
More Information
Submitted: 08 April 2020 | Approved: 28 April 2020 | Published: 29 April 2020
How to cite this article: Bano G, Phillips C, Tang S, Sharma A, Beharry N. Diagnostic imaging in congenital adrenal hyperplasia – how does it help? Ann Clin Endocrinol Metabol. 2020; 4: 007-010.
DOI: 10.29328/journal.acem.1001013
ORCiD: orcid.org/0000-0003-0470-0461
Copyright License: © 2020 Bano G, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

Diagnostic imaging in congenital adrenal hyperplasia – how does it help?
Gul Bano1*, Claudette Phillips2, Sarah Tang2, Anup Sharma2 and Nigel Beharry3
1Department of Diabetes and Endocrinology, St George’s University Hospital, London, UK
2Department of Endocrine Surgery, St George’s University Hospital, London, UK
3Department of Radiology, St George’s University Hospital, London, UK
*Address for Correspondence: Gul Bano, Department of Diabetes and Endocrinology. St George’s University Hospital, London, UK, Tel: 447779628865; Email: gbano@sgul.ac.uk; gul.bano@nhs.net
The phenotypic manifestation of congenital adrenal hyperplasia (CAH) is variable, and this largely depends on the extent of 21-Hydroxylase enzyme deficiency. In non- classic CAH (NCCAH), the clinical features predominantly reflect the androgen excess rather than adrenal insufficiency. In boys, the condition may not present until much later in childhood, where the diagnosis is made following presentation with precocious puberty, features of aldosterone insufficiency, or this condition may be detected during fertility workup Imaging is generally not used in the evaluation of CAH, but may be helpful for the diagnosis, management, and follow-up of these patients. CAH can result in adrenal enlargement in both classic and non-classic forms of adrenal hyperplasia. The so-called adrenal rest tissue may be seen at several sites throughout the body, including the celiac plexus region, broad ligaments, normal ovaries, and testes. Sustained elevation of adrenocorticotropic hormone (ACTH) in patients with CAH has been postulated to cause adrenal rest cells to grow and become functionally active. The discovery of bilateral adrenal enlargement during radiologic evaluation for unrelated disease processes might serve as a mode of presentation for clinically not apparent or non- classical congenital adrenal hyperplasia (NCCAH).
Congenital adrenal hyperplasia (CAH) is a collection of autosomal recessive genetic disorders affecting cortisol biosynthesis. 1 in 18,000 live births in the United Kingdom is affected by the condition. The classification of this condition is complex and dependent on the specific enzyme mutation, as well as the clinical presentation. Two major genotypic variants are described, the most frequent (95%) involves a deficiency of 21-hydroxylase enzyme, and a small minority (5%) are accounted for by 11-hydroxylase deficiency [1, 2].
The phenotypic manifestation of CAH is variable, and this largely depends on the extent of 21-Hydroxylase enzyme deficiency. The most severely affected individuals with classic CAH due to 21-hydroxylase deficiency present during the neonatal period and early infancy with adrenal insufficiency and salt wasting, or in the first few years of life with virilization. Females have ambiguous genitalia. In non- classic CAH (NCCAH), the clinical features predominantly reflect the androgen excess rather than adrenal insufficiency. In boys, the condition may not present until much later in childhood, where the diagnosis is made following presentation with precocious puberty, features of aldosterone insufficiency, or this condition may be detected during fertility workup [3].
Imaging is generally not used in the evaluation of CAH but may be helpful for the diagnosis, management, and follow-up of these patients. CAH can result in adrenal enlargement in both classic and non-classic forms of adrenal hyperplasia. The so-called adrenal rest tissue may be seen at several sites throughout the body, including the celiac plexus region, broad ligaments, normal ovaries, and testes [4]. Sustained elevation of adrenocorticotropic hormone (ACTH) in patients with congenital adrenal hyperplasia has been postulated to cause adrenal rest cells to grow and become functionally active [5]. The discovery of bilateral adrenal enlargement during radiologic evaluation for unrelated disease processes might serve as a mode of presentation for clinically not apparent or non- classical congenital adrenal hyperplasia (NCCAH) [6]. We present some of the radiological features in 2 of our non-classical CAH cases.
A 41-year-old female, presented with intermittent vaginal bleeding after five years of amenorrhea. She had features of hyperandrogenism that included male pattern hair loss with marked facial hirsutism, thick coarse skin, acne, and clitoromegaly on examination. She was a full-term baby born to non-consanguinous parents. Her first hospital admission was at the age of 2 with vomiting, and she was treated with intravenous fluids. She had numerous hospital admissions between the ages of 7-8 years. She was finally diagnosed with CAH and was treated with Hydrocortisone 10mgs three times a day and Fludrocortisone 100 mcg. At the age of 30, she stopped all her medications as she was gaining weight and had developed abdominal striae. She also stopped attending her hospital appointments. Her investigations showed high serum testosterone 14.3 nmol/l (0.2-2.86) with suppressed gonadotrophins luteinizing hormone (LH) < 0.2 IU/l (1-13) and Follicular stimulating hormone (FSH) < 0.8 IU/l (1-8)]. Her 17-Hydroxyprogesterone (17-OHP) was significantly elevated at > 152 nmol/l (Follicular phase 1-8 and luteal phase < 18.8). Her androstenedione was also high at 45 nmol/l (2-5.4). She had polycythemia with high hemoglobin of 18 g/l and a hematocrit (HCT) of 0.56 l/l, most likely secondary to testosterone-driven erythropoiesis.
Her CT scan adrenals demonstrated hypertrophy of both adrenal glands with a 2.5 cm nodule in the lateral limb of the right gland and a 6cm mass in the medial limb of the left adrenal with heterogeneous enhancement and foci of calcification. This was reported as being suspicious for carcinoma of the adrenal gland or phaeochromocytoma. There was a further dense 2.2 cm nodule in the limb of the left adrenal gland (Figure 1). She had CT Chest/Abdomen/Pelvis. This showed a large fibroid uterus, and no other lesions were identified (Figure 2). A pelvic ultrasound identified morphologically normal appearing ovaries lying behind the uterus. Both medical and surgical treatment options were discussed with the patient, and she opted for adrenalectomy. She had laparoscopic bilateral adrenalectomy followed by hysterectomy for her large uterine fibroid. Her histology confirmed bilateral florid adrenal cortical hyperplasia with multiple myelolipomas and bilateral adrenal cortical tumors. The larger left-sided tumor showed some necrosis, but there was no evidence of malignancy. Following adrenalectomy, she resumed her periods. Her current testosterone is 0.8 nmol/l with LH of 6.6 IU/L and FSH 8.8IU/L. Her 17 OHP is 4.3 nmol/l and androstenedione 1.2 nmol/l. She is on Hydrocortisone 10, 10 and 5 mg and fludrocortisone 100mcg replacement. She remains very well.
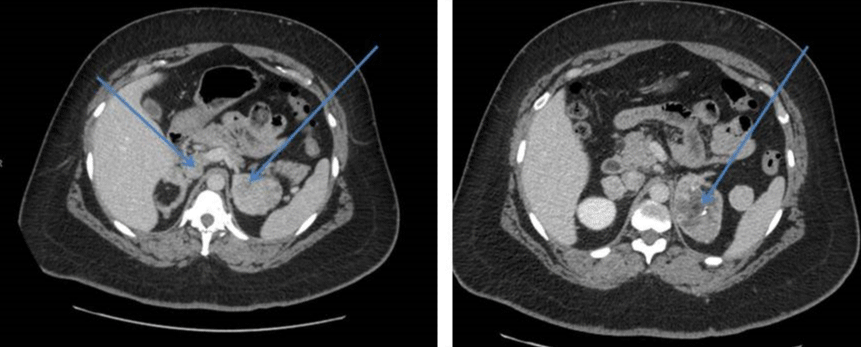
Figure 1: Hypertrophy of both adrenal glands with a nodule in the lateral limb of the right gland and a large mass in the medial limb of the left adrenal with heterogeneous enhancement and foci of calcification.
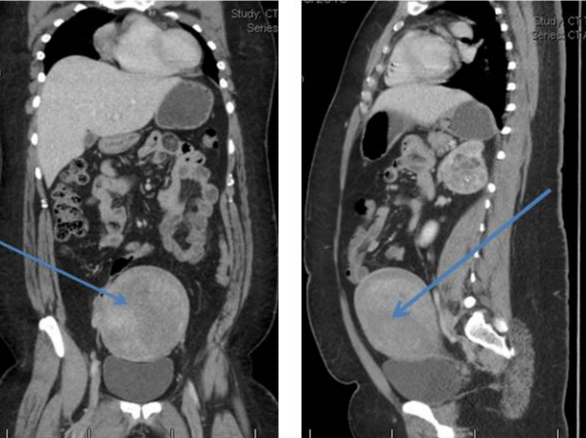
Figure 2: Large fibroid uterus.
A 29-year-old male presented to the endocrine clinic with a history of erectile dysfunction. He was not on any medication. He had short stature with a height of 156.5 cm, whereas his estimated mid parental height was 170 cm. He was at Tanner stage 5 of puberty. His testes were 12ml and were soft with no palpable masses. He had bilateral gynecomastia. His initial investigations showed suppressed gonadotrophins: LH < 0.2 (1-9 IU/L), FSH < 0.8 (1-10 IU/L) and testosterone of 22.4 (9-24 nmol/l). His prolactin, thyroid function, and insulin like growth (IGF-1) were normal. He had suboptimal cortisol response on the synacthen test with a 30-minute value of 219 (expected peak of 550 nmol/l) and no increment from the baseline. Cortisol deficiency was confirmed on the insulin tolerance test. He had a high 17 hydroxyprogesterone (17-OHP) of 677 and high androstenedione of 80nmol/l, suggesting the diagnosis of non-classical CAH. His urinary steroid profile confirmed CAH due to 21 hydroxylase deficiency by excess of 17-OHP metabolites. His CT adrenal showed markedly enlarged adrenal glands bilaterally (Figure 3). Ultrasound scan of the testes showed lobulated areas of increased density associated with decreased vascularity in both testes consistent with testicular adrenal rest tumors (Figure 4). His genetic test confirmed mutation in CYP 21 gene.
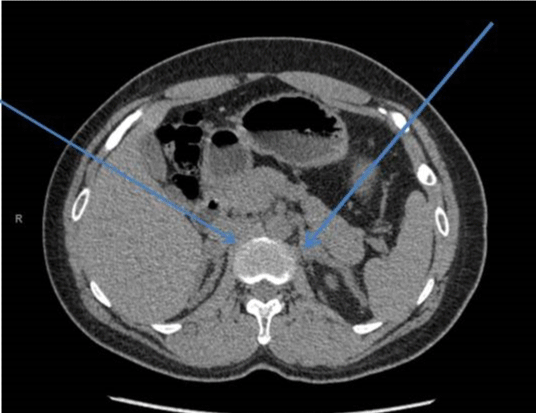
Figure 3: Bilaterally adrenal Hypertrophy.
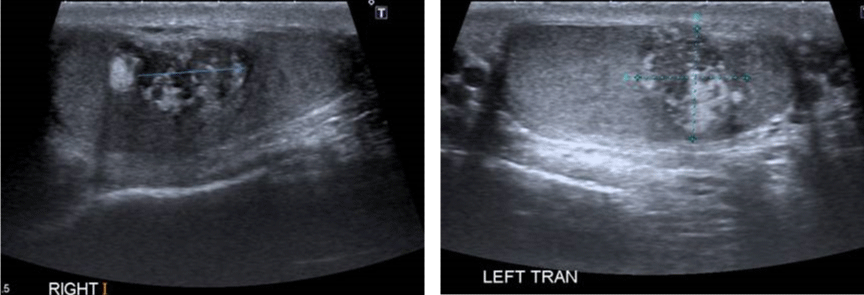
Figure 4: Ultrasound scan showing bilateral testicular adrenal rest tumors.
Imaging techniques used for various clinical indications are able to detect incidental adrenal enlargement, and this may alert clinicians to the underlying subclinical conditions. Adrenal enlargement can be present in both classic and non-classic forms of adrenal hyperplasia. Non-classical cases of congenital adrenal hyperplasia have been reported in family studies investigating the inheritance of the disorder that is first suspected on adrenal imaging. 7 Imaging defining the adrenal size and presence of testicular masses is useful in alerting the clinician to the possible diagnosis of clinically non-classical forms of CAH. It may also help in defining the adequacy and compliance of treatment in patients with a diagnosis of CAH. Adrenocortical tumors are not rare in patients with CAH. Up to 83% of homozygous CAH patients have been reported to have adrenocortical masses. Adrenocortical masses in CAH are most likely to be benign, and malignant lesions are rare. Several reports in the literature have described CT scans showing nodules that may regress with adequate therapy. Other adrenal lesions described in CAH include adenomas, myelolipomas and the typical pattern of diffuse enlargement with a heterogeneous enhancement [8].
Ectopic adrenocortical tissue can be an incidental finding during abdominal and inguinal surgery without clinical implications. Typical locations include within the testes, the celiac plexus, and along the spermatic cord [9]. In adult patients with CAH, ectopic adrenal rest tumors can be present outside the testicular region. Some rare locations described are the liver and the spinal canal [10-19]. The Adrenal glands in embryological period develop in the vicinity of the gonads. The Adrenocortical cells may nest within the testes or descend together with the testes in the retroperitoneal region. The commonest location of adrenal rests is within the testes or along the spermatic cord. Pro-opiomelanocortin (POMC) is a precursor polypeptide. It is cleaved to produce multiple peptide hormones, including a-MSH, adrenocorticotrophin (ACTH), and b-endorphins. In the presence of high plasma Pro-opiomelanocortin (POMC) concentrations, these cells can proliferate, leading to the development of adrenal rest tumors [12].
The development of testicular tumors is one of the most frequently detected complications in male CAH patients, particularly those who are not adequately treated. These tumors were first reported in 1940 by Wilkins, et al. [13]. These tumors have morphological and functional resemblance with adrenal tissue and are called “testicular adrenal rest tumors” (TART). TART resembles adrenocortical tissue on histology [17-19]. It is sometimes difficult to distinguish between Leydig cell tumors and TART. The distinguishing feature between the two is that TART is bilateral in more than 80%, whereas Leydig cell tumors are bilateral in only 3% of the cases. 10% of Leydig cell tumors can show Malignant degeneration but has not been reported in patients with TART.
The reported prevalence of TART is variable between 0 and 94%. It depends on the selection of the patients (age, hormonal control) and the method of tumor detection [10-16]. Only tumors of more than 2 cm are usually detectable by palpation because of their location within the rete testis [14]. These tumors can be easily missed when imaging such as ultrasound or magnetic resonance imaging (MRI) is not performed. Screening of TART is not routinely done in male CAH patients. TART can be associated with infertility. Adequate and intensification of glucocorticoid treatment can lead to regression in the size of the tumor by suppression of ACTH. This can result in a successful pregnancy. Ovarian adrenal rest tumors (OART) in female CAH patients are very rare [15]. A systematic review of the literature by Tiosano, et al. reveals only 11 confirmed cases to date. All these patients exhibited high levels of ACTH and the age range was from 2 to 41 years [16]. There are no defined criteria for a radiologic diagnosis of OART [17]. Bilateral adrenalectomy for CAH is a reasonable alternative to conventional medical therapy for carefully selected patients who have had unsatisfactory outcomes with conventional medical management. This treatment requires managing adrenal insufficiency without the requirement for HPA axis suppression. The indications for the surgery include severe virilisation, infertility with rest tumors and refractory hypertension. Patients have to be warned about the adrenal crisis and importance of adherence to treatment [18].
The diagnosis of CAH is mainly based on clinical features, hormonal, and genetic analysis. Imaging has an important role in the diagnosis and management of these patients. It provides important information for the diagnosis, follow-up, compliance with treatment, and surgical planning. Ectopic adrenocortical masses should be considered in the differential diagnosis of other tumors, particularly when associated with hyperandrogenism in females. The detection of TART should be done as early as possible, as the patients can be monitored and treated more intensively in order to prevent fertility impairment and testicle damage.
Screening for CAH or carrier status should be undertaken in patients presenting with multiple adrenal adenomas (AIs) with hyperandrogenism or Middle East origin [19]. Persons with CAH may be at increased risk of developing adrenal myelolipomas, particularly if their CAH is poorly controlled [20].
Learning points
• The non- classic CAH is more prevalent and less severe than classic CAH as 20% - 50% of 21-hydroxylase enzyme activity is still retained.
• Individuals with NCCAH may remain asymptomatic or may present with androgen excess in both childhood and adulthood.
• Adrenal enlargement can be present in both classic and non-classic forms of adrenal hyperplasia. It may be reported as an incidental finding on imaging and may lead to the diagnosis of NCCAH.
• Other adrenal lesions that may alert to the diagnosis of CAH include adenomas, myelolipomas and the typical pattern of diffuse enlargement with a heterogeneous enhancement
• The presence of testicular masses can be another clue to the diagnosis of CAH. These masses may be missed without imaging if < 2 cm.
• Imaging may also be helpful in defining the adequacy and compliance of treatment in patients with a diagnosis of CAH
• Bilateral adrenalectomy for CAH is a reasonable alternative to conventional medical therapy for carefully selected patients who have had unsatisfactory outcomes with conventional medical management.
- Huynh T, McGown I, Cowley D, Nyunt O, Leong GM, et al. The clinical and biochemical spectrum of congenital adrenal hyperplasia secondary to 21- hydroxylase deficiency. Clin Biochem Rev 2009; 30: 75-86. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/19565027
- Khalid JM, Oerton JM, Dezateux C, Hindmarsh PC, Kelnar CJ, et al. Incidence and clinical features of congenital adrenal hyperplasia in Great Britain. Arch Dis Child. 2012; 97: 101-106. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/22241917
- Kanaka GC, Chrousos GP, Dacou VC. The spectrum of clinical, hormonal and molecular findings in 280 individuals with nonclassical congenital adrenal hyperplasia caused by mutations of the CYP21A2 gene. Clin Endocrinol (Oxf). 2015; 82: 543-549. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25041270
- Avilal NA, Premkumar A, Merke DP. TesticularAdrenal RestTissue in Congenital Adrenal Hyperplasia: Comparison of MR Imaging and Sonographic Findings. AJR. 1999; 172: 1003-1006. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/10587136
- White PC, Speiser PW. Congenital Adrenal Hyperplasia due to21-Hydroxylase Deficiency. Endocrine Reviews. 2000; 21: 245-291. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28450075
- Witchel SM, Azziz R. Nonclassic Congenital Adrenal Hyperplasia. Int J Pediatr Endocrinol. 2010; 2010: 625105.
- Georgitis WJ. Clinically Silent Congenital Adrenal Hyperplasia Masquerading as Ectopic Adrenocorticotropic Hormone syndrome. The American Journal of Medicine. 1986; 80: 703-708. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/3008554
- Teixeira SR, Elias PL, Andrade MS, Melo AF, Elias JJ. The role of imaging in congenital adrenal hyperplasia. Arq Bras Endocrinol Metab. 2014; 58: 702-708. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25372578
- Souverijns G, Peene P, Keuleers H, Vanbockrijck M. Ectopic localization of adrenal cortex. European Radiology, 2000; 10: 1165-1168.
- Tajima T, Funakoshi A, Ikeda Y, Hachitanda Y, Yamaguchi M, et al. Nonfunctioning adrenal rest tumour of the liver: radiologic appearance. J Comput Assist Tomogr. 2001; 25: 98-101. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/11176302
- Kepes JJ, O’Boynick P, Jones S, Baum D, McMillan J. Adrenocortical adenoma in the spinal canal of an 8-year old girl. Am J Surg Pathol 1990; 14: 481-484. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/2327553
- Val P, Jeays-Ward K, Swain A. Identification of a novel population of adrenal-like cells in the mammalian testis. Developmental Biology 2006; 299: 250-256. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16949566
- Wilkins L, Fleishmann W, Howard JE. Macrogenitosomia precox associated with hyperplasia of the androgenic tissue of the adrenal and death from corticoadrenal insufficiency. Endocrinology. 1940; 26: 385-395.
- Claahsen-van der Grinten HL, Hermus ARMM, Otten BJ. Testicular Adrenal Rest Tumours in Congenital Adrenal Hyperplasia. International Journal of Pediatric Endocrinology. 2009; 2009: 624823.
- Claahsen-van der Grinten HL, Hulsbergen-van de Kaa CA, Otten BJ. Overian adrenal rest tissue in congenital adrenal hyperplasia – a patient report. Journal of Pediatric Endocrinology and Metabolism. 2006; 19: 177-182.
- Tiosano D, Vlodavsky E, Filmar S, Weiner Z, Goldsher D. Ovarian adrenal rest tumor in a congenital adrenal hyperplasia patient with adrenocorticotropin hypersecretion following adrenalectomy. Horm Res Paediatr. 2010; 74: 223-2238. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/20431278
- Stikkelbroeck NM, Hermus A, Schouten D. Prevalence of ovarian adrenal rest tumours and polycystic ovaries in females with congenital adrenal hyperplasia: results of ultrasonography and MR imaging. Eur Radiol. 2004; 14: 1802-1806. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15322809
- Diana Mackay, Anna Nordenstro, Henrik Falhammar. Bilateral Adrenalectomy in Congenital Adrenal Hyperplasia: Systematic Review and Meta-Analysis. The journal of Clinical Endocrinology& Metabolism. 2018; 103: 1767-1778. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29554355
- Falhammar H. Non-functioning adrenal incidentalomas caused by 21-hydroxylase deficiency or carrier status? Endocrine. 2014; 47: 308-314. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24452876
- German-Mena E, Zibari GB, Levine SN. Adrenal myelolipomas in patients with congenital adrenal hyperplasia: review of the literature and a case report. Endocr Pract. 2011; 17: 441-447. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21324823