


Le syndrome des antiphospholipides (SAP) est une maladie auto-immune caractérisée par la production d’autoanticorps et est associé à la thrombose veineuse, artérielle ou des petits vaisseaux et à des complications obstétricales.
Il faut envisager un dosage des anticorps antiphospholipides (anticoagulants lupiques, anticorps anticardiolipine et anti-β2 glycoprotéine-1) chez les patients qui subissent des événements thrombotiques inhabituels ou récurrents inexpliqués, surtout s’ils sont jeunes, s’ils présentent une maladie auto-immune systémique sous-jacente ou chez les femmes qui ont connu 3 fausses-couches précoces ou des pertes fœtales tardives inexpliquées.
De nombreux patients atteints de SAP présentent une maladie auto-immune systémique sous-jacente; le plus souvent, il s’agit du lupus érythémateux disséminé.
L’acide acétylsalicylique à faible dose, des mesures rigoureuses pour corriger les facteurs de risque cardiovasculaires modifiables et la maîtrise de toute maladie inflammatoire sousjacente peuvent réduire le risque d’événements thrombotiques chez les patients porteurs d’anticorps antiphospholipides.
Des essais contrôlés avec répartition aléatoire ont montré que la warfarine est supérieure aux anticoagulants oraux directs chez les patients atteints de SAP thrombotique.
Un homme de 22 ans a consulté son médecin de famille pour une douleur intense au pied droit, accompagnée d’une coloration sombre et d’une hypoesthésie. Il avait des antécédents de migraines et, depuis 2 ans, un léger malaise intermittent au mollet en faisant de l’exercice, mais n’avait pas consulté pour ce problème. Le jeune homme avait des antécédents familiaux de coronaropathie précoce. L’échographie en mode duplex des artères périphériques a montré une réduction du débit sanguin dans les 2 artères tibiales et une thrombose veineuse profonde à la jambe droite. Étant donné l’anomalie du débit artériel, une angiotomodensitométrie a été demandée et a révélé une occlusion chronique de l’artère fémorale superficielle droite et une occlusion de l’aorte abdominale infrarénale avec développement d’un vaisseau collatéral (figure 1). On a également noté une splénomégalie et une adénopathie axillaire. Le patient a été traité par apixaban et orienté vers l’équipe de chirurgie vasculaire.
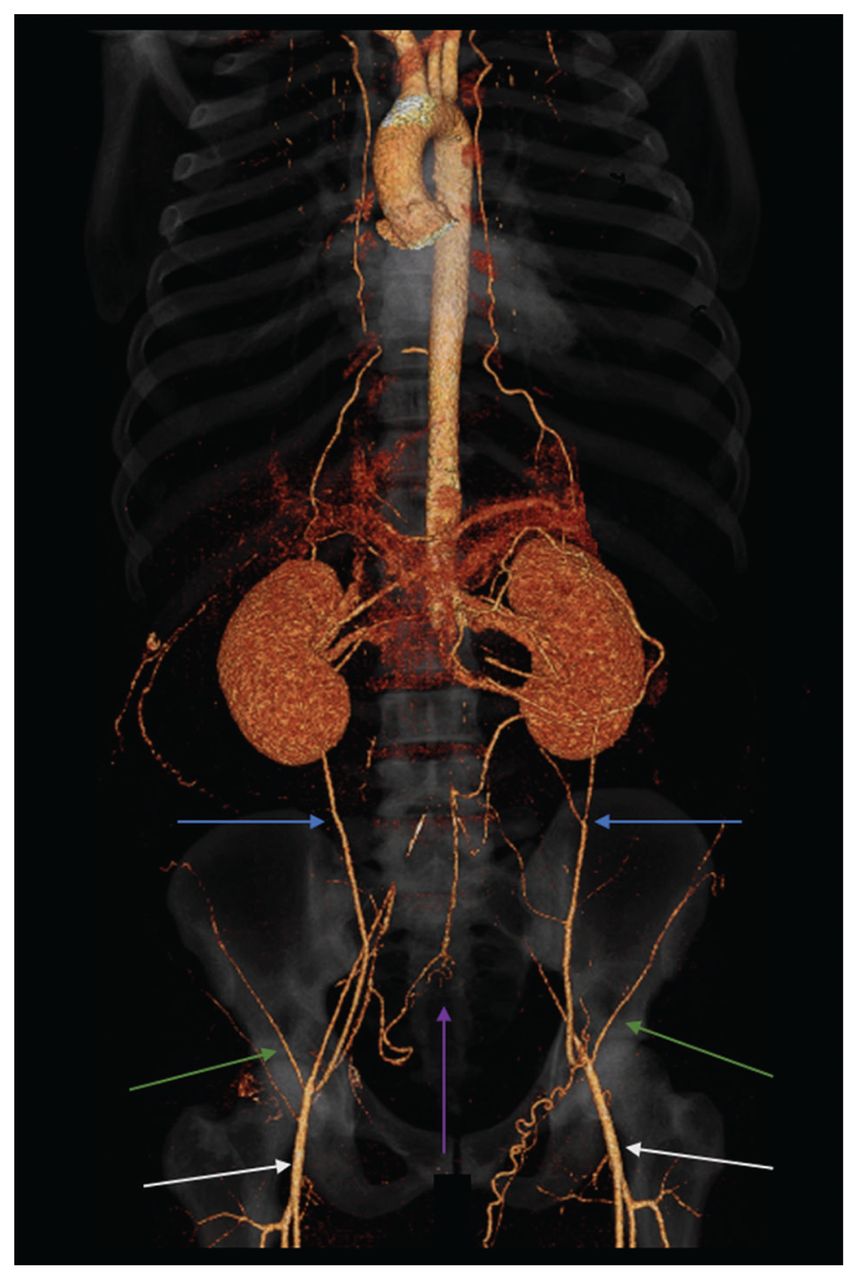
Vue coronale d’une angiotomodensitométrie en projection d’intensité maximale chez un homme de 22 ans atteint de lupus érythémateux disséminé et d’un syndrome des antiphospholipides secondaire, montrant l’occlusion complète de l’aorte abdominale infrarénale et de l’artère iliaque commune, avec reconstitution de la perfusion des 2 artères fémorales par de nombreux vaisseaux collatéraux, y compris une ramification des artères épigastrique inférieure, intercostales et mésentériques supérieure et inférieure. Les flèches bleues indiquent les vaisseaux collatéraux de l’artère épigastrique inférieure, la flèche mauve indique les vaisseaux collatéraux de l’artère rectale supérieure, les flèches vertes indiquent les artères circonflexes profondes (avec circulation collatérale par les artères intercostales, non illustrée) et les flèches blanches indiquent le débit reconstitué dans les artères fémorales par des vaisseaux collatéraux proximaux.
L’équipe de chirurgie vasculaire a envisagé d’effectuer un pontage aorto-bifémoral et a orienté le patient vers l’équipe de médecine interne pour une évaluation préopératoire. Étant donné que le patient ne présentait aucun facteur manifeste (néoplasique, par exemple) pouvant expliquer ses thromboses artérielles et veineuses inhabituelles, un dosage des anticorps antiphospholipides a été demandé et s’est révélé positif à l’égard des anticoagulants lupiques, avec des taux élevés d’anticorps IgG anticardiolipine et antiβ2 glycoprotéine-1 (aβ2GP1). Le patient a été orienté vers nos cliniques d’hématologie et de rhumatologie. Il ne prenait aucun autre médicament à part l’apixaban.
Le patient s’est rappelé avoir éprouvé pendant 6 mois une insidieuse arthralgie migratoire, avec une raideur articulaire matinale prolongée et des symptômes apparentés au phénomène de Raynaud. Quelques années avant la consultation initiale, il avait présenté une douleur pleurétique attribuée à l’époque à une costochondrite, mais la douleur ne s’est jamais tout à fait résorbée. Lorsque nous avons examiné le patient, il avait un indice de masse corporelle de 31, une tension artérielle de 140/85 mm Hg, ainsi qu’une rougeur déclive et une alopécie à la jambe droite. Plusieurs de ses articulations étaient sensibles. Nous n’avons pas observé d’œdème articulaire, d’adénopathie de la tête ou du cou, ni d’éruption cutanée.
Étant donné que les symptômes, les thromboses et les résultats d’analyses de laboratoire du patient évoquaient un lupus érythémateux disséminé (LÉD), une batterie de tests immunosérologiques a été demandée et a montré un titre d’anticorps antinucléaires supérieur à 1:640, la présence d’anticorps antichromatine, anti-Smith et antiribonucléoprotéiques, un titre élevé d’anticorps anti-ADN double brin, une hypocomplémentémie et un résultat positif au test de Coombs direct, sans signe d’hémolyse (tableau 1).
Analyses de laboratoire chez un homme de 22 ans atteint d’une grave maladie aorto-iliaque occlusive
Nous avons diagnostiqué un LÉD et un syndrome des antiphospholipides (SAP) secondaire, et mis en route un traitement par hydroxychloroquine (400 mg/j). Nous avons cessé l’administration de l’apixaban et commencé un traitement de longue durée par warfarine (ratio international normalisé [RIN] cible 2–3). La claudication et l’hypoesthésie à la jambe du patient se sont améliorées sans autre intervention. En raison de sa douleur pleurétique chronique, nous avons demandé une angiotomodensitométrie pulmonaire pour débusquer une possible embolie pulmonaire. Nous n’avons observé aucun embole, mais noté un volumineux épanchement péricardique, probablement causé par son lupus. Nous avons administré des corticostéroïdes intraveineux, suivis d’un cycle dégressif de prednisone. Nous avons ajouté du mycophénolate mofétil (1 g, b.i.d.) comme agent d’épargne des corticostéroïdes; la douleur thoracique et l’arthralgie du patient ont alors rapidement disparu.
Malgré l’immunosuppression, le patient a ensuite présenté un malaise croissant à la poitrine et au cou, différent de sa douleur pleurétique antérieure, laissant croire à une cause non inflammatoire. Étant donné son SAP sous-jacent et sa maladie artérielle connue, nous avons craint une coronaropathie et l’avons adressé en cardiologie. Une scintigraphie myocardique a révélé une grave ischémie du territoire irrigué par la descendante antérieure. Le cardiologue a prescrit du clopidogrel (75 mg/j), du bisoprolol (2,5 mg/j) et de l’atorvastatine (20 mg/j). Le patient a ensuite subi une coronarographie, qui a révélé une grave occlusion de 3 coronaires (figure 2).

Figure en anglais. Coronarographies préopératoires des coronaires gauche et droite chez un homme de 22 ans atteint de lupus érythémateux disséminé avec syndrome des antiphospholipides secondaire montrant une atteinte diffuse de l’ensemble des artères coronaires. Les flèches indiquent une sténose à 50 % du tronc commun coronaire gauche (TC = LM), une oblitération à 100 % de l’interventriculaire antérieure proximale (IVAp = pLAD), une sténose à 90 % de la lésion de la circonflexe proximale (CXp = pLCx), une sténose à 40 % et à 50 % de la coronaire droite proximale et moyenne (CDp et CDm [pRCA et mRCA], respectivement), ainsi qu’une sténose dans le double rameau moyen (RI [Double RI = Dual RI]) (60 % dans la branche antérieure et 80 % dans la branche postérieure). Les zones de sténose ont été confirmées par imagerie multiplanaire.
Il a donc subi un triple pontage aortocoronarien à l’âge de 24 ans. Pendant cette intervention, son péricarde s’est révélé adhérent au cœur, probablement en raison d’épisodes antérieurs de péricardite lupique.
Discussion
Le syndrome des antiphospholipides se caractérise par la présence d’autoanticorps qui se fixent aux phospholipides de la membrane cellulaire (ou protéines de liaison phospholipidiques) et par des signes de thrombose artérielle, veineuse ou des petits vaisseaux ou par des complications obstétricales1. Les complications thrombotiques sont variées et peuvent comprendre l’AVC, l’infarctus du myocarde, la thromboembolie périphérique ou des sièges inhabituels de thrombose, comme une thrombose des veines splanchniques ou cérébrales1. Les complications obstétricales comprennent fausses-couches précoces à répétition, perte fœtale tardive, prééclampsie précoce ou prématurité due à une insuffisance placentaire. Le syndrome catastrophique des antiphospholipides est une forme rare et gravissime de SAP qui s’accompagne de plusieurs complications thrombotiques quasi simultanées1.
L’incidence du SAP a été estimée à 1–2 pour 100 000 habitants et sa prévalence est d’environ 40–50 cas pour 100 000 habitants2. Le syndrome des antiphospholipides cause plus de 20 % des AVC chez les jeunes patients2. Les critères diagnostiques du SAP comprennent présence persistante d’anticorps antiphospholipides (anticoagulants lupiques, anticorps anticardiolipine ou aβ2GP1) en association avec des complications thrombotiques ou obstétricales (voir critères de Sapporo révisés, encadré 1)3. La présence des anticorps peut être transitoire et due à différents facteurs, notamment à des comorbidités, et, par conséquent, il faut donc la vérifier 2 fois à au moins 12 semaines d’intervalle. Les patients peuvent aussi présenter d’autres anomalies, telles que thrombocytopénie, hémorragie alvéolaire diffuse ou néphropathie, qui ne font pas partie des critères de Sapporo révisés1.
Encadré 1: Critères de Sapporo révisés pour le diagnostic du syndrome des antiphospholipides
Le syndrome des antiphospholipides est présent si au moins 1 des critères cliniques et au moins 1 des critères de laboratoire suivants sont présents:
Critères cliniques
Thrombose vasculaire
Un épisode clinique ou plus de thrombose artérielle, veineuse ou des petits vaisseaux concernant tout tissu ou organe. La thrombose doit être confirmée au moyen de critères objectifs validés.
Morbidité obstétricale
Un décès inexpliqué ou plus d’un fœtus morphologiquement normal à partir de la 10e semaine de gestation.
Une naissance prématurée ou plus d’un nouveau-né morphologiquement normal avant la 34e semaine de gestation en raison d’éclampsie ou de prééclampsie grave ou de caractéristiques reconnues d’insuffisance placentaire.
Trois avortements spontanés consécutifs inexpliqués ou plus avant la 10e semaine de grossesse, à l’exclusion de toute anomalie anatomique ou hormonale chez la mère ou cause génétique chez le père et la mère.
Critères de laboratoire
Anticoagulants lupiques à 2 occasions ou plus, à au moins 12 semaines d’intervalle.
Titre moyen ou élevé d’anticorps IgG ou IgM anticardiolipine à 2 occasions ou plus, à au moins 12 semaines d’intervalle.
Titre moyen ou élevé d’anticorps IgG ou IgM anti-β2 glycoprotéine-1 à 2 occasions ou plus, à au moins 12 semaines d’intervalle.
D’après Miyakis et coll.3 avec l’autorisation de John Wiley and Sons.
Le syndrome des antiphospholipides peut être primaire, mais plus du tiers des cas sont associés à une maladie autoimmune systémique, le plus souvent le LÉD4. Environ 40 % des patients atteints de LÉD portent ces anticorps et environ 20 %–50 % de ceux qui en sont porteurs finissent par présenter le syndrome1,4.
Dosage des anticorps antiphospholipides
Les anticorps antiphospholipides doivent être vérifiés chez les patients qui subissent des thromboses veineuses ou artérielles récurrentes ou inhabituelles, particulièrement chez les patients jeunes ou qui ont une maladie auto-immune systémique connue ou présumée. Les femmes devraient subir un dosage des anticorps antiphospholipides si elles font des fausses-couches précoces à répétition ou dans les cas de mort fœtale qui demeurent inexpliqués (encadré 1).
Il est possible de mesurer les anticorps anticardiolipine et aβ2GP1, même si le patient est sous anticoagulothérapie. Les anticoagulants lupiques sont mesurés au moyen de tests de coagulation dépendants des phospholipides qui varient d’un laboratoire à l’autre. Les résultats ne peuvent pas être interprétés chez les patients qui prennent des anticoagulants oraux directs (AOD) ou de l’héparine et ils sont difficiles à interpréter chez les patients sous warfarine, surtout lorsque le RIN est élevé. Il existe des recommandations internationales pour le dosage et l’interprétation des taux anticoagulants lupiques5.
Nous suggérons que tous les patients atteints de SAP fassent l’objet d’une surveillance des signes et symptômes de maladie auto-immune systémique sous-jacente, tels qu’alopécie, photosensibilité, éruptions cutanées laissant des cicatrices, arthrite inflammatoire, hématurie ou protéinurie inexpliquées et cytopénies. Si des patients atteints de SAP présentent des signes évoquant une maladie auto-immune systémique, il est recommandé de les orienter en rhumatologie. Les patients atteints de LÉD et d’autres maladies des tissus conjonctifs devraient se soumettre à un dépistage des anticorps antiphospholipides, particulièrement avant une grossesse.
Prise en charge du SAP thrombotique (encadré 2)
Encadré 2: Principes généraux de la prise en charge du syndrome des antiphospholipides (SAP)
La prophylaxie primaire au moyen d’acide acétylsalicylique à faible dose peut être envisagée chez les patients qui présentent un profil d’anticorps à risque élevé, par exemple qui ont une triple positivité à l’égard des anticorps antiphospholipides (c.-à-d., anticoagulants lupiques, anticorps anticardiolipine et anti-β2 glycoprotéine-1).
Chez les patients atteints d’un SAP thrombotique, un essai contrôlé avec répartition aléatoire appuie l’anticoagulothérapie par antivitamine K (p. ex., warfarine), plutôt que par anticoagulants oraux directs.
Une maîtrise rigoureuse des facteurs de risque cardiovasculaires modifiables (p. ex., hypertension, glycémie, dyslipidémie, maladie inflammatoire sousjacente) est nécessaire chez les patients atteints de SAP afin d’atténuer davantage le risque d’événements thrombotiques.
On utilise l’immunosuppression pour traiter les patients qui présentent une maladie inflammatoire sous-jacente, un syndrome catastrophique des antiphospholipides et certaines manifestations microvasculaires et non thrombotiques des anticorps antiphospholipides (comme une hémorragie alvéolaire diffuse, la néphropathie ou des cytopénies).
La prise en charge périopératoire requiert la participation d’experts pour gérer les risques thrombotiques et hémorragiques et surveiller adéquatement l’héparine non fractionnée chez les patients qui ont un temps de céphaline activée prolongé au départ en raison de la présence des anticoagulants lupiques.
On peut envisager de recourir à l’acide acétylsalicylique (AAS) à faible dose en prévention primaire des thromboses, particulièrement chez les patients atteints d’une maladie auto-immune, qui ont des profils d’anticorps antiphospholipides à haut risque (p. ex., triple positivité à l’égard des anticorps antiphospholipides [c.-à-d., anticoagulants lupiques, anticorps anticardiolipine et aβ2GP1]) ou qui présentent d’autres facteurs de risque cardiovasculaires6. Les données à l’appui du recours à l’AAS dans d’autres circonstances ne sont pas concluantes, par exemple chez les personnes en bonne santé indemnes de maladie auto-immune.
Les thromboses sont habituellement traitées par anti-vitamine K, comme la warfarine, la vie durant6,7. Les anticoagulants oraux directs ne sont pas recommandés, car des essais contrôlés avec répartition aléatoire ont montré un taux accru d’événements thrombotiques (artériels, particulièrement) chez les patients traités par AOD plutôt que par warfarine8,9; la supériorité apparente de la warfarine par rapport aux AOD est encore mal élucidée. En ce qui concerne les patients présumément atteints de SAP (p. ex., qui ont un 1 seul résultat positif à un test d’anticorps antiphospholipides n’ayant pas été répété), il est raisonnable de prescrire de la warfarine ou de les adresser vers un spécialiste des thromboses. La correction des facteurs de risque cardiovasculaires modifiables (p. ex., hypertension, glycémie, dyslipidémie, maladie inflammatoire sous-jacente) chez des patients ayant ou non subi une thrombose pourrait réduire davantage le risque d’événements thrombotiques.
Nous suggérons un seuil bas pour investiguer toute nouvelle complication thrombotique veineuse ou artérielle chez les patients atteints de SAP qui en présentent les signes annonciateurs. Chez les patients qui présentent des événements récurrents alors qu’ils sont sous warfarine, il est possible de hausser le RIN cible, d’ajouter un antiplaquettaire ou de passer à une héparine de bas poids moléculaire. Certains médicaments peuvent être ajoutés au traitement antithrombotique chez les patients qui ne répondent pas à l’anticoagulothérapie habituelle, y compris des statines ou de l’hydroxychloroquine (même en l’absence de LÉD), mais les données probantes concernant ces agents sont limitées, et la recherche devra être approfondie10. On peut ajouter des immunosuppresseurs à l’anticoagulothérapie chez les patients qui ont d’autres manifestations de SAP, telles que la néphropathie, des cytopénies ou l’hémorragie alvéolaire diffuse10. Chez les patients qui présentent un syndrome catastrophique des antiphospholipides, on utilisera l’héparine, la plasmaphérèse ou les immunoglobulines intraveineuses, les corticostéroïdes et, parfois même, le rituximab (un agent biologique anti-CD20) ou l’éculizumab (un agent biologique qui inhibe la voie du complément)10.
Les hormones exogènes utilisées pour la contraception ou l’hormonothérapie substitutive sont généralement à éviter chez les patientes qui présentent des anticorps antiphospholipides ou un SAP, en raison de leur un effet prothrombotique. Les exceptions à cet égard sont les stérilets à progestatif seul et les contraceptifs oraux à progestatif seul, qui n’accroissent pas le risque de thrombose.
La prise en charge des patients atteints de SAP thrombotique requiert une collaboration multidisciplinaire, particulièrement durant la grossesse et les périodes périopératoires, lorsqu’il est important de gérer les risques thrombotiques et hémorragiques. Étant donné qu’un résultat positif au dépistage des anticoagulants lupiques peut prolonger faussement le temps de céphaline activée (PTT) au départ, l’anticoagulothérapie par héparine non fractionnée pourrait devoir être vérifiée au moyen d’autres tests, comme les taux d’anti-Xa ou un PTT insensible aux phospholipides.
Le SAP et le LÉD peuvent se manifester de manière insidieuse. Tout retard dans leur diagnostic et leur traitement peut entraîner une atteinte irréversible aux organes. Les cliniciens doivent maintenir un fort degré de suspicion à l’égard du SAP, surtout chez les jeunes patients qui présentent des thromboses non provoquées ou inhabituelles ou en cas de fausses-couches précoces ou tardives récurrentes. Le syndrome des antiphospholipides peut être la première manifestation d’une maladie autoimmune systémique sous-jacente, souvent le LÉD. La prise en charge requiert des soins multidisciplinaires comprenant l’anticoagulothérapie, la modification des facteurs de risque cardiovasculaires et l’identification et le traitement de toute maladie inflammatoire sous-jacente.
Remerciements
Les auteurs remercient sincèrement pour son expertise l’équipe multidisciplinaire ayant participé à la prise en charge de ce cas, y compris les chirurgiens, les internistes, intensivistes, cardiologues et anesthésiologistes pour la période périopératoire, de même que le médecin de famille et toute l’équipe soignante. Les auteurs remercient la Dre Elizabeth Chan pour son aide à interpréter l’angiographie cardiaque, la Dre Teresa Kieser pour ses commentaires constructifs et le Dr Mathew Li pour son aide à interpréter l’angiotomodensitométrie et l’acquisition des images.
Footnotes
Intérêts concurrents: Megan Barber est cochercheuse au projet APSACTION (Antiphospholipid Syndrome Alliance for Clinical Trials and InternatiOnal Networking) et a reçu des honoraires de GSK en tant que modératrice, de même que des honoraires pour sa participation à des comités consultatifs pour Janssen, Sanofi-Genzyme, AstraZeneca et Abbvie. Ann Clarke est membre d’APS-ACTION et fait état d’honoraires versés par AstraZeneca, Bristol Myers Squibb et GSK, de même que d’un financement de recherche de GSK. Leslie Skeith est membre d’APS-ACTION et du Réseau CanVECTOR (Canadian Venous Thromboembolism Research Network) et déclare des honoraires de Leo Pharma et de Sanofi. Aucun autre intérêt n’a été déclaré.
Cet article a été révisé par des pairs.
Les auteurs ont obtenu le consentement du patient.
Collaborateurs: Tous les auteurs ont contribué à l’élaboration et à la conception des travaux. Megan Barber a rédigé l’ébauche du manuscrit. Tous les auteurs ont révisé de façon critique son contenu intellectuel important, ont donné leur approbation finale pour la version destinée à être publiée et assument l’entière responsabilité de tous les aspects du travail.
This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY-NC-ND 4.0) licence, which permits use, distribution and reproduction in any medium, provided that the original publication is properly cited, the use is noncommercial (i.e., research or educational use), and no modifications or adaptations are made. See: https://creativecommons.org/licenses/by-nc-nd/4.0/
In this issue

{kind=link}
{kind=link}
Article tools






Jump to section
Related Articles
Cited By...
- No citing articles found.
More in this TOC Section
Similar Articles

