Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
La physiopathologie du métabolisme protéique

2
La physiopathologie des protéines plasmatiques
Hypoprotéinémies 1. Hypoprotéinémies apparentes (fausses) Il s’agit d’une hypoprotéinémie associée à une hyperhydratation avec hémodilution consécutive.
 Il s’agit d’une hypoprotéinémie associée à une hyperhydratation avec hémodilution consécutive.")
3
La physiopathologie des protéines plasmatiques
Hypoprotéinémies 2. Hypoprotéinémies réelles La carence d’apport en protéines Le syndrome de maldigestion – malabsorption Le défaut de synthèse La fuite anormale des protéines au niveau : cutané, rénal, intestinal.

4
La physiopathologie des protéines plasmatiques
Hyperprotéinémies 1. Hyperprotéinémies apparentes (fausses) Il s’agit d’une hyperprotéinemie associée à une hypohydratation avec hémoconcentration consécutive.
 Il s’agit d’une hyperprotéinemie associée à une hypohydratation avec hémoconcentration consécutive.")
5
La physiopathologie des protéines plasmatiques
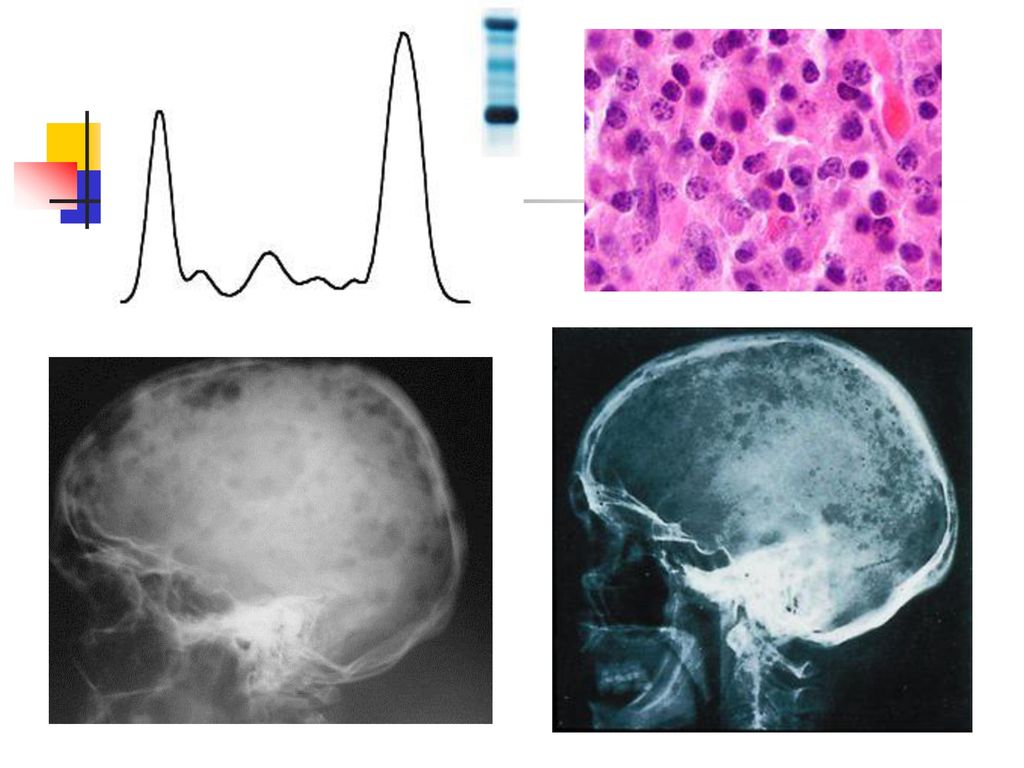
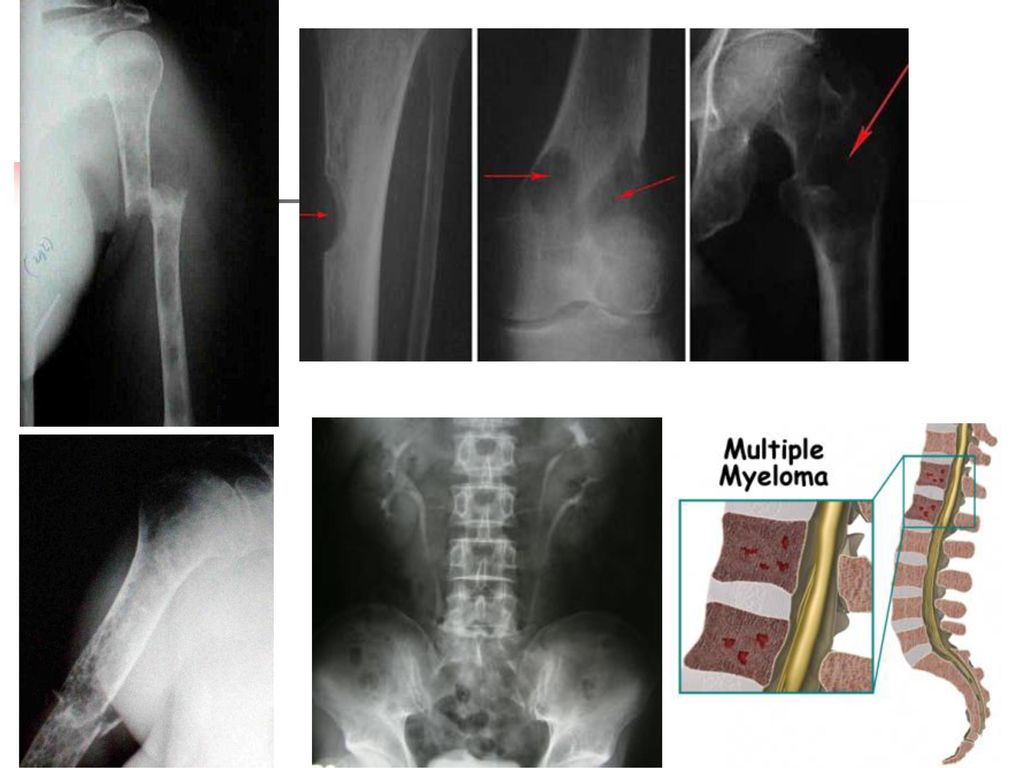
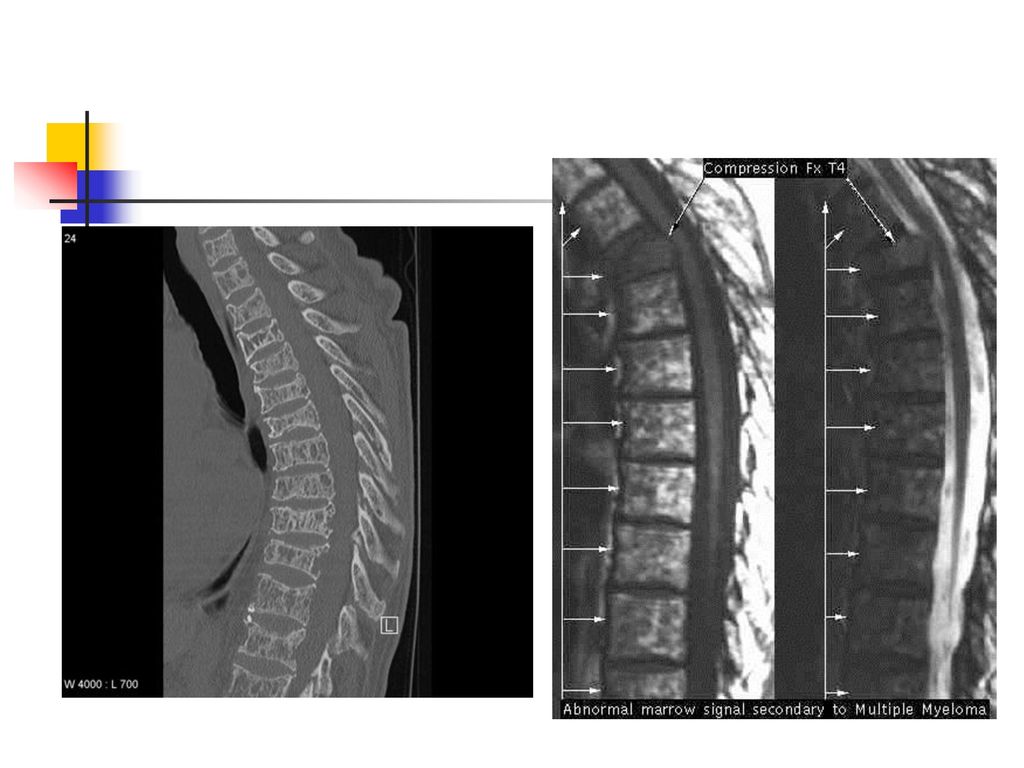
Hyperprotéinémies 2. Hyperprotéinémies réelles inflammation chronique, gammapathie monoclonale : le myélome multiple, la maladie de Waldenström, les leucémies lymphoïdes chroniques, la maladie des chaînes lourdes, etc.

6
La physiopathologie des protéines plasmatiques
Dysprotéinémies L'électrophorèse des protéines sériques permet la séparation des protéines du sérum en 5 fractions : Albumine % soit g /l α1 -globulines 1 - 4 % soit g /l α2 -globulines % soit g/l β-globulines % soit g /l γ-globulines % soit g /l

7
La physiopathologie des protéines plasmatiques
Dysprotéinémies La dysprotéinémie de l’inflammation aiguë l’augmentation des α1 et α2 globulines α1 – antitrypsine, α1 – glycoprotéine acide, α2 – haptoglobine, α2 – ceruloplasmine, C2 , C3 , C4 , fibrinogène, la diminution (modérée) des albumines, de la transferrine, de la pseudo-cholinestérase et des β – lipoprotéines. IL1, IL6, TNF.
 des albumines, de la transferrine, de la pseudo-cholinestérase et des β – lipoprotéines. IL1, IL6, TNF.")
8
L’électrophorèse sérique dans une inflammation aiguë

9
La physiopathologie des protéines plasmatiques
Dysprotéinémies La dysprotéinémie de l’inflammation chronique large

10
La physiopathologie des protéines plasmatiques
Dysprotéinémies La dysprotéinémie des gammapathies monoclonales étroite

14
La physiopathologie des protéines plasmatiques
Dysprotéinémies La dysprotéinémie des affections hépatiques chroniques

15
La physiopathologie des protéines plasmatiques
Dysprotéinémies La dysprotéinémie du syndrome néphrotique

16
La physiopathologie des protéines plasmatiques
Dysprotéinémies Les dysprotéinémies par l’absence d’une protéine plasmatique L’analbuminemie Maladie héréditaire rare, à transmission autosomique récessive dont l’anomalie génétique se trouve sur le bras long du chromosome 4 (4q1).
.")
17
La physiopathologie des protéines plasmatiques
Dysprotéinémies Les dysprotéinémies par l’absence d’une protéine plasmatique L’afibrinogénémie C'est une maladie excessivement rare transmise autosomale récessive caractérisée par un défaut de synthèse du fibrinogène provoquant des hémorragies à répétition apparaissant dès la naissance de l’enfant ou lors de la chute du cordon ombilical. Ces hémorragies se situent quelquefois à l’intérieur du crâne, ou bien ce sont des hématomes ou plus rarement des hémarthroses

18
La physiopathologie des protéines plasmatiques
Dysprotéinémies Les dysprotéinémies par l’absence d’une protéine plasmatique Le déficit d’alpha 1- antitrypsine L’alpha 1-antitrypsine est un inhibiteur de la sérine protéase (serpine). Elle protège les tissus des enzymes des cellules inflammatoires (élastase et la protéinase 3 des polynucléaires neutrophiles). C’est un déficit héréditaire avec la transmission autosomale récessive caractérisé par : emphysème panlobulaire, apparaissant chez l’adulte jeune, atteinte hépatique: cholestase, évolution inconstante vers la cirrhose,
. Elle protège les tissus des enzymes des cellules inflammatoires (élastase et la protéinase 3 des polynucléaires neutrophiles). C’est un déficit héréditaire avec la transmission autosomale récessive caractérisé par : emphysème panlobulaire, apparaissant chez l’adulte jeune, atteinte hépatique: cholestase, évolution inconstante vers la cirrhose,")
19
La physiopathologie des protéines plasmatiques
Dysprotéinémies Les dysprotéinémies par l’absence d’une protéine plasmatique Le déficit de transferrine l’atransferrinemie héréditaire est une maladie autosomale récessive liée à une mutation du gène de la transferrine sur le chromosome 3.

20
La physiopathologie des protéines plasmatiques
Dysprotéinémies Les dysprotéinémies par l’absence d’une protéine plasmatique Le déficit héréditaire en céruloplasmine La céruloplasmine joue un rôle dans le transport plasmatique du cuivre. Dans la maladie de Wilson ou la dégénérescence hépato-lenticulaire il existe une insuffisance de synthèse de la céruloplasmine, un défaut de l’excrétion biliaire du cuivre, une affinité importante des protéines des différents tissus de l’organisme pour le cuivre.

21
La physiopathologie des protéines plasmatiques
Dysprotéinémies Les dysprotéinémies par l’absence d’une protéine plasmatique Le déficit héréditaire en céruloplasmine La maladie de Wilson associe : des lésions hépatiques et rénales, des lésions cérébrales au niveau des ganglions de la base caractérisées par : mouvements involontaires (tremblements, spasmes ou athétose), rigidité, irritabilité, un anneau brun verdâtre péricornéen de Kayser-Fleischner.
, rigidité, irritabilité, un anneau brun verdâtre péricornéen de Kayser-Fleischner.")
25
La physiopathologie des protéines plasmatiques
Dysprotéinémies Les dysprotéinémies par l’absence d’une protéine plasmatique L’agammaglobulinémie ou maladie de Bruton C’est une maladie due à des mutations du gène XLA (Xq21.3-q22) codant pour une protéine-tyrosine kinase spécifique de la lignée B. Il résulte un blocage de la maturation du lymphocyte au stade pré-B et donc de l’absence des lymphocytes B, des IgG, IgA, IgM, IgE et IgD (agammaglobulinémie)
 codant pour une protéine-tyrosine kinase spécifique de la lignée B. Il résulte un blocage de la maturation du lymphocyte au stade pré-B et donc de l’absence des lymphocytes B, des IgG, IgA, IgM, IgE et IgD (agammaglobulinémie)")
26
La physiopathologie du métabolisme des acides aminés
Hyperaminoaciduries sans altération des systèmes de transfert Ces affections se caractérisent en général par un déficit enzymatique (héréditaire ou acquis) bloquant une des premières étapes du catabolisme d’un ou de plusieurs acides aminés. Les acides aminés non catabolisés augmentent dans le sang et passent en plus grandes quantités dans l’urine primaire.
 bloquant une des premières étapes du catabolisme d’un ou de plusieurs acides aminés. Les acides aminés non catabolisés augmentent dans le sang et passent en plus grandes quantités dans l’urine primaire.")
27
Hyperaminoaciduries par des blocages métaboliques du groupe phénylalanine – tyrosine

28
FEN TIR mélanine Ac. Parahidroxi fenilpiruvic Ac. 2,3 dihidroxi Ac. homogentizinic Fumaril acetoacetat

29
TIR FEN troubles neurologiques graves : retard mental, troubles du comportement, psychoses, spasmes en flexion, etc. Ac. phénylpyruvique Ac. phénylacétique Ac. phényllactique Ac. oxyphénylacétique

30
FEN TIR mélanine Ac. Parahidroxi phénylpyruvique Ac. 2,3 dihidroxi Ac. homogentisique Fumaril acétoacétate

35
FEN TIR Precursori de melanină Catecolamine Ac. Parahidroxi phénylpyruvique Ac. 2,3 dihidroxi Ac. homogentisique Fumaril acétoacétate Alcaptonurie

36
Coloration foncée des urines laissées à la température ambiante (appelée également: maladie de l'urine noire)
")
37
Pigmentation inhabituelle de la peau
(de gris brun à noir) : Ochronose
38
Arthrite de la colonne vertébrale et des grosses articulations par dépôt d’acide homogentisique
40
La physiopathologie du métabolisme des acides aminés
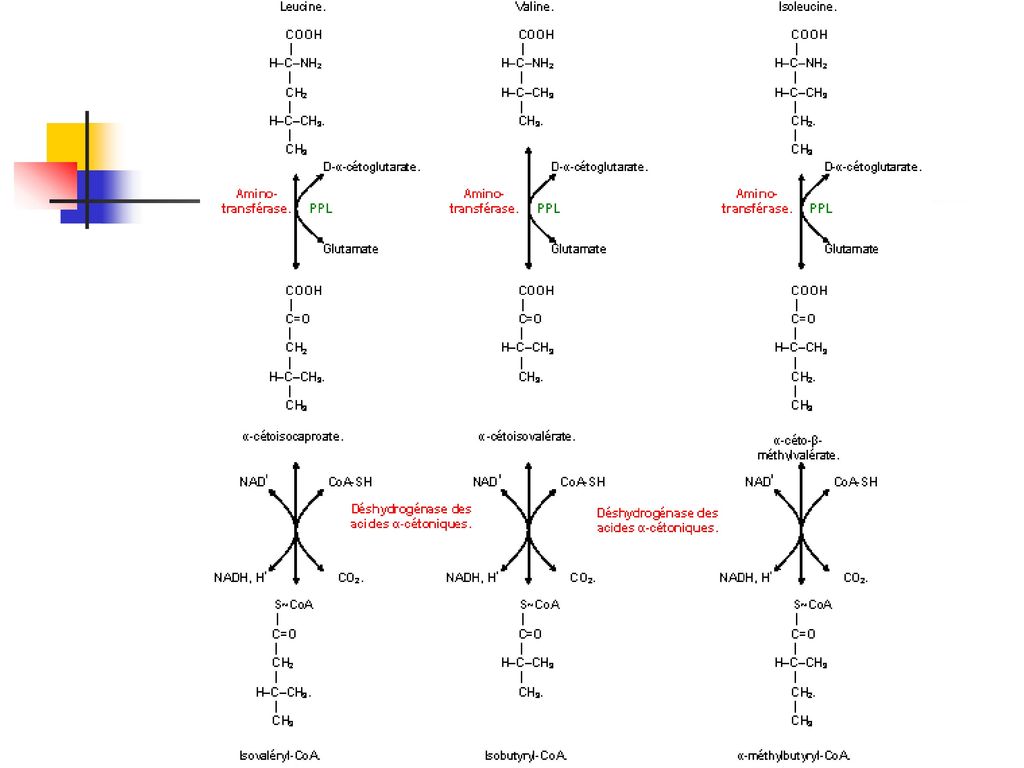
Hyperaminoaciduries sans altération des systèmes de transfert La leucinose ou maladie des urines à odeur de sirop d'érable C’est une maladie métabolique héréditaire (sur le mode récessif autosomique) due à un déficit des alpha-cétodécarboxylases des acides aminés ramifiés (valine, leucine, isoleucine). Il résulte une augmentation plasmatique et urinaire de valine, leucine et isoleucine.
 due à un déficit des alpha-cétodécarboxylases des acides aminés ramifiés (valine, leucine, isoleucine). Il résulte une augmentation plasmatique et urinaire de valine, leucine et isoleucine.")
42
La physiopathologie du métabolisme des acides aminés
Hyperaminoaciduries sans altération des systèmes de transfert La leucinose ou maladie des urines à odeur de sirop d'érable Les manifestations cliniques sont : somnolence, hypotonie, troubles de succion, troubles respiratoires, intolérance digestive avec vomissements.

43
La physiopathologie du métabolisme des acides aminés
Hyperaminoaciduries avec altération des systèmes de transfert Il s’agit des affections déterminées par des anomalies des transporteurs protéiques spécifiques des acides aminés. Les anomalies peuvent se localiser strictement au tubule rénal ou bien affecter aussi les cellules intestinales intervenant dans l’absorption des acides aminés.

44
La physiopathologie du métabolisme des acides aminés
Hyperaminoaciduries avec altération des systèmes de transfert La maladie d’Hartnup C’est une affection héréditaire à transmission autosomique récessive caractérisée par un trouble du transport des acides aminés neutres et aromatiques, avec une absorption intestinale diminuée et une élimination urinaire augmentée. Entraîne une carence en tryptophane

45
une éruption cutanée après exposition au soleil,
une ataxie cérébelleuse intermittente (manque de coordination fine des mouvements volontaires), des troubles du comportement, du caractère, délire sérotonine, mélatonine nicotinamide NAD NADP
, des troubles du comportement, du caractère, délire. sérotonine, mélatonine. nicotinamide. NAD. NADP.")
47
La physiopathologie du métabolisme des acides aminés
Hyperaminoaciduries avec altération des systèmes de transfert Le syndrome de De Toni – Debré – Fanconi Il s’agit d’une tubulopathie primaire complexe, d’origine héréditaire caractérisée par : hyperaminoacidurie non-sélective, hyperphosphaturie, glycosurie, ostéomalacie, acidose tubulaire rénale.

48
La physiopathologie du métabolisme des acides aminés
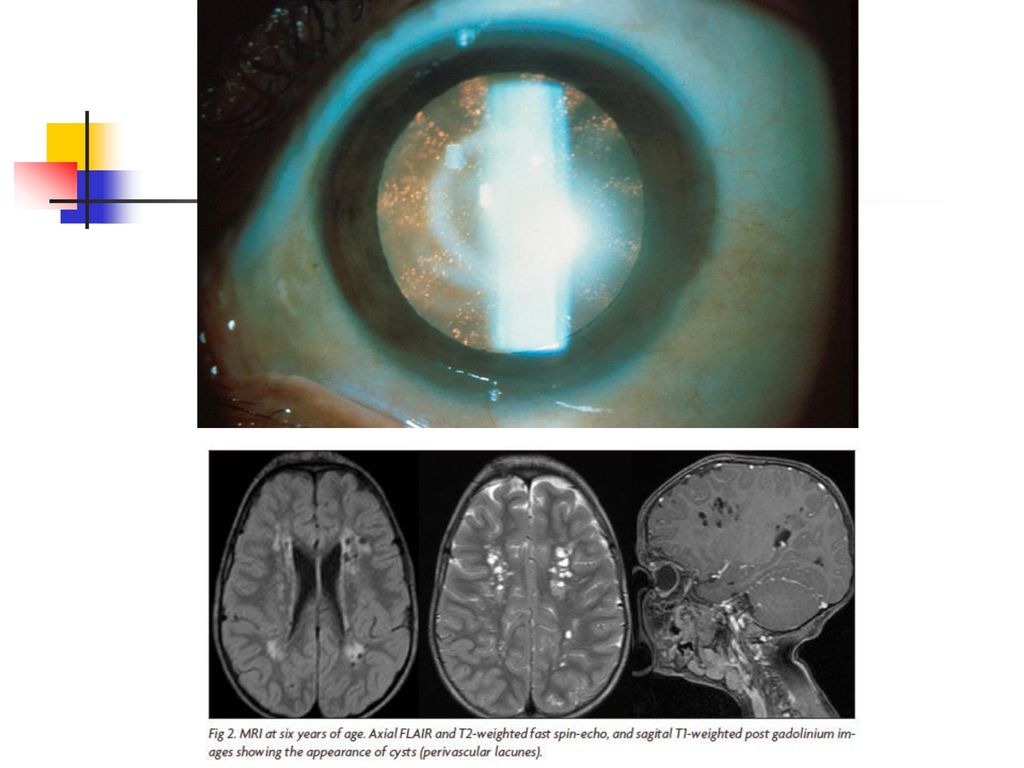
Hyperaminoaciduries avec altération des systèmes de transfert Le syndrome oculo-cérébro-rénal de Lowe C’est une maladie génétique et héréditaire (liée à l’X) caractérisée par : diabète phospho-gluco-aminé avec insuffisance rénale cataracte, glaucome, strabisme, lésions de la cornée, troubles neurologiques graves,
 caractérisée par : diabète phospho-gluco-aminé avec insuffisance rénale. cataracte, glaucome, strabisme, lésions de la cornée, troubles neurologiques graves,")
50
La physiopathologie du métabolisme des acides aminés
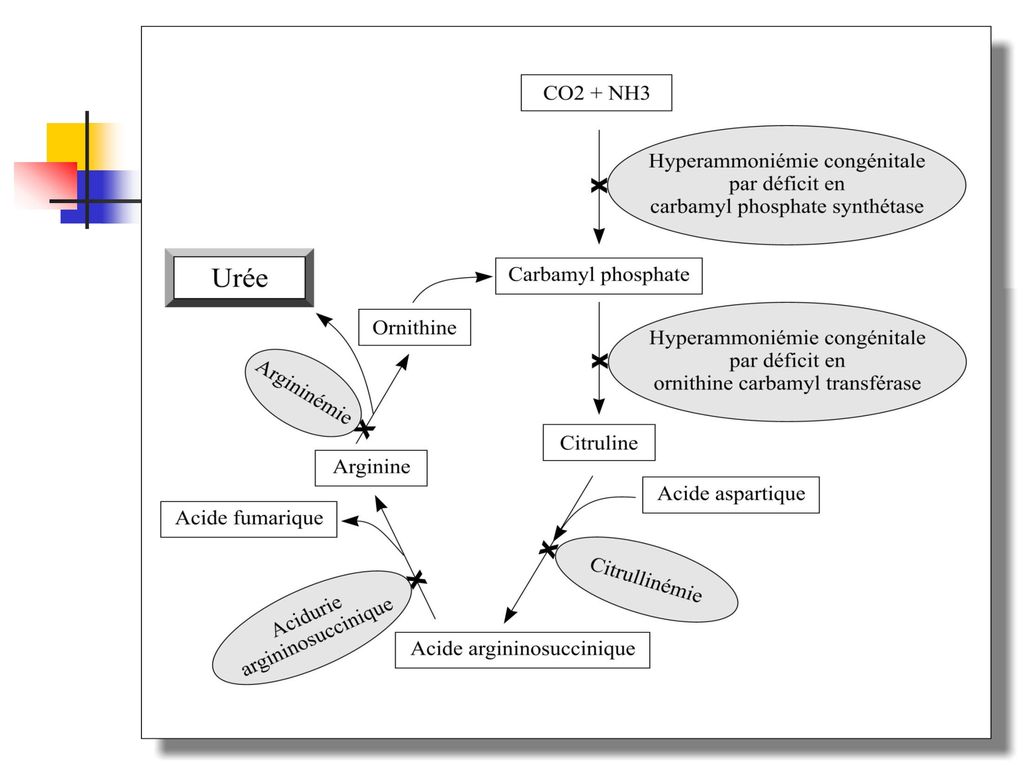
Les désordres du cycle de l'urée Le cycle de l’urée est la voie métabolique par laquelle l’ammoniaque (toxique) est transformé en urée (non toxique). Les déficits principaux sont en : carbamylphosphate synthétase →→ hyperammoniémie congénitale de type I ornithine transcarbamylase →→ hyperammoniémie congénitale de type II argininosuccinate synthétase →→ citrullinémie argininosuccinate lyase →→ acidurie argininosuccinique arginase →→ argininémie
 est transformé en urée (non toxique). Les déficits principaux sont en : carbamylphosphate synthétase →→ hyperammoniémie congénitale de type I. ornithine transcarbamylase →→ hyperammoniémie congénitale de type II. argininosuccinate synthétase →→ citrullinémie. argininosuccinate lyase →→ acidurie argininosuccinique. arginase →→ argininémie.")
52
La physiopathologie du métabolisme des nucléoprotéines
Acides nucléiques Protamines Histones

53
La physiopathologie du métabolisme des nucléoprotéines

54
La physiopathologie du métabolisme des purines
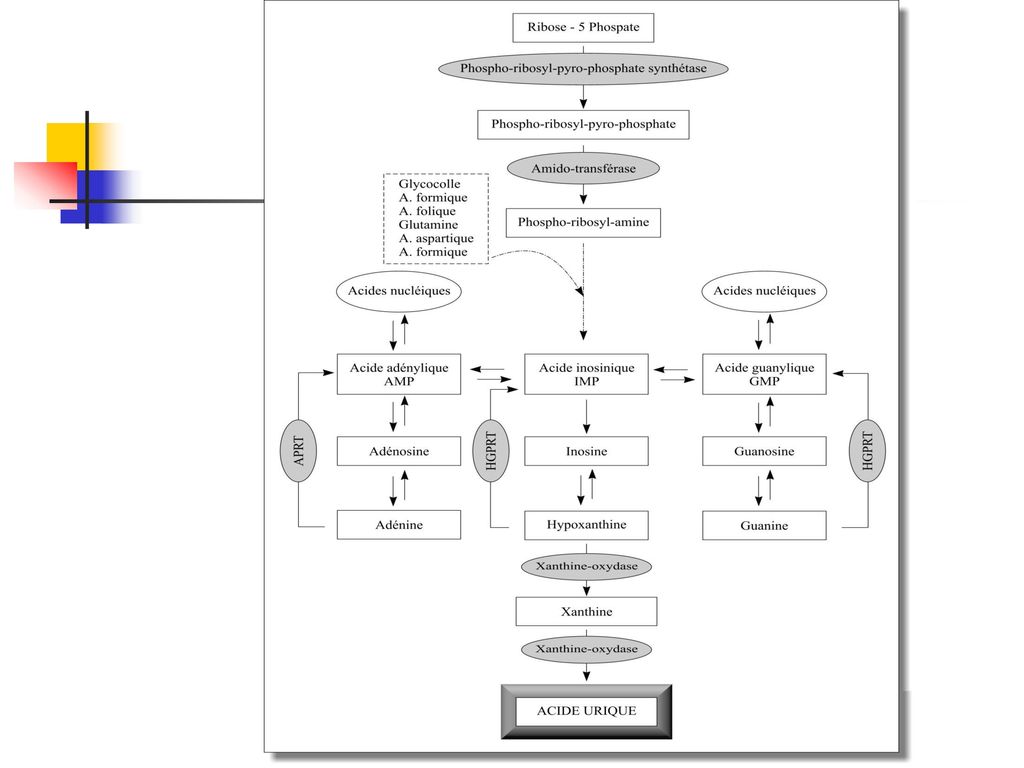
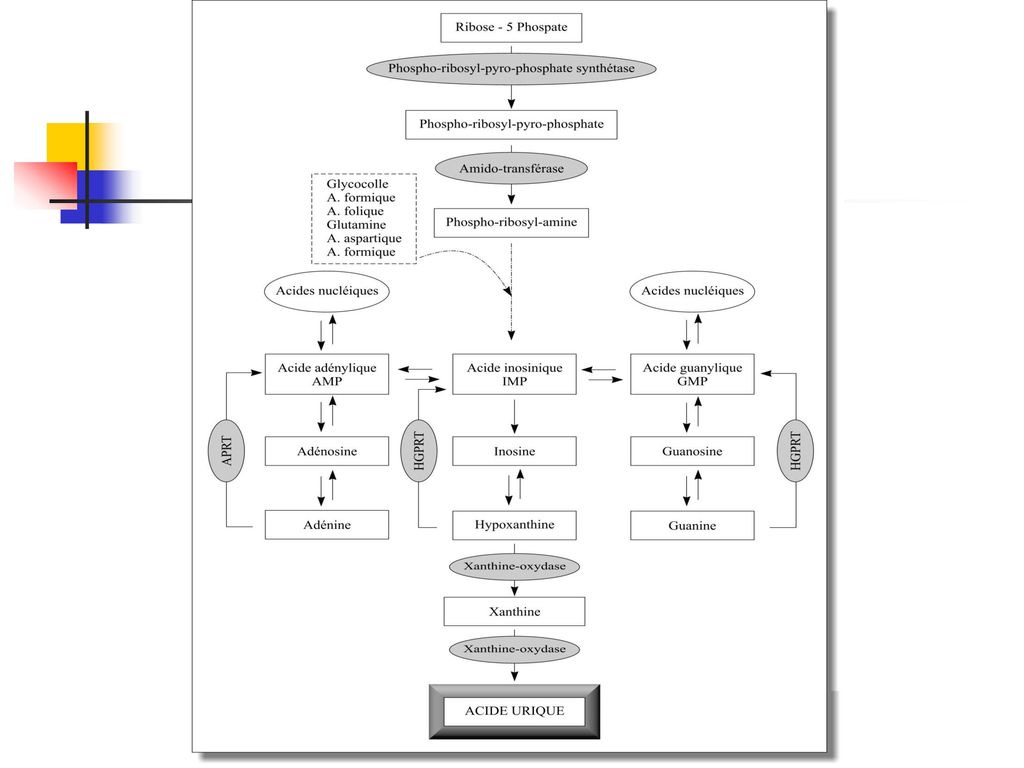
Les sources de l’acide urique La purinosynthèse de novo à partir de nucléotides puriniques (la plus importante) Le catabolisme des acides nucléiques cellulaires Le catabolisme des purines alimentaires.
 Le catabolisme des acides nucléiques cellulaires. Le catabolisme des purines alimentaires.")
55
La purinosynthèse de novo

56
La physiopathologie du métabolisme des purines
L’élimination de l'acide urique La voie urinaire : la filtration glomérulaire (l’acide urique a une clearance voisine de celle de la créatinine), la réabsorption tubulaire proximale, la sécrétion tubulaire distale. Par uricolyse intestinale
, la réabsorption tubulaire proximale, la sécrétion tubulaire distale. Par uricolyse intestinale.")
57
Les hyperuricémies Les hyperuricémies idiopathiques
Ces hyperuricémies représentent 95% des gouttes. L’hérédité et la suralimentation ont des rôles importants. Les mécanismes d’apparition sont : l’hyperpurinosynthèse de novo isolée, l’hypoexcrétion rénale isolée l’association deux mécanismes précédents (70%).
.")
58
Les hyperuricémies Les hyperuricémies enzymopathiques
Le syndrome Lesch Nyhan – l’encéphalopathie hyperuricémique héréditaire Ce syndrome est une maladie génétique récessive liée à l’X déterminée par un déficit enzymatique complet en HGPRT

60
Les hyperuricémies Les hyperuricémies enzymopathiques
Le syndrome Lesch Nyhan – l’encéphalopathie hyperuricémique héréditaire Elle associe une : hyperuricémie avec hyperuricosurie, encéphalopathie avec spasticité, retard mental (la déficience intellectuelle est sévère chez les personnes atteintes) et auto-mutilation (le grattage excessif des doigts, des orteils et des paupières ainsi que la morsure des extrémités des membres et de la langue), lithiase rénale urique, anémie par déficit en acide folique.
 et auto-mutilation (le grattage excessif des doigts, des orteils et des paupières ainsi que la morsure des extrémités des membres et de la langue), lithiase rénale urique, anémie par déficit en acide folique.")
62
Les hyperuricémies Les hyperuricémies enzymopathiques
Le déficit incomplet en HGPRT C’est une maladie génétique liée à l’X avec un début précoce par une : goutte sévère, hyperuricémie, hyperuricosurie déterminant des lithiases uratiques. Il n'y a pas de retard mental.

63
Les hyperuricémies Les hyperuricémies secondaires
Elles représentent % des hyperuricémies. Les causes principales et les mécanismes sont: l’insuffisance rénale chronique par un défaut d'excrétion rénale, les médicaments : les diurétiques (thiazidiques, furosémide, acide éthacrynique) entraînent une hyperuricémie par hypo-uricurie, l’aspirine diminue l’uricurie par action déprimante sur l’urico-sécrétion tubulaire, pyrazinamide (antituberculeux), l’éthambutol, la ciclosporine,
 entraînent une hyperuricémie par hypo-uricurie, l’aspirine diminue l’uricurie par action déprimante sur l’urico-sécrétion tubulaire, pyrazinamide (antituberculeux), l’éthambutol, la ciclosporine,")
64
Les hyperuricémies Les hyperuricémies secondaires
Les causes principales et les mécanismes sont: les hémopathies par un catabolisme exagéré des acides nucléiques globulaires : les hémopathies myéloïdes ou lymphoïdes, les leucoses aiguës, etc., les traitements cytolytiques des leucoses aiguës et des lymphomes par un catabolisme également exagéré des acides nucléiques globulaires, Le psoriasis par l’hypercatabolisme des nucléoprotéines cellulaires,

65
Les hyperuricémies Les hyperuricémies secondaires
Les causes principales et les mécanismes sont: la glycogénose hépatique I (déficit en G-6-phosphatase) par un trouble du métabolisme du glucose qui détermine une synthèse excessive de ribose-5-phosphate et une purinosynthèse de novo augmentée déficit en G-6-phosphatase pentose ↑↑↑ G-6-phosphate
 par un trouble du métabolisme du glucose qui détermine une synthèse excessive de ribose-5-phosphate et une purinosynthèse de novo augmentée. déficit en G-6-phosphatase. pentose. ↑↑↑ G-6-phosphate.")
67
Les hyperuricémies Les hyperuricémies secondaires
Les causes principales et les mécanismes sont: les hyperlactacidémies par une inhibition compétitive de l’éxcrétion urinaire d’acide urique : l’effort musculaire intense, l’ingestion exagérée d’alcool, etc., l’hypertension artérielle par un défaut de filtration glomérulaire et par la prise de diurétiques,

68
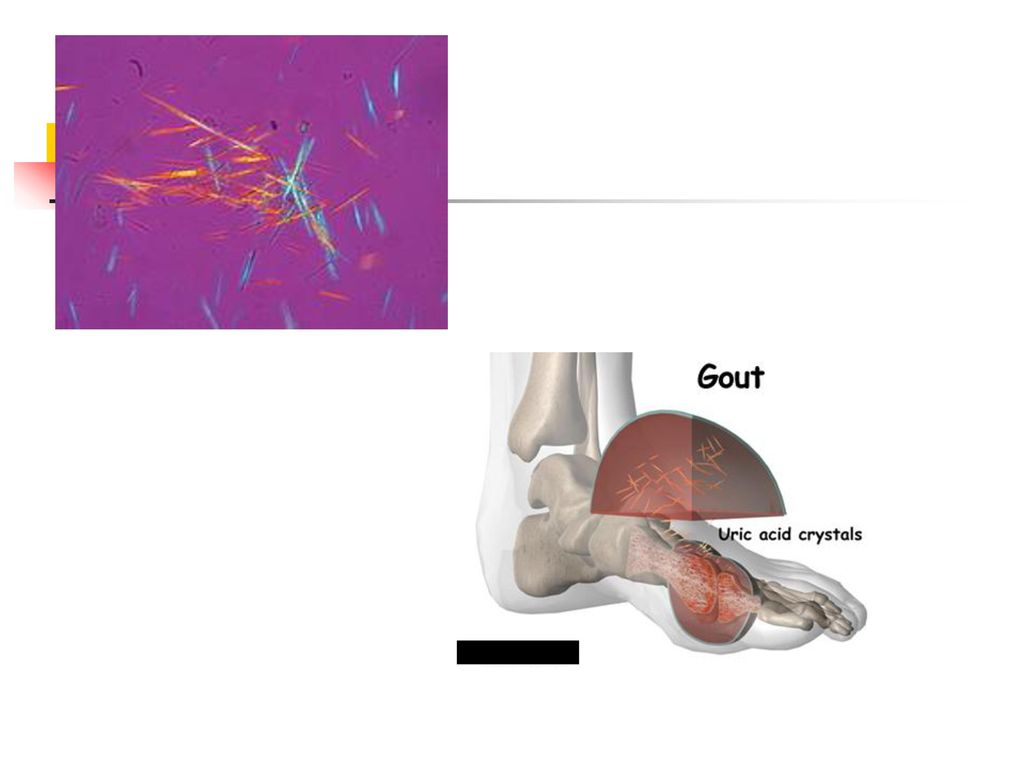
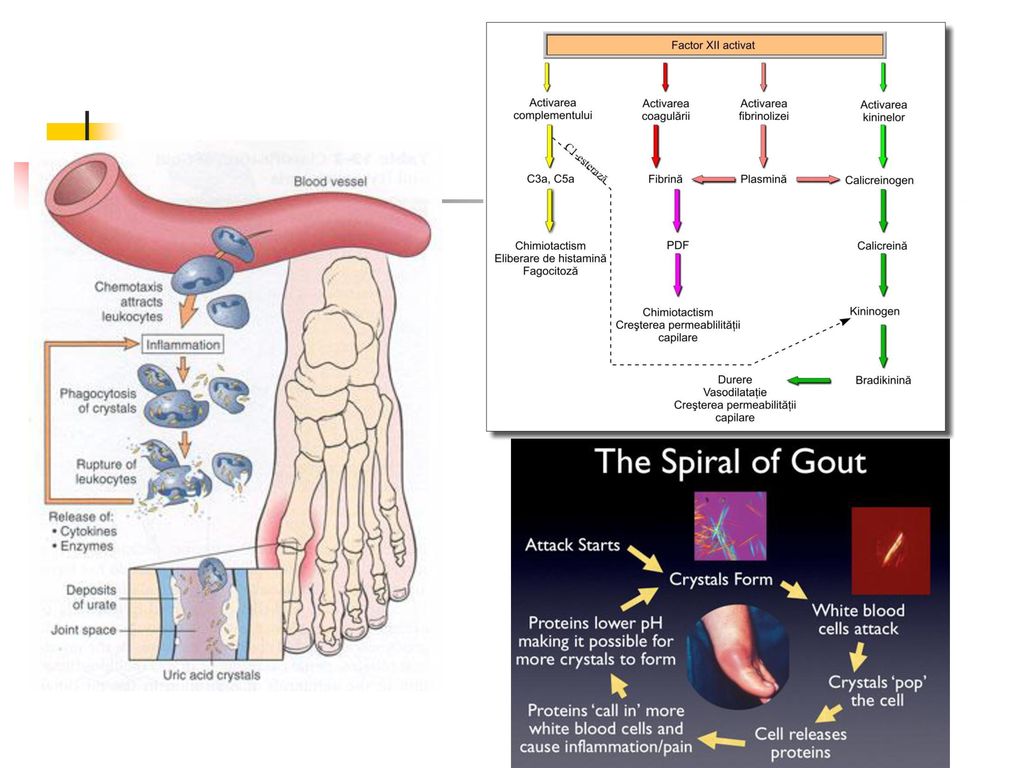
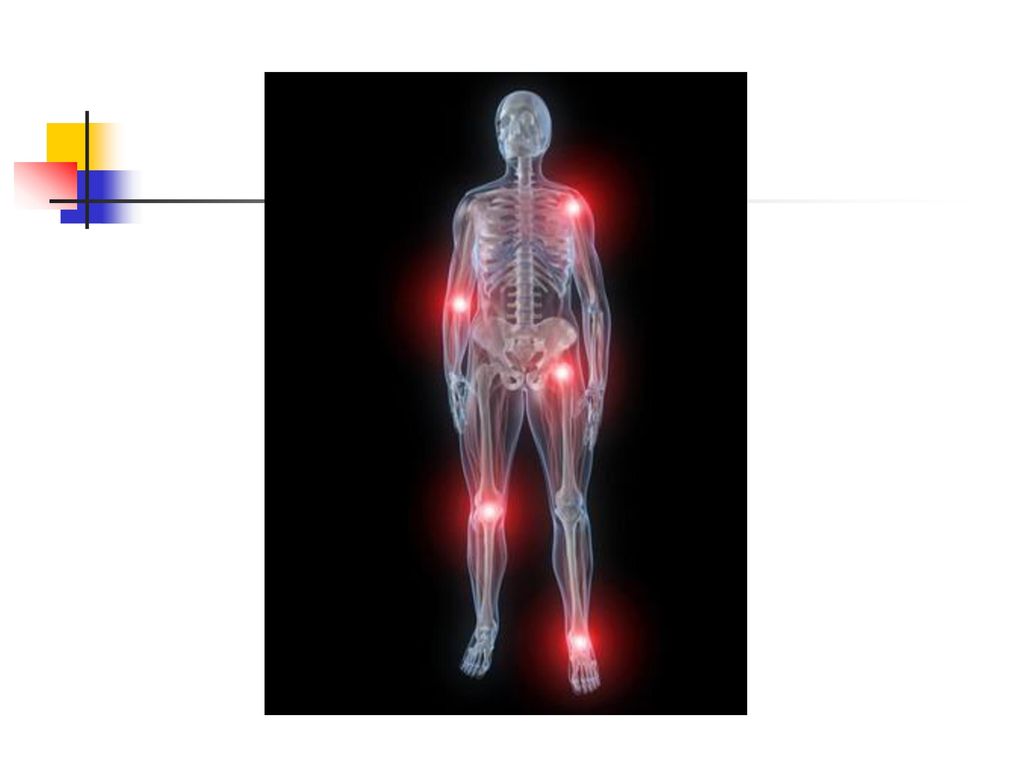
La physiopathologie de l'accès aigu de goutte
La goutte résulte par une accumulation d'urates dans divers tissues. Il existe un état de sursaturation en urates, qui cristallisent. La réaction inflammatoire articulaire est déclenchée par la présence de cristaux d’urate de sodium dans la cavité articulaire. Les cristaux infiltrent premièrement la synoviale, puis ils sont libérés dans la cavité synoviale. Les cristaux activent : le complément : C3a et C5a sont des anaphylatoxines avec propriétés vasodilatatrices et chimiotactiques, le complexe C567 est chimiotactique pour les polynucléaires, le facteur Hageman (XII).
.")
69
La physiopathologie de l'accès aigu de goutte
Les cellules synoviales activées synthétisent des cytokines (TNFa, IL1, IL6, IL8) qui vont attirer et activer les polynucléaires neutrophiles. L’opsonisation des cristaux par C3 et IgG favorise leur phagocytose par les polynucléaires neutrophiles qui libèrent : des enzymes lysosomiales, des radicaux libres, des prostaglandines et lencotriènes qui augmentent la réaction inflammatoire articulaire. La répétition des crises se fait selon des intervalles libres très variables avec extension à d’autres articulations. D’une façon générale, l’intensité des crises diminue tout en augmentant en fréquence et en durée. Dans ces conditions, l’évolution aboutit à la goutte chronique.
 qui vont attirer et activer les polynucléaires neutrophiles. L’opsonisation des cristaux par C3 et IgG favorise leur phagocytose par les polynucléaires neutrophiles qui libèrent : des enzymes lysosomiales, des radicaux libres, des prostaglandines et lencotriènes qui augmentent la réaction inflammatoire articulaire. La répétition des crises se fait selon des intervalles libres très variables avec extension à d’autres articulations. D’une façon générale, l’intensité des crises diminue tout en augmentant en fréquence et en durée. Dans ces conditions, l’évolution aboutit à la goutte chronique.")
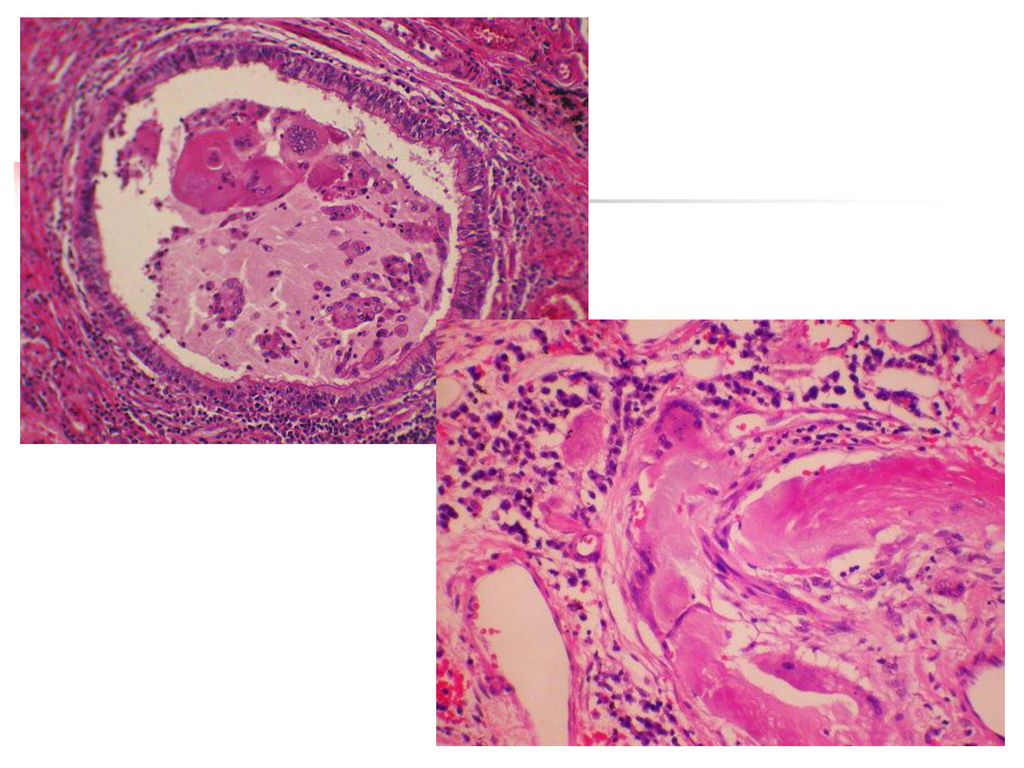
75
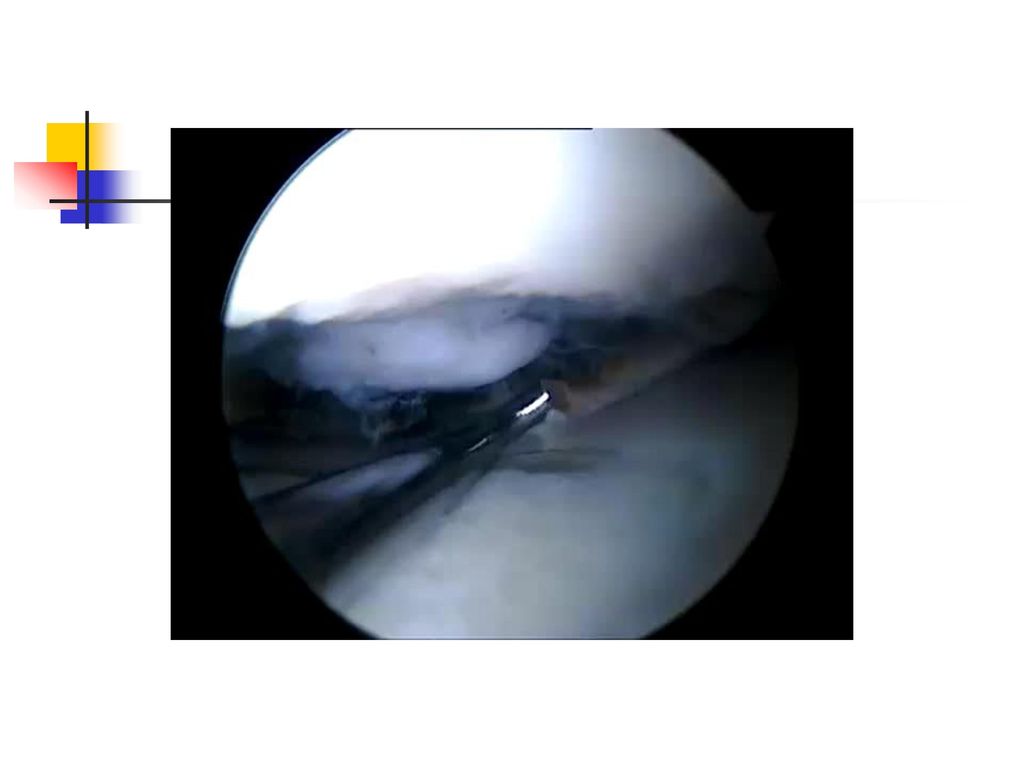
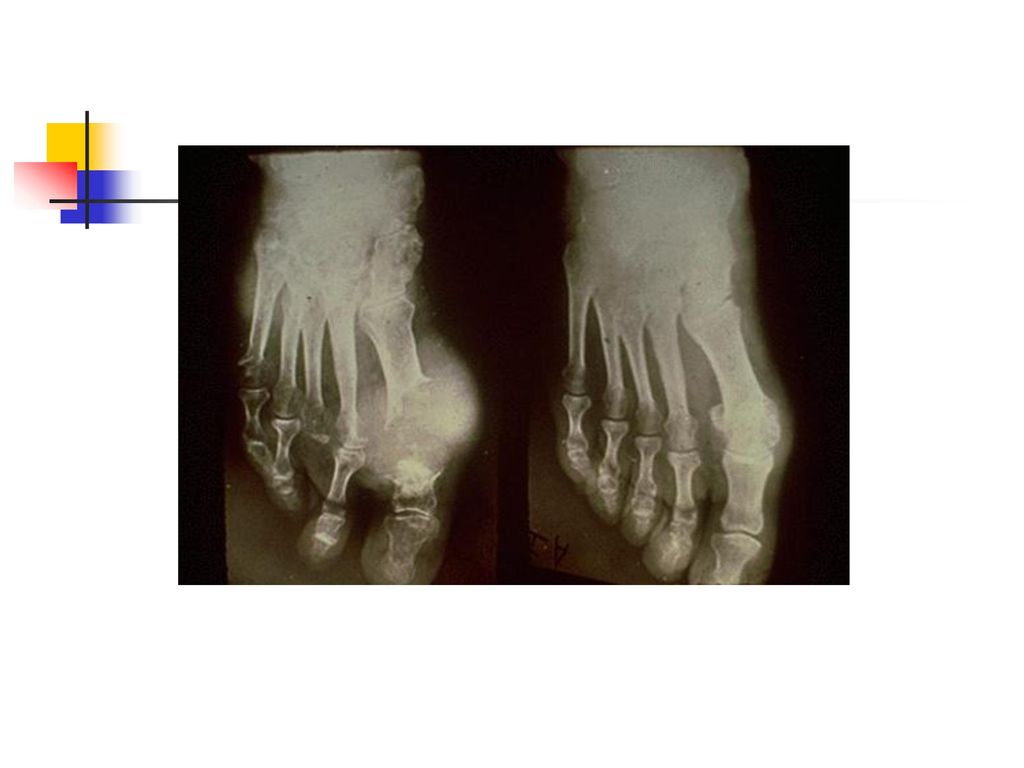
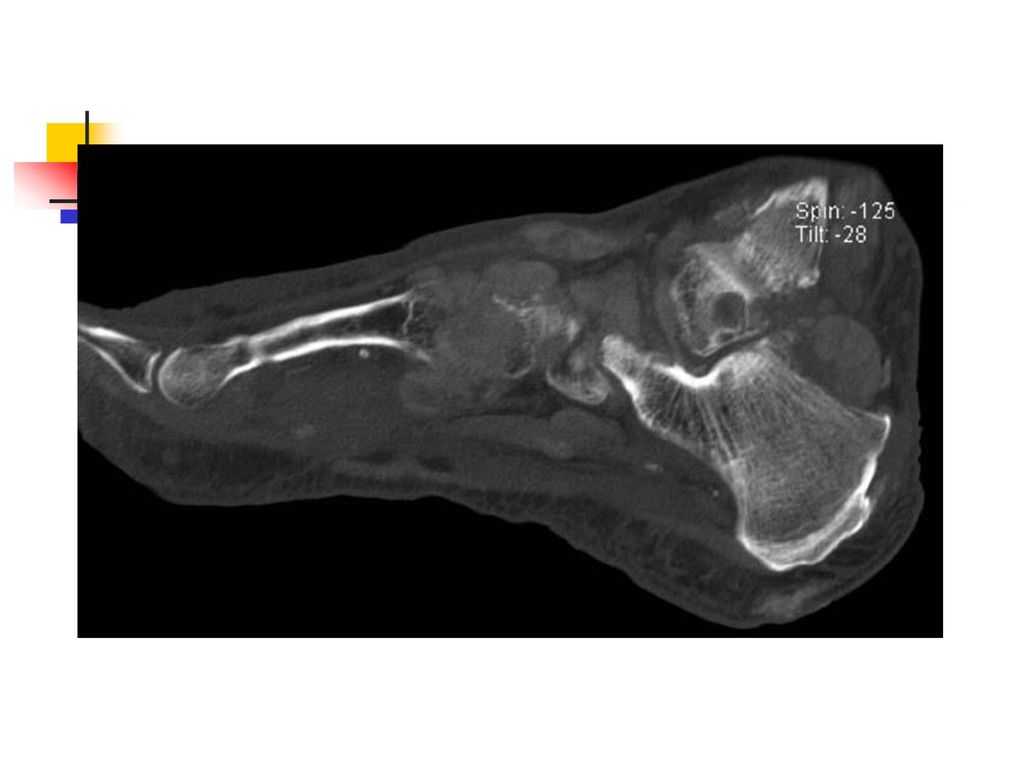
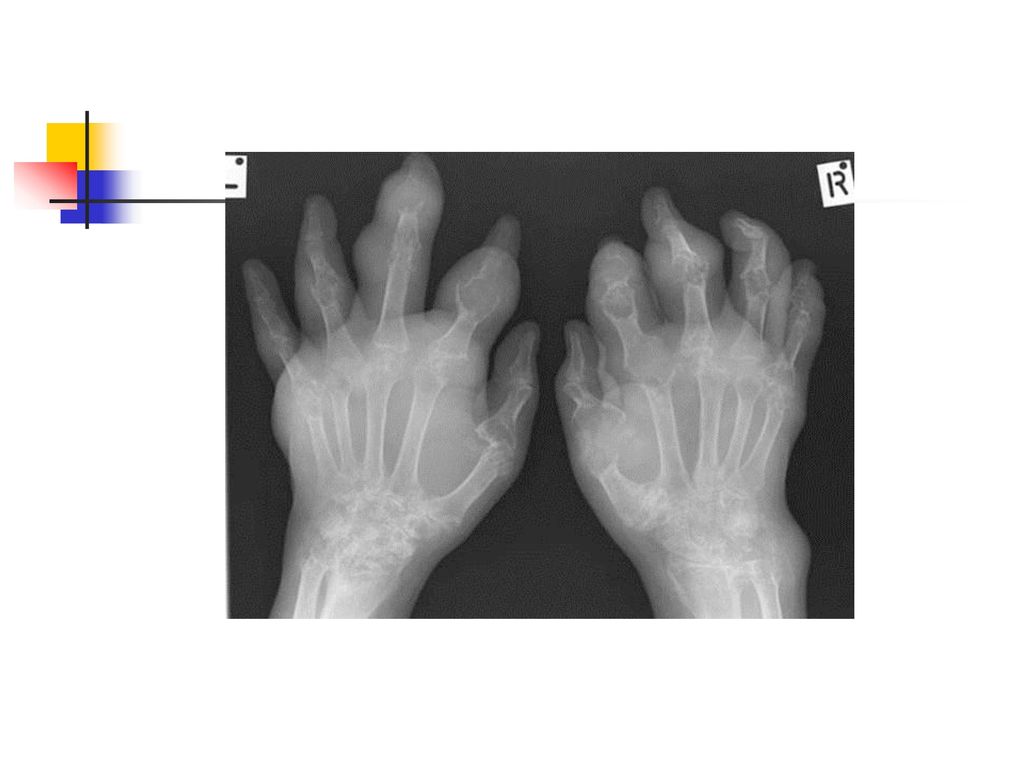
La goutte chronique Elle a une installation progressive en 5 à 10 ans.
Le tophus reste la lésion fondamentale et il se localise au niveau : des articulations (détermine une véritable invalidité) du tégument, de l'oreille, des mains, des tendons, de l'olécràne. L’atteinte rénale est la complication majeure et elle se manifeste par une : lithiase urinaire urique, néphropathie goutteuse avec des lésions : glomérulaires, interstitielles insuffisance rénale.
 du tégument, de l oreille, des mains, des tendons, de l olécràne. L’atteinte rénale est la complication majeure et elle se manifeste par une : lithiase urinaire urique, néphropathie goutteuse avec des lésions : glomérulaires, interstitielles. insuffisance rénale.")
Présentations similaires