the thallus and developmental stages, but were segregated into a new order after phylogenies of SSU and LSU rDNA showed that the genera were not related to Chytridiales (Mozley-Standridge et al. 2009). These data corroborated the differences in zoospore ultrastructure found for these two groups (Lucarotti 1981; Barr 1986; Barr et al. 1987). Currently, classical morphological characters such as operculation, septation on rhizomycelial swellings (turbinate cells), apophysis and catenulations are no longer considered reliable because they are shared by independent lineages (James et al. 2000, 2006b; Powell et al. 2018b; Jerônimo et al. 2019).
The ultrastructural characters of the zoospore have proven to be useful and, in some cases, more informative than morphology. However, in Cladochytriales, they still present some obstacles for their use. The necessity of an enormous production of zoospores in pure cultures is a limitation for the polycentric species (e.g. Nowakowskiella J. Schrot. and Cladochytrium Nowak.), which account for the majority of the order and in general do not produce sufficient zoospores in culture medium (Powell et al. 2018b; Jerônimo et al. 2019). In Cladochytriales, only six species (Allochytridium expandens Salkin, Allochytridium luteum Barr, Catenochytridium hemicysti J.S. Knox, Cladochytrium replicatum Karling, Nephrochytrium sp. JEL125 and Nowakowskiella elegans (Nowak.) J. Schrot.), have been investigated by transmission electron microscopy. A unifying zoospore ultrastructural character of the group is the presence and configuration of the microtubular root, in which fibrous linkers connect the microtubules (Mozley-Standridge et al. 2009). This feature and the absence of a paracrystalline inclusion in the zoospore distinguish members of the Cladochytriales from Chytridiales. Mozley-Standridge et al. (2009) hypothesized that variation in configuration of the fenestrated cisterna may be diagnostic at the generic level, and although it appears to be constant, the fenestrae’s morphology and number of tiers of fenestrae may represent character states. Fenestrae in Allochytridium expandens, Cladochytrium replicatum and Nowakowskiella elegans are elongate and single-tiered. In Catenochytridium hemicysti they are compact and single-tiered, while in Allochytridium luteum they are compact and multi-tiered (Mozley-Standridge et al. 2009). However, additional studies with a broadly generic sampling are warranted before assessing the value of ultrastructure for taxonomic delineation. For some researchers of zoosporic fungi, molecular data, which are universally obtainable from pure isolates, have been used for making taxonomic revisions without zoospore ultrastructure, especially for taxa that produce zoospores rarely or with extreme difficulty. For example, Steiger et al. (2011) removed Cylindrochytrium jonhstonii Karling from Chytridiales and transferred it to Cladochytriales, and Jerônimo et al. (2019) established a new genus (Karlingiella G.H. Jerônimo, A.L. Jesus & Pires-Zottar.) without use of ultrastructural characters.
As the majority of the taxa were described exclusively on morphology, morphology still represents an important starting point for species recognition in Cladochytriales. An incorrect morphological identification can cause profound impacts on the systematics of the group in question, since it is highly dependent on both a correct type-species recognition and accurate assignment of names. Although many of the classic morphological characters have been shown to be homoplastic, and morphological knowledge and training has gradually dissipated among new researchers, it is essential for complementation of the molecular studies and taxonomic reviews. Powell et al. (2018b) identified a rare chytrid named Cladochytrium polystomum (Zopf 1884) in water bodies from North America. This chytrid is an interesting example of how species are delineated in Cladochytriales, since Sparrow (1960) and Karling (1977) questioned whether there was a distinction between C. polystomum and C. replicatum. Powell et al. (2018b) compared thallus morphology and observed numerous distinctions between those two species. In addition, they also discussed the phylogenetic relationship of C. polystomum with the type species of the genus (Cladochytrium tenue). According to these authors, C. polystomum diverges from C. replicatum based on the lack of internal proliferation of zoosporangia, morphology of rhizomycelial swellings, and differences in zoospore ultrastructure in addition to molecular evidence. Phylogenetic analysis demonstrated that C. polystomum and C. tenue are members of Chytridiales instead of Cladochytriales, however, and thallus development and molecular data supported the distinction of C. polystomum from C. tenue. Consequently, the new genus Zopfochytrium M.J. Powell, Longcore & Letcher was erected to accommodate the new combination, Z. polystomum (Powell et al. 2018b). The phylogenetic placement of C. tenue within Chytridiales brings to light an important debate on Cladochytriales systematics, and according to Powell et al. (2018b), the order needs to be reconsidered in the future when zoospore morphology of C. tenue is determined.
Although, Z. polystomum differs from C. replicatum for the reasons described above, Jerônimo et al. (2019) suggested that C. replicatum still represents a complex of multiple species according to the phylogenies of partial SSU and LSU regions of rDNA. Despite the amount of study afforded C. replicatum, certain aspects of its morphology need additional clarification. The first point is related to the nature of the resting spores, which were not observed in the original strain (Karling 1931). In subsequent observations, Karling described a similar strain, which produced smooth resting spores, even though spines were present on zoosporangia (Karling 1935). Subsequently, Karling (1937) investigated a strain, which produced both smooth and spiny-walled resting spores, and later, he reported that only 10% of the resting spores in another strain were smooth, the great majority being spiny (Karling 1941). Since none of these studies was made from single-spore isolates, it is entirely possible, as suggested by Sparrow (1960), that there are several distinct species involved. One line of evidence that supports this hypothesis, was the description of two similar species, C. aureum (Karling 1949) and C. aurantiacum (Richards 1956), which were distinguished from C. replicatum by production of spines on resting spores, tubes on the zoosporangium and the coloration of turbinate cells. However, these two species were rarely cited in subsequent studies (Czeczuga et al. 2005), while C. replicatum is considered one of the most common and widespread chytrids around the world (Hassan and Shoulkamy 1991; Marano et al. 2008; Jerônimo et al. 2015; Davis et al. 2018), suggesting there may be a consistent misidentification of cultures or observations as C. replicatum. From the conflicting evidence concerning morphology and phylogeny, it is possible, even likely, that C. replicatum is a species complex, however a careful morphological circumscription, based on comparative analysis of the original descriptions, and molecular characterization are essential for species delineation. This taxonomic problem highlights the complexity of modern taxonomy when the history of the discipline relies on characteristics we now know to be subject to great homoplasy and molecular methods cannot provide a solution because of the absence of type material. Nephridiophagales was recently found to form a well-supported monophyletic group within the Chytridiomyota, possibly as a sister to the Cladochytriales (Strassert et al. 2021). However, the assignment to the Cladochytriales is uncertain and awaits further investigation. The ultrastructure of nephridiophagid spores as revealed by transmission electron microscopy does not imply any evidence for flagellae and the kinetosome apparatus required for flagellate movement.
Species concepts in Spizellomycetales
The Spizellomycetales was the first order segregated from Sparrow’s (1960) broad concept of Chytridiales based on differing ultrastructural characteristics (Barr 1980). Until that time, morphological comparisons in situ were most often used to define familial and generic rankings. However, morphological variation, most prevalently observed once a fungus was in pure culture, made many investigators question the basis on which these taxonomic divisions were being made. In the 1960s and 1970s, Donald J. S. Barr, together with his colleagues, and his students, began to examine the ultrastructural features of the single-celled, posteriorly-flagellated zoospore of these fungi to determine if more constant, synapomorphic characters were to be found in these propagules. The zoospore is explicitly necessary for reproduction and was thus hyopthesised to be more evolutionarily constrained and conservative. Barr and others observed ultrastructural characters that were used as the defining features for the order Spizellomycetales. Among the most conspicuous characters that separated the Spizellomycetales from the Chytridiales that had been studied at that time were (1) ribosomes scattered throughout the cytoplasm, rather than enclosed in a membrane, as in the Chytridiales, (2) a non-flagellated centriole generally at an angle to the kinetosome, (3) a proximity of a portion of the nucleus to the posterior of the zoospore near the kinetosome, (4) microtubules emanating, possibly in multiple directions, from a spur on the kinetosome, and (5) multiple lipid globules permeating the cytoplasm.
Spizellomycetales originally included four new genera at the time of its description that would be the cornerstones of the new order. These genera were delineated from each other based on variations, or character states, of the features that distinguished the order from the Chytridiales. More specifically, these genera possessed different character states of the kinetosome spurs and the associated microtubules, ranging from a simple spur seemingly connecting the kinetosome and microtubules, to an elongated spur extending over half the length of the zoospore and encasing a subset of microtubules. The order also included previously described genera, e.g. Rhizophlyctis (≡ Karlingia), Rozella, and Olpidium, which exhibited a high degree of variation in their character states, indicative of their phylogenetic unrelatedness to the Spizellomycetales, as has now been recapitulated by molecular methods (James et al. 2006a; Sekimoto et al. 2011). When the Chytridiomycota was initially investigated by large-scale molecular phylogenies (James et al. 2000, 2006b), the cornerstone genera of the Spizellomycetales were upheld as monophyletic, supporting the group’s delineation based on shared zoosporic characteristics.
Additionally, the new genus Powellomyces was described in Spizellomycetales while Entophlyctis, previously transitioned to Spizellomycetales, was reassigned to the Chytridiales (Longcore et al. 1995) based on type species host preference. Powellomyces differed morphologically from other spizellomycetalean genera in that it developed exogenously, meaning the zoosporangium was produced exterior to the initial encysted zoospore, rather than the zoospore cyst enlarging to produce the zoosporangium, as in endogenous development. Within the last decade, further study of Powellomyces has led to the segregation of three additional genera within this morphological group (Simmons and Longcore 2012), now recognized as the Powellomycetaceae (Simmons 2011). These genera were recognized for their molecular monophyly from other Spizellomycetales taxa, their development method, and intergeneric ultrastructural character states.
Still, morphology, in correlation with phylogenetic species concepts, is necessary for species designations and identifications to be accessible to a wider mycological audience, which is generally limited to analyses by light microscopy. When the Spizellomycetales was described, Barr (1980) made this same argument and highlighted the emphasis on species delimitation by the “abundance of characters” represented by morphology, development, and physiology of these fungi in pure culture. The problem was and remains in determining which characters best predict the molecularly-recognized taxa and are thus phylogenetically useful. To that end, Simmons and Longcore (2012) used a principal components analysis of genera and species in the Powellomycetaceae to determine the morphological characters that were most useful for taxon description. Though this analysis did indicate useful features, the study was limited in scope, and the method was not completely accurate in species assignment, given intraspecific morphological variation. Presently, phylogenetic species concepts are thus the gold standard for zoosporic fungi species recognition, but it is necessary for specialists in these groups to provide as many lines of evidence as possible when delimiting new taxa.
As in all fungi, new genera of the Spizellomycetales continue to be found by the production of molecular phylogenies, which indicate cryptic lineages of interest within the larger groupings. When zoospores are examined by electron microscopy, these lineages have shown morphological evidence to support their separation from the previously characterized taxon. This has so far proven to be true of both the exogenously-developing (Powellomycetaceae) and endogenously-developing (Spizellomycetaceae) members of the Spizellomycetales. On the other hand, typically the number of strains examined by electron microscopy is not the same as the number investigated using DNA sequencing, and the level of intraspecific (or lab-to-lab) variation in ultrastructure is largely unexplored. After the description of the Powellomycetaceae, the remaining Spizellomycetales taxa, which undergo endogenous development, remained in the Spizellomycetaceae. In the last five years, the Spizellomycetaceae has also shown increased zoosporic variability with further sampling, isolation, and reevaluation, providing evidence for four more genera (Letcher and Powell 2018; Powell et al. 2018a). Though the Powellomycetaceae remains monophyletic in a large-scale phylogeny of the order (Simmons et al. 2020), there is molecular evidence that the endogenous Spizellomycetales could be split to separate Brevicalcar into its own family, though there is no noteworthy morphological feature, by light or electron microscopy, that would be indicative of this distinction. As convergent thallus morphology has done before, convergent ultrastructural evolution can mask these divergent lineages until an adequate technique is able to observe the differences. As electron microscopy once opened the door to better classification methods, molecular phylogenetics and further techniques will undoubtedly continue to question or reaffirm our current classifications.
Species concepts in Zygophlyctidales
How obligate parasitic chytrids infect specific hosts and expand their host ranges vary depending on species (Sparrow 1960). In traditional chytrid taxonomy, species delineation of chytrids was based primarily on thallus morphological characters, and host specificity was not considered as a taxonomic criterion for species in many cases. For example, Micromyces zygogonii was originally described as an endoparasite of Zygogonium sp. (Zygnematophyceae) by Dangeard (1889), but many researchers found similar chytrids infecting other genera of zygnematophycean algae (Mougeotia and Spirogyra) and identified them as M. zygogonii (Sparrow 1960). Canter (1963) described Zygorhizidium cystogenum parasitic on cysts of Dinobyrion and Uroglena (Chrysophyceae). Although the chytrids on two host algae differ in their rhizoidal system, they were identified as the same species based on similarity of zoosporangial shape and resting spore with unique covering. Exceptionally, in the plant pathogenic genus Synchytrium, which includes > 200 species (Karling 1964), host specificity was often used as a criterion for species identification. Because Synchytrium spp. have been considered to have a certain host specificity, parasites occurring on different host plants tended to be described as new species. Actually, more than 50 new species were described based primarily on host plants during the decade of 1950s–1960s (Karling 1964).
However, our knowledge of host specificity of parasitic chytrids largely relies on light microscopic observation and identification of each species. Only a few parasitic chytrids were examined for their host specificity under controlled conditions due to difficulty of cultivation of obligate parasites that cannot grow as pure cultures in general. Therefore, there is a possibility that a “species” infecting multiple host species actually represents a species complex of parasites infecting different hosts with similar morphology. Earlier experimental studies on host specificity of algal parasites using dual cultures revealed that parasitic chytrids are highly (genus or species) specific (Canter and Jaworski 1978, 1986; Canter et al. 1992; Doggett and Porter 1995) in most cases, but Gromov et al. (1999) showed that Rhizophydium algavorum had quite a broad host range; it could infect 5 genera and 20 species of green algae as well as the xanthophycean algae Tribonema gayanum. In Synchytrium, host specificities of some species have been examined by inoculation experiments (summarized in Karling 1964). In many cases, Synchytrium spp. were revealed to infect several genera or species of plants, but a few species such as S. macrosporum (Karling 1964) and S. fulgens (Hartmann 1958) had wider host ranges. Karling (1964) noted that many Synchytrium spp. need to be re-examined for their morphology, life cycle, and host specificity.
Difficulty of cultivation also hinders the molecular phylogenetic examination of obligately parasitic chytrids. While saprotrophic chytrids have been well studied in the recent taxonomic revision of zoosporic fungi, most known parasitic species remain to be examined with molecular phylogenetic and zoospore ultrastructural analysis. Recent efforts to establish dual cultures of algal parasites and their taxonomic examination revealed unexpected phylogenetic diversity of parasitic chytrids. Some known as well as newly described parasitic species represented new orders in Chytridiomycota (Karpov et al. 2014a; Seto et al. 2020). In other cases, new families or genera of parasitic chytrids were described in the well-studied orders: Collimycetaceae (Seto and Degawa 2018a), Dinomycetaceae (Lepelletier et al. 2014), Staurastromycetaceae (Van den Wyngaert et al. 2017) in Rhizophydiales and Pendulichytrium in Chytridiales (Seto and Degawa 2018b).
The Zygophlyctidales is one of the recently described orders represented by parasitic taxa (Seto et al. 2020). This order currently includes three species of diatom parasites, Zygophlyctis asterionellae, Zygop. melosirae, and Zygop. planktonica, which formerly belonged to the genus Zygorhizidium. In these, Zygop. asterionellae infecting Asterionella formosa and Zygop. planktonica infecting Ulnaria spp. (formerly Synedra, (Williams 2011)) were known as Zygorhizidium planktonicum, which has been extensively studied in the context of ecology and evolution of parasitic chytrids (Van Donk and Ringelberg 1983; de Bruin et al. 2004, 2008; Kagami et al. 2007). However, the species concept of Zygor. planktonicum was controversial. Zygor. planktonicum was originally described by Canter and Lund (1953) as a chytrid infecting both Asterionella and Ulnaria. In contrast, Pongratz (1966) suggested that Zygor. planktonicum on Asterionella and Ulnaria, respectively, are host specific based on the observation of phytoplankton in the same lake throughout the year and erected Zygor. asterionellae for the chytrid on Asterionella. Later, dual cultures of Zygor. planktonicum infecting Asterionella (Canter and Jaworski 1986) and Ulnaria (Canter et al. 1992) were established and cross inoculation experiments were conducted. The results showed that Zygor. planktonicum on Asterionella infects only Asterionella and could not infect Ulnaria spp., and vice versa. Canter et al. (1992) concluded that these two host-specific chytrids are the single species Zygor. planktonicum, but proposed formae speciales for two host specific variants. Afterward, the name Zygor. planktonicum has been used for chytrids on Asterionella (Van Donk and Ringelberg 1983; de Bruin et al. 2004, 2008; Kagami et al. 2007) and Ulnaria (Doggett and Porter 1995, 1996).
Recently, the species concept of Zygor. planktonicum was re-examined based on molecular phylogeny as well as host specificity (Seto et al. 2020). Two dual cultures of Zygor. planktonicum on Asterionella and Ulnaria, respectively, were revealed to be highly specific to their original host as with earlier studies (Canter and Jaworski 1986; Canter et al. 1992), and they were clearly phylogenetically distinguished. Additionally, the two cultures differed in zoospore ultrastructure, specifically in the contents of a fibrillar vesicle associated with the fenestrated cisterna. Seto et al. (2020) concluded that two host specific variants of Zygor. planktonicum warranted separation into distinct species (transferred to the genus Zygophlyctis), Zygop. asterionellae on Asterionella and Zygop. planktonica on a species of diatom, Ulnaria.
Interestingly, Zygop. planktonica can infect more than one species of Ulnaria but it prefers a single species and infects other species weakly (Canter et al. 1992; Doggett and Porter 1995). A similar phenomenon was observed in Zygop. melosirae infecting Aulacoseira spp. (Seto et al. 2020). Two strains of Zygop. melosirae were examined, one parasitizing A. granulata, and the other on A. ambigua. Although the two chytrids were almost identical in rDNA sequences, they could be distinguished based on host preference. Both chytrids could parasitize the two species of Aulacoseira, but heavily on the original host species and weakly on the other species. These two chytrids were regarded as intraspecific variants in a single species Zygop. melosirae based on the identity of rDNA sequences. From these results, zygophlyctidaleean chytrids are specific to a single diatom genus, but each chytrid species includes several intraspecific variants preferring a certain diatom species. Therefore, in Zygophlyctidales, host genus could be an important taxonomic criterion for species delineation, and is thus far supported by molecular phylogenetics.
Although host specificity has tended to be disregarded as a criterion for species identification, it could be important in pathogenic lineages as discussed above. For the well-studied Zygor. planktonicum, species concepts were revised by a careful re-examination of host range and morphology with molecular techniques (Seto et al. 2020). The host genus is important for species delineation in Zygophlyctidales, but it is necessary to say that the degree of host specificity differs depending on lineages of chytrids, ranging from family to species specific (Lepelletier et al. 2014; Seto and Degawa 2018b, a; Ding et al. 2018; Van den Wyngaert et al. 2018a, b; Seto et al. 2020). Most descriptions of parasitic chytrid species are based solely on light microscopic observation. It is necessary to re-examine these species concepts by both cross-inoculation experiments and molecular phylogenetic analyses. Ideally, dual culture-based studies are the best method for examination of species concepts in parasitic chytrids, but it is still a difficult task to establish such cultures. Some culture-independent techniques such as single-cell PCR methods (Kagami et al. in press; Ishida et al. 2015) could be helpful to examine both the host specificity and phylogeny of parasitic chytrids (Fig. 3).

Morphological similarity of Chytridiomycetes masks genetic divergence between species and orders. a Zoosporangium of Zygophlyctis asterionellae (Zygophlyctidales) (black arrow) parasitizing the diatom Asterionella. b Zoosporangium of Zygophlyctis planktonicum (Zygophlyctidales) (black arrow) parasitizing the diatom Ulnaria. c Immature zoosporangium and zoospore cyst (white arrow) of Thoreauomyces humboldtii JEL095 (Spizellomycetales) in PmTG agar medium. d Immature zoosporangium and zoospore cyst (white arrow) of Entophlyctis luteolus JEL0129 (Chytridiales) in PmTG agar medium. e Zoosporangium of Cladochytrium replicatum CCIBt4014 (Cladochytriales) on onion skin. f Resting spores of Cladochytrium replicatum CCIBt4014 (Cladochytriales) on onion skin. g Resting spores and intercalary cell of Cladochytrium tenue CCIBt4013 (Chytridiales) on onion skin. h Different zoosporangial stages of Cladochytrium tenue CCIBt4013 (Chytridiales) in liquid PmTG medium. Scale bars = 10 µm. Photo credits, a–b Kensuke Seto; c–d Joyce Longcore, e–h Gustavo Jerônimo Alves
Species concepts in Aphelidiomycota
Aphelidiomycota was recently raised to phylum by Karpov et al. (2014b) (as Aphelida) and described as a fungal phylum by Tedersoo et al. (2018). Recent studies either accept aphelids as Fungi or consider them separately in Opisthosporidia Karpov, Aleoshin Et Mikhailov (Karpov et al. 2014a, b; James et al. 2020). Within aphelids, four genera have been described (Wijayawardene et al. 2018, 2020): Aphelidium, Amoeboaphelidium, Paraphelidium, and Pseudaphelidium containing a total of 15 described species (Wijayawardene et al. 2018, 2020; Letcher and Powell 2019). The morphology, life cycle, ecology, and taxonomy of aphelids have been explored in several studies (Karpov et al. 2014a, b, 2016; Naranjo-Ortiz and Gabaldón 2019; Letcher and Powell 2019; Hurdeal et al. 2021). All described representatives of aphelids are phagotrophic parasites of algae. The life cycle and morphology of aphelids resemble those of rozellids, however the two can be differentiated based on the host (Karpov et al. 2014a, b; Torruella et al. 2018; Letcher and Powell 2019). Aphelids include a characteristic food vacuole with an excretory body, lamellar or tubular cristae, and ciliated or amoeboid cells.
Morphology
Species delineation of aphelids using exclusively light microscopy is insufficient, as microscopic features do not provide adequate resolution. For example, Paraphelidium is genetically distinct, but morphologically indistinguishable from other aphelid genera (Karpov et al. 2017). All known aphelids share similar life cycle stages, which include zoospores, cysts, and multinuclear plasmodia. Zoospore morphology is important in distinguishing species among genera. Specifically, the zoospores of Amoeboaphelidium are amoeboid with highly reduced flagellae. The zoospores of Paraphelidium and Aphelidium are also amoeboid forming filopodia and lamellipodia. However, the zoospores of Paraphelidium can produce subfilopodia from lamellipodia compared to members of Aphelidium who do not produce these structures. The zoospores of marine Pseudaphelidium, encyst immediately upon release and subsequently release motile zoospores from these cysts. Informative taxonomic characters to distinguish genera include spore size and shape, length of the flagellum, nature of pseudopodia, morphology of sporocyst, size of the residual body in the plasmodium, and presence or absence of a resting spore (Letcher and Powell 2019).
Differences in the morphology within species of the same genus have also been documented (Letcher and Powell 2019). These are taxonomically informative and can be used in species delimitation. Flagellum length and size of cyst, spore, and resting spore are used to characterize species within aphelid genera. For instance, the spore size of Aphelidium ranges from 1.5 to 4 μm (Letcher and Powell 2019; Karpov et al. 2020), with the exception of Aphelidium melosirae, which produces larger spores (4 × 6 µm). Regarding flagellum length, in A. tribonematis it ranges from 6 to 8 µm, while in other members, the flagellum is longer ranging from 9 to 14 µm. For Paraphelidium, until now only two species have been discovered and documented (Karpov et al. 2017; Letcher and Powell 2019). Spore size of Paraphelidium is around 2–2.5 µm, but differences in spore shape length of flagellum and number of lamellipodia and subfilopodia have also been noted. The flagellum of Paraphelidium tribonematis is around 7 µm long, while that of Paraphelidium letcheri ranges from 8 to 10 µm. Given that only a few species are known in this genus and in aphelids in general, it is difficult to advocate which morphological characters are taxonomically informative. As more species are discovered and described, it is likely that additional informative characters will be discovered.
Molecular phylogeny
The use of molecular phylogenetics has increased significantly in recent fungal taxonomic studies. Molecular phylogeny has helped improve and facilitate taxonomic classification of numerous taxa. Many recent taxonomic studies have incorporated a polyphasic approach using molecular phylogeny, as well as, other criteria (Karpov et al. 2016; Tcvetkova et al. 2019; Seto et al. 2020). For aphelids, molecular phylogenetic analysis is based on SSU rRNA sequences. Using taxa with available molecular sequence data, phylogenetic hypotheses to discriminate species have been proposed. All recently described aphelids have been clearly discriminated using molecular phylogenetics. Genetic distance of DNA sequences can also be informative; however, this should be assessed on a case-by-case basis. For instance, the divergence rate in the SSU rRNA between species in Aphelidium is around 15%, Amoeboaphelidium 25%, and among the genera Aphelidium, Amoeboaphelidium and Paraphelidium is around 20–25% (Letcher et al. 2017).
Species concepts in Rozellomycota
Rozellomycota species are intracellular parasites that grow as naked protoplasts within their hosts. Designation of this phylum was accepted by Tedersoo et al (2018). The Rozellomycota consists of Rozella species that are mainly endoparasites of water moulds, and Paramicrosporidium and Nucleophaga that are endonuclear parasites of amoebae. Rozellomycota groups in some phylogenies together with the Aphelidiomycota, endoparasites of algae, and the Microsporidia, mainly endoparasites of animals, at the base of the fungal tree of life (Corsaro et al. 2020), or in other analyses only with Microsporidia, while Aphelidiomycota diverges later (Torruella et al. 2018; Galindo et al. 2021). Nonetheless, their inclusion within the fungal kingdom is debated. Karpov et al. (2014a, b) included Rozellomycota within the Opisthosporidia together with aphelids. Environmental surveys have implied a rich biodiversity, which remains poorly characterised (Jones et al. 2011a, b; James et al. 2020). All known species are parasites of algae, water molds (e.g. Rozella), crustaceans (e.g. Mitospodidium), and amoebae (e.g. Nucleophaga, Paramicrosporidium) (Corsaro et al. 2020). Identification of Rozellomycota and species delimitation often relies on a combination of morphology and molecular phylogeny using the SSU rDNA gene region. Microsporidia, a well-known group of intracellular parasites has also been placed within Rozellomycota, though this is debated (Tedersoo et al. 2018; Wijayawardene et al. 2018, 2020; Adl et al. 2012; James et al. 2020).
Morphology
Morphological characters are informative and crucial for identification of Rozellomycota. Use of ultrastructural characters, such as the zoospores, polar filaments, and sporangium among others is common practice. True Microsporidia are distinguished by lacking a mitochondrion and by having a spore with a well-developed polar filament (Corsaro et al. 2014; Quandt et al. 2017). Species of Microsporidia are difficult to distinguish based on morphological features and host information needs to be considered (Keeling 2009). There are currently over 1300 described species in this group. Herein, Amphiacantha, an aquatic genus that parasitizes gregarine protists, is used to illustrate how morphology has been used to describe microsporidian species. Three species are accepted within Amphiacantha (Wijayawardene et al. 2018, 2020). Two of these (Amphiacantha ovalis and Amphiacantha attenuate) were described and introduced by Stubblefield (1955) based on morphological observations. Stubblefield used the characters of the cysts to classify Amphiacantha in Metchnikovellidae (now the genus is classified in Amphiacanthidae). Both species were described as having closely similar gametocysts as A. longa and were isolated from the same or closely related species of gregarines. Small differences in the dimensions of the cysts of A. ovalis and A. attenuate were also noted. Amphiacantha ovalis had a higher size average than A. attenuate. Another significant observation was the number of gametocysts in the cyst. Amphiacantha attenuate had 32–60, while A. ovalis had 14–50 and that of A. longa was 100. The latter also had significant bigger cysts (70–80 × 4.5 µm). Thus, cyst size can be significant in species delimitation of Amphiacantha. However, since only very few species of Amphiacantha are known the value of this character may change as more taxa are introduced later on.
Rozella species have uniflagellated zoospores, which germinate intracellularly to form a wall-less thallus. Thallus morphology and ultrastructure are used to distinguish Rozella species. This genus is differentiated from others by holocarpic, endobiotic, unwalled and inoperculate thallus, one or more discharge papillae (zoospores) with a single posterior flagellum and resting spores, which are thick-walled, smooth and spiny (Letcher et al. 2017; Sparrow 1960). Intrageneric species differentiation takes into account the size of the zoospores, parasite sporangial morphology, and presence of host hypertrophy. For example, the hosts of Rozella rhizoclosmatii comprise enlarged host sporangia, while there is absence of hypertrophy in some species including Rozella apodyae brachynematis. The morphology of the resting spores also has some value as a taxonomically informative character (Letcher et al. 2017). However, identification based exclusively on morphology can be difficult and at times uninformative. This has been shown with the use of modern molecular phylogeny based on DNA-based sequences.
Molecular phylogeny
The SSU rRNA gene is used in molecular phylogenetic analysis of Rozellomycota (Corsaro et al. 2016, 2020). This includes the introduction of new species. In most taxonomic research of zoosporic basal fungi, sequence data of related environmental samples are included. This is done in order to account for the low number of taxa currently described, to show the diversity of these fungi and to see if they have already been picked up in environmental surveys. Similarly, to the aphelids, the lack of available data highlights the need for more taxonomic work on these organisms. So far, the large number of sequence data available from environmental samples depicts a great diversity yet to be explored (Corsaro et al. 2016).
Species concepts in Neocallimastigomycota
Although the zoospores of anaerobic fungi were observed in the earliest studies of the rumen ecosystem (Gruby and Delafond 1843) and several were named (as flagellate protozoa) more than a century ago (Braune 1913; Liebetanz 1910), it was only in the 1970s that these cells were recognised as the fungal propagules of fungi, linked to rhizomycelial systems, which grow on ingested feed particles (Orpin 1975, 1976). The anaerobic fungi are classified in the phylum Neocallimastigomycota M. J. Powell (Hibbett et al. 2007), class Neocallimastigomycetes M. J. Powell (Hibbett et al. 2007). This class contains a single order (Neocallimastigales) and a single family (Neocallimastigaceae). Nineteen genera are currently recognised within Neocallimastigaceae (Table 2), with an additional new genus (Paucimyces) has been currently published (Hanafy et al. 2021). All but six of these genera have been named since 2015, the result of an upsurge in research activity relating to these fungi over the past decade, primarily driven by the biotechnological potential of these fungi. These activities have led to the publication of ca. 10 genome sequences and 30 transcriptomes (Solomon et al. 2016; Murphy et al. 2019; Wang et al. 2019).
Stable and distinctive morphological characters permitting differentiation of the anaerobic fungi are meagre, specifically: the formation of a bulbous holdfast (Caecomyces and Cyllamyces) or a rhizoidal thallus (all other genera); the formation of a polycentric thallus bearing several sporangia with nuclei present in the rhizoids (Anaeromyces, Orpinomyces, Paucimyces) or more limited monocentric anucleate thallus bearing a single sporangium (all other genera); multiflagellate zoospores (bearing > 16 flagella; Aestipascuomyces, Feramyces, Ghazallomyces, Neocallimastix, Orpinomyces) or uniflagellate zoospores (rarely 2–4 flagella; all other genera). It is noteworthy that the formation of multiflagellate zoospores in these genera appears to be a unique trait not found elsewhere amongst opisthokonts, with phylogenetic reconstruction suggesting that this trait arose once within the anaerobic fungi, with a single reversion to uniflagellate zoospores (Pecoramyces) (Hanafy et al. 2017, 2018, 2020b; Stabel et al. 2020). Morphological features of anaerobic fungi (Neocallimastigomycota) are illustrated in Fig. 4.

Anaerobic fungi (Neocallimastigomycota). a Buwchfawromyces eastonii exhibiting monocentric growth morphology (inset image of the sporangium stained with DAPI to show the nuclei of the developing zoospores within the zoosporangium). b, c Piromyces sp. sporangia emerging from a fragment of wheat straw. d Thalli of Caecomyces sp. growing from the end of a forage particle. e Detailed view of a thallus of Caecomyces sp. showing the multiple bulbous holdfasts. f, g Multiflagellate zoospores of Neocallimastix frontalis. Scale bars = 20 µm. Images by Gareth Griffith and Tony Callaghan
The majority (11) of the currently recognised genera form monocentric thalli and uniflagellate zoospores. The first of these was isolated and characterised by Orpin (1977) and later named as Piromyces by Gold et al. (1988). However, it is likely that some of the nine species later named within Piromyces prior to the advent of DNA barcoding should more correctly have been placed in one of the other 10 morphologically indistinguishable genera. However, the absence of physical type material or ex-type cultures for six of these species prevents exploration of this possibility.
In contrast to other eukaryotes inhabiting the mammalian digestive tracts, notably the ciliate protozoa (Cedrola et al. 2017; Newbold et al. 2015), the anaerobic fungi show remarkably little host specificity. Possible exceptions include Oontomces anksri (pseudoruminant camellids) (Dagar et al. 2015), Piromyces finnis (horse) (Hanafy et al. 2020b) and Ghazallomyces constrictus (axis deer) (Hanafy et al. 2020a). With the exception of Liggenstoffer et al. (2010) and Hanafy et al. (2020a), who used DNA metabarcoding to assess rumen fungal populations in diverse wild, captive and domesticated animals, the distribution of anaerobic fungi has mostly focused on domesticated hosts. The lack of host specificity is likely due to the efficiency with which anaerobic fungi disperse between hosts (Becker 1929). These are known to survive for considerable periods under aerobic conditions (McGranaghan et al. 1999) but the aerotolerant propagules, which are occasionally observed have hitherto defied detailed investigation (Brookman et al. 2000).
DNA-based identification of anaerobic fungi initially focused on the ITS1 spacer region but the high level of intragenomic variation, sometimes > 10% sequence divergence between the ITS1 repeats of a single isolate (Callaghan et al. 2015), necessitated the cloning of amplicons prior to sequencing, as well as problematic sequence alignment. However, sequencing of the D1/D2 subunits of the LSU locus has permitted robust phylogenetic reconstruction and provides clear species delimitation (Dagar et al. 2011). Most recently, the use of PacBio SMRT technology has permitted DNA metabarcoding of gut fungal communities from environmental samples using a combined ITS1/2 and D1/D2 LSU amplicon (Hanafy et al. 2020a). Not only does this permit reliable linkage of sequences to species names via the LSU region but also the identification of published sequences from unidentified environmental sequences, which comprise mostly ITS1 only. Phylogenetic reconstruction based on ITS1 and informed by predicted secondary structure had previously identified a number of hitherto unidentified clades of anaerobic fungi (Koetschan et al. 2014). Several of these have since been named from pure cultures and based on the PacBio sequencing approach developed by Hanafy et al. (2020a), at least 10 additional genera remain unnamed beyond the 20 hitherto formally named. There is no reason to believe that these additional taxa are refractory to axenic cultivation.
There are currently 45 named species but only 32 of these are clearly correct at present. Some additional names exist in the literature, most puzzlingly Piromyces equi for which no formal description exists beyond its name but which has been studied in numerous physiological publications [e.g. (Nagy et al. 2007; Poidevin et al. 2009)]. A phylogeny representing evolutionary relationships within Neocallimastigomycota is shown in Fig. 5.

Phylogenetic reconstruction using Maximum Likelihood based on partial sequences of 28S rRNA locus showing evolutionary relationships within Neocallimastigomycota. Blue indicates bulbous clades and red indicates clade with multiflagellate zoospores (except Feramyces). Scale bar indicates substitutions per site and salient bootstrap support values (1000 replicates) are shown at nodes. The outgroup (Quaeritorhiza) is a chytrid parasite of Haematococcus pluvialis (Chlorophyta)
Ecology and distribution of Mucoromycota
Ecology
The first taxonomic studies of the phylum Mucoromycota, whose species were treated within the traditional phylum Zygomycota, were in the late nineteenth century, mainly in France (van Tieghem and Le Monnier 1873; van Tieghem 1875, 1878; Bainier 1882), Germany (Fischer 1892; Schröter 1889) and the USA (Sumstine 1910). In the early twentieth century, subsequent studies were conducted in Switzerland (Lendner 1908), USA (Blakeslee 1905; Thaxter 1922) and Norway (Hagem 1907, 1910), followed by studies in Germany (Zycha 1935; Zycha et al. 1969) and Pakistan (Mirza et al. 1979). However, since the 1960s, there has been a considerable increase in information regarding the morphological taxonomy of the phylum Mucoromycota, which is mainly attributed to taxonomic revisions by Hesseltine and Fennell (1955) (USA, genus Circinella), Hesseltine and Ellis (1961, 1964, 1966) (USA, Absidia), Schipper (1973, 1975, 1976, 1978a, 1978b, 1984) (The Netherlands, Mucor, Rhizomucor, and Rhizopus), Benny and Benjamin (1975, 1976), Benny et al. (1985) (USA, Thamnidiaceae), Benjamin (1979) (USA, Endogonaceae), Zheng and Chen (2001) (China, Cunninghamella), Meyer and Gams (2003) (Australia/The Netherlands, Umbelopsis), Zheng et al. (2007) (China, Rhizopus), Walther et al. (2013) (Germany, Mucorales), Alastruey-Izquierdo et al. (2010) (Spain, Lichtheimia), and Wagner et al. (2019) (Germany, Mucor circinelloides complex).
In most previous studies, the classification of Zygomycota has been performed according to morphological characteristics; however, the advent of molecular biology, which was a major milestone in science and the taxonomic characterization of this phylum, has permitted the regrouping of zygomycotan taxa into new phyla, classes, orders, and families (Voigt 2012a, b; Hoffmann et al. 2013; Spatafora et al. 2016; Tedersoo et al. 2018). Among the seven phyla of zygosporic fungi, Mucoromycota is by far the most studied and comprises the largest number of species (354 spp.) that are distributed among the orders Mucorales (303 spp.), Endogonales (34 spp.), and Umbelopsidales (17 spp.) (Wijayawardene et al. 2020; www.speciesfungorum.org).
Benny and Benjamin (1991) recognized the order Mucorales and the families Choanephoraceae, Cunninghamellaceae, Gilbertellaceae, Mucoraceae, Mycotyphaceae, Phycomycetaceae, Radiomycetaceae and Thamnidiaceae based on their morphological characteristics. At that time, Endogonales only covered the family Endogonaceae, and Umbelopsidales (Spatafora et al. 2016) was not yet proposed; moreover, their species were allocated in the Mortierellales (Cavalier-Smith 1998). However, molecular analyses conducted by O’Donnell et al. (2001), Voigt and Wöstemeyer (2001), Meyer and Gams (2003), Voigt (2012a, b), and Hoffmann et al. (2013) provided new insights into the classification of Mucoromycota along with the establishment of new families, while others were disregarded. Presently, the following 3 orders and 17 families are accepted in the phylum Mucoromycota: (1) Mucorales: Backusellaceae, Choanephoraceae, Cunninghamellaceae, Lentamycetaceae, Lichtheimiaceae, Mucoraceae, Mycocladaceae, Mycotyphaceae, Phycomycetaceae, Pilobolaceae, Radiomycetaceae, Rhizopodaceae, Saksenaeaceae, and Syncephalastraceae; (2) Endogonales: Densosporaceae, and Endogonaceae; and (3) Umbelopsidales: Umbelopsidaceae (Wijayawardene et al. 2020; Hoffmann et al. 2013; Desirò et al. 2017). Most species belonging to the Mucoromycota are saprobic and found in soil (Benny et al. 2016), although some taxa of Mucorales and Umbelopsidales are endophytes (Bezerra et al. 2018; Sarsaiya et al. 2019; Rashmi et al. 2019). Endogonales, in addition to being saprobic, are ectomycorrhizal symbionts of plants as well as endophytes (Desirò et al. 2017; Bonafante and Venice 2020). Species of Mucorales can also be commonly isolated from the excrement of animals, such as herbivores and rodents (Benny 2008, 2012; Santiago et al. 2011; Melo et al. 2020), dead vegetables, and stored grains (Hoffmann et al. 2013). Moreover, the presence of these fungi in different substrates reflects their ecological importance in biodegradation processes, primarily in decomposition and nutrient cycling (Dix and Webster 1995; Bills et al. 2004; Richardson 2009). Facultative parasitic species of plants (Benny et al. 2014) and animals, including humans, are known to cause mucormycosis (Kamei 2000; Jacobsen 2019). Some genera are facultative mycoparasites, such as Chaetocladium, Dicranophora, Spinellus, and Syzygites (Zycha et al. 1969; Voglmayr and Krisai-Greilhuber 1996; Kovacs and Sundberg 1999; Benny 2005), whereas Sporodiniella is a facultative insect parasite (Evans and Samson 1977).
In Mucoromycota, Pilobolus is the unique obligatory coprophilous genus and, therefore, exhibits morphological characteristics that allow its growth on excrement as well as its dispersion from this substrate. These include positive phototropism mechanisms with active expulsion of the adhesive sporangia, sporangiospores that survive the digestion of animals and the capacity to grow under relatively high pH conditions (Dix and Webster 1995). As obligate coprophiles, Pilobolus species are difficult to grow on artificial media, even on dung agar or culture media supplemented with hemin. We have been trying to cultivate different species of Pilobolus over the past few years and have observed low growth and smaller morphological structures compared to those that grow directly on dung. In addition, most of the strains that can be grown on artificial media do not survive through sequential transfers to other plates (Foos et al. 2011). Other genera, such as Pilaira, Benjaminiella, Chaetocladium, Cokeromyces, Ellisomyces, Phascolomyces, Phycomyces, Thamnostylum, Utharomyces, and Zychaea, are mostly coprophilous, but some may include one or more non-coprophilic species or records (Krug et al. 2004).
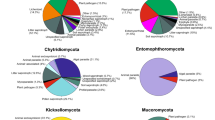
Unfortunately, data on the ecology of Mucoromycota are scarce. Considering the abundance of fungi in the soil environment at some stage in their life-cycle (Bridge and Spooner 2001), it is not surprising that this substrate is preferred by researchers for the isolation of Mucoromycota. For example, according to the Index Fungorum database (http://www.indexfungorum.org), of the 74 newly identified species of Mucoromycota described for the first time from 2015 to 2020 (until October 16), 45 (60.8%) were isolated from either soil or leaf/litter, followed by some from animal excrements, along with the reports from other fungi, insects, and water. Only a few species were isolated from decayed wood, human patients, fruits, air, Zea mays, and as laboratory contaminants (Fig. 6). Thus, the information regarding the taxonomy and ecology of Mucoromycota could advance considerably if there were more taxonomists willing to do inventories of these fungal species, specifically in poorly or unexplored habitats/hosts (Hawksworth and Rossman 1997; Hawksworth 2001a, b; Aime and Brearley 2012), thereby revealing unforeseen ecological interactions. For example, the well-known saprotrophic fruit parasite, Gilbertella persicaria (Mehrotra 1963a, 1963b; Guo et al. 2012; Pinho et al. 2014), was found infecting the black tiger shrimp (Penaeus monodon) (Karthikeyan and Gopalakrishnan 2014), while Actinomucor elegans, a saprotrophic soil fungus, which is occasionally involved in causing mucormycosis, has been reported to infect chafer beetle (Karimi et al. 2015). This fungus has also been found in a mutualistic association with Abutilon theophrasti roots (Kia et al. 2014). Interestingly, a high number of specialized operational taxonomic units of Mucor has been detected in decomposing Picea abies wooden blocks (Gómez-Brandón et al. 2020). In general, since mucoralean species do not use cellulose or lignin but use less complex and soluble sources of carbon and nitrogen (Benny 2012), they are probably using simple sugars produced by other fungi decomposers of cellulose or lignin. Interestingly, Gómez-Brandón et al. (2020) demonstrated the association of Mucor spp. with bacterial endosymbionts of the order Burkholderiales. This type of endosymbiotic interaction has previously been reported in Rhizopus microsporus, a well-known plant pathogen, which hosts Mycetohabitans rhizoxinica and M. endofungorum. These bacteria supplement the fungus with toxins that are the causative agent of plant disease and are involved in the regulation of asexual and sexual reproduction in the fungus (Partida-Martinez et al. 2007a,b). Other interesting and unusual ecological findings with respect to Mucoromycota that require further studies are as follows: pathogenic nature of Syncephalastrum sp. in gardens of leaf-cutting ants (Barcoto et al. 2017); Mucor racemosus as part of the lichen-associated mycobiota (Tripathi et al. 2014; Lagarde et al. 2018); and frequent and occasional human mucormycosis-causing agents, Apophysomyces variabilis (Prakash et al. 2016) and Actinomucor elegans (Dorin et al. 2017), respectively, which have been observed to be a part of the green turtle nest mycobiota, where both the fungal species can be considered novel pathogens for the turtles (Candan 2018). Mucor abundans, Umbelopsis angularis, U. isabelina, U. ramanniana and a new putative species of Apophysomyces have been found to be associated with the millipede fungivore Brachycybe lecontii (Macias et al. 2019). Additionally, zygospores of the two new mucoralean endoparasites, Mucor lilianae and M. rudolphii, have been found to be present in the basidiomas of Hysterangium spp. (Voglmayr and Clémençon 2015).

Number of Mucoromycota species described for the first time from 2015 to 2020 (until October 16) in different substrates
Distribution
Although several studies on the taxonomy and classification of Mucoromycota have been conducted (including the description of new genera and species), the species inventories of this phylum in different domains and substrates existing worldwide are not only limited but also temporally and spatially distributed. Some of these studies have been conducted in Brazil (Schoenlein-Crusius et al. 2006; de Souza et al. 2008, 2017; Santiago et al. 2011, 2013; Lima et al. 2015, 2016, 2018; Melo et al. 2020), Chile (Oscar Martínez and Eduardo Valenzuela 2003), China (Chen et al. 2007), France (Mousavi et al. 2018), Indonesia [Boedijn 1958], Iran (Ziaee et al. 2016), Malaysia (Loh et al. 2001; Lee et al. 2018), Mexico (Cruz-Lachica et al. 2018), Pakistan (Mirza et al. 1979), the UK (Campbell 1938), and the USA (Christenberry 1940). Therefore, whether the majority of the known species belonging to the phylum Mucoromycota are cosmopolitan cannot be confirmed. The genera commonly isolated from soil and decomposing vegetables, those causing food spoilage (e.g. Absidia, Cunninghamella, Rhizopus, Gongronella, Syncephalastrum, and Umbelopsis), plant pathogens (e.g. Rhizopus, Gilbertella, and Choanephora), and the causal agents of mucormycosis (e.g. Apophysomyces, Rhizopus, Lichtheimia, Mucor, Rhizomucor, and Saksenaea) are more frequently mentioned, and in most cases, they can be considered cosmopolitan. However, even within these genera, the distribution of some species remains poorly documented. For example, Cunninghamella septata has only been isolated in China (Zheng and Chen. 2001), and C. clavata (Zheng and Chen. 1998) has only been reported to occur in China and Brazil (Alves et al. 2017). Absidia idahoensis, and A. macrospora have been reported once in the USA (Hesseltine et al. 1990) and Czech Republic (Váňová 1968), respectively, and Isomucor trufemiae has only been reported in Brazil (de Souza et al. 2012; de Lima et al. 2020). The monospecific Halteromyces and Chlamydoabsidia are rare. While the former species has only been reported in Australia (Shipton and Schipper 1975), Chlamydoabsidia has been reported both in the USA (Hesseltine and Ellis 1966) and India (Behera and Mukerji 1985). Hyphomucor seems to be restricted to India, Malaysia, Sri Lanka (Schipper 1986), Nepal (Mikawa 1988), and Japan, while Ambomucor has been recently isolated from China (Zheng and Liu 2013) and the USA (MGnify 2020). Rhizopodopsis is also rare and has only been isolated in Indonesia (Boedijn 1958), while Dicranophora, Spinellus, and Syzygites seem to have diverse global distributions. While Dicranophora was especially reported from North America and Europe (Benny 2005), Spinellus was reported all across the Northern hemisphere (Ueda 2020; van Tieghem 1875; Ling-Young 1930; Indoh 1961; Ellis and Hesseltine 1962). Syzygites has been shown to be distributed worldwide (GBIF 2019). The facultative insect parasite, Sporodiniella, has only been reported in the warmer regions ranging from Ecuador (Evans and Samson 1977), Taiwan (Chien and Huang 1997), Japan (Orihara 2020) to Papua New Guinea (CABI 2020). Endogone is the best-known cosmopolitan species (Cannon and Kirk 2007). Peridiospora spinosa seems to be restricted to Brazil (Goto and Maia 2006), and Taiwan (Wu and Lin 1997). Sclerogone is restricted to Australia (Warcup 1990). Jimgerdemannia has been found in Australia, Japan, Europe and North and Central America (Desirò et al. 2017; Yamamoto et al. 2020). Vinositunica is a newly described genus reported in Japan (Yamamoto et al. 2020).
Many genera of Mucoromycota grow only or are more common on animal dung, and therefore, data on these coprophilous fungi are still insufficient, which is probably because animal dung may not be “attractive” for many researchers. Additionally, many of these coprophilous fungi do not exhibit any economic potential, and therefore, they may be of minimal interest to researchers. Pilobolus, Pilaira and Thamnostylum have been reported worldwide (Benny and Benjamin 1975; Cannon and Kirk 2007), while Utharomyces was reported in Africa, Bahamas, Brazil, Ghana, France, India, Indonesia, Mexico, Panama, Republic of China, the USA, and Venezuela (Alves et al. 2020; MGnify 2017, 2019). Benjaminiella (India, Mexico, Spain, and the USA) (Kirk 1989), Ellisomyces (the USA) (Hesseltine and Anderson 1956; Benny and Benjamin 1975), Fennellomyces (Australia, India, Malaysia, and the USA) (Benny and Benjamin 1975; Mirza et al. 1979; CABI 2020), Phascolomyces (China, Indonesia, and Panama) (Boedijn 1958; Benny and Benjamin 1976), and Zychaea Benny & RK Benj. (Mexico) (Benny and Benjamin 1975) exhibit restricted distribution. Chaetocladium, a parasitic genus of other Mucorales, grows better at low temperatures, and therefore, it is more commonly found in temperate countries, such as the Austria, China, Germany, Norway, Portugal, Spain, Sweden, the Netherlands, and United Kingdom (Zycha et al. 1969; Benny and Benjamin 1976). According to Benny (2005), this genus can be found during winters only in the hottest parts of the USA.
Estimates of the number of fungal species on a global scale range between 712,000 to 13.2 million species (Hawksworth 1991, 2001a, b; Schmit and Mueller 2007; Blackwell 2011; Wu et al. 2019). Hawksworth and Lücking (2017) estimated between 2.2 and 3.8 million fungal species, of which only 135,110 have been described up to 2019 (Catalog of Life: http://www.catalogueoflife.org/annual-checklist/2019/). Regrettably, considering the limited number of fungal taxonomists worldwide and the average rate of 2000 species described per year (Hawksworth and Lücking 2017), the task of cataloging all fungal species may take approximately 1430 years at the current rate of progress (Lücking 2020). Undoubtedly, the majority of known fungal species belong to the sister phyla Ascomycota and Basidiomycota, with only a small percentage of basal fungal species described, including the ones belonging to Mucoromycota (www.indexfungorum.org). Therefore, an important question that needs to be addressed is whether the Mucoromycota has been more neglected than other phyla of fungi sensu stricto with respect to descriptions of new species. Prior to 2008, nearly 98,000 fungal species were described (Kirk et al. 2008), of which 245 spp. belonged to Mucoromycota (a ratio of 400:1). The same ratio was observed eleven years later, based on the Catalogue of Life (2019) that estimated 135,110 known species of fungi along with 338 species belonging to the phylum Mucoromycota.
To accurately address the aforementioned question, we expanded our research and retrieved data regarding all new fungal species that were described for the first time between 1950 and 2019 using the Index Fungorum database (http://www.indexfungorum.org). Only valid species were considered, and new combinations were excluded (Fig. 7). In the 1960s and 1970s, 40 and 48 species of Mucoromycota were described, with other phyla: Mucoromycota ratios of 250:1 and 230:1, respectively. However, the number of new species identified in this phylum markedly reduced in the 1980s (16 spp; ratio of 720:1), followed by a successive increase in the following three decades. The 2010s should be highlighted for the identification of the largest number of mucoromycotan species described since the 1950s (64 spp. until 2019) with other phyla: Mucoromycota ratio of 300:1. Moreover, the number of species of other phyla described was also the highest (19,421 spp.). This can probably be attributed to the increased use of molecular biology techniques for species identification (Hawksworth and Lücking 2017; Cheek et al. 2020). Interestingly, considering the average number of species described in the past 70 years, we could estimate on other phyla: Mucoromycota ratio of 350:1 (data not shown), which is lower than the ratio projected in 2019 (400:1), based on the Catalog of Life (2019). Therefore, it appears that Mucoromycota have not been neglected more than other fungal phyla in terms of description of new species, at least in the past 70 years. So why is the knowledge of distribution of this phylum still so limited? A possible reason is that studies on this phylum have been conducted by restricted groups of taxonomists belonging to just a few countries. Therefore, the number of known species could probably be markedly higher and distribution data could be more accurate than at present if more expert taxonomists become interested in surveying these basal fungi. In addition, description of many new species of Mucoromycota as well as information regarding distribution of some taxa are based only on the sources provided by culture collections and clinical studies (Walther et al. 2019). Interestingly, recent data seem to be encouraging since increasing number of taxonomists have dedicated their research to describing new species of Mucoromycota over the last few years. For example, between 2015 and 2020 (until October 16), 13,218 species of other fungi and 74 species of Mucoromycota have been described (ratio of 178:1); however, these studies are still restricted to only a few countries. Figure 8 shows that among the 74 newly described species of Mucoromycota between 2015 and 2020 (until October 16), 54 (74%) were isolated from Brazil, South Korea, Australia, and China, while the rest were identified from 11 other countries. This confirms that only a few prolific taxonomists are concentrated in some countries, and in many cases, they are concentrated in one region (state or province) of a single country. For example, all of the 17 new species of Mucoromycota described in Brazil in the last five years were isolated by the same research group in the state of Pernambuco. The same has been observed in South Korea and Australia. This alarming situation demands attention for the urgent need to train new taxonomists to discover new species of Mucoromycota before they become extinct.

Number of species of Mucoromycota and other phyla described for the first time per decade from 1950 to 2019. Only valid species are considered, thereby excluding new combinations. The bars indicate the ratio of other phyla: Mucoromycota species described in each decade

Number of Mucoromycota species described for the first time from 2015 to 2020 (until October 16) in different countries
In addition to the poorly investigated or unexplored ecosystems, such as tropical areas and diversity hotspots, new species of Mucoromycota are also expected to be delimited from species complexes, specifically in the non-revised genera, such as Absidia, Mucor and Syncephalastrum, based on their morphological and molecular features. Wagner et al. (2019), for example, identified five new Mucor species after reviewing the Mucor circinelloides complex. Moreover, information regarding the distribution of Mucoromycota may be significantly enhanced by using data generated from environmental sequence studies. This may also reveal a covert diversity, as observed by Tedersoo et al. (2017), who analysed global soil DNA samples and fungal ITS2 dataset from 365 sites in 38 countries, following which they identified unclassified fungal species phylogenetically related to Endogonales and Umbelopsidales.
Impact of Chytridiomycota
Phytoplankton-infecting chytrids
Next to saprotrophic chytrids, numerous facultative and obligate parasitic forms, which infect diverse plants and microalgae, can be found in a broad range of terrestrial and aquatic environments (Voigt 2012b; Voigt et al. 2013; Frenken et al. 2017). Moreover, chytrids were found to dominate fungal communities in high-elevation soils and link aquatic and terrestrial ecosystems in alpine regions unsuspectedly (Freeman et al. 2009). Spread and propagation take place mainly asexually via free-swimming zoospores, that settle on a host, penetrate the cell and develop rhizoids to extract its nutrients while developing into sporangia, which again release new zoospores upon maturation (e.g. Ibelings et al. 2004).
In studies investigating marine and freshwater plankton communities, chytrids remained almost unrecognized for many decades. This was not only due to the small size of their infectious stages (zoospores), which is in the range of nanoplankton, but also due to their inconspicuous morphological features, which caused several misidentifications as bacterivorous flagellate protozoa (Lefèvre et al. 2007; 2008). In recent years, however, molecular culture-independent surveys have revealed an unexpected widespread occurrence and high abundance of several chytrid lineages (Lefèvre et al. 2012; Wurzbacher et al. 2014; Comeau et al. 2016; Hassett and Gradinger 2016; Hassett et al. 2017). Based on high-throughput sequencing of the hypervariable region V4 of the SSU rRNA gene, chytrids have been documented for example in temperate and polar marine ecosystems, where they made between 38 and 93% of all fungal sequences (Comeau et al. 2016). Comparable results were obtained for freshwater environments using full-length rRNA operon amplicon metabarcoding (Heeger et al. 2018).
Those findings raised the interest of plankton researchers and today many chytrids from diverse orders (e.g. Chytridiales, Gromochytriales, Lobulomycetales, Mesochytriales, Polyphagales, Rhizophydiales, Synchytriales) are morphologically identified as lethal parasites across all the major phytoplankton groups (e.g. cyanobacteria, diatoms, dinoflagellates, chlorophytes; Sparrow 1960; Lepelletier et al. 2014; Gutiérrez et al. 2016; Van den Wyngaert et al. 2018a, b). So far, studies quantifying the ecological significance of phytoplankton-infecting chytrids in aquatic environments are scarce (e.g. Rasconi et al. 2009, Taube et al. 2019). Yet, it is generally assumed that the parasites play not only an important role in their host population dynamics causing a significant impact on the planet’s carbon cycle (e.g. by terminating algal blooms; Gleason et al. 2015; Frenken et al. 2016; Jephcott et al. 2016) but constitute also a highly nutritional food source for zooplankton (Agha et al. 2016; Frenken et al. 2017). Being rich in fatty acids and sterols (Gerphagnon et al. 2018), which are absent for instance in cyanobacteria, parasitic chytrids may create trophic links between low-quality phytoplankton and zooplankton (so-called ‘mycoloop’; Kagami et al. 2014). Also, chytrid infection can boost carbon availability when large inedible diatoms or poorly edible filamentous cyanobacteria dominate phytoplankton communities (e.g. Kagami et al. 2007; Frenken et al. 2020).
Batrachochytrium as the cause of amphibian population declines
Frogs and salamander populations in many parts of the world are rapidly disappearing. Infection of amphibian hosts by fungal pathogens in the order Chytridiales, Batrachochytrium dendrobatidis (denoted Bd) and B. salamandrivorans (Bsal), has been implicated in many population declines. Upon infecting the skin of susceptible amphibians, these chytrids can cause clinical signs of the disease chytridiomycosis. In some species, infected individuals face near certain death. In others, sublethal effects of infection may result in life-history changes that reduce population growth. Over the past 50 years, population declines in more than 500 species and extinctions of at least 90 species have been attributed to chytridiomycosis (Scheele et al. 2019; but see Lambert et al. 2020).
The disease was first recognized in the late 1990s. Zoopathologists were puzzled by deaths in captive colonies of several amphibian species. Histological examination revealed organisms in the skin of preserved frogs and toads that were associated with severe skin dermatitis. The organisms initially were identified as fungal-like protists (Nichols et al. 1996). Similar structures in the skin of dying Australian frogs were described as Perkinsus-like protists (L. Berger, pers. comm.). Only when Nichols consulted Joyce Longcore was the error recognized. First from electron micrographs and then upon culturing the fungus, Longcore recognized taxonomically distinctive features of the pathogen that allowed her to identify it as a member of the Chytridiales. The development of the thallus and the ultrastructure of the zoospores differed sufficiently from those of other chytridialean genera that B. dendrobatidis was described as a new species in a new genus (Longcore et al. 1999). Later, from dying fire salamanders (Salamandra salamandra) in The Netherlands, Martel et al. (2013) isolated another chytrid pathogen, B. salamandrivorans (Bsal).
Bd and Bsal have motile, asexual zoospores with a single flagellum. Infection begins as zoospores infiltrate and colonize the superficial layers of hosts’ skin or other keratinized tissues such as larval mouthparts. Zoospores minimally require aqueous films to disperse, but infection can occur even in terrestrial species as they require moist microhabitats. Once in contact with the epidermis, zoospores inhabit the skin and develop a germ tube, which grows through epidermal layers (Fig. 9). The germ tube swells to produce a thallus and smooth-walled zoosporangia (Berger et al. 2005). When the zoosporangia mature, new zoospores are released through discharge papillae that project out of sloughing skin into the environment, continuing the cycle of infection. Bd typically causes hyperplasia and hyperkeratosis of superficial epidermal layers, whereas Bsal infection often is associated with superficial epidermal erosion and deeper ulcerations (Martel et al. 2013).

Adapted from Fu and Waldman (2019)
Skin histology of infected Litoria caerulea. Histological sections from BdAsia-1 treated groups. a Less infected skin region, note thickened epidermis. b–d Severe skin infection in individuals with high Bd infection loads. 1 a nearly empty zoosporangium. 2 a mature zoosporangium with zoospores in sloughing skin. 3 an empty zoosporangium that already has released zoospores in sloughed skin. 4 a mature zoosporangium with eight zoospores, about to be released. All four images are at the same magnification. Scale bar = 20 μm.
Curiously, amphibian population declines and species extinctions attributed to Bd were found in Central America and Australia around the same time (Berger et al. 1998). The puzzle of how Bd could simultaneously emerge as a pathogen on opposite sides of the world remains unsolved. Nonetheless, shortly after these epizootics were reported, Bd was identified in dying frogs in New Zealand (Waldman et al. 2001), North America (Bradley et al. 2002), and Europe (Garner et al. 2005). Bd now has been found on every continent except Antarctica, and has been associated with population declines or extirpations everywhere except Asia.
Where the fungus became endemic, some amphibian hosts have evolved effective defenses against it including secretion of antimicrobial peptides (Rollins-Smith et al. 2005; Woodhams et al. 2007), adaptive immune responses (Rollins-Smith 2020; Zamudio et al. 2020), and commensal bacteria that mitigate fungal effects (Harris et al., 2006). Bd has been associated mostly with declines in anurans, while Bsal’s effects so far seem limited to urodeles (Farrer 2019). Coinfection of some salamanders by Bd and Bsal suggests that the pathogens may act synergistically to increase morbidity and mortality (McDonald et al. 2020). Acquired immunity may be possible, as pre-infection by less virulent Bd variants can confer resistance to more virulent ones in frogs and some salamanders (Greener et al. 2020).
Analyses of host transcription following infection may point to Bd and Bsal’s different modes of action (Farrer et al. 2017). The genome size of Bd (23.7 Mb) is smaller than that of Bsal (32.6 Mb). Bsal contains three times as many genes in the M36 metalloprotease family and CBM18 genes (cell-surface proteins) that are thought to degrade host tissue and contribute to pathogenicity (Farrer et al. 2013, 2017; Joneson et al. 2011). When Bd infects hosts, large transcriptional responses ensue, including both up-regulated innate and adaptive immunity genes and down-regulated mucin genes. Salamanders, however, fail to show similar responses to Bsal infection (Farrer et al. 2017).
Diagnosis of chytridiomycosis
Initial surveys for infection by Bd required laborious histological methods, and owing to poor preservation of specimens, diagnoses based on necropsies sometimes generated less than certain results. Other fungi (e.g. Basidiobolus ranarum) were identified in amphibian skin that appeared to cause similar superficial skin infections (Taylor et al. 1999). Unambiguous identification of Bd infection was facilitated by staining with polyclonal antibodies (Berger et al. 2002). Soon thereafter, molecular identification of Bd was made possible by real-time polymerase chain reaction (PCR) (Boyle et al. 2004) and nested PCR protocols (Goka et al. 2009). For Bsal diagnosis, real-time PCR protocols also have been established (Blooi et al. 2013). Today, virtually all surveys for Bd and Bsal are based on PCR assays of skin swabs. Surveys of infection prevalence can be more expeditiously conducted using these methods, but subjects with low chytrid loads sometimes are erroneously inferred to be free of infection (Shin et al. 2014). PCR techniques have been refined to permit retrospective studies of infection in museum specimens, revealing that amphibians may have been infected by Bd in many parts of the world over the past 100 years or longer (Goka et al. 2009; Rodriguez et al. 2014; Fong et al. 2015; Talley et al. 2015).
Asian origin
The simultaneous emergence of chytridiomycosis as an infectious disease around the world might suggest that the virulence of amphibian chytrid pathogens, already geographically widely distributed and colonizing amphibian skin, was triggered by global environmental changes. Alternatively, hosts may have evolved resistance to, or tolerance of, endemic chytrid lineages. Then, as novel variants of the pathogens spread, facilitated by growth in the international amphibian trade, hosts were unable to respond efficaciously to them. Recent molecular evidence favors the latter hypothesis.
Early genetic studies, based on multilocus sequencing, failed to find significant variation among Bd isolates from African, Australian, Panamanian, and North American host species. This suggested that the pathogen represented a recently emerged clone (Morehouse et al. 2003). However, further studies revealed substantial genetic variation among Bd lineages, some endemic to particular geographic regions where effects on hosts were not apparent. Disease outbreaks followed by population extirpations and species extinctions more often occurred when host species were naïve to Bd or became infected by novel variants.
The highest diversity of Bd variants has been found on the Korean peninsula (Bataille et al. 2013), presumably resulting from the pathogen’s long evolutionary history there. Full genome sequencing of Bd isolated from fire-bellied toads (Bombina orientalis) supports this hypothesis (O’Hanlon et al. 2018). Moreover, the endemic lineage BdAsia-1 infecting Korean amphibians appears to be in mutation-drift equilibrium, as expected of long established host–pathogen interactions (O’Hanlon et al. 2018). Phylogenetic analysis reveals that BdAsia-1 is basal to other Bd lineages and overlaps extensively with global Bd lineages. These findings suggest that Bd originated in Asia and then radiated around the world between 10,000 and 40,000 years ago (Rosenblum et al. 2013). Byrne et al. (2019) found another basal lineage, BdAsia-3, in Southeast Asia using a custom amplicon sequencing assay they developed that genotypes assorted regions of the Bd genome. Other potential basal lineages may remain to be discovered in Asia. Bsal also appears widespread in Asia, having little if any effect on sympatric hosts, again suggesting an Asian origin (Laking et al. 2017; Fisher and Garner 2020).
Six major Bd lineages have been identified (Byrne et al. 2019; O’Hanlon et al. 2018) (Table 3). Initial focus was on areas where population dieoffs occurred. A “global pandemic Bd lineage” (BdGPL) is associated with most epizootics in Australia, Europe and the Americas (Byrne et al. 2019; O’Hanlon et al. 2018). The hypervirulence of this lineage initially was hypothesized to arise from a hybridization event (Farrer et al. 2011) but this no longer seems likely (Fisher and Garner 2020). BdGPL is the most recent derived lineage, and dominates in most parts of the world except Asia and eastern Africa (O’Hanlon et al. 2018).
BdCape was isolated from frogs in South Africa and was subsequently identified in other parts of Africa as well as Great Britain and Central America (Farrer et al. 2011; O’Hanlon et al. 2018). Xenopus frogs, which were routinely used for human pregnancy tests beginning in the 1930s, were thought to have spread Bd around the world (Weldon et al. 2004), but genetic evidence is not consistent with this hypothesis. BdCH initially was isolated from the midwife toad (Alytes obstetricans) in Switzerland (Farrer et al. 2011) but clusters closely with BdAsia-1 (O’Hanlon et al. 2018).
American bullfrogs (Rana catesbeiana) farmed on several continents, are tolerant of Bd so may spread the pathogen to native species where they are introduced. In Korea, infection loads of R. catesbeiana are higher than those of native species, and they appear to carry principally the lineage BdAsia-2 (Bataille et al. 2013; O’Hanlon et al. 2018). Similarly, in the USA and Brazil, R. catesbeiana serves as a reservoir of BdBrazil, which apparently has spread to native species (Schloegel et al. 2012). BdBrazil, also found on frogs in Japan, clusters with BdAsia-2 into one distinct lineage (O’Hanlon et al. 2018). Genetic sequencing thus provides supportive evidence that trade in bullfrogs played a significant role in spreading Bd around the world before reestablishing itself in Asia (Fisher and Garner 2020).
In addition to the six major lineages, recombinants or hybrids of BdAsia-2/Brazil and BdGPL, as well as other undetermined lineages, have been found (Byrne et al. 2019; Fisher and Garner 2020). Sexual recombination in Bd was first reported via internal transcribed spacer (ITS) region sequencing (Schloegel et al. 2012) and was later supported by whole genome sequencing (O’Hanlon et al. 2018). Recombination between lineages may produce high virulence, as evidenced by studies of a BdBrazil/BdGPL cross (Greenspan et al. 2018).
Bd lineages vary not only in genomic information but also in virulence and morphology. For example, BdCH has larger sporangia than BdCape or BdGPL (Farrer et al. 2011). BdBrazil zoospores are smaller than those of BdGPL (Becker et al. 2017). In addition, zoospore and zoosporangium characteristics vary even among nearby, genetically connected populations, suggesting local host adaptation or phenotypic plasticity (Fisher et al. 2009; Farrer et al. 2011; Lambertini et al. 2016). As BdAsia-1 appears hypervirulent to susceptible species outside Asia (Fu and Waldman 2019), further characterization of this basal lineage is necessary to delineate the association between virulence and morphology (Fisher et al. 2009).
Taxonomy of nephridiophagids
The fungal kingdom adopts the obligate insect-pathogenic nephridiophagids (Nephridiophagidae): Nephridiophagales
The nephridiophagids are unicellular obligate parasites that infect the Malpighian tubules of arthropods where especially in the lumen different life cycle stages can be densely packed (Fig. 10). Their life cycle includes a vegetative phase with multinucleate plasmodia that divide into oligo- and uninucleate cells, and a generative phase, in which the vegetative plasmodia transform into sporogenic plasmodia that form uninucleate spores. Mature spores with a thick chitinous wall are released with the host feces facilitating infection of further individuals by oral uptake (e.g. Woolever 1966; Radek et al. 2002). So far, nephridiophagids have been found mostly in cockroaches and beetles (Radek and Herth 1999) but are also known from other, more distantly related hosts such as the European earwig Forficula auricularia (Ormières and Manier 1973) or the honey bee Apis mellifera (formal description of the genus Nephridiophaga; Ivanić 1937). Although generally assumed to coevolve with their hosts, transmission of nephridiophagids between only distantly related cockroaches has recently been documented in a study, in which, however, the different cockroach species were kept together in the same cultural area (Strassert et al. 2021).

Life cycle stages of Nephridiophaga blattellae (Chytridiomycota). a Extracted gut of Blattella germanica (German cockroach) with Malpighian tubules (M) at the border of midgut (mg) and hindgut (hg) as habitat for Nephridiophaga blattellae. c, caeca; fg, foregut; t, trachea. b Infected Malpighian tubule full of parasite stages. Differential inferference contrast (DIC). c Three sporogenic plasmodia of different sizes. DIC. d Spores released from ruptured sporogenic plasmodia. DIC. f Giemsa stained smear of infected Malpighian tubules. Residual vegetative nuclei (arrows) in sporogenic plasmodium (vp); vegetative plasmodium (vp) with numerous nuclei. e Spores labelled with the fluorescent stain Calcofluor White reveal the presence of chitin in the spore walls. g, h Paraffin sections of B. germanica stained wirh hemalaun eosin. Rarely, intracellular plasmodia (ip) are found in epithel cells of the Malpighian tubules. Mostly, vegetative and sporogenic plasmodia develop in the lumen of the tubules, generally attached to the epithelium. i Ultra-thin section of Malpighian tubule. The plasmodia of Nephriophaga attach to the microvilli border (mv) or are free in the lumen. Sporogenic plasmodia with mature spores (s) and residual vegetative nuclei (vn) in the mother cell cytoplasm. Vegetative plasmodia with nuclei (n). mi, mitochondria. j Freeze-etch sample of a mature spore shows a little central cap at the spore opening (arrow). Ultrathin cross-section of mature spore with thick spore wall at the border and thin spore wall at the flat upper and lower sides. Scale bars: a = 1 mm, b = 50 µm, c–g, i = 10 µm, h = 100 µm, j, k = 1 µm
Being poor in morphological characteristics, the phylogenetic affiliation of nephridiophagids remained controversial for many decades (e.g. Perrin 1906; Ivanić 1937; Sprague 1970; Purrini and Rohde 1988; Lange 1993). Based on molecular phylogenetic analyses of the SSU rRNA gene of the nephridiophagid Nephridiophaga blattellae (from the German cockroach Blattella germanica), an assignment to the fungi was first been proposed by Wylezich et al. (2004). The fungal nature of nephridiophagids was then later confirmed with statistical support by adding the SSU rRNA gene sequences of two further Nephridiophaga species to the phylogenetic analysis (Radek et al. 2017). Most recently, molecular tree inferences from a concatenated alignment of SSU and LSU rRNA genes of nine nephridiophagid species have finally uncovered their robust assignment to the phylum Chytridiomycota (Strassert et al. 2021). Whereas these analyses showed a clear distinction of nephridiophagids from other order-level clades within the Chytridiomycota, confirming the order Nephridiophagales, their closer relationship to one of these clades remains uncertain (a sister relationship to the Cladochytriales is hypothesized; Strassert et al. 2021).
Nephridiophagales Doweld 2014 emend. Strassert and Radek 2021
Thalli reduced to reproductive structures comprising sporogenic plasmodia; filamentous and flagellated stages absent or not observed; Spores thick-walled, flattened, oval to elongate, uninucleate or binucleate, resembling spore sori; vegetative and sporogenic life cycles stages generally develop in the lumen of Malpighian tubules and rarely intracellular in their epithelium; in extremely high infections of Blattella germanica, stages also occurred in the fat bodies and the male accessory glands; obligate parasitic/commensal or mutualistic in arthropods.
Genus: Nephridiophaga Ivanić 1937
Existing type species: N. apis Ivanić 1937
Proposed type species: N. blattellae (Crawley 1905; Woolever 1966; Fig. 10).
Habitat: Malpighian tubules of Blattella germanica (Fig. 10a, b, h). Sporogenic plasmodia contain 10–30 spores and residual vegetative nuclei (Fig. 10c, f, i). Native mature spores are oval, measuring 5.5 (5.0–6.0) × 3.2 (2.5–3.5) µm, and are flattened (about 2.5 µm thick) (Fig. 10d, j, k). Central spore opening on flat side (Fig. 10j); chitin-containing cell wall (Fig. 10e). Intra- and extracellular, multinucleate vegetative plasmodia of different sizes (e.g. 20 µm) (Fig. 10f, g–i).
The illustration of N. apis by Ivanić (1937) can be interpreted as the holotype but an epitype should be ‘justifiable’, e.g., from the same host (as these organisms are generally host specific). However, N. apis was never re-isolated from honey bees. Although Plischuk and Lange (2011) reported light microscopic stages of N. apis from Argentinian honey bees, we could not prove Nephridiophaga by transmission electron microscopy or molecular analysis in a sample provided (but only microsporidia). If the relatedness of N. apis to the other species of Nephridiophaga is doubtful, all the other species of Nephridiophaga would require a new generic name and published combinations. This would be highly destabilizing and thus the only option to maintain stability in the naming of the majority of these species is a conserved type. The first and most numerous descriptions of nephridiophagids are from cockroaches (Lutz and Splendore 1903; Crawley 1905; Wijayawardene et al. 2020). Therefore, and because N. blattellae (Fig. 10) is the best investigated species of the whole group (Crawley 1905; Léger 1909; Woolever 1966; Radek and Herth 1999; Radek et al. 2017; Strassert et al. 2021), we propose this species as the new type species.
Short history of proven nephridiophagids and given genus names
The genus Nephridiophaga was introduced for N. apis from the honey bee Apis mellifera by Ivanić (1937). The name refers to the detrimental effect of an infection to the Malpighian tubules (‘feeders of nephridia’). However, nephridiophagids have been described earlier under different genus names, mainly because their affiliation to known groups of spore-forming unicellular pathogens was unknown. The first report was by Lutz and Splendore (1903) for nephridiophagids from the American cockroach Periplaneta americana. The authors interpreted the stages as microsporidia and thus described them as Plistophora periplanetae. In 1905, Crawley found a similar infection in the German cockroach Blattella germanica. Since he believed in a haplosporidian nature of the parasite, he named it Coelosporidium blattellae. In 1909, Léger created a new genus Peltomyces (referring to mycetozoans) for new nephridiophagids from the beetle Olocrates hyalinus and the earwig Forficula auricularia. Morphology and life cycle stages of the two new species closely resemble that of Nephridiophaga and this later led to a synonymization with the genus Nephridiophaga (Woolever 1966; Radek and Herth 1999). Molecular data would be necessary to justify the recognition of the genus Peltomyces. In the following years, an intense discussion was held concerning the true affiliation of the nephridiophagids, the genus and family names they should be given, and which spore formers really belong to this group (e.g. Sprague 1970; Toguebaye et al. 1986; Woolever 1966; Purrini and Weiser 1990; Lange 1993; Radek and Herth 1999). In addition to the genus Nephridiophaga, two further (monotypic) genera most probably represent nephridiophagids: Coleospora (binucleata) from the beetle Gonocephalum arenarium (Gibbs 1955), and Oryctospora (alata) from the beetle Oryctes monoceros (Purrini and Weiser 1990). These genera differ morphologically from Nephridiophaga either by possessing elongate mature spores with two nuclei (Coleospora), or by having spores with lateral protrusions and a polar opening.
Other species of Nephridiophaga:
N. archimandrita R. Radek, Wellmanns, A. Wolf 2011
N. blaberi Fabel, (Radek et al. 2002)
N. blattellae (H. Crawley) P. Woolever 1966
N. forficulae (Léger 1909) Ormières & Manier 1973
N. javanicae J.F.H. Strassert & R. (Strassert et al. 2021)
N. meloidorum (Purrini & Rhode 1988) Lange 1993
N. lucihormetica R. Radek, Wellmanns, A. Wolf 2011
N. maderae R. Radek, Owerfeldt, Gisder & Wurzbacher 2017
N. ormieresi (Toguebaye et al. 1986) Purrini & Weiser 1990
N. periplanetae (Lutz and Splendore 1903) Lange 1993
N. postici J.F.H. Strassert & (Strassert et al. 2021)
N. schalleri (Purrini & Rhode 1988) Lange 1993
N. tangae (Purrini et al. 1988) Lange 1993
N. xenoboli P.N. Ganapati & C.C. Narasimhamurti 1960
For the reason of a diverging spore type (spherical with two valves) N. xenoboli probably has to be removed from the genus Nephridiophaga according to Radek and Herth (1999).
Genomes and the rise of phylogenomic studies
Currently, a total of 2845 fungal genomes are available (GenBank, accessed on 19th April, 2021). The species-genome ratio among basal fungi is comparable to that of the derived Dikarya meaning a catch up for genome projects among basal fungi which was highly encouraged by Shelest and Voigt (2014). However, a lack of genome projects of the missing link and parasitic taxa still hampers phylogenomic analyses with sufficient statistical clade stability supports. We can conclude that an acceleration of both a search for missing link species in rarely sampled habitats and an increase of fungal genome projects of evolutionary link taxa will be beneficial for the phylogenetic reconstruction of basal fungi (Fig. 11).

Species counts versus genome counts among the most predominant fungal phyla (accessed at SpeciesFungorum and GenBank, respectively, as of 19th April, 2021). The terms ‘Chytridiomycota’ and ‘Zygomycota’ were used in a colloquial sense to depict the basal lineages of zoosporic fungi (Chytridiomycota and Blastocladiomycota) and zygosporic fungi (Entomophthoromycotina, Glomeromycotina, Kickxellomycotina, Mortierellomycotina, Mucoromycotina, Zoopagomycotina)
Conclusions and prospectives
This paper provides an overview of current species concepts, ecology, phylogeny, and the distribution of basal fungal lineages. We summarize existing knowledge on the phylogeny and review the taxonomy of selected groups. The diversity and ecology of basal fungal lineages is obscure but new groups are being described at a fast rate. We highlight the impact of two chytrid species belonging to Chytridiomycota on amphibian populations. It is increasingly important to know the roles of more basal fungi in global ecosystems. Discovery of new taxa has led to the dramatic changes in the phylogeny and diversity of fungi in basal clades, but the taxonomy is still debated. Additional taxonomic studies of diverse genera in the basal lineages are required in the future.
References
Adl SM, Simpson AGB, Farmer MA, Andersen RA, Anderson OR, Barta JR et al (2012) The revised classification of eukaryotes. J Eukaryot Microbiol 59:429–493. https://doi.org/10.1111/j.1550-7408.2012.00644.x
Agha R, Saebelfeld M, Manthey C, Rohrlack T, Wolinska J (2016) Chytrid parasitism facilitates trophic transfer between bloom-forming cyanobacteria and zooplankton (Daphnia). Sci Rep 6:28098
Aime CA, Brearley FQ (2012) Tropical fungal diversity: closing the gap between species estimates and species Discovery. Biodivers Conserv 21:2177–2180
Alves ALSDM, de Souza CAF, de Oliveira RJV, Cordeiro TRL, Santiago ALCMDA (2017) Cunninghamella clavata from Brazil: a new record for the western hemisphere. Mycotaxon 132:381–389
Alves MA, da Cruz MO, Santiago ALCMDA (2020) The first record of the coprophylous fungi Utharomyces epallocaulus Boedijn ex P.M. Kirk & Benny (Mucoromycotina, Mucorales, Pilobolaceae) in Brazil. Check List 16(3):737–741
Alastruey-Izquierdo A, Hoffmann K, de Hoog GS, Rodriguez-Tudela JL, Voigt K, Bibashi E, Walther G (2010) Species recognition and clinical relevance of the zygomycetous genus Lichtheimia (syn. Absidia pro parte, Mycocladus). J Clin Microbiol 48:2154–2170
Bainier G (1882) Etude sur les Mucorinees. These. F. Pichon et A. Cotillon, Imprimeurs, Paris, France
Barr DJS (1980) An outline for the reclassification of the Chytridiales, and for a new order, the Spizellomycetales. Can J Bot 58:2380–2394
Barr DJS (1986) Allochytridium expandens rediscovered: morphology, physiology and zoospore ultrastructure. Mycologia 78:439–448. https://doi.org/10.1080/00275514.1986.12025267
Barr DJS, Désaulniers NL, Knox JS (1987) Catenochytridium hemicysti n. sp.: morphology, physiology and zoospore ultrastructure. Mycologia 79:587–594. https://doi.org/10.1080/00275514.1987.12025428
Barr DJS (2001) Chytridiomycota. In: McLaughlin DJ, McLaughlin EG, Lemke PA (eds) The mycota VIIA, systematics and evolution. Springer-Verlag, Berlin, pp 93–112
Barr DJS, Kudo H, Jakober KD, Cheng KJ (1989) Morphology and development of rumen fungi—Neocallimastix sp., Piromyces communis, and Orpinomyces bovis gen. nov., sp. nov. Can J Bot 67:2815–2824
Barcoto MO, Pedrosa F, Bueno OC, Rodrigues A (2017) Pathogenic nature of Syncephalastrum in Atta sexdens rubropilosa fungus gardens. Pest Manag Sci 3:999–1009
Bataille A, Fong JJ, Cha M, Wogan GOU, Baek HJ, Lee H, Min MS, Waldman B (2013) Genetic evidence for a high diversity and wide distribution of endemic strains of the pathogenic chytrid fungus Batrachochytrium dendrobatidis in wild Asian amphibians. Mol Ecol 22:4196–4209
Becker ER (1929) Methods of rendering the rumen and reticulum of ruminants free from their normal infusorian fauna. Proc Natl Acad Sci USA 15:435–438
Becker CG, Greenspan SE, Tracy KE, Dash JA, Lambertini C, Jenkinson TS, Leite DS, Toledo LF, Longcore JE, James TY et al (2017) Variation in phenotype and virulence among enzootic and panzootic amphibian chytrid lineages. Fungal Ecol 26:45–50
Behera N, Mukerji KG (1985) Seasonal variation and distribution on microfungi in forest soils of Delhi. Folia Geobot Phytotax 20(3):291–311
Benjamin RK (1979) Zygomycetes and their spores. In: Kendrick B (ed) The whole fungus: the sexual-asexual synthesis, vol 2. National Museums of Canada, Ottawa
Benny GL, Benjamin RK (1975) Observations on Thamnidiaceae (Mucorales). New taxa, new combinations, and notes on selected species. Aliso 8:301–351
Benny GL, Benjamin RK (1976) Observations on Thamnidiaceae (Mucorales). II. Chaetocladium, Cokeromyces, Mycotypha, and Phascolomyces. Aliso 8:391–424
Benny GL, Benjamin RK (1991) The Radiomycetaceae (Mucorales: Zygomycetes. III. A new species of Radiomyces, and cladistic analysis and taxonomy of the family: with a discussion of evolutionary ordinal relationships on Zygomycotina. Mycologia 83:713–735
Benny GL, Kirk PM, Samson RA (1985) Observations on Thamnidiaceae (Mucorales). III. Mycotyphaceae fam. nov. and a re-evaluation of Mycotypha sensu Benny & Benjamin illustrated by two new species. Mycotaxon 22:119–148
Benny GL (2005) Zygomycetes. http://www.zygomycetes.org
Benny GL (2008) The methods used by Dr. R.K. Benjamin, and other mycologists to isolate zygomycetes. Aliso 26(1):37–61
Benny GL (2012) Current systematics of zygomycota with a brief review of their biology. In: Misra JK, Tewari JP, Deshmukh SK (eds) Systematics and evolution of fungi. Science Publishers, USA, pp 56–105
Benny GL, Humber RA, Voigt K (2014) Zygomycetous fungi: phylum entomophthoromycota and subphyla kickxellomycotina, mortierellomycotina, mucoromycotina, and zoopagomycotina. In: Mclaughlin DJ, Spatafora JW (eds) The mycota VII part A, systematics and evolution, 2a edn. Springer-Verlag, Berlin, pp 209–250
Benny GL, Smith ME, Kirk PM, Tretter ED, White MM (2016) Challenges and future perspectives in the systematics of Kickxellomycotina, Mortierellomycotina, Mucoromycotina, and Zoopagomycotina. In: Li D-W (ed) Biology of microfungi, fungal biology. Springer International Publishing, Switzerland, pp 65–126
Berbee ML, Taylor JW (2001) Fungal molecular evolution: gene trees and geologic time. In: McLaughlin DJ, McLaughlin EG, Lemke PA (eds) The mycota: systematics and evolution, vol VII. Part B. Springer, Berlin, pp 229–245
Berger L, Hyatt AD, Olsen V, Hengstberger SG, Boyle D, Marantelli G, Humphreys K, Longcore JE (2002) Production of polyclonal antibodies to Batrachochytrium dendrobatidis and their use in an immunoperoxidase test for chytridiomycosis in amphibians. Dis Aquat Org 48:213–220
Berger L, Speare R, Daszak P, Green DE, Cunningham AA, Goggin CL, Slocombe R, Ragan MA, Hyatt AD, Mcdonald KR et al (1998) Chytridiomycosis causes amphibian mortality associated with population declines in the rain forests of Australia and Central America. Proc Natl Acad Sci USA 95:9031–9036
Berger L, Speare R, Skerratt LF (2005) Distribution of Batrachochytrium dendrobatidis and pathology in the skin of green tree frogs Litoria caerulea with severe chytridiomycosis. Dis Aquat Org 68:65–70
Bezerra JDP, Machado AR, Firmino AL, Rosado AWC, de Souza CAF, Souza-Motta CM, Freire KTLDS, Paiva LM, Magalhães OMC, Pereira OL, Crous PW, de Oliveira TGL, de Abreu VP, Fan X (2018) Mycological diversity description I. Acta Bot Bras 32(4):656–666
Bills GF, Christensen M, Powel MJ, Thorn RG (2004) Saprobic soil fungi. In: Mueller GM, Bills GF, Foster MS (eds) Inventory and monitoring methods. Biodiversity fungi. Elsevier Academic Press, San Diego, pp 271–302
Blackwell M (2011) The fungi. 1, 2, 3,…5.1 million species? Am J Bot 98:426–438. https://doi.org/10.3732/ajb.1000298
Blakeslee AF (1905) Two conidia-bearing fungi. Cunninghamella and Thamnocephalis n. gen. Bot Gaz 40(3):161–170
Blooi M, Pasmans F, Longcore JE, Spitzen-Van Der Sluijs A, Vercammen F, Martel A (2013) Duplex real-time PCR for rapid simultaneous detection of Batrachochytrium dendrobatidis and Batrachochytrium salamandrivorans in amphibian samples. J Clin Microbiol 51:4173–4177
Boedijn KB (1958) Note on the Mucorales of Indonesia. Sydowia 12:321–362
Boyle DG, Boyle DB, Olsen V, Morgan JAT, Hyatt AD (2004) Rapid quantitative detection of chytridiomycosis (Batrachochytrium dendrobatidis) in amphibian samples using real-time Taqman PCR assay. Dis Aquat Org 60:141–148
Bradley GA, Rosen PC, Sredl MJ, Jones TR, Longcore JE (2002) Chytridiomycosis in native Arizona frogs. J Wildl Dis 38:206–212
Braune R (1913) Untersuchungen über die im Wiederkäuermagen vorkommenden Protozoen. Arch Protistenk 32:111–170
Breton A, Bernalier A, Dusser M, Fonty G, Gaillard-Martinie B, Guillot J (1990) Anaeromyces mucronatus nov. gen., nov. sp. A new strictly anaerobic rumen fungus with polycentric thallus. FEMS Microbiol Lett 58:177–182
Bridge P, Spooner B (2001) Soil fungi: diversity and detection. Plant Soil 232:147–154
Brookman JL, Ozkose E, Rogers S, Trinci APJ, Theodorou MK (2000) Identification of spores in the polycentric anaerobic gut fungi which enhance their ability to survive. FEMS Microbiol Ecol 31:261–267
Byrne AQ, Vredenburg VT, Martel A, Pasmans F, Bell RC, Blackburn DC, Bletz MC, Bosch J, Briggs CJ, Brown RM et al (2019) Cryptic diversity of a widespread global pathogen reveals expanded threats to amphibian conservation. Proc Natl Acad Sci U S A 116:20382–20387
CABI - Centre for Agriculture and Biosciences International (2020) CABI bioscience genetic resource collection. Occurrence dataset. https://doi.org/10.15468/yr757j. https://www.gbif.org/occurrence/42636125. Accessed 12 Oct 2020
Callaghan TM, Podmirseg SM, Hohlweck D, Edwards JE, Puniya AK, Dagar SS, Griffith GW (2015) Buwchfawromyces eastonii gen. nov., sp. nov.: a new anaerobic fungus (Neocallimastigomycota) isolated from buffalo faeces. MycoKeys 9:11–28
Campbell ME (1938) An investigation of the Mucorales in the soil. Rans Roy Soc Edin, vol 59, Part II, No. 16.
Candan ED (2018) Molecular identification of fungal isolates and hatching success of green turtle (Chelonia mydas) nests. Arch Microbiol 20(6):911–919
Cannon PF, Kirk PM (2007) Fungal families of the world. CAB International, Wallingford
Canter (1963) Studies on British chytrids: XXIII. New species on chrysophycean algae. Trans Brit Mycol Soc 46:305–320. https://doi.org/10.1016/S0007-1536(63)80022-4
Canter HM, Jaworski GHM (1978) The isolation, maintenance and host range studies of a chytrid Rhizophydium planktonicum Canter emend., parasitic on Asterionella formosa Hassall. Ann Bot 42:967–979. https://doi.org/10.1093/oxfordjournals.aob.a085536
Canter HM, Jaworski GHM (1986) A study on the chytrid Zygorhizidium planktonicum Canter, a parasite of the diatoms Asterionella and Synedra. Nova Hedwigia 43:269–298
Canter HM, Jaworski GHM, Beakes GW (1992) Formae speciales differentiation in the chytrid Zygorhizidium planktonicum Canter, a parasite of the diatoms Asterienella and Synedra. Nova Hedwigia 55:437–455
Canter HM, Lund (1953) Studies on plankton parasites: II. The parasitism of diatoms with special reference to lakes in the English Lake District. Trans Brit Mycol Soc 36:13–37. https://doi.org/10.1016/S0007-1536(53)80038-0
Cavalier-Smith T (1998) A revised six-kingdom system of life. Biol Rev Camb 73:203–266
Cedrola F, Dias R, Martinele I, D’Agosto M (2017) Description of Diploplastron dehorityi sp. nov. (Entodiniomorphida, Ophryoscolecidae), a new rumen ciliate from Brazilian sheep. Zootaxa 4258:581–585
Chang Y, Wang S, Sekimoto S, Aerts AL, Choi C, Clum A, LaButti KM, Lindquist EA, Yee Ngan C, Ohm RA, Salamov AA, Grigoriev IV, Spatafora JW, Berbee ML (2015) Phylogenomic analyses indicate that early fungi evolved digesting cell walls of algal ancestors of land plants. Genome Biol Evol 14:1590–1601
Chang Y, Rochon D, Sekimoto S, Wang Y, Chovatia M, Sandor L, Salamov A, Grigoriev IV, Stajich JE, Spatafora JW (2021) Genome-scale phylogenetic analyses confirm Olpidium as the closest living zoosporic fungus to the non-flagellated, terrestrial fungi. Sci Rep 11(1):3217. https://doi.org/10.1038/s41598-021-82607-4
Chen C-C, Liou G-Y, Lee F-W (2007) Rhizopus and related species from peka in Taiwan. Fung Sci 22(1, 2):51–57
Cheek M, Lughadha EN, Kirk P, Lindon H, Carretero J, Looney B, Douglas B, Haelewaters D, Gaya E, Llewwllyn T, Ainsworth AM, Gafforov Y, Hyde K, Crous P, Hughes M, Walker BE, Forzza RC, Wong KM, Niskanen T (2020) New scientific discoveries: plants and fungi. Plants People Planet 2:371–388
Chien C-Y, Huang B-C (1997) First record of the occurence of Sporonidiella umbelata (Mucorales) in Taiwan. Mycoscience 38:343–346
Christenberry GA (1940) A taxonomic study of the Mucorales in the Southeastern United States. J Elisha Mitch Sci S 56(2):333–366
Comeau AM, Vincent WF, Bernier L, Lovejoy C (2016) Novel chytrid lineages dominate fungal sequences in diverse marine and freshwater habitats. Sci Rep 6:30120
Corsaro D, Michel R, Walochnik J, Venditti D, Müller K-D, Hauröder B, Wylezich C (2016) Molecular identification of Nucleophaga terricolae sp. nov. (Rozellomycota), and new insights on the origin of the Microsporidia. Parasitol Res 115(8):3003–3011. https://doi.org/10.1007/s00436-016-5055-9
Corsaro D, Walochnik J, Danielle V, Hauröder B (2020) Solving an old enigma: Morellospora saccamoebae gen. nov., sp. nov. (Rozellomycota), a Sphaerita-like parasite of free-living amoebae. Parasitol Res 119:925–934. https://doi.org/10.1007/s00436-020-06623-5
Corsaro D, Walochnik J, Venditti D, Steinmann J, Müller K-D, Michel R (2014) Microsporidia-like parasites of amoebae belong to the early fungal lineage Rozellomycota. Parasitol Res 113:1909–1918. https://doi.org/10.1007/s00436-014-3838-4
Crawley H (1905) Coelosporidium blattellae, a new sporozoan parasite of Blattella germanica. Acad Nat Sci Phil 57:158–161
Cruz-Lachica I, Marquez-Zequera I, Allende-Molar R, Sañudo-Barajas JÁ, Leon-Felix J, Ley-Lopez N, Garcia-Estrada RS (2018) Diversity of mucoralean fungi in soils of papaya (Carica papaya L.) producing regions in Mexico. Fungal Biol 122(8):810–816
Czeczuga B, Mazalska B, Godlewska A, Muszyńska E (2005) Aquatic fungi growing on dead fragments of submerged plants. Limnologica 35:283–297. https://doi.org/10.1016/j.limno.2005.07.002
Dangeard PS (1889) Mémoire sur les Chytridinées. Le Botaniste 1:39–74
Dagar SS, Kumar S, Griffith GW, Edwards JE, Callaghan TM, Singh R, Nagpal AK, Puniya AK (2015) A new anaerobic fungus (Oontomyces anksri gen. nov., sp. nov.) from the digestive tract of the Indian camel (Camelus dromedarius). Fungal Biol 119(8):731–737
Dagar SS, Kumar S, Mudgil P, Singh R, Puniya AK (2011) D1/D2 domain of large-subunit ribosomal DNA for differentiation of Orpinomyces spp. Appl Environ Microbiol 77:6722–6725
Davis WJ, Picard KT, Antonetti J et al (2018) Inventory of chytrid diversity in two temporary forest ponds using a multiphasic approach. Mycologia. https://doi.org/10.1080/00275514.2018.1510725
De Bruin A, Ibelings BW, Kagami M et al (2008) Adaptation of the fungal parasite Zygorhizidium planktonicum during 200 generations of growth on homogeneous and heterogeneous populations of its host, the diatom Asterionella formosa. J Eukaryot Microbiol 55:69–74. https://doi.org/10.1111/j.1550-7408.2008.00306.x
de Bruin A, Ibelings BW, Rijkeboer M et al (2004) Genetic variation in Asterionella formosa (Bacillariophyceae): is it linked to frequent epidemics of host-specific parasitic fungi? J Phycol 40:823–830. https://doi.org/10.1111/j.1529-8817.2004.04006.x
Desirò A, Rimington WR, Jacob A, Pol NV, Smith ME, Trappe JM, Bidartondo MI, Bonito G (2017) Multigene phylogeny of Endogonales, an early diverging lineage of fungi associated with plants. IMA Fungus 8(2):245–257
de Lima CLF, Lima DX, da Cunha GCL, Leitão JDA, de Freitas LWS, Gurgel LMS, Santiago ALCMDA (2020) Isomucor trufemiae J.I. de Souza, Pires-Zottarelli & Harakava (Mucorales, Mucoromycota): the second report wordwide and first from soil in northearstern Brazil. Check List 16(5):1103–1107
de Souza JI, Pires-Zottarelli CL, Dos Santos JF, Costa JP, Harakava R (2012) Isomucor (Mucoromycotina): a new genus from a Cerrado reserve in state of Sao Paulo, Brazil. Mycologia 104:232–241
de Souza JI, Schoenlein-Crusius IH, dos Oliveira LH, S, (2008) Selected species of Mucorales from soil contaminated with toxic metals in São Paulo State, Brazil. Mycotaxon 106:273–288
de Souza CAF, Lima DX, Gurgel LSM, Santiago ALCMA (2017) Coprophilous Mucorales (ex Zygomycota) from three areas in the semi-arid of Pernambuco, Brazil. Braz J Microbiol 48(1):79–86
Ding Y, Peng X, Wang Z et al (2018) Occurrence and characterization of an epibiotic parasite in cultures of oleaginous microalga Graesiella sp. WBG-1. J Appl Phycol 30:819–830. https://doi.org/10.1007/s10811-017-1302-4
Dix NJ, Webster J (1995) Fungal ecology. Chapman & Hall, London
Doggett MS, Porter D (1995) Further evidence for host-specific variants in Zygorhizidium planktonicum. Mycologia 87:161–171. https://doi.org/10.1080/00275514.1995.12026517
Doggett MS, Porter D (1996) Sexual reproduction in the fungal parasite, Zygorhizidium planktonicum. Mycologia 88:720–732. https://doi.org/10.1080/00275514.1996.12026709
Dorin J, D’Áveni M, Debourgogne A, Cuenin M, Guillaso M, Rivier A, Gallet P, Lecoanet G, Machouart M (2017) Update on Actinomucor elegans, a mucormycete infrequently detected in human specimens: how combined microbiological tools contribute efficiently to a more accurate medical care. Int J Med Microbiol 307(8):435–442
Doweld AB (2013) Nomenclatural novelties. Index Fungorum 43:1
Doweld AB (2014) Nomenclatural novelties. Index Fungorum 49:1
Ellis JJ, Hesseltine CW (1962) Rhopalomyces and Spinellus in pure cultures and the parasitism of Rhopalomyces on nematode eggs. Nature 193:699–670
Evans HC, Samson RA (1977) Sporodiniella umbelata, an entomogenous fungos of the Mucorales from cocoa farms in Ecuador. Can J Bot 55:2981–2984
Fabel P, Radek R, Storch V (2000) A new spore-forming protist, Nephridiophaga blaberi sp. nov., in the death’s head cockroach Blaberus craniifer. Eur J Protistol 36:387–395
Farrer RA (2019) Batrachochytrium salamandrivorans. Trends Microbiol 27:892–893
Farrer RA, Henk DA, Garner TW, Balloux F, Woodhams DC, Fisher MC (2013) Chromosomal copy number variation, selection and uneven rates of recombination reveal cryptic genome diversity linked to pathogenicity. PLoS Genet 9:e1003703
Farrer RA, Martel A, Verbrugghe E, Abouelleil A, Ducatelle R, Longcore JE, James TY, Pasmans F, Fisher MC, Cuomo CA (2017) Genomic innovations linked to infection strategies across emerging pathogenic chytrid fungi. Nat Commun 8:14742
Farrer RA, Weinert LA, Bielby J, Garner TWJ, Balloux F, Clare F, Bosch J, Cunningham AA, Weldon C, du Preez L et al (2011) Multiple emergences of genetically diverse amphibian-infecting chytrids include a globalized hypervirulent recombinant lineage. Proc Natl Acad Sci USA 108:18732–18736
Fischer A (1892) Rabenhorst’s Kryptogamen-flora, pilze - phycomycetes, 2nd edn. Eduard Kummer, Leipzig
Fisher MC, Ghosh P, Shelton JMG, Bates K, Brookes L, Wierzbicki C, Rosa GM et al (2018) Development and worldwide use of non-lethal, and minimal population-level impact, protocols for the isolation of amphibian chytrid fungi. Sci Rep 8:7772
Fisher MC, Bosch J, Yin Z, Stead DA, Walker J, Selway L, Brown AJ, Walker LA, Gow NA, Stajich JE et al (2009) Proteomic and phenotypic profiling of the amphibian pathogen Batrachochytrium dendrobatidis shows that genotype is linked to virulence. Mol Ecol 18:415–429
Fisher MC, Garner TW (2020) Chytrid fungi and global amphibian declines. Nat Rev Microbiol 18:332–343
Flad V, Young D, Seppälä S, Hooker C, Youssef N, Podmirseg SM, Nagler M, Reilly M, Li Y, Fliegerová K (2020) Chapter 17: the biotechnological potential of anaerobic gut fungi, genetics and biotechnology. Springer, New York, pp 413–437
Fliegerova K, Mrazek J, Voigt K (2012) 1. Gut fungi: classification, evolution, life style and application. In: Silva AP, Sol M (eds) Fungi: types, environmental impact and role in disease. Series: environmental health—physical, chemical and biological factors. Nova Science Publisher Inc, Hauppauge, NY, pp 3–20
Fliegerova K, Kaerger K, Kirk PM, Voigt K (2015) Rumen fungi. In: Puniya AK, Singh R, Kamra DN (eds) Rumen microbiology: from evolution to revolution. Part II: rumen microbial diversity, chapter 7. Springer, Heidelberg, pp 97–112
Foos KM, May NL, Beach DL, Pomper M, Sheerhan KB, Ruch DG (2011) Phylogeny of Pilobolaceae. Mycologia 103(1):36–44
Fong JJ, Cheng TL, Bataille A, Pessier AP, Waldman B, Vredenburg VT (2015) Early 1900s detection of Batrachochytrium dendrobatidis in Korean amphibians. PLoS ONE 10:e0115656
Freeman KR, Martin AP, Karki D, Lynch RC, Mitter MS, Meyer AF, Longcore JE, Simmons DR, Schmidt SK (2009) Evidence that chytrids dominate fungal communities in high-elevation soils. PNAS 106(43):18315–18320
Frenken T, Alacid E, Berger SA, Bourne EC, Gerphagnon M et al (2017) Integrating chytrid fungal parasites into plankton ecology: research gaps and needs. Environ Microbiol 19:3802–3822. https://doi.org/10.1111/1462-2920.13827
Frenken T, Velthuis M, de Senerpont Domis LN, Stephan S, Aben R, Kosten S, van Donk E, Van de Waal DB (2016) Warming accelerates termination of a phytoplankton spring bloom by fungal parasites. Glob Chang Biol 22:209–309
Frenken T, Wolinska J, Tao Y, Rohrlack T, Agha R (2020) Infection of filamentous phytoplankton by fungal parasites enhances herbivory in pelagic food webs. Limnol Oceanogr 65:2618–2626
Fu M, Waldman B (2019) Ancestral chytrid pathogen remains hypervirulent following its long coevolution with amphibian hosts. Proc R Soc B 286:20190833
Galindo LJ, López-García P, Torruella G, Karpov S, Moreira D (2021) Phylogenomics of a new fungal phylum reveals multiple waves of reductive evolution across Holomycota. Nat Commun 12:4973. https://doi.org/10.1038/s41467-021-25308-w
Garner TW, Walker S, Bosch J, Hyatt AD, Cunningham AA, Fisher MC (2005) Chytrid fungus in Europe. Emerg Infect Dis 11:1639–1640
GBIF (2019) Syzygites ehrenb. In: GBIF Secretariat. GBIF backbone taxonomy. Checklist dataset. https://doi.org/10.15468/39omei. Accessed 19 Oct 2020
Gerphagnon M, Agha R, Martin-Creuzburg D, Bec A, Perriere F, Rad-Menéndez C, Gachon CMM, Wolinska J (2018) Comparison of sterol and fatty acid profiles of chytrids and their hosts reveals trophic upgrading of nutritionally inadequate phytoplankton by fungal parasites. Environ Microbiol 21:949–958
Gleason FH, Jephcott TG, Küpper FC, Gerphagnon M, Sime-Ngando T, Karpov SA, Guillou L, van Ogtrop FF (2015) Potential roles for recently discovered chytrid parasites in the dynamics of harmful algal blooms. Fungal Biol Rev 29:20–33
Goka K, Yokoyama J, Une Y, Kuroki T, Suzuki K, Nakahara M, Kobayashi A, Inaba S, Mizutani T, Hyatt AD (2009) Amphibian chytridiomycosis in Japan: distribution, haplotypes and possible route of entry into Japan. Mol Ecol 18:4757–4774
Gold JJ, Heath IB, Bauchop T (1988) Ultrastructural description of a new chytrid genus of caecum anaerobe, Caecomyces equi gen. nov. sp. nov. assigned to the Neaocallimasticaceae. BioSystems 21:403–415
Gómez-Brandón M, Probst MS, Siles JÁ, Peintner U, Bardelli T, Egli M, Insam E, Ascher-Jenull J (2020) Fungal communities and their association with nitrogen-fixing bacteria affect early decomposition of Norway spruce deadwood. Sci Rep 10:8025
Goto BT, Maia LC (2006) Contribution to the study of Endogonales in Brazil: the first record of Peridiospora tatachia. Mycotaxon 96:327–332
Greener MS, Verbrugghe E, Kelly M, Blooi M, Beukema W, Canessa S, Carranza S, Croubels S, De Troyer N, Fernandez-Giberteau D et al (2020) Presence of low virulence chytrid fungi could protect European amphibians from more deadly strains. Nat Commun 11:5393
Greenspan SE, Lambertini C, Carvalho T, James TY, Toledo LF, Haddad CFB, Becker CG (2018) Hybrids of amphibian chytrid show high virulence in native hosts. Sci Rep 8:9600
Gromov BV, Plujusch AV, Mamkaeva KA (1999) Morphology and possible host range of Rhizophydium algavorum sp. nov. (Chytridiales) an obligate parasite of algae. Protistology 1:62–65
Grossart HP, Wurzbacher C, James TY, Kagami M (2016) Discovery of dark matter fungi in aquatic ecosystems demands a reappraisal of the phylogeny and ecology of zoosporic fungi. Fungal Ecol 19:28–38. https://doi.org/10.1016/j.funeco.2015.06.004
Gruby D, Delafond HMO (1843) Recherches sur les animalcules se devellopant en grand nombre dans l’estomac et dans les intestines pendant la digestion des animaux herbivores et carnivores. Extrait d’une note. Comptes Rendus Hebdomaire Des Sciences De L’academie Des Sciences Paris 17:1304–1308
Guo LW, Wu YX, Mao ZC, Ho HH, He YQ (2012) Storage rot of dragon fruit caused by Gilbertella persicaria. Plant Dis 96:1826
Gutiérrez MH, Jara AM, Pantoja S (2016) Fungal parasites infect marine diatoms in the upwelling ecosystem of the Humboldt current system off central Chile. Environ Microbiol 18:1646–1653
Hagem O (1907) Untersuchungen uber Norwegische Mucorineen, 1, Vidensk. Selsk. 1. Math Natuvr KL 7:1–50
Hagem O (1910) Untersuchungen uber Norwegische Mucorineen 2, Vidensk. Selsk. 1. Math Naturv KL 4:1–152
Haitjema CH, Solomon KV, Henske JK, Theodorou MK, O’Malley MA (2014) Anaerobic gut fungi: advances in isolation, culture, and cellulolytic enzyme discovery for biofuel production. Biotechnol Bioeng 111:1471–1482
Hanafy RA, Elshahed MS, Liggenstoffer AS, Griffith GW, Youssef NH (2017) Pecoramyces ruminantium, gen. nov., sp. nov., an anaerobic gut fungus from the feces of cattle and sheep. Mycologia 109:231–243
Hanafy RA, Elshahed MS, Youssef NH (2018) Feramyces austinii, gen. nov., sp. nov., an anaerobic gut fungus from rumen and fecal samples of wild Barbary sheep and fallow deer. Mycologia 110(3):513–525
Hanafy RA, Johnson B, Youssef NH, Elshahed MS (2020a) Assessing anaerobic gut fungal diversity in herbivores using D1/D2 large ribosomal subunit sequencing and multi-year isolation. Environ Microbiol 22:3883–3908
Hanafy RA, Lanjekar VB, Dhakephalkar PK, Callaghan TM, Dagar SS, Griffith GW, Elshahed MS, Youssef NH (2020b) Seven new Neocallimastigomycota genera from wild, zoo-housed, and domesticated herbivores greatly expand the taxonomic diversity of the phylum. Mycologia 112:1212–1239
Hanafy RA, Youssef NH, Elshahed MS (2021) Paucimyces polynucleatus gen. nov, sp. nov., a novel polycentric genus of anaerobic gut fungi from the feces of a wild blackbuck antelope. Int J Syst Evol Microbiol 71(6). https://doi.org/10.1099/ijsem.0.004832
Harris RN, James TY, Lauer A, Simon MA, Patel A (2006) Amphibian pathogen Batrachochytrium dendrobatidis is inhibited by the cutaneous bacteria of amphibian species. EcoHealth 3:53–56
Hartmann GC (1958) Susceptibility of clarkia to Synchytrium fulgens. Mycologia 50:562–569. https://doi.org/10.2307/3756118
Hassan SKM, Shoulkamy MA (1991) Chytridiaceous fungi from water streams in Upper Egypt. Zentralbl Mikrobiol 146:509–523. https://doi.org/10.1016/S0232-4393(11)80239-8
Hassett BT, Ducluzeau A-LL, Collins RE, Gradinger R (2017) Spatial distribution of aquatic marine fungi across the western Arctic and sub-arctic. Environ Microbiol 19:475–484
Hassett BT, Gradinger R (2016) Chytrids dominate arctic marine fungal communities. Environ Microbiol 18:2001–2009
Hawksworth DL (1991) The fungal dimension of biodiversity: magnitude, significance, and conservation. Mycol Res 95:641–655
Hawksworth DL (2001a) The magnitude of fungal diversity: the 1.5 million species estimate revisited. Mycol Res 105(12):1422–1432
Hawksworth DL, Rossman AY (1997) Where are all the undescribed fungi? Phytopathology 87:888–891
Hawksworth DL, Lücking R (2017) Fungal diversity revisited 2.2 to 3.8 million species. Microbiol Spectr 5(4). https://doi.org/10.1128/microbiolspec.FUNK-0052-2016
Hawksworth DL (2001b) The magnitude of fungal diversity: the 1.5 million species estimate revisited. Mycol Res 105:1422–1432
Hawksworth DL, Sutton BC, Ainsworth GC (1983) Ainsworth & Bisby’s dictionary of the fungi, 7th edn. Commonwealth Mycological Institute, Kew, Surrey
Hawksworth DL, Kirk PM, Sutton BC, Pegler DN (1995) Ainsworth & Bisby’s dictionary of the fungi, 8th edn. International Mycological Institute, Kew, Surrey
Heath IB, Bauchop T, Skipp RA (1983) Assignment of the rumen anaerobe Neocallimastix frontalis to the Spizellomycetales (Chytridiomycetes) on the basis of its polyflagellate zoospore ultrastructure. Can J Bot 61:295–307
Heeger F, Bourne EC, Baschien C, Yurkov A, Bunk B, Spröer C, Overmann J, Mazzoni CJ, Monaghan MT (2018) Long-read DNA metabarcoding of ribosomal RNA in the analysis of fungi from aquatic environments. Mol Ecol Resour 18:1500–1514
Hesseltine CW, Anderson P (1956) The genus Thamnidium and a study of the formation of its zygospores. Am J Bot 43:696–703
Hesseltine CW, Ellis JJ (1961) Notes on Mucorales, especially Absidia. Mycologia 53:406–426
Hesseltine CW, Ellis JJ (1964) The genus Absidia: Gongronella and cylindrical-spored species of Absidia. Mycologia 56:568–601
Hesseltine CW, Ellis JJ (1966) Species of Absidia with ovoid sporangiospores. I. Mycologia 58:761–785
Hesseltine CW, Fennell DI (1955) The genus Circinella. Mycologia 47:193–212
Hesseltine CW, Mahoney MK, Peterson SW (1990) A new species of Absidia from alkalai bee brood chamber. Mycologia 82:523–526
Hibbett DS, Binder M, Bischoff JF, Blackwell M, Cannon PF, Eriksson O, Huhndorf S, James T, Kirk PM, Lücking R, Lumbsch T, Lutzoni F, Matheny PB, McLaughlin DJ, Powell MJ, Redhead S, Schoch CL, Spatafora JW, Stalpers JA, Vilgalys R, Aime MC, Aptroot A, Bauer R, Begerow D, Benny GL, Castlebury LA, Crous PW, Dai Y-C, Gams W, Geiser DM, Griffith GW, Gueidan C, Hawksworth DL, Hestmark G, Hosaka K, Humber RA, Hyde K, Köljalb U, Kurtzman CP, Larsson K-H, Lichtwardt R, Longcore J, Miadlikowska J, Miller A, Moncalvo J-M, Mozley-Standridge S, Oberwinkler F, Parmasto R, Reeb V, Rogers JD, Roux C, Ryvarden L, Sampaio JP, Schuessler A, Sugiyama J, Thorn RG, Tibell L, Untereiner WA, Walker C, Wang A, Weir A, Weiss M, White M, Winka K, Yao Y-J, Zhang N (2007) A higher-level phylogenetic classification of the Fungi. Mycol Res 111:509–547
Hoffmann K, Pawłowska J, Walther G, Wrzosek M, de Hoog GS, Benny GL, Kirk PM, Voigt K (2013) The family structure of the Mucorales: a synoptic revision based on comprehensive multigene-genealogies. Persoonia 30:57–76
Hoffmann K, Voigt K, Kirk PM (2011) Mortierellomycotina subphyl. nov. based on multi-gene genealogies. Mycotaxon 115:353–363
Hurdeal VG, Gentekaki E, Hyde KD et al (2021) Where are the basal fungi? Current status on diversity, ecology, evolution, and taxonomy. Biologia 76:421–440. https://doi.org/10.2478/s11756-020-00642-4
Hyde KD, Al-Hatmi AM, Andersen B, Boekhout T, Buzina W, Dawson TL, Eastwood DC, Jones EG, de Hoog S, Kang Y, Longcore JE (2018) The world’s ten most feared fungi. Fungal Divers 93:161–194
Ibelings BW, De Bruin A, Kagami M, Rijkeboer M, Brehm M, Van DE (2004) Host parasite interactions between freshwater phytoplankton and chytrid fungi (Chytridiomycota). J Phycol 40:437–453
Indoh H (1961) On the genus Spinellus, one of fungicolous Phycomycetes found in Japan. J Japan Bot 36:201–204
Ishida S, Nozaki D, Grossart HP, Kagami M (2015) Novel basal, fungal lineages from freshwater phytoplankton and lake samples. Environ Microbiol Rep 7:435–441. https://doi.org/10.1111/1758-2229.12268
Ivanić M (1937) Die Entwicklungsgeschichte und die parasitäre Zerstörungsarbeit einer in den Zellen der Malpighischen Gefäße der Honigbiene (Apis mellifera) schmarotzenden Haplosporidie Nephridiophaga apis n. g. n. sp. Cellule 45:291–324
Jacobsen ID (2019) Animal models to study mucormycosis. J Fungi 5(2):27
James TY, Kauff F, Schoch CL, Matheny PB, Hofstetter V, Cox CJ, Celio G, Gueidan C, Fraker E, Miadlikowska J, Lumbsch HT, Rauhut A, Reeb V, Arnold AE, Amtoft A, Stajich JE, Hosaka K, Sung GH, Johnson D, O’Rourke B, Crockett M, Binder M, Curtis JM, Slot JC, Wang Z, Wilson AW, Schüler A, Longcore JE, O’Donnell K, Mozley-Standridge S, Porter D, Letcher PM, Powell MJ, Taylor JW, White MM, Griffith GW, Davies DR, Humber RA, Morton JB, Sugiyama J, Rossman AY, Rogers JD, Pfister DH, Hewitt D, Hansen K, Hambleton S, Shoemaker RA, Kohlmeyer J, Volkmann-Kohlmeyer B, Spotts RA, Serdani M, Crous PW, Hughes KW, Matsuura K, Langer E, Langer G, Untereiner WA, Lücking R, Büdel B, Geiser DM, Aptroot A, Diederich P, Schmitt I, Schultz M, Yahr R, Hibbett DS, Lutzoni F, McLaughlin DJ, Spatafora JW, Vilgalys R (2006a) Reconstructing the early evolution of the fungi using a six-gene phylogeny. Nature 443:818–822
James TY, Letcher PM, Longcore JE et al (2006b) A molecular phylogeny of the flagellated fungi (Chytridiomycota) and description of a new phylum (Blastocladiomycota). Mycologia 98:860–871
James TY, Porter D, Leander CA et al (2000) Molecular phylogenetics of the Chytridiomycota supports the utility of ultrastructural data in chytrid systematics. Can J Bot 78:336–350
James TY, Berbee ML (2012) No jacket required—new fungal lineage defies dress code: recently described zoosporic fungi lack a cell wall during trophic phase. BioEssays 34(2):94–102
James TY, Pelin A, Bonen L, Ahrendt S, Sain D, Corradi N, Stajich JE (2013) Shared signatures of parasitism and phylogenomics unite Cryptomycota and microsporidia. Curr Biol 23(16):1548–1553
James TY, Litvintseva AP, Vilgalys R, Morgan JAT et al (2009) Rapid global expansion of the fungal disease chytridiomycosis into declining and healthy amphibian populations. PLoS Pathog 5:e10000458
James TY, Stajich JE, Hittinger CT, Rokas A (2020) Toward a fully resolved fungal tree of life. Annu Rev Microbiol 74:291–313. https://doi.org/10.1146/annurev-micro-022020-051835
Jephcott TG, Alves-de-Souza C, Gleason FH, van Ogtrop FF, Sime-Ngando T, Karpov SA, Guillou L (2016) Ecological impacts of parasitic chytrids, syndiniales and perkinsids on populations of marine photosynthetic dinoflagellates. Fungal Ecol 19:47–58
Jerônimo GH, de Jesus AL, Marano AV et al (2015) Diversity of Blastocladiomycota and Chytridiomycota from Parque Estadual da Ilha do Cardoso, Cananéia, São Paulo State, Brazil. Hoehnea 42:135–163
Jerônimo GH, Jesus AL, Simmons DR et al (2019) Novel taxa in Cladochytriales (Chytridiomycota): Karlingiella (gen. nov.) and Nowakowskiella crenulata (sp. nov.). Mycologia 111:506–516. https://doi.org/10.1080/00275514.2019.1588583
Jeewon R, Hyde K (2016) Establishing species boundaries and new taxa among fungi: recommendations to resolve taxonomic ambiguities. Mycosphere 7:1669–1677. https://doi.org/10.5943/mycosphere/7/11/4
Jones MD, Richards TA, Hawksworth DL, Bass D (2011a) Validation and justification of the phylum name Cryptomycota phyl. nov. IMA Fungus 2(2):173–175
Jones MDM, Forn I, Gadelha C, Egan MJ, Bass D, Massana R, Richards TA (2011b) Discovery of novel intermediate forms redefines the fungal tree of life. Nature 474:200–203
Joneson S, Stajich JE, Shiu S-H, Rosenblum EB (2011) Genomic transition to pathogenicity in chytrid fungi. PLoS Pathog 7:e1002338
Joshi A, Lanjekar VB, Dhakephalkar PK, Callaghan TM, Griffith GW, Dagar SS (2018) Liebetanzomyces polymorphus gen. et sp. nov., a new anaerobic fungus (Neocallimastigomycota) isolated from the rumen of a goat. MycoKeys 40:89–110
Kagami M, Miki T, Takimoto G (2014) Mycoloop: chytrids in aquatic food webs. Front Microbiol 5:166
Kagami M, Seto K, Nozaki D et al (2021) Single dominant diatom can host diverse parasitic fungi with different degree of host specificity. Limnol Oceanogr. https://doi.org/10.1002/lno.11631
Kagami M, von Elert E, Ibelings BW et al (2007) The parasitic chytrid, Zygorhizidium, facilitates the growth of the cladoceran zooplankter, Daphnia, in cultures of the inedible alga, Asterionella. Proc R Soc B 274:1561–1566. https://doi.org/10.1098/rspb.2007.0425
Karling JS (1977) Chytridiomycetarum Iconographia. Lubrecht and Cramer, Monticello, NY
Karling JS (1931) Studies in the Chytridiales VI. the Occurrence and life history of a new species of Cladochytrium in cells of Eriocaulon septangulare. Am J Bot 18:526–557. https://doi.org/10.2307/2435907
Karling JS (1935) A further study of Cladochytrium replicatum with special reference to its distribution, host range, and culture on artificial media. Am J Bot 22:439–452. https://doi.org/10.2307/2436115
Karling JS (1941) Texas chytrids. Torreya 41:105–108
Karling JS (1964) Synchytrium. Academic Press, New York
Karling JS (1937) The cytology of the Chytridiales with special reference to Cladochytrium replicatum. Mem Torrey Bot Club 19:1–92
Karling JS (1949) Nowakowskiella crassa sp. nov., Cladochytrium aureum sp. nov., and other polycentric chytrids from Maryland. Bull Torrey Bot Club 76:294–301. https://doi.org/10.2307/2482324
Karpov SA, Kobseva AA, Mamkaeva MA et al (2014a) Gromochytrium mamkaevae gen. & sp. nov. and two new orders: gromochytriales and mesochytriales (Chytridiomycetes). Persoonia 32:115–126. https://doi.org/10.3767/003158514x680234
Karpov SA, Mamkaeva MA, Aleoshin VV et al (2014b) Morphology, phylogeny, and ecology of the aphelids (Aphelidea, Opisthokonta) and proposal for the new superphylum Opisthosporidia. Front Microbiol 5:112. https://doi.org/10.3389/fmicb.2014.00112
Karpov S, Lopez-Garcia P, Mamkaeva MA et al (2016) Chytridiomycete Polyphagus parasiticus: molecular phylogeny supports the erection of a new Chytridiomycete order. Mikol Fitopatol 50:362–366
Karpov SA, Tcvetkova VS, Mamkaeva MA, Torruella G, Timpano H et al (2017) Morphological and genetic diversity of Opisthosporidia: new aphelid Paraphelidium tribonemae gen. et sp. nov. J Eukaryot Microbiol 64(2):204–212. https://doi.org/10.1111/jeu.12352
Karpov SA, Vishnyakov A, López-García P, Zorina N, Ciobanu M, Tcvetkova VS, Moreira D (2020) Morphology and molecular phylogeny of Aphelidium insulamus sp. nov. (Aphelida, Opisthosporidia). Protistology 14:191–203
Kamei K (2000) Animal models of Zygomycosis—Absidia, Rhizopus, Rhizomucor, and Cunninghamella. Mycopathologia 152:5–13
Karimi K, Arzanlou M, Ahari AB, Ghazi MM (2015) Phenotypic and molecular characterization of the causal agent of chafer beetle mortality in the wheat fields of the Kurdistan province, Iran. J Plant Prot Res 55(3):227–234
Karthikeyan V, Gopalakrishnan A (2014) A novel report of phytopathogenic fungi Gilbertella persicaria infection on Penaeus monodon. Aquaculture 340:224–229
Keeling P (2009) Five questions about Microsporidia. PLoS Pathog 5(9):e1000489. https://doi.org/10.1371/journal.ppat.1000489
Kia SH, Schulz M, Ayah E, Schouten A, Müllenborn C, Paetz C, Schneider B, Hofmann D, Disko U, Tabaglio V, Marocco A (2014) Abutilon theophrasti’s defense against the allelochemical benzoxazolin-2(3H)-one: support by Actinomucor elegans. J Chem Ecol 40:1286–1298
Kirk PM (1989) A new species of Benjaminiella (Mucorales: Mycotyphaceae). Mycotaxon 35(1):121–125
Kirk PM, Cannon PF, Minter DH, Stalpers JA (2008) Ainsworth & Bisby’s dictionary of the fungi, 10th edn. CABI Europe, UK
Koetschan C, Kittelmann S, Lu J, Al-Halbouni D, Jarvis GN, Müller T, Wolf T, Janssen PH (2014) Internal transcribed spacer 1 secondary structure analysis reveals a common core throughout the anaerobic fungi (Neocallimastigomycota). PLoS ONE 9:e91928
Kirk PM, Cannon PF, David JC, Stalpers JA (2001) Ainsworth & Bisby’s dictionary of the fungi, 9th edn. CABI Publishing, Wallingford
Kovacs RL, Sundberg WL (1999) Syzygites megalicarpus (Mucorales, Zygomycetes) in Illinois. Trans Illinois State Acad Sci 92:181–190
Kõljalg U, Larsson KH, Abarenkov K, Nilsson RH, Alexander IJ, Eberhardt U, Erland S, Høiland K, Kjøller R, Larsson E, Pennanen T, Sen R, Taylor AF, Tedersoo L, Vrålstad T, Ursing BM (2005) UNITE: a database providing web-based methods for the molecular identification of ectomycorrhizal fungi. New Phytol 166(3):1063–1068
Krug JC, Benny GL, Keller HW (2004) Coprophilous fungi. In: Mueller GM, Bills GF, Foster MS (eds) Biodiversity of fungi. Inventory and monitoring methods. Elsevier Academic Press, London, pp 467–499
Lagarde A, Jargeat P, Roy M, Girardot M, Imbert C, Millot M, Mambu L (2018) Fungal communities associated with Evernia prunastri, Ramalina fastigiata and Pleurosticta acetabulum: Three epiphytic lichens potentially active against Candida biofilms. Microbiol Res 211:1–12
Laking AE, Ngo HN, Pasmans F, Martel A, Nguyen TT (2017) Batrachochytrium salamandrivorans is the predominant chytrid fungus in Vietnamese salamanders. Sci Rep 7:4443
Lambert MR, Womack MC, Byrne AQ, Hernández-Gómez O, Noss CF, Rothstein AP, Blackburn DC, Collins JP, Crump ML, Koo MS et al (2020) Comment on “Amphibian fungal panzootic causes catastrophic and ongoing loss of biodiversity.” Science 367:e1838
Lambertini C, Becker CG, Jenkinson TS, Rodriguez D, da Silva LD, James TY, Zamudio KR, Toledo LF (2016) Local phenotypic variation in amphibian-killing fungus predicts infection dynamics. Fungal Ecol 20:15–21
Lange CE (1993) Unclassified protists of arthropods: the ultrastructure of Nephridiophaga periplanetae (Lutz & Splendore, 1903) n. comb., and the affinities of the Nephridiophagidae to other protists. J Eukaryot Microbiol 40:689–700
Lara E, Moreira D, López-García P (2010) The environmental clade LKM11 and Rozella form the deepest branching clade of fungi. Protist 161:116–121
Lee SH, Nguyen TTT, Lee HB (2018) Isolation and characterization of two rare mucoralean species with specific habitats. Mycobiology 46(3):205–214
Lefèvre E, Bardot C, Noël C, Carrias J-F, Viscogliosi E, Amblard C, Sime-Ngando T (2007) Unveiling fungal zooflagellates as members of freshwater picoeukaryotes: evidence from a molecular diversity study in a deep meromictic lake. Environ Microbiol 9:61–71
Lefèvre E, Roussel B, Amblard C, Sime-Ngando T (2008) The molecular diversity of freshwater picoeukaryotes reveals high occurrence of putative parasitoids in the plankton. PLoS ONE 3(6):e2324
Lefèvre E, Letcher PM, Powell MJ (2012) Temporal variation of the small eukaryotic community in two freshwater lakes: emphasis on zoosporic fungi. Aquat Microb Ecol 67:91–105
Léger L (1909) Sur un Mycetozoaire nouveau endoparasi te des insects. CR Acad Sci Paris 149:239–241
Lepelletier F, Karpov SA, Alacid E, Le Panse S, Bigeard E, Garcés E, Jeanthon C, Guillou L (2014) Dinomyces arenysensis gen. et sp. nov. (Rhizophydiales, Dinomycetaceae fam. nov.), a chytrid infecting marine dinoflagellates. Protist 165:230–244. https://doi.org/10.1016/j.protis.2014.02.004
Lendner A (1908) Les Mucorinées de la Suisse. Beiträge zur Kryptogamenflora der Schweiz. III(I). K.-J. Wyss, Bern, Switzerland
Letcher PM, Powell MJ, Barr DJS et al (2008a) Rhizophlyctidales-a new order in Chytridiomycota. Mycol Res 112:1031–1048. https://doi.org/10.1016/j.mycres.2008.03.007
Letcher PM, Velez CG, Barrantes ME et al (2008b) Ultrastructural and molecular analyses of Rhizophydiales (Chytridiomycota) isolates from North America and Argentina. Mycol Res 112:759–782. https://doi.org/10.1016/j.mycres.2008.01.025
Letcher PM, Powell MJ, Chambers JG et al (2005) Ultrastructural and molecular delineation of the Chytridiaceae (Chytridiales). Can J Bot 83:1561–1573. https://doi.org/10.1139/b05-115
Letcher PM, Powell MJ, Churchill PF, Chambers JG (2006) Ultrastructural and molecular phylogenetic delineation of a new order, the Rhizophydiales (Chytridiomycota). Mycol Res 110:898–915. https://doi.org/10.1016/j.mycres.2006.06.011
Letcher PM, Longcore JE, Quandt CA, Leite DdS, James TY, Powell MJ (2017) Morphological, molecular, and ultrastructural characterization of Rozella rhizoclosmatii, a new species in Cryptomycota. Fungal Biol 121(1):1–10. https://doi.org/10.1016/j.funbio.2016.08.008
Letcher PM, Powell MJ (2018) A taxonomic summary and revision of Rozella (Cryptomycota). IMA Fungus 9:383–399. https://doi.org/10.5598/imafungus.2018.09.02.09
Letcher PM, Powell MJ (2019) A taxonomic summary of Aphelidiaceae. IMA Fungus 10(1):4. https://doi.org/10.1186/s43008-019-0005-7
Liebetanz E (1910) Die parasitischen Protozoen des Wiederkäuermagen. Arch Protistenkunde 19:19–80
Liggenstoffer AS, Youssef NH, Couger MB, Elshahed MS (2010) Phylogenetic diversity and community structure of anaerobic gut fungi (phylum Neocallimastigomycota) in ruminant and non-ruminant herbivores. ISME J 4:1225–1235
Lima DX, Santiago ALCMDA, Souza-Motta CM (2015) Diversity of Mucorales in natural and degraded semi-arid soils. Braz J Bot 38(1):1–9
Lima DX, Santiago ALCMDA, Souza-Motta CM (2016) Diversity of Mucorales in natural and degraded semi-arid soils. Braz J Bot 39(4):1127–1133
Lima DX, Cordeiro TRL, de Souza CAF, Santiago ALCMDA, Souza-Motta CM (2018) Diversity of basal fungal order Mucorales (Mucoromycota) in a remaining area of the Brazilian Atlantic Rainforest. Nova Hedwigia 107(3–4):459–471
Ling-Young M (1930) Étude biologique des phénomènes de la sexualité chez les Mucorinées. Rev Gén Bot 42:722–752
Liu YJJ, Hodson MC, Hall BD (2006) Loss of the flagellum happened only once in the fungal lineage: phylogenetic structure of Kingdom Fungi inferred from RNA polymerase II subunit genes. BMC Evol Biol 6:74. https://doi.org/10.1186/1471-2148-6-74
Liu XY, Voigt K (2010) Molecular characters of zygomycetous fungi. In: Gherbawy Y, Voigt K (eds) Molecular identification of fungi. Part 2. Springer Verlag, Berlin, pp 461–488
Loh LS, Nawawi A, Kuthubutheen AJ (2001) Mucoraceous fungi from Malaysia. University of Malaysia, Malaysia, Institute of Biological Sciences
Longcore JE, Barr DJS, Desaulniers N (1995) Powellomyces, a new genus in the Spizellomycetales. Can J Bot 73:1385–1390
Longcore JE, Qin S, Simmons DR, James TY (2020) Quaeritorhiza haematococci is a new species of parasitic chytrid of the commercially grown alga, Haematococcus pluvialis. Mycologia 112:606–615. https://doi.org/10.1080/00275514.2020.1730136
Longcore JE, Pessier AP, Nichols DK (1999) Batrachochytrium dendrobatidis gen. et sp. nov., a chytrid pathogenic to amphibians. Mycologia 91:219–227
Lucarotti CJ (1981) Zoospore ultrastructure of Nowakowskiella elegans and Cladochytrium replicatum (Chytridiales). Can J Bot 59:137–148
Lücking R (2020) Three challenges to contemporaneous taxonomy from a licheno-mycological perspective. Megataxa 1(1):8–103
Lutz A, Splendore A (1903) Über Pebrine und verwandte Mikrosporidien. Ein Beitrag zur Kenntnis der brasilianischen Sporozoen. Centr Bakt Parasitenkd I Abt Orig 33:150–157
Macias AM, Marek PE, Morrissey EM, Brewer MS, Short DPG, Stauder CM, Wickert KL, Berger MC, Metheny AM, Stajich JE, Boyce G, Rio RVM, Panaccione DG, Wong V, Jones TH, Kasson MT (2019) Diversity and function of fungi associated with the fungivorous millipede, Brachycybe lecontii. Fungal Ecol 41:187–197
Marano AV, Steciow MM, Arellano ML (2008) New records of chytridiaceous fungi (Chytridiomycota) from the Reserva Natural Selva Marginal Punta Lara (Argentina) with comments on some previously reported species. Nord J Bot 26:248–254. https://doi.org/10.1111/j.1756-1051.2008.00214.x
Martel A, Spitzen-van der Sluijs A, Blooi M, Bert W, Ducatelle R, Fisher MC, Woeltjes A, Bosman W, Chiers K, Bossuyt F (2013) Batrachochytrium salamandrivorans sp. nov. causes lethal chytridiomycosis in amphibians. Proc Natl Acad Sci U S A 110:15325–15329
McDonald CA, Longo AV, Lips KR, Zamudio KR (2020) Incapacitating effects of fungal coinfection in a novel pathogen system. Mol Ecol 29:3173–3186
Mcgranaghan P, Davies JC, Griffith GW, Davies DR, Theodorou MK (1999) The survival of anaerobic fungi in cattle faeces. FEMS Microbiol Ecol 29:293–300
Mehrotra MD [1963a (1964)] Fruit rot of pear caused by Gilbertella persicaria var. indica. Sydowia 17:124–125
Mehrotra MD [(1963b) (1964)]. Fruit rot of tomato caused by Gilbertella persicaria. Sydowia 17:17–19
Melo RFR, Gondim NH de B, Santiago, ALCM de A, Maia LC, Miller NA (2020) Coprophilous fungi from Brazil: updated identification keys to all recorded species. Phytotaxa 436:104–124
Mendoza L, Vilela R, Voelz K, Ibrahim AS, Voigt K, Lee SC (2014) Human fungal pathogens of Mucorales and Entomophthorales, Chapter 27. In: Casadevall A, Mitchell AP, Berman J, Kwon-Chung KJ, Perfect JR, Heitman J (eds) Cold Spring Harbor Perspectives: fungal pathogens. Cold Spring Harbor Press, New York
Meyer W, Gams W (2003) Delimitation of Umbelopsis (Mucorales, Umbelopsidaceae fam. nov.) based on ITS sequence and RFLP data. Mycol Res 107:339–350
MGnify (2017) Inter-comparison of Marine Plankton metagenome analysis methods. Sampling event dataset. https://doi.org/10.15468/re7eoi. https://www.gbif.org/occurrence/2027088237. Accessed 05 Nov 2020
MGnify (2019) Water and sediment samples targeted loci environmental. Sampling event dataset. https://doi.org/10.15468/oputoa. https://www.gbif.org/occurrence/2022869673. Accessed 05 Nov 2020
MGnify (2020) Raw sequence reads from soil relic DNA study. Sampling event dataset. https://doi.org/10.15468/r6mbte. https://www.gbif.org/occurrence/2870940604. Accessed 22 Oct 2020
Mikawa T (1988) The Mucorales of Kathmandu, Nepal (1). In: Watanabe M, Malla SB (eds) Cryptogams of the Himalayas, vol 1. National Science Museum, Tsukuba, Japan
Mirza JH, Khan SM, Begum S, Shagufta S (1979) Mucorales of Pakistan. University of Agriculture, Faisalabad, Pakistan
Morehouse EA, James TY, Ganley AR, Vilgalys R, Berger L, Murphy PJ, Longcore JE (2003) Multilocus sequence typing suggests the chytrid pathogen of amphibians is a recently emerged clone. Mol Ecol 12:395–403
Mousavi B, Costa JM, Arne P, Guillot J, Chermette R, Botterel F, Dannaoui E (2018) Occurrence and species distribution of pathogenic Mucorales in unselected soil samples from France. Med Mycol 56:315–321
Mozley-Standridge SE, Letcher PM, Longcore JE et al (2009) Cladochytriales-a new order in Chytridiomycota. Mycol Res 113:498–507. https://doi.org/10.1016/j.mycres.2008.12.004
Murphy CL, Youssef NH, Hanafy RA, Couger M, Stajich JE, Wang Y, Baker K, Dagar SS, Griffith GW, Farag IF, Callaghan TM, Elshahed MS (2019) Horizontal gene transfer as an indispensable driver for Neocallimastigomycota evolution into a distinct gut-dwelling fungal lineage. Appl Environ Microbiol 85(15):e00988-e01019
Nagy LG, Petkovits T, Kovács GM, Voigt K, Vágvölgyi C, Papp T (2011) Where is the unseen fungal diversity hidden? A study of Mortierella reveals a large contribution of reference collections to the identification of fungal environmental sequences. New Phytol 191:789–794
Nagy T, Tunnicliffe RB, Higgins LD, Walters C, Gilbert HJ, Williamson MP (2007) Characterization of a double dockerin from the cellulosome of the anaerobic fungus Piromyces equi. J Mol Biol 373:612–622
Naranjo-Ortiz MA, Gabaldón T (2019) Fungal evolution: diversity, taxonomy and phylogeny of the Fungi. Biol Rev 94:2101–2137
Newbold CJ, de la Fuente G, Belanche A, Ramos-Morales E, McEwan NR (2015) The role of ciliate protozoa in the rumen. Front Microbiol 6:1313
Nichols D, Smith A, Gardiner C (1996) Dermatitis of anurans caused by fungal-like protists. Proc Am Assoc Zoo Vet Puerto Vallerta, Mexico, pp 220–221
Nishida H, Nishiyama M, Kobashi N, Kosuge T, Hoshino T, Yamane H (1999) A prokaryotic gene cluster involved in synthesis of lysine through the amino adipate pathway: a key to the evolution of amino acid biosynthesis. Genome Res 9:1175–1183. https://doi.org/10.1101/gr.9.12.1175
O’Donnell K, Lutzoni FM, Ward TJ, Benny GL (2001) Evolutionary relationships among mucoralean fungi (Zygomycota): evidence for family polyphyly on a large scale. Mycologia 93:286–297
O’Hanlon SJ, Rieux A, Farrer RA, Rosa GM, Waldman B, Bataille A, Kosch TA, Murray KA, Brankovics B, Fumagalli M et al (2018) Recent Asian origin of chytrid fungi causing global amphibian declines. Science 360:621–627
Ormières R, Manier J-F (1973) Observations sur Nephridiophaga forficulae (Léger, 1909). Ann Parasitol Hum Comparée 48:1–10
Orpin CG (1975) Studies on the rumen flagellate Neocallimastix frontalis. J Gen Microbiol 91:249–262
Orpin CG (1976) Studies on the rumen flagellate Sphaeromonas communis. J Gen Microbiol 94:270–280
Orpin CG (1977) Rumen flagellate Piromonas communis - Its life history and invasion of plant material in rumen. J Gen Microbiol 99:107–117
Oscar Martínez V, Eduardo Valenzuela F (2003) Zygomycota citados para Chile (Zygomycota cited in Chile). Boletín Micológico 18:67–74
Partida-Martinez LP, Monajembashi S, Greulich K-O, Hertweck C (2007a) Endosymbiont-dependent host reproduction maintains bacterial-fungal mutualism. Curr Biol 17:773–777
Partida-Martinez LP, Groth I, Schmitt I, Richter W, Roth M, Hertweck C (2007b) Burkholderia rhizoxinica sp. nov. and Burkholderia endofungorum sp. nov., bacterial endosymbionts of the plant-pathogenic fungus Rhizopus microsporus. Int J Syst Evol Microbiol 57:2583–2590
Perrin WS (1906) Observations on the structure and life-history of Pleistophora periplanetæ, Lutz and Splendore. J Cell Sci 49:615–633
Pinho DB, Pereira OL, Soares DJ (2014) First report of Gilbertella persicaria as the cause of soft rot of fruit of Syzygium cumini. Australas Plant Dis Notes 9:143
Plischuk S, Lange CS (2011) Registro de Nephridiophaga sp. (Protista: Nephridiophagidae) en Apis mellifera (Hymenoptera: Apidae) del Sur de la región Pampeana. Rev Soc Entomol Argent 70:357–361
Poidevin L, Levasseur A, Paes G, Navarro D, Heiss-Blanquet S, Asther M, Record E (2009) Heterologous production of the Piromyces equi cinnamoyl esterase in Trichoderma reesei for biotechnological applications. Lett Appl Microbiol 49:673–678
Pongratz E (1966) De quelques champignons parasites d’organismes planctoniques du Léman. Schweiz Z Für Hydrol 28:104–132
Powell MJ (1976) Ultrastructure and isolation of glyoxysomes (microbodies) in zoospores of the fungus Entophlyctis sp. Protoplasma 89:1–27
Powell MJ (1984) Fine structure of the unwalled thallus of Rozella polyphagi in its host Polyphagus euglenae. Mycologia 76:1039–1048
Powell MJ, Letcher PM (2014) Chytridiomycota, Monoblepharidomycota, and Neocallimastigomycota. In: McLaughlin DJ, Spatafora JW (eds) Systematics and evolution. The mycota VII part A, 2nd edn. Springer, Berlin, pp 141–175
Powell MJ, Letcher PM, Chen S-F (2018a) Phylogeny and taxonomic revision of the soil chytrid, Gaertneriomyces, and description of the new genus Barromyces (Spizellomycetaceae, Chytridiomycota). Nova Hedwigia 107:205–228. https://doi.org/10.1127/nova_hedwigia/2018/0465
Powell MJ, Letcher PM, Longcore JE, Blackwell WH (2018b) Zopfochytrium is a new genus in the Chytridiales with distinct zoospore ultrastructure. Fungal Biol 122:1041–1049. https://doi.org/10.1016/j.funbio.2018.08.005
Prakash H, Ghosh AK, Sm R, Paul RA, Gupta S, Negi V, Chakrabarti A (2016) The environmental source of emerging Apophysomyces variabilis infection in India. Med Mycol 54(6):567–575
Purrini K, Rohde M (1988) Light and electron microscope studies on two new protists, Coelosporidium schalleri n. sp. and Coelosporidium meloidorum n. sp. (Protista), infecting natural populations of the flea beetle, Podagrica fusciocornis, and flower beetle. Mylabris Maculiventris Zool Anz 220:323–333
Purrini K, Weiser J (1990) Light and electron microscope studies on a protozoan, Oryctospora alata n. gen., n. sp. (Protista: Coelosporidiidae), parasitizing a natural population of the rhinoceros beetle, Oryctes monoceros Oliv. (Coleoptera, Scarabaeidae). Zool Beitr 33:209–220
Quandt CA, Beaudet D, Corsaro D, Walochnik J, Michel R, Corradi N, James TY (2017) The genome of an intranuclear parasite, Paramicrosporidium saccamoebae, reveals alternative adaptations to obligate intracellular parasitism. Elife 6:e29594
Radek R, Herth W (1999) Ultrastructural investigation of the spore-forming protist Nephridiophaga blattellae in the Malpighian tubules of the German cockroach Blattella germanica. Parasitol Res 85:216–231
Radek R, Klein G, Storch V (2002) The spore of the unicellular organism Nephridiophaga blattellae: ultrastructure and substances of the spore wall. Acta Protozool 41:169–181
Radek R, Wellmanns D, Wolf A (2011) Two new species of Nephridiophaga (Zygomycota) in the Malpighian tubules of cockroaches. Parasitol Res 109(2):473−482
Radek R, Wurzbacher C, Gisder S, Nilsson RH, Owerfeldt A, Genersch E, Kirk PM, Voigt K (2017) Morphologic and molecular data help adopting the insect-pathogenic nephridiophagids (Nephridiophagidae) among the early diverging fungal lineages, close to the Chytridiomycota. MycoKeys 25:31–50. https://doi.org/10.3897/mycokeys.25.12446
Rasconi S, Jobard M, Jouve L, Sime-Ngando T (2009) Use of calcofluor white for detection, identification, and quantification of phytoplanktonic fungal parasites. Appl Environ Microbiol 75(8):2545−2553
Rashmi M, Kushveer JS, Sarma VV (2019) A worldwide list of endophytic fungi with notes on ecology and diversity. Mycosphere 10(1):798–1079
Richards M (1956) Some inoperculate chytrids from South Wales. Trans Brit Mycol Soc 39:261. https://doi.org/10.1016/S0007-1536(56)80005-3
Richardson MJ (2009) The ecology of the Zygomycetes and its impact on environmental exposure. Clin Microbiol Infect 15:2–9
Rodriguez D, Becker CG, Pupin NC, Haddad CF, Zamudio KR (2014) Long-term endemism of two highly divergent lineages of the amphibian-killing fungus in the Atlantic Forest of Brazil. Mol Ecol 23:774–787
Rollins-Smith LA (2020) Global amphibian declines, disease, and the ongoing battle between Batrachochytrium fungi and the immune system. Herpetologica 76:178–188
Rollins-Smith LA, Reinert LK, O’Leary CJ, Houston LE, Woodhams DC (2005) Antimicrobial peptide defenses in amphibian skin. Integr Comp Biol 45:137–142
Rosenblum EB, James TY, Zamudio KR, Poorten TJ, Ilut D, Rodriguez D, Eastman JM, Richards-Hrdlicka K, Joneson S, Jenkinson TS et al (2013) Complex history of the amphibian-killing chytrid fungus revealed with genome resequencing data. Proc Natl Acad Sci U S A 110:9385–9390
Santiago ALCMDA, Trufem SFB, Malosso E, dos Santos PJP, Cavalcanti MAQ (2011) Zygomycetes from herbivore dung in the ecological reserve of dois Irmãos, Northeast Brazil. Braz J Microbiol 42:89–95
Santiago ALCMDA, dos Santos PJP, Maia LC (2013) Mucorales from the semiarid of Pernambuco, Brazil. Braz J Microbiol 44(1):299–305
Sarsaiya S, Shi J, Chen J (2019) A comprehensive review on fungal endophytes and its dynamics on Orchidaceae plants: current research, challenges, and future possibilities. Bioengineered 10(1):316–334
Schaffner JH (1909) The classification of plants, IV. Ohio Naturalist 9:446–455
Scheele BC, Pasmans F, Skerratt LF, Berger L, Martel A, Beukema W, Acevedo AA, Burrowes PA, Carvalho T, Catenazzi A et al (2019) Amphibian fungal panzootic causes catastrophic and ongoing loss of biodiversity. Science 363:1459–1463
Schloegel L, Picco A, Kilpatrick A, Davies AJ, Hyatt AD, Daszak P (2009) Magnitude of the US trade in amphibians and presence of Batrachochytrium dendrobatidis and ranavirus infection in imported North American bullfrogs (Rana catesbeiana). Biol Conserv 142:1420–1426
Schloegel LM, Toledo LF, Longcore JE, Greenspan SE, Vieira CA, Lee M, Zhao S, Wangen C, Ferreira CM, Hipolito M et al (2012) Novel, panzootic and hybrid genotypes of amphibian chytridiomycosis associated with the bullfrog trade. Mol Ecol 21:5162–5177
Schmit JP, Mueller GM (2007) An estimate of the lower limit of global fungal diversity. Biodivers Conserv 16:99–111
Schipper MAA (1986) Hyphomucor, a new genus in the Mucorales for Mucor assamensis. Mycotaxon 27:83–86
Schipper MAA (1973) A study on variability in Mucor hiemalis and related species. Stud Mycol 4:1–40
Schipper MAA (1975) Mucor mucedo, Mucor flavus and related species. Stud Mycol 10:1–33
Schipper MAA (1976) On Mucor circinelloides, Mucor racemosus and related species. Stud Mycol 12:1–40
Schipper MAA (1978a) On certain species of Mucor with a key to all accepted species. Stud Mycol 17:1–52
Schipper MAA (1978b) On the genera Rhizomucor and Parasitella. Stud Mycol 17:53–71
Schipper MAA (1984) A revision of the genus Rhizopus. I. The Rhizopus stolonifer-group and Rhizopus oryzae. Stud Mycol 25:1–19
Schoch CL, Seifert KA, Huhndorf S et al (2012) Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc Natl Acad Sci U S A 109:6241–6246. https://doi.org/10.1073/pnas.1117018109
Schoenlein-Crusius IH, Milanez AI, Trufem SFB, Pires-Zottarelli CLA, Grandi RAP, Santos ML, Giustra KC (2006) Microscopic fungi in the Atlantic Rainforest in Cubatão, São Paulo, Brazil. Braz J Microbiol 7:267–275
Schröter J (1889) VIII. Ordn. Zygomycetes. Brefeld, 111. Gatt. Endogone link 1809. In: Cohn F (ed) Kryptogamen-Flora von Schleisen III Band, 1 Hälfte. Pilze J. U. Kern’s Verlag (Max Müller), Breslau, Germany, pp 197–225
Sekimoto S, Rochon D, Long JE et al (2011) A multigene phylogeny of Olpidium and its implications for early fungal evolution. BMC Evol Biol 11:331. https://doi.org/10.1186/1471-2148-11-331
Seto K, Degawa Y (2018a) Collimyces mutans gen. et sp. nov. (Rhizophydiales, Collimycetaceae fam. nov.), a new chytrid parasite of Microglena (Volvocales, clade Monadinia). Protist 169:507–520. https://doi.org/10.1016/j.protis.2018.02.006
Seto K, Degawa Y (2018b) Pendulichytrium sphaericum gen. et sp. nov. (Chytridiales, Chytriomycetaceae), a new chytrid parasitic on the diatom. Aulacoseira Granulata Mycosci 59:59–66. https://doi.org/10.1016/j.myc.2017.08.004
Seto K, Van Den Wyngaert S, Degawa Y, Kagami M (2020) Taxonomic revision of the genus Zygorhizidium: Zygorhizidiales and Zygophlyctidales ord. nov. (Chytridiomycetes, Chytridiomycota). Fungal Syst Evol 5:17–38. https://doi.org/10.3114/fuse.2020.05.02
Shelest E, Voigt K (2014) Genomics to study basal lineage fungal biology: phylogenomics suggests a common origin. In: Nowrousian M (ed) The Mycota. Vol. XIII: fungal genomics, 2nd edn. Springer Verlag, Berlin, pp 31–60
Simmons DR (2011) Phylogeny of Powellomycetaceae fam. nov and description of Geranomyces variabilis gen. et comb. nov. Mycologia 103:1411–1420. https://doi.org/10.3852/11-039
Simmons DR, Bonds AE, Castillo BT et al (2020) The Collection of Zoosporic Eufungi at the University of Michigan (CZEUM): introducing a new repository of barcoded Chytridiomyceta and Blastocladiomycota cultures. IMA Fungus 11:20. https://doi.org/10.1186/s43008-020-00041-z
Simmons DR, James TY, Meyer AF, Longcore JE (2009) Lobulomycetales, a new order in the Chytridiomycota. Mycol Res 113:450–460. https://doi.org/10.1016/j.mycres.2008.11.019
Simmons DR, Longcore JE (2012) Thoreauomyces gen. nov., Fimicolochytrium gen. nov. and additional species in Geranomyces. Mycologia 104:1229–1243. https://doi.org/10.3852/12-015
Shin J, Bataille A, Kosch TA, Waldman B (2014) Swabbing often fails to detect amphibian chytridiomycosis under conditions of low infection load. PLoS ONE 9:e111091
Shipton WA, Schipper MAA (1975) Halteromyces, a new genus in the Mucorales. Antonie Van Leeuwenhoek Ned Tijdschr Hyg 41:337–342
Solomon KV, Haitjema CH, Henske JK, Gilmore SP, Borges-Rivera D, Lipzen A, Brewer HM, Purvine SO, Wright AT, Theodorou MK, Grigoriev IV, Regev A, Thompson DA, O’Malley MA (2016) Early-branching gut fungi possess a large, comprehensive array of biomass-degrading enzymes. Science 351:1192–1195
Sparrow FK (1960) Aquatic phycomycetes, 2nd edn. University of Michigan Press, Ann Arbor, MI
Spatafora JW, Chang Y, Benny GL, Lazarus K, Smith ME, Berbee ML, Bonito G, Corradi N, Grigoriev I, Gryganskyi A, James TY, O’Donnell K, Roberson R, Taylor TN, Uehling J, Vilgalys R, White MM, Stajich JE (2016) A phylum-level phylogenetic classification of zygomycete fungi based on genome-scale data. Mycologia 108(5):1028–1046
Sprague V (1970) Recent problems of taxonomy and morphology of Haplosporidia. J Parasitol 56:327–328
Stabel M, Hanafy RA, Schweitzwe T, Greif M, Aliyu H, Flad V, Young D, Lebuhn M, Elshahed MS, Ochsenreither K (2020) Aestipascuomyces dupliciliberans gen. nov, sp. nov., the first cultured representative of the uncultured SK4 clade from aoudad sheep and alpaca. Microorganisms 8:1734
Stajich JE, Berbee ML, Blackwell M, Hibbett DS, James TY, Spatafora JW, Taylor JW (2009) Primer—the fungi. Curr Biol 19(18):R840–R845. https://doi.org/10.1016/j.cub.2009.07.004
Steiger RA, Simmons DR, Longcore JE (2011) Cylindrochytridium johnstonii is a member of the Cladochytriales. Mycotaxon 118:293–302. https://doi.org/10.5248/118.293
Strassert JFH, Wurzbacher C, Hervé V, Antany T, Brune A, Radek R (2021) Long rDNA amplicon sequencing of insect-infecting nephridiophagids reveals their affliation to the Chytridiomycota and a potential to switch between hosts. Sci Rep 11:396
Stubblefield JW (1955) The morphology and life history of Amphiacantha ovalis and A. attenuata, two new Haplosporidian parasites of gregarines. J Parasitol 41(5):443–459. https://doi.org/10.2307/3273803
Sumstine DR (1910) The North American Mucorales–I. Family Mucoracaeae Mycologia 2:125–154
Talley BL, Muletz CR, Vredenburg VT, Fleischer RC, Lips KR (2015) A century of Batrachochytrium dendrobatidis in Illinois amphibians (1888–1989). Biol Conserv 182:254–261
Taube R, Fabian J, Van den Wyngaert S, Agha R, Baschien C, Gerphagnon M, Kagami M, Krüger A, Premke K (2019) Potentials and limitations of quantification of fungi in freshwater environments based on PLFA profiles. Fungal Ecol 41:256–268
Taylor JW, Jacobson DJ, Kroken S et al (2000) Phylogenetic species recognition and species concepts in fungi. Fungal Genet Biol 31:21–32. https://doi.org/10.1006/fgbi.2000.1228
Taylor SK, Williams ES, Mills KW (1999) Mortality of captive Canadian toads from Basidiobolus ranarum mycotic dermatitis. J Wildl Dis 35:64–69
Tcvetkova V, Zorina N, Mamkaeva M, Karpov S (2019) Molecular phylogeny of Aphelidium arduennense sp. nov.—new representative of Aphelida (Opisthosporidia). Protistology. https://doi.org/10.21685/1680-0826-2019-13-4-2
Tedersoo L, Bahram M, Puusepp R, Nilsson RH, James TY (2017) Novel soil-inhabiting clades fill gaps in the fungal tree of life. Microbiome 5(1):42
Tedersoo L, Sánchez-Ramírez S, Kõljalg U, Bahram M, Döring M, Schigel D et al (2018) High-level classification of the Fungi and a tool for evolutionary ecological analyses. Fungal Divers 90(1):135–159. https://doi.org/10.1007/s13225-018-0401-0
Thaxter R (1922) A revision of the Endogoneae. P Am Acad Arts Sci 57:291–351
Theodorou MK, Williams BA, Dhanoa MS, McAIlan AB, France I (1994) A new gas production method using a pressure transducer to determine the fermentation kinetics of ruminant feeds. Anim Feed Sci Technol 48:185–197
Toguebaye BS, Manier JF, Bouix G, Marchand B (1986) Nephridiophaga ormieresi n. sp., Protiste parasite d´Aspidiomorpha cincta Fabricius, 1781 (Insecte Coléoptère: Chrysomelidae). Étude Ultrastructurale Protistologica 22:317–325
Torruella G, Grau-Bové X, Moreira D, Karpov SA, Burns JA, Sebé-Pedrós A, Völcker E, López-García P (2018) Global transcriptome analysis of the aphelid Paraphelidium tribonemae supports the phagotrophic origin of fungi. Commun Biol 1:231. https://doi.org/10.1038/s42003-018-0235-z
Tripathi M, Joshi Y, Gupta RC (2014) Assessment of endolichenic fungal diversity in some forests of Kumaun Himalaya. Curr Sci 107:745–748
Ueda K (2020) iNaturalist research-grade observations. iNaturalist.org. Occurrence dataset. https://doi.org/10.15468/ab3s5x. https://www.gbif.org/occurrence/2603461203. Accessed 12 Oct 2020
Van den Wyngaert S, Rojas-Jimenez K, Seto K et al (2018a) Diversity and hidden host specificity of chytrids infecting colonial Volvocacean algae. J Eukaryot Microbiol 65:870–881. https://doi.org/10.1111/jeu.12632
Van den Wyngaert S, Seto K, Rojas-Jimenez K et al (2017) A new parasitic chytrid, Staurastromyces oculus (Rhizophydiales, Staurastromycetaceae fam. nov.), infecting the freshwater desmid Staurastrum sp. Protist 168:392–407. https://doi.org/10.1016/j.protis.2017.05.001
Van den Wyngaert S, Rojas-Jimenez K, Seto K, Kagami M, Grossart H-P (2018b) Diversity and hidden host specificity of chytrids infecting colonial volvocacean algae. J Eukaryot Microbiol 65:870–881
Van Donk E, Ringelberg J (1983) The effect of fungal parasitism on the succession of diatoms in Lake Maarsseveen I (The Netherlands). Freshw Biol 13:241–251. https://doi.org/10.1111/j.1365-2427.1983.tb00674.x
Váňová M (1968) Contribution to the taxonomy of the genus Absidia (Mucorales) I. Absidia macrospora sp. Nov. Česká. Mykologie 22(4):296–300
van Tieghem P (1875) Nouvelles recherches sur les Mucorinées. Ann Sci Nat Bot Sér VI 1:5–175
van Tieghem P (1878) Troisième mémoire sur les Mucorinées. Ann Sci Nat Bot Sér VI 4:312–399
van Tieghem P, Le Monnier G (1873) Recherches sur les Mucorinées. Ann Sci Nat Bot Sér V 17:261–399
Voigt K, Wöstemeyer J (2001) Phylogeny and origin of 82 Zygomycetes from all 54 genera of the Mucorales and Mortierellales based on combined analysis of actin and translation elongation factor EF-1a genes. Gene 270:113–120
Voigt K (2012a) Zygomycota Moreau. In: Frey W (ed) Syllabus of plant families. A. Engler’s syllabus der Pflanzenfamilien, Blue-green algae, Myxomycetes and Myxomycete-like organisms, Phytoparasitic protists, heterotrophic Heterokontobiota and Fungi, 13ª edn. Borntraeger Science Publishers, Berlin.
Voigt K (2012b) Chytridiomycota. In: Engler A, Frey W (eds) Syllabus of Plant Families, Vol 1/1: blue-green algae, myxomycetes and myxomycete-like organisms, phytoparqasitic protists, heterotrophic Heterokontobionta and Fungi pp. Borntraeger, Stuttgart, pp 106–129
Voigt K, Kirk PM (2014) 136. FUNGI | Classification of the Zygomycetes: reappraisal as coherent class based on a comparison between traditional versus molecular systematics. In: Batt CA, Tortorello ML (eds) Encyclopedia of food microbiology, vol 2. Academic Press, Elsevier Ltd, pp 54–67
Voigt K, Marano AV, Gleason F (2013) Ecological and economical importance of parasitic and zoosporic true Fungi. In: Kempken F (ed) The mycota vol XI: agricultural applications, 2nd edn. Springer Verlag, Berlin, pp 243–270
Voglmayr H, Krisai-Greilhuber I (1996) Dicranophora fulva, a rare mucoraceous fungus growing on boletes. Mycol Res 100(5):583–590
Voglmayr H, Clémençon H (2015) Identification and taxonomic position of two mucoralean endoparasites of Hysterangium (Basidiomycota) based on molecular and morphological data. Mycol Prog 15(9):8–17
Wagner L, Stielow JB, de Hoog GS, Bensch K, Schwartze VU, Voigt K, Alastruey-Izquierdo A, Kurzai O, Walther G (2019) A new species concept for the clinically relevant Mucor circinelloides complex. Persoonia 44:67–97
Waldman B, Van de Wolfshaar KE, Klena JD, Andjic V, Bishop PJ, Norman RJDB (2001) Chytridiomycosis in New Zealand Frogs. Surveillance 38(3):9–11
Walther G, Pawłowska J, Alastruey-Izquierdo A, Wrzosek M, Rodriguez-Tudela J, Dolatabadi S, Chakrabarti A, de Hoog GS (2013) DNA barcoding in Mucorales: an inventory of biodiversity. Persoonia 30:11–47
Walther G, Wagner G, Kurzai O (2019) Updates on the taxonomy of Mucorales with an emphasis on clinically important taxa. J Fungi 5(4):106
Walther G, Wagner L, Kurzai O (2020) Outbreaks of Mucorales and the species involved. Mycopathologia 185:765–781
Wang Y, Youssef N, Couger M, Hanafy R, Elshahed M, Stajich JE (2019) Comparative genomics and divergence time estimation of the anaerobic fungi in herbivorous mammals. bioRxiv. https://doi.org/10.1128/mSystems.00247-19
Warcup JH (1990) Taxonomy, culture and mycorrhizal associations of some zygosporic Endogonaceae. Mycol Res 94(2):173–178
Weldon C, Du Preez LH, Hyatt AD, Muller R, Speare R (2004) Origin of the amphibian chytrid fungus. Emerg Infect Dis 10:2100–2105
Woodhams DC, Ardipradja K, Alford R, Marantelli G, Reinert L, Rollins-Smith L (2007) Resistance to chytridiomycosis varies among amphibian species and is correlated with skin peptide defenses. Anim Conserv 10:409–417
Woolever P (1966) Life history and electron microscopy of a haplosporidian, Nephridiophaga blattellae (Crawley) n. comb, in the Malphigian tubules of the German Cockroach, Blattella germanica (L.). J Protozool 13:622–642
Wijayawardene NN, Pawłowska J, Letcher PM, Kirk PM, Humber RA, Schussler A, Wrzosek M, Muszewska A, Okrasinska A, Istel L, Gesiorska A, Mungai P, Lateef AA, Rajeshkumar KC, Singh RV, Radek R, Walther G, Wagner L, Walker C, Wijesundara DSA, Papizadeh M, Dolatabadi S, Shenoy BD, Tokarev YS, Lumyong S, Hyde KD (2018) Notes for genera: basal clades of Fungi (including Aphelidiomycota, Basidiobolomycota, Blastocladiomycota, Calcarisporiellomycota, Caulochytriomycota, Chytridiomycota, Entomophthoromycota, Glomeromycota, Kickxellomycota, Monoblepharomycota, Mortierellomycota, Mucoromycota, Neocallimastigomycota, Olpidiomycota, Rozellomycota and Zoopagomycota). Fungal Divers 92:43–129
Wijayawardene NN, Hyde KD, Al-Ani LKT, Tedersoo L, Haelewaters D, Rajeshkumar KC, Zhao RL, Aptroot A, Leontyev DV, Saxena RK, Tokarev YS, Dai DQ, Letcher PM, Stephenson SL, Ertz D, Lumbsch HT, Kukwa M, Issi IV, Madrid H, Phillips AJL, Selbmann L, Pfliegler WP, Horváth E, Bensch K, Kirk PM, Kolaříková K, Raja HA, Radek R, Papp V, Dima V, Ma J, Malosso E, Takamatsu S, Rambold G, Gannibal PB, Triebel D, Gautam AK, Avasthi S, Suetrong S, Timdal E, Fryar SC, Delgado G, Réblová M, Doilom M, Dolatabadi S, Pawłowska J, Humber RA, Kodsueb R, Sánchez-Castro I, Goto BT, Silva DKA, de Souza FA, Oehl F, da Silva GA, Silva IR, Błaszkowski J, Jobim K, Maia LC, Barbosa FR, Fiuza PO, Divakar PK, Shenoy BD, Castañeda-Ruiz RF, Somrithipol S, Lateef AA, Karunarathna SC, Tibpromma S, Mortimer PE, Wanasinghe DN, Phookamsak R, Xu J, Wang Y, Tian F, Alvarado P, Li DW, Kušan I, Matočec N, Maharachchikumbura SSN, Papizadeh M, Heredia G, Wartchow F, Bakhshi M, Boehm E, Youssef N, Hustad VP, Lawrey JD, Santiago ALCMA, Bezerra JDP, Souza-Motta CM, Firmino AL, Tian Q, Houbraken J, Hongsanan S, Tanaka K, Dissanayake AJ, Monteiro JS, Grossart HP, Suija A, Weerakoon G, Etayo J, Tsurykau A, Vázquez V, Mungai P, Damm U, Li QR, Zhang H, Boonmee S, Lu YZ, Becerra AG, Kendrick B, Brearley FQ, Motiejūnaitė J, Sharma B, Khare R, Gaikwad S, Wijesundara DSA, Tang LZ, He MQ, Flakus A, Rodriguez-Flakus P, Zhurbenko MP, McKenzie EHC, Stadler M, Bhat DJ, Liu JK, Raza M, Jeewon R, Nassonova ES, Prieto M, Jayalal RGU, Erdogdu M, Yurkov A, Schnittler M, Shchepin ON, Novozhilov YK, Silva-Filho AGS, Gentekaki E, Liu P, Cavender JC, Kang Y, Mohammad S, Zhang LF, Xu RF, Li YM, Dayarathne MC, Ekanayaka AH, Wen TC, Deng CY, Pereira OL, Navathe S, Hawksworth DL, Fan XL, Dissanayake LS, Kuhnert E, Grossart HP, Thines M (2020) Outline of fungi and fungus-like taxa. Mycosphere 11(1):1060–1456
Williams DM (2011) Synedra, Ulnaria: definitions and descriptions—a partial resolution. Null 26:149–153. https://doi.org/10.1080/0269249X.2011.587646
Wu B, Hussain M, Zhang W, Stadler M, Liu X, Xiang M (2019) Current insights into fungal species diversity and perspective on naming the environmental DNA sequences of fungi. Mycology 10:127–140
Wu C-G, Lin S-J (1997) Endogonales in Taiwan: a new genus with unizygosporic sporocarps and a hyphal mantle. Mycotaxon 64:179–188
Wurzbacher C, Rosel S, Rychla A, Grossart H-P (2014) Importance of saprotrophic freshwater fungi for pollen degradation. PLoS ONE 9:e94643
Wylezich C, Radek R, Schlegel M (2004) Phylogenetische Analyse der 18S rRNA identifiziert den parasitischen Protisten Nephridiophaga blattellae (Nephridiophagidae) als Vertreter der Zygomycota (Fungi). Denisia 13:435–442
Yamamoto K, Degawa Y, Yamada A (2020) Taxonomic study of Endogonaceae in the Japanese islands—new species of Endogone, Jimgerdemannia, and Vinositunica, gen. nov. Mycologia 112(2):309–328
Zamudio KR, McDonald CA, Belasen AM (2020) High variability in infection mechanisms and host responses: a review of functional genomic studies of amphibian chytridiomycosis. Herpetologica 76:189–200
Zheng R-Y, Chen GQ (1998) Cunninghamella clavata sp. nov., a fungus with an unusual type of braching of sporophore. Mycotaxon 69(1):187–198
Zheng R-Y, Chen G-Q (2001) Monograph of Cunninghamella. Mycotaxon 80:1–75
Zheng R-Y, Chen G-Q, Hyang H, Liu X-Y (2007) A monograph of Rhizopus. Sydowia 59(2):273–372
Zheng R-Y, Liu X-Y (2013) Ambomucor gen. & spp. nov. from China. Mycotaxon 126:97–108
Ziaee A, Zia M, Bayat M, Hashemi J (2016) Identification of Mucorales isolates from soil using morphological and molecular methods. Curr Med Mycol 2(1):13–19
Zopf W (1884) Zur kenntniss der phycomyceten. I. Zur Morphologie und Biologie der Ancylisteen und Chytridiaceen. Nova Acta Der Kaiserlichen Leopoldinisch Carolinischen Deutschen Akademie Der Naturforscher 47:143–236
Zycha H (1935) Mucorineae. In: Kryptogamenflora der Mark Brandenburg. Band VIa. Gebrüder Borntraeger, Leipzig, Germany, pp 1–264
Zycha H, Siepmann R, Linnemann G (1969) Mucorales eine Beschreibung aller Gattungen und Arten dieser Pilzgruppe. J. Cramer, Lehre, Lichtenstein.
Acknowledgements
HBL was supported by the Graduate Program for the Undiscovered Taxa of Korea (NIBR202130202), and by the Project on Survey and Discovery of Indigenous Fungal Species of Korea funded by NIBR, and by the Project on Discovery of Fungi from Freshwater and Collection of Fungarium funded by NNIBR of the Ministry of Environment (MOE) of the Republic of Korea. TYJ and DRS were supported by the US National Science Foundation Grants tDBI-1756202, DBI-1910720, and DEB-1929738. GHJ thank FAPESP (2018/24915-1 and 2019/17237-0). KS was supported by a Grant-in-Aid for JSPS Overseas Research Fellowship from the Japan Society for the Promotion of Science. BW was supported by grants from the National Research Foundation of Korea funded by the Ministry of Education (2015R1D1A01057282) and the Ministry of Science, ICT, and Future Planning (2018R1A2B6006833) of the Republic of Korea. ALS was supported by the Fundação de Amparo à Ciência do Estado de Perbambuco and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (FACEPE/CNPq-APQ-0842-2.12/14.). We thank the National Council for Scientific and Technological Development (CNPq) for the research scholarship Granted to ALS. RR and JFHS were supported by Grants of the German Research Foundation (DFG; RA850/6-1 Project No. 397619173 and STR1349/2-1 Project No. 432453260, respectively). KV was supported by the German Research Foundation (DFG) through the Collaborative Research Center/Transregio FungiNet 124 ‘Pathogenic fungi and their human host: Networks of Interaction’, DFG Project Number 210879364, Project A6.
Author information
Authors and Affiliations
Corresponding author
Additional information
Handling Editor: Jian-Kui Liu.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Voigt, K., James, T.Y., Kirk, P.M. et al. Early-diverging fungal phyla: taxonomy, species concept, ecology, distribution, anthropogenic impact, and novel phylogenetic proposals. Fungal Diversity 109, 59–98 (2021). https://doi.org/10.1007/s13225-021-00480-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13225-021-00480-y