Key Facts
Musculoskeletal System
- •
Radiographs can demonstrate the characteristic features of systemic sclerosis including calcinosis, loss of normal distal soft tissue and skin creases, and osteolysis that particularly involves the distal tufts.
- •
Computed tomography (CT) is useful in demonstrating paraspinal calcifications.
- •
Magnetic resonance imaging (MRI) is valuable for assessing paraspinal calcifications and their effect on neurologic structures, as well as for identifying areas of inflammatory myopathy.
- •
Pulmonary Involvement
- •
Radiographs may be normal in early interstitial lung disease.
- •
High-resolution computed tomography (HRCT) is more sensitive than radiography in the assessment of early interstitial lung disease and is able to differentiate active inflammation from fibrosis.
- •
Bronchoalveolar lavage (BAL) abnormalities may precede HRCT findings.
- •
Peripheral Vascular System
- •
Arteriography: Vasospasm and pruning of the small digital vessels are characteristic findings.
- •
Pulmonary Hypertension and Cardiac Involvement
- •
On posteroanterior erect chest radiographs, the transverse diameter of the right interlobar artery should ≤16 mm. Enlargement occurs in pulmonary artery hypertension.
- •
Right heart catheterization optimally evaluates pulmonary arterial hypertension.
- •
Myocardial disease can be evaluated using radionuclide imaging, coronary angiography, or cardiac MRI.
- •
Renal Involvement
- •
Ultrasound and biopsy are used for exclusion of other causes of renal disease.
- •
Nuclear scans can demonstrate decreased renal blood flow and glomerular filtration rate.
- •
Gastrointestinal System
- •
Radiographs and CT may show obstruction, pseudoobstruction, volvulus, intestinal perforation, and pneumatosis cystoides intestinalis.
- •
Barium swallow: Esophageal dilatation, diminished or absent peristalsis in the distal two thirds of the esophagus, and a patulous gastroesophageal junction are typical features. Stricture occurs secondary to chronic gastroesophageal reflux.
- •
Barium follow through: Delay in transit time with decreased motility, pseudodiverticula, and hidebound appearance.
- •
Barium enemas are usually avoided because they may result in impaction.
- •
Scintigraphy: Delayed esophageal transit time and gastric emptying and gastroesophageal reflux are typical features. Delayed scans are useful for detection of pulmonary aspiration.
- •
The term scleroderma is derived from the Greek for “hard” (skleros) and “skin” (derma) and describes a connective tissue disorder of unknown etiology, characterized by thickening and fibrosis of the skin. Although the cutaneous manifestations are the most easily recognized, generalized scleroderma is a systemic disorder affecting virtually every organ system and is more appropriately referred as systemic sclerosis .
Scleroderma can affect virtually every organ system or can be localized.
It is more prevalent in women than men and has a mean onset around age 40 years.
Scleroderma can be classified into disease restricted to the skin (localized scleroderma, including the morphea and linear forms) and systemic disease with visceral organ involvement (systemic sclerosis) ( Box 21-1 ). Systemic sclerosis can be further subclassified based on the extent of involvement. Two main subsets of systemic sclerosis are typically identified: diffuse cutaneous systemic sclerosis and limited cutaneous systemic sclerosis. Diffuse cutaneous systemic sclerosis is characterized by skin thickening of the proximal and distal extremities, the face, and often the trunk; these patients are at increased risk for the development of significant pulmonary, cardiac, and renal disease. Limited cutaneous systemic sclerosis is characterized by skin thickening restricted to the hands and possibly the extremities (distal to the elbows or knees) and may involve the face and neck. Visceral involvement occurs late; hence the clinical course is relatively benign. The term limited cutaneous systemic sclerosis is preferable to CREST ( c alcinosis, R aynaud’s, e sophageal dysphagia, s clerodactyly, t elangiectasia syndrome), because cutaneous manifestations often extend beyond sclerodactyly.
LOCALIZED
Morphea
Localized
Generalized
Linear
en coup de saber *
* French term that means “cut of the sword.” Midline or parasagittal variant of linear scleroderma that manifests in childhood and may occur with defects in underlying facial and skeletal structures
SYSTEMIC SCLEROSIS
Limited cutaneous systemic sclerosis
Diffuse cutaneous systemic sclerosis
Overlap syndromes
Systemic sclerosis sine scleroderma †
† Vascular or fibrotic visceral features without skin changes (< 1% cases)
RAYNAUD’S PHENOMENON
Autoimmune (secondary) Raynaud’s phenomenon
Primary Raynaud’s phenomenon
Patients with scleroderma express a variety of serologic markers. These autoantibodies are useful in defining clinical subsets of the disease and provide important prognostic information. For example, anticentromere antibodies are most often seen with limited cutaneous involvement, and affected individuals have a lower frequency of pulmonary fibrosis and a lower mortality despite an increased risk of pulmonary hypertension.
IMAGING IN SYSTEMIC SCLEROSIS
Cutaneous and Musculoskeletal Findings
Cutaneous Involvement
Involvement of the skin is the hallmark of scleroderma, and skin changes usually proceed through three phases: (1) edematous, (2) indurative, and (3) atrophic.
Changes limited to the fingers (sclerodactyly) consist of soft tissue resorption of the fingertips and, later, flexion contractures. Soft tissue resorption is frequently accompanied by calcific deposits and bone resorption. Loss of the normal skin folds at the proximal interphalangeal (PIP) joints can be detected on oblique or lateral radiographs.
Calcinosis
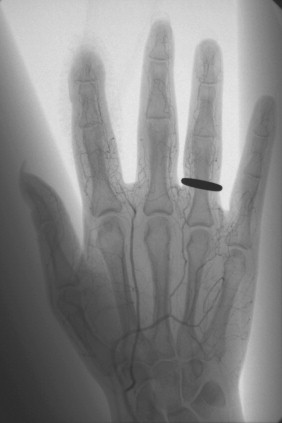
Calcinosis (abnormal deposition of calcium salts in body tissues) occurs in about 25% of patients with scleroderma, usually those with limited cutaneous scleroderma. The cutaneous, subcutaneous, and periarticular calcification is composed of calcium hydroxyapatite (HA) crystal deposits. Calcifications are usually seen at the sites of recurrent microtrauma, such as the finger pads, buttocks, and extensor surfaces of the elbows and forearms ( Figure 21-1 ). Large periarticular calcific deposits can simulate those seen in renal osteodystrophy or tumoral calcinosis.
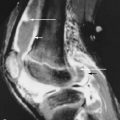
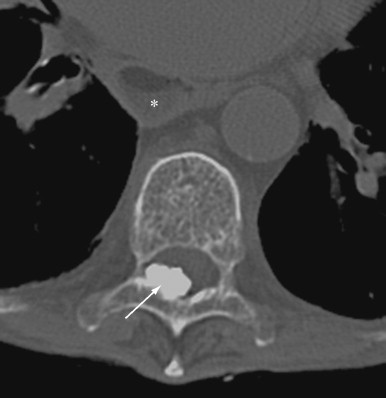
Clinically, the calcific deposits often present as bothersome subcutaneous lumps that may cause local inflammation due to the release of HA crystals. Superficial calcinosis may result in ulceration of the overlying skin leading to secondary infection. Massive paraspinal calcifications have been reported, leading to pain, stiffness, dysphagia, and spinal cord or nerve root compression.
Radiographs can detect and delineate the extent of soft tissue calcifications. Paraspinal calcifications and their effect on neurologic structures are better evaluated by computed tomography (CT) scanning and magnetic resonance imaging (MRI) ( Figure 21-2 ).
Osteolysis
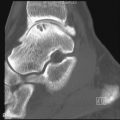
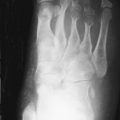
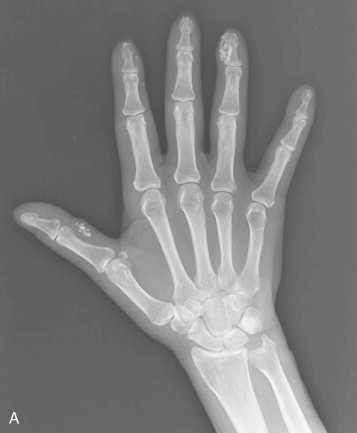
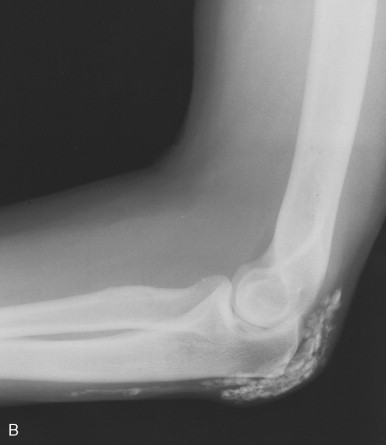
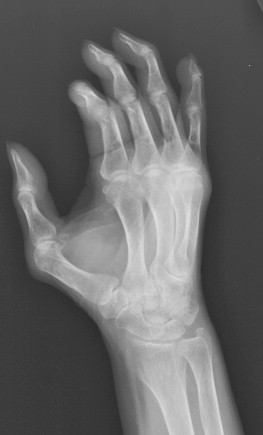
Bony resorption of the phalanges (acroosteolysis) in the hand occurs in 40% to 80% of patients with scleroderma. Resorption involves particularly the palmar aspect of the distal phalangeal tufts, leading to penciling or conical deformity of the phalanges ( Figure 21-3 ). In advanced cases, the entire distal phalanx can be destroyed. Resorption of the middle and proximal phalanges, although less common, also occurs. Other sites of bone resorption include the distal ends of the ulna and radius, the clavicle, and mandible, ribs, and humeri.
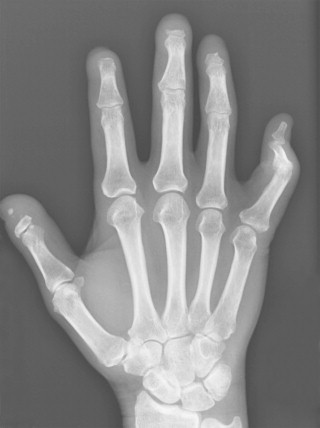
The combination of acroosteolysis and distal soft tissue calcification is typical of scleroderma.
Articular Involvement
Articular abnormalities are very common in patients with systemic sclerosis. The presentation of joint involvement is quite variable, ranging from arthralgia to frank polyarthritis.
Articular changes are usually manifest on radiographs as narrowing of the cartilage space, juxtaarticular osteoporosis, flexion contractures, and thickening of the periarticular soft tissue. Occasionally, radiographic changes may be indistinguishable from those seen in rheumatoid arthritis (RA) and patients may have a detectable rheumatoid factor, frequently believed to represent an overlap syndrome. More than one third of the patients with scleroderma have a positive serologic test for rheumatoid factor. Despite the large percentage of patients with scleroderma who manifest clinical, physical, and laboratory signs of RA, very few show the typical erosive changes of RA on radiographs. This discrepancy in clinical and radiologic findings is most likely due to the underlying pathology of the disease. Early in the clinical course of scleroderma, synovial biopsies show inflammatory reaction with infiltration by lymphocytes and plasma cells—a picture similar to that of RA. In patients with longer disease duration, the synovium is covered and replaced by fibrous tissue. Pannus, which leads to the destructive changes of RA, is notably absent.
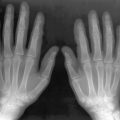
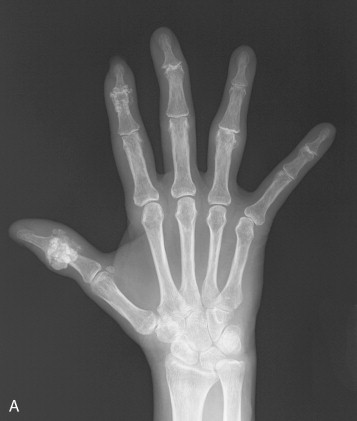
A minority of patients with scleroderma develops erosive changes resembling those occurring in psoriasis or erosive osteoarthritis ( Figure 21-4 ). Radiographic abnormalities include erosions involving primarily the distal interphalangeal (DIP) and PIP joints with relative sparing of the metacarpophalangeal (MCP) and wrist joints. Bony ankylosis and pencil-in-cup deformity or ball-in-socket erosions can be observed. Selective involvement of the first carpometacarpal joint with bony resorption and radial subluxation of the first metacarpal has also been reported as a distinctive feature of scleroderma. Associated findings may include intraarticular calcification.
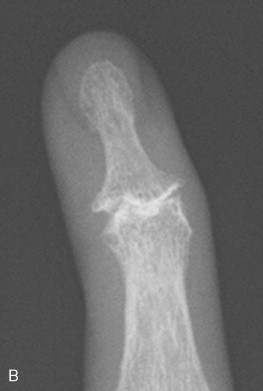
Flexion contractures may occur as a result of fibrosis of the skin, ligaments, and joint capsules, restricting joint mobility. Radiographs of these patients show contractures and periarticular osteopenia without erosions ( Figure 21-5 ). Flexion contractures are particularly seen in the fingers, wrists, elbows, and knees. The fibrosis can also affect the tendons and tendon sheaths, causing tendon friction rubs.
Muscle Involvement
Muscle involvement occurs in most patients with scleroderma and may occur in both the diffuse and limited cutaneous forms of systemic sclerosis.
The patients usually present with one of three forms of myopathy. First, and most commonly, the muscle weakness and wasting are the result of disuse from joint contractures and chronic disease. Second, in about 20% of patients, a chronic myopathy (also called simple myopathy ) occurs that is characterized by mild muscle weakness and atrophy, minimal elevation of creatine phosphokinase, few or no changes on electromyelography (EMG), and subtle histologic features showing focal replacement of myofibrils with collagen and fibrosis without inflammatory change. This form of myopathy is often unresponsive to antiinflammatory medication. Third, a minority of patients has an inflammatory myopathy resembling dermatomyositis with significant elevation of creatine phosphokinase and positive EMG changes. Muscle biopsies usually show mononuclear cell infiltrates. In contrast to the noninflammatory myopathy, which is unresponsive to corticosteroid therapy, inflammatory myositis may require treatment with corticosteroids and immunosuppressive drugs.
MRI has been demonstrated to be useful in the diagnosis of inflammatory muscle disorders. Areas of increased signal (brightness) on T2-weighted images corresponding to muscle inflammation and fascial thickening have been reported, especially in the hamstring and quadriceps muscles in scleroderma patients with inflammatory myositis. Utilization of MRI for localization of muscle abnormalities prior to muscle biopsy, could decrease sampling errors encountered in “blind” biopsies.
Dental Manifestations
Uniform thickening of the periodontal membrane is a relatively specific finding of scleroderma. The thickened membrane creates lucency between the teeth and mandible that is best detected on dental films. The posterior teeth are involved more often than the anterior teeth.
Thickening of the periodontal membrane on dental radiographs is a relatively specific feature of scleroderma.
Peripheral Vascular System
Raynaud’s phenomenon is the earliest and most common clinical manifestation of systemic sclerosis and is characterized by episodes of pallor followed by cyanosis of the distal digits, provoked by cold or emotion. Associated findings include ischemic necrosis, digital ulcerations, and gangrene of digits of the hand or foot. Progressive deficiency in vasodilatory capacity is proposed as a mechanism of Raynaud’s phenomenon. The superimposed intimal hyperplasia could result in complete obstruction of small arteries. Similar histopathologic changes are evident in the small arteries and arterioles of affected internal organs.
When a patient presents with Raynaud’s phenomenon, it is necessary to determine whether there is an underlying connective tissue disease or whether the disorder is primary (idiopathic). Examining the serum autoantibodies and nail fold capillaroscopy are simple, noninvasive procedures that are reliable for detecting patients with secondary Raynaud’s phenomenon. The capillaroscopy shows characteristic abnormalities in scleroderma-spectrum disorders, which are enlarged capillary loops, with areas of avascularity and disruption of the normal capillary bed. Areas of hemorrhage may be seen, especially in association with widened capillaries and “bushy” capillaries. They appear much like the head of Medusa and are consistently observed in dermatomyositis and in psoriasis.
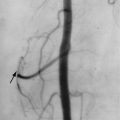
Arteriography is not usually performed in patients with scleroderma. The characteristic arteriographic findings of Raynaud’s phenomenon include vasospasm and pruning of the small digital vessels. Other arteriographic findings are incomplete, poorly formed, or absent palmar arterial arches and ulnar artery. These latter changes may be related to developmental or acquired factors ( Figure 21-6 ).
Pulmonary Involvement
Lung disease is the primary cause of mortality in patients with systemic sclerosis. Pulmonary manifestations of scleroderma include interstitial lung disease (ILD), pulmonary hypertension, alveolar hemorrhage, aspiration pneumonitis, bronchiectasis, spontaneous pneumothorax, bronchogenic carcinoma, and pleural disease.
Interstitial Lung Disease
ILD with progressive fibrosis is the most common pulmonary complication of systemic sclerosis with a prevalence of 74% to 100% reported at autopsy. Interstitial pulmonary fibrosis occurs more often in patients with diffuse cutaneous systemic sclerosis than in those with the limited form of cutaneous involvement.
Histopathologic features of scleroderma pulmonary fibrosis resemble idiopathic pulmonary fibrosis. Alveolar inflammation has been recognized as a primary event. In early stages, alveolar edema with lymphocytic and granulocytic infiltrates occurs. Later, fibrosis is detected involving the interstitium, alveolar septa, and bronchial walls, causing obliteration of alveolar spaces and capillaries.
Patients with pulmonary involvement present with dyspnea, initially with exertion and later at rest and a nonproductive cough. Chest pain, hemoptysis, pleurisy, and fever are uncommon. Pulmonary function tests will be markedly impaired, typically demonstrating a restrictive pattern with proportionate reduction of carbon monoxide transfer factor (DLCO) and forced vital capacity (FVC).
Radiologic Findings
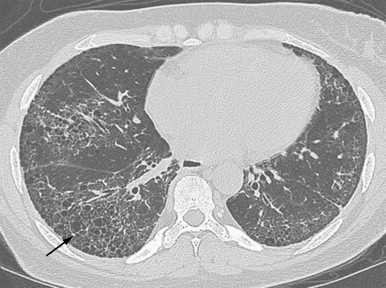
The chest radiograph is abnormal in 25% to 65% of patients with established scleroderma. As with other fibrosing lung diseases, the interstitial fibrosis of systemic sclerosis can be present despite a normal chest radiograph. The most common radiographic abnormality is a widespread, symmetric, basally predominant reticulonodular pattern that typically starts as a very fine reticular pattern and progresses to coarser reticulation ( Figure 21-7 ). Cystic honeycomb lesions commonly develop in the areas of fibrosis. Serial radiographs may show progressive loss of lung volume manifested by elevation of the diaphragm.
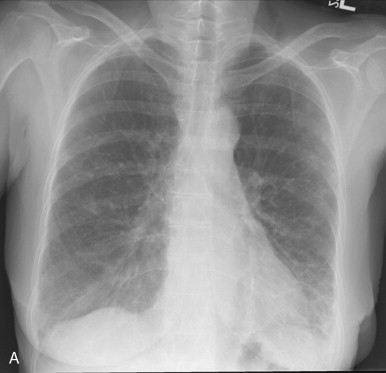
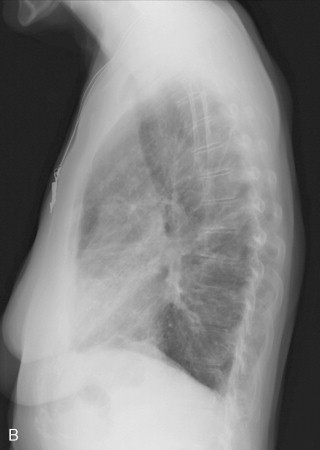
The chest radiograph is abnormal in as many as 65% of patients with established scleroderma, but interstitial fibrosis may be present even when the radiograph appears normal.
High-resolution computed tomography (HRCT) is more sensitive than radiography in assessing the subtle pulmonary involvement in patients with systemic sclerosis. HRCT not only allows better visualization of the pulmonary parenchyma but also allows differentiation of active inflammation from fibrosis. The most common abnormalities seen on HRCT are (1) “ground glass” opacification; (2) a fine reticular pattern, often posterior and subpleural, that is usually associated with traction bronchiectasis and bronchiolectasis; (3) honeycombing with subpleural cysts (1 to 3 cm) ( Figure 21-8 ); (4) lines of various types (septal, subpleural, nonseptal parenchymal lines); and (5) subpleural micronodules.