Emerging cellular themes in leukodystrophies
 Joseph C. Nowacki†
Joseph C. Nowacki†  Meng Meng Fu
Meng Meng Fu- NINDS (National Institute of Neurological Disorders and Stroke), National Institutes of Health, Bethesda, MD, United States
Leukodystrophies are a broad spectrum of neurological disorders that are characterized primarily by deficiencies in myelin formation. Clinical manifestations of leukodystrophies usually appear during childhood and common symptoms include lack of motor coordination, difficulty with or loss of ambulation, issues with vision and/or hearing, cognitive decline, regression in speech skills, and even seizures. Many cases of leukodystrophy can be attributed to genetic mutations, but they have diverse inheritance patterns (e.g., autosomal recessive, autosomal dominant, or X-linked) and some arise from de novo mutations. In this review, we provide an updated overview of 35 types of leukodystrophies and focus on cellular mechanisms that may underlie these disorders. We find common themes in specialized functions in oligodendrocytes, which are specialized producers of membranes and myelin lipids. These mechanisms include myelin protein defects, lipid processing and peroxisome dysfunction, transcriptional and translational dysregulation, disruptions in cytoskeletal organization, and cell junction defects. In addition, non-cell-autonomous factors in astrocytes and microglia, such as autoimmune reactivity, and intercellular communication, may also play a role in leukodystrophy onset. We hope that highlighting these themes in cellular dysfunction in leukodystrophies may yield conceptual insights on future therapeutic approaches.
Introduction
White matter development initiates in utero in the fetus, continues into late adolescence and adulthood, and culminates to constitute ∼40% of adult human brains. White matter contains both neuronal axons and myelin sheaths that wrap concentrically in many layers around axons like paper towels. White matter is so named, because myelin is lipid rich and has a white, fatty appearance. Myelin functions to insulate axons, to facilitate saltatory conduction, and to increase axon potential velocity. During human development, myelin production begins slowly during the third trimester of pregnancy, and rapidly accelerates postnatally and during early childhood (Barkovich et al., 1988; Jakovcevski et al., 2009; Tomassy, Dershowitz, and Arlotta 2016). By 2 years of age, the majority of myelination is complete, as visualized by MRI scans (magnetic resonance imaging) and postmortem silver staining (Flechsig 1920; Wolf et al., 1993). Although white matter volume continues to increase into adulthood, its rate of formation is significantly slower (Fields 2010; Groeschel et al., 2010).
Developmental white matter diseases generally fall under the category of leukodystrophies. The term leukodystrophy can be broken down into its Greek origins: “leuko” for white, “dys'' for bad or abnormal, and “trophy” for growth. Together, these root words define leukodystrophies as a broad spectrum of neurological disorders that are characterized primarily by deficiencies in myelin formation that are usually not secondary to neuronal defects. Incidence estimates range from one in 8,000 to lower estimates of one in 50,000 or one in 80,000 (Vanderver et al., 2012). Symptom onset typically occurs during childhood, when neural pathways and associated white matter tracts involved in speech, motor coordination, and memory are refined. Thus, common symptoms of leukodystrophies include regression or loss of developmental abilities, such as speech and walking, poor motor control, and cognitive defects. Though the majority of leukodystrophies present during childhood, some are adult-onset. This disparity in age of onset likely reflects diverse etiologies and disease mechanisms and, therefore, is an ongoing topic of research.
Though some cases of leukodystrophy have unclear etiology (see Box 1), many types have a genetic cause with diverse patterns of inheritance, including autosomal recessive, autosomal dominant, or X-linked recessive. These modes of inheritance dictate the appropriate types of therapeutic approaches that have been attempted in translational studies. Recessive leukodystrophies are often addressed through replacement therapies that aim to restore loss of function. These approaches include nutrient supplementation, enzymatic replacement via viruses, and transplantation of stem cells. For example, iPSCs (induced pluripotent stem cells) derived from human skin cells can be induced to become specialized cells, such as oligodendrocytes (Chanoumidou et al., 2020). Autosomal dominant leukodystrophies are often addressed through therapies that aim to suppress the mutant gene, mRNA, or protein. Traditional small molecule drug screens have been performed, typically on cellular models of leukodystrophy. In addition, a new class of drug called ASOs (antisense oligonucleotides) can bind to RNA targeting sequences to achieve gene silencing. ASOs for the treatment of SMA (spinal muscular atrophy) are FDA-approved and several ongoing ASO clinical trials target ALS (amyotrophic lateral sclerosis) and other genetic neurological diseases (Amado and Davidson 2021). Thus, multiple therapeutic approaches are available to address the diverse types of leukodystrophies.
BOX 1Non-genetic factors, particularly neonatal white matter injury, can adversely affect neurodevelopment in long-lasting ways. Abnormal MRI findings in premature infants are very strong predictors of unfavorable neurodevelopmental outcomes, including cognitive delays, motor delays, and cerebral palsy (Woodward et al., 2006). Congenital heart disease is the most common major birth defect, and can place infants at risk of hypoxia (Marelli et al., 2007). Hypoxic insult causes oligodendrocyte death and delayed oligodendrocyte differentiation, thus leading to abnormalities in developmental myelination (Back et al., 2001; Jablonska et al., 2012). Interestingly, hypoxic newborn brains may share some common molecular markers with adult multiple sclerosis (MS) brains. Indeed, the remyelination regulator Axin2 is present in white matter lesions that are found in both human newborn brains with hypoxic damage and active MS lesions in adults (Fancy et al., 2011).
Non-genetic FactorsNon-genetic factors, particularly neonatal white matter injury, can adversely affect neurodevelopment in long-lasting ways. Abnormal MRI findings in premature infants are very strong predictors of unfavorable neurodevelopmental outcomes, including cognitive delays, motor delays, and cerebral palsy (Woodward et al., 2006). Congenital heart disease is the most common major birth defect, and can place infants at risk of hypoxia (Marelli et al., 2007). Hypoxic insult causes oligodendrocyte death and delayed oligodendrocyte differentiation, thus leading to abnormalities in developmental myelination (Back et al., 2001; Jablonska et al., 2012). Interestingly, hypoxic newborn brains may share some common molecular markers with adult multiple sclerosis (MS) brains. Indeed, the remyelination regulator Axin2 is present in white matter lesions that are found in both human newborn brains with hypoxic damage and active MS lesions in adults (Fancy et al., 2011).
Pathogens and the immune responses that they instigate can also have effects on white matter development. Perinatal inflammatory insult subsequent to maternal infection is associated with cerebral palsy as well as low scores on a number of development indexes (Stoll et al., 2004). For example, E. coli infection reduces the expression of oligodendrocyte-differentiation promoting transcripts and leads to myelin loss during a critical period of peak myelination (Favrais et al., 2011; Lieblein-Boff et al., 2013). Thus, the etiology of leukodystrophies are diverse and genetic causes are not the only consideration in white matter development and disease.
In this review, we provide an updated overview of 35 hypomyelinating leukodystrophies and home in on cellular mechanisms for disease. We find many common mechanisms affecting oligodendrocytes, the brain cells responsible for making myelin sheaths. These mechanisms often affect specialized oligodendrocyte functions, such as defects in myelin protein production, lipid processing, and peroxisome health. Additional oligodendrocyte-centric mechanisms include metabolic dysfunction, transcriptional and translational alterations, cytoskeletal dysregulation, and defects in cell junctions. Other glial cells, including astrocytes and microglia, may also be involved in certain leukodystrophies (Figure 1; Table 1). By identifying these emerging themes in cellular dysfunction, we hope to highlight conceptual insights that can contribute to the development of future therapeutic strategies for leukodystrophies.
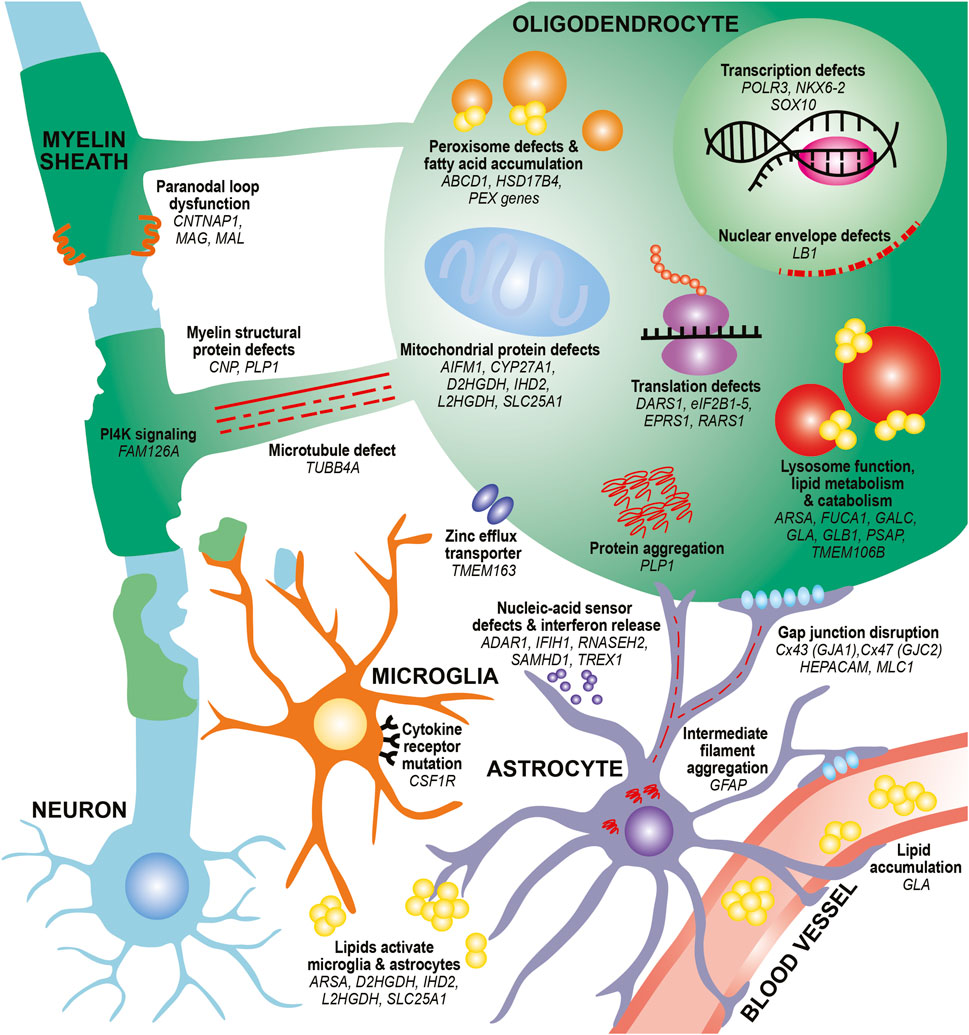
FIGURE 1. Diverse mechanisms and cell types contribute to hypomyelination in leukodystrophies. In oligodendrocytes, mutations in myelin structural proteins (CNP, PLP1) can lead to thinner myelin sheaths and paranodal loop malformation (CNTNAP1, MAG, MAL). PLP mutations can also lead to aberrant protein aggregation. Other mutations can affect the function of organelles that participate in lipid metabolism and catabolism, such as peroxisomes (ABCD1, HSD17B4, PEX family genes) and lysosomes (ARSA, FUCA1, GALC, GLA, GLB1, PSAP). These defects can also lead to toxic lipid accumulation that builds up in the bloodstream (GLA) or activate astrocytes and microglia (ARSA). Finally, mutations affecting transcription factors (NKX6-2, SOX10), transcriptional machinery (POLR3 genes) and translation machinery (DARS1, eIF2B genes, EPRS1, RARS1) cause impair differentiation of oligodendrocytes and reduce production of myelin proteins. Defects in microtubules that project toward and within the myelin sheath (TUBB4A) as well as in PI4K signaling pathways that contribute to wrapping (FAM126A) can affect myelin sheath growth. Mutations in mitochondrial genes can lead to lipid processing defects (CYP27A1), aberrant activation of apoptotic pathways (AIFM1), and metabolic issues (D2HGDH, IHD2, L2HGDH, SLC25A1). Other pathways, such as nuclear envelope integrity (LB1) and zinc efflux (TMEM163) can also affect oligodendrocyte health. In astrocytes, intermediate filament protein mutations (GFAP) can lead to its aggregation and formation of Rosenthal fibers. Mutations affecting gap junctions can disrupt cell junctions between astrocytes and oligodendrocytes (GJA1, GJC2) and between astrocytes and vasculature (HEPACAM/GLIALCAM, MLC1). Mutations affecting nucleic-acid sensors (ADAR1, IFIH1, RNASEH2 genes, SAMHD1, TREX1) can activate an immune response and release of interferons from astrocytes. In microglia, mutations in cytokine receptors (CSF1R) can affect microglial proliferation, differentiation, and activation. Finally, mutations affecting lipid processing (ARSA, D2HGDH, IHD2, L2HGDH, SLC25A1) can lead to aberrant lipid accumulation, which can activate microglia and astrocytes.
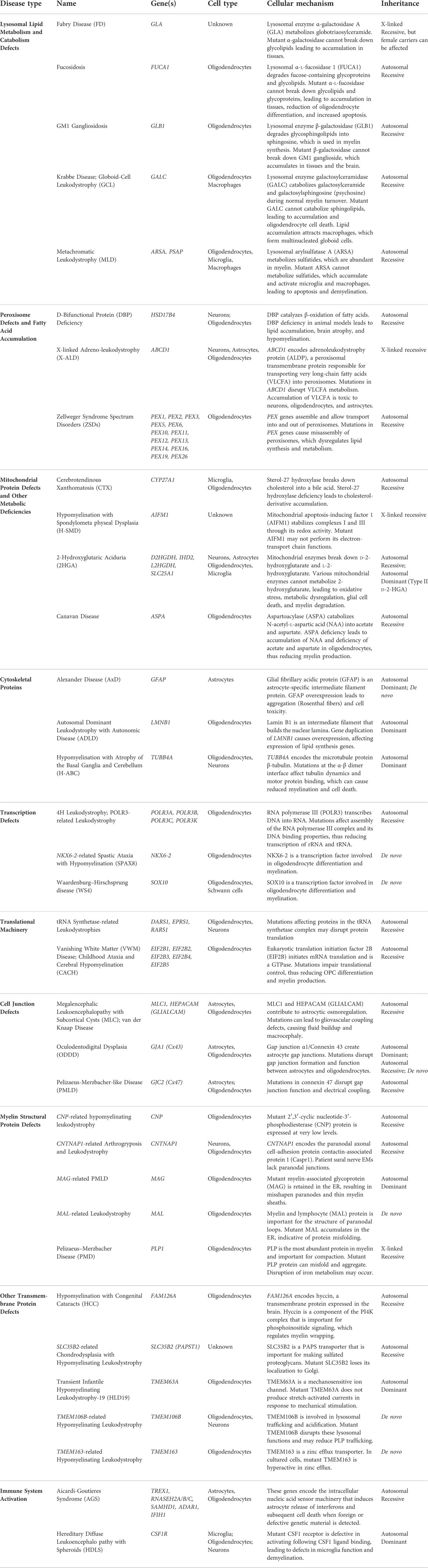
TABLE 1. Leukodystrophy-causing mutations and their cellular mechanisms.
Lysosomal lipid metabolism and catabolism defects
Oligodendrocytes are extraordinary producers of membranes and each oligodendrocyte is capable of forming multiple myelin sheaths. For example, oligodendrocytes from the cortex region of the brain can make as many as 50 myelin sheaths (Hughes et al., 2013). Each oligodendrocyte can make an estimated 2 mm2 of membrane surface area, which is 200 times higher than a typical epithelial cell (Poitelon, Kopec, and Belin 2020). Thus, oligodendrocytes are one of the most lipid-rich cell types in the body and, therefore, perhaps more vulnerable to mutations affecting lipid production.
Many mutations associated with leukodystrophies affect pathways involved in the production or breakdown of myelin lipids. The three major classes of lipids found in the myelin sheath include cholesterol, phospholipids (e.g., plasmalogen, phosphatidylcholine), glycolipids (e.g., galactosylceramide and its sulfated form, sulfatide), and sphingomyelin. Early studies found CNS myelin to contain molar percentages of 40–46% cholesterol, 26% phospholipid, 19–20% glycolipid, and 4–6% sphingomyelin (O’Brien 1965; Norton and Poduslo 1973). Leukodystrophy mutations affect lysosomal enzymes that participate in both lipid metabolism and catabolism. Defects in lipid metabolism can lead to aberrant lipid accumulation and dysfunctional myelin growth. Myelin is dynamic and, in addition to developmental myelin growth, myelin turnover replaces old membranes. This involves the modification of myelin and lipid breakdown as well as the formation of organelles called myelinoid bodies within the myelin sheath (Meschkat et al., 2022). Though it is still unclear whether lysosomal function is necessary for myelin clearance, myelin turnover may explain why mutations in lysosomal enzymes affecting lipid catabolism can lead to disease. Thus, understanding the nuanced relationship between lysosomal lipid metabolism and catabolism could be crucial to understanding the following leukodystrophies.
Fabry disease (FD)
Fabry disease is a lysosomal storage disorder characterized by deposits of glycosphingolipids in blood vessels and smooth muscle tissue (Sweeley and Klionsky 1963; Aerts et al., 2008). Patients present with clusters of dark red spots on the skin (angiokeratomas), cloudy corneas, episodes of pain localized to hands and feet (acroparesthesias), gastrointestinal symptoms, and hearing loss. Symptoms eventually progress to affect the heart and kidneys, with patient death usually attributed to renal failure (Schiffmann et al., 2009). The most severe cases of FD begin between 3 and 10 years of age (Hopkin et al., 2008), but large-scale metabolic screening of newborns revealed that undiagnosed later-onset FD is about 11 times more common (Spada et al., 2006).
FD is caused by mutations in the GLA gene encoding α-galactosidase A. This enzyme is involved in the catabolism of multiple glycoproteins, glycolipids, and polysaccharides (Brady et al., 1967). The majority of the nearly 600 mutations associated with FD are thought to cause a complete loss of function (Gal 2010). GLA is located on the X chromosome, and thus FD-causing mutations are inherited in an X-linked pattern. Interestingly, women heterozygous for FD mutations are not unaffected but in fact also experience significant disease burden, including renal and cardiovascular involvement (Wang et al., 2007; Waldek et al., 2009). Indeed, women carriers experience lifespan reduction of ∼10–15 years while men with FD experience lifespan reduction of ∼20 years (MacDermot et al., 2001; 2001).
Hypomyelination is one facet of progressive FD. Both affected men and women over the age of 35 display white-matter lesions (WML) in the central nervous system (CNS) (Fellgiebel et al., 2005), but the mechanism for this is unclear. Changes in cerebral blood flow and vascular changes due to glycolipid deposits have been suggested to contribute to glial cell death and demyelination. However it is unclear if cerebral blood flow increases or decreases in FD. One MRI study found that FD patients have increased cerebral blood flow and reduced glucose metabolism (Moore et al., 2003; Vagli et al., 2020). However, both a Gla knockout mouse model of FD and human imaging studies found reduced cerebral blood flow (Itoh et al., 2001; Hilz et al., 2004). Thus, the mechanism for hypomyelination in FD remains a mystery.
There are two standard treatment courses for FD: enzyme replacement therapy and oral chaperone therapy. First, enzyme replacement consists of infusions of recombinant human GLA enzyme (Eng et al., 2001), which reduces sphingolipid levels in the urine, plasma, and tissues (Rombach et al., 2013) and has been shown to stabilize WML progression. However, it is time-consuming and does not cure FD (Fellgiebel et al., 2005). Second, oral chaperone therapy introduces a small molecule ligand that aims to stabilize mutant GLA and restore its enzymatic activity. In cultured patient fibroblasts, this ligand increased baseline GLA levels and reduced accumulated sphingolipids (Yam, Zuber, and Roth 2005; Benjamin et al., 2009). Oral chaperone therapy presents some benefits over enzyme replacement, because patients display improvements in cardiac and digestive symptoms and do not develop antibodies to the ligand (Wilcox et al., 2012; Lenders et al., 2016). However, OCT is only effective in patients with certain mutations, thus limiting its use (Germain et al., 2016; Hughes et al., 2017).
Very recently, five men with FD received stem cell therapy through transfusions of hematopoietic stem cells (HSCs) transduced with human GLA cDNA. All patients who received this treatment reached healthy reference levels of circulating GLA enzyme and remained above baseline levels indefinitely (Khan et al., 2021). Thus, stem cell therapy is a promising therapeutic for FD patients, but additional clinical studies are required before this option can become widely available to patients.
Fucosidosis
Fucosidosis is a progressive lysosomal storage disorder with autosomal recessive inheritance pattern. Patients with fucosidosis present with aberrant facial features (enlarged head, lips, and tongue, small or malformed teeth, and flattened nose), impaired physical growth and intellectual ability, recurrent respiratory infections, progressive skeletal dysplasia, and neurodegeneration (Stepien, Ciara, and Jezela-Stanek 2020). The mean age of onset is ∼1 year old (Willems et al., 1991). Fucosidosis presents with myelin loss in the cerebellum and cerebrum that is visible by MRI (Steenweg et al., 2010; Jain et al., 2012). Some patients display rapidly progressing neurodegeneration resulting in death before the age of 10 (Loeb et al., 1969) while others have a life expectancy into young adulthood (Kousseff et al., 1976).
Fucosidosis is linked to mutations in FUCA1 (α-l-fucosidase 1), that lead to enzymatic deficiency (Willems et al., 1988; 1991). This results in the inability to metabolize certain sugar compounds (e.g., fucose-containing glycolipids and fucose-containing glycoproteins), which then accumulate (Van Hoof and Hers 1968). A canine model of fucosidosis displayed reduced expression of key oligodendrocyte differentiation genes, including CNP, PLP (proteolipid protein), MAG, MAL, MRF, OPALIN) as well as oligodendrocyte death that was confirmed with apoptosis markers. Because these defects were observed during developmental stages, they support a mechanism of hypomyelination rather than demyelination (Fletcher et al., 2011; 2014). In addition, a Fuca1 knockout mouse exhibited lysosomal dysregulation, lipid accumulation, neuroinflammation, and behavioral deficits (e.g., impaired coordination and spatial learning) that parallel the motor and intellectual deficits observed in fucosidosis patients (Stroobants et al., 2018). Thus, both canine and murine disease models indicate that many downstream effects of FUCA1 deficiency contribute to myelin defects in fucosidosis.
Most potential fucosidosis treatment strategies aim to ameliorate symptoms. Canine studies have shown success with enzyme replacement, which reduces vacuolation, neuron death, and neuroinflammation. However, these therapies only reduce symptoms and do not stop neurological progression (Kondagari et al., 2011; 2015). Bone marrow transplantation has shown success in both canine models and one human patient, but the risks and side effects of this procedure may outweigh the benefits (Taylor, Farrow, and Stewart 1992; Vellodi et al., 1995; Miano et al., 2001). Thus, additional preclinical studies are needed in order for any of these approaches to move forward as viable therapies for fucosidosis.
GM1 gangliosidosis
GM1 gangliosidosis is an autosomal recessive lysosomal storage disorder ranging from infantile-onset to chronic presentation. Patients experience intrauterine growth restriction, abnormal fluid accumulation in serous cavities, and placental vacuolization (Regier et al., 1993) coupled with the rapid progression of CNS dysfunction including blindness, deafness, seizures, and feeding difficulties. Pathologically, multiple brain regions including caudate, putamen, corpus callosum, basal ganglia, and cerebellar white matter are atrophied (Nestrasil et al., 2018). MRI scans and postmortem brain analyses revealed hypomyelination and atrophy of the caudate, putamen, corpus callosum, and basal ganglia (King et al., 2020; Uchino et al., 2020).
GM1 gangliosidosis is a result of homozygous or compound heterozygous (biallelic) mutations in the GLB1 gene (Morrone et al., 2000; Bidchol et al., 2015). GLB1 encodes for the ubiquitous lysosomal enzyme β-galactosidase (β-GAL), which is responsible for the degradation of GM1 ganglioside, a type of glycosphingolipid composed of a ceramide lipid tail linked to a glycan headgroup containing a sialic acid residue (Regier et al., 1993; Sipione et al., 2020). The breakdown of GM1 gangliosides creates the downstream catabolite sphingosine, which is used in sphingomyelin synthesis and ultimately myelin formation. Mutations in GLB1 cause reduced or null β-GAL enzymatic activity, which leads to toxic and progressive accumulation of GM1 gangliosides and other glycoproteins (Kajihara et al., 2020).
Several studies have revealed cellular pathways involved in GM1 gangliosidosis pathogenesis, such as an activated unfolded protein response (Tessitore et al., 2004), calcium signaling in the endoplasmic reticulum (ER) (Sano et al., 2009), autophagy (Takamura et al., 2008), and inflammasome activation (Son et al., 2015). Additional studies could elucidate molecular and cellular mechanisms of hypomyelination in GM1 gangliosidosis.
Recent studies have focused on two potential therapeutic approaches. First, multiple studies have used viruses, such as AAV9 (adeno-associated virus), a virus with broad CNS tropism, to deliver a functional copy of the GLB1 gene. In one study, Glb1 knockout mice received intravenous injections of AAV9 containing the GLB1 gene, which increased β-GAL activity in various regions of the brain and in peripheral tissues. Interesting sex-specific effects were observed, including more viral copies in females and a significant difference in median survival from ∼100 to ∼577 days (in females) and ∼398 days (in males) (Weismann et al., 2015). In a more recent study, researchers used human iPSCs to create GLB1 knockout cerebral organoids that recapitulated GM1 gangliosidosis by exhibiting progressive accumulation of gangliosides at 10 and 20 weeks in culture. Delivering AAV9 containing the GLB1 gene significantly increased β-GAL activity and reduced GM1 accumulation in these organoids (Latour et al., 2019). Thus, enzyme replacement via stem cell therapy is a viable strategy for the treatment of GM1 gangliosidosis.
Researchers have also taken a drug screening approach. iPSCs from GM1 gangliosidosis patients were differentiated into neural stem cells, which model disease biochemistry by exhibiting accumulated GM1 gangliosides. High-content screening identified 25 small molecule compounds that reduced the accumulation of GM1 gangliosides. Of these, amodiaquine (a heme polymerase inhibitor) and thiethylperazine (an adrenergic antagonist) were the most effective. When administered in vivo to β-GAL null mice, these two compounds decreased GM1 ganglioside accumulation in the brain (Kajihara et al., 2020). Thus, researchers are exploring both stem cell-based enzymatic replacement and drug therapies as therapeutic strategies to treat GM1 gangliosidosis.
Krabbe disease
Krabbe disease, also known as globoid-cell leukodystrophy (GCL), is a rapidly progressive autosomal recessive neurodegenerative disorder. Patients have rapid demyelination resulting in near total loss of myelin, severe astrogliosis, and the presence of multinucleated globoid cells in the white matter (Suzuki and Suzuki 1970). Hypomyelination in the CNS leads to muscle spasticity, paralysis, irritability, loss of vision and hearing, seizures, peripheral neuropathy, and premature death (Hagberg, Sourander, and Svennerholm 1963; Morse and Rosman 2006; Sakai 2009; Brodsky and Hunter 2011; Beltran-Quintero et al., 2019). The median survival is 1.5 and 9.5 years for early- and late-infantile Krabbe disease, respectively, whereas juvenile- and adult-onset subtypes have projected survival beyond 30 years of age (Komatsuzaki et al., 2019).
Krabbe disease patients have mutations in the GALC (galactosylceramidase) gene that result in loss of function of this lysosomal enzyme (Wenger and Luzi 2020). GALC removes the sugar galactose from galactosylceramide, galactosylsphingosine (psychosine), and other galactolipids (Suzuki and Suzuki 1970; Beltran-Quintero et al., 2019; Komatsuzaki et al., 2019). The inability to catabolize galactosylceramide and galactosylsphingosine leads to their accumulation. On the one hand, galactosylceramide accumulation is postulated to attract macrophages, which phagocytose it, then form multinucleated globoid cells (Komatsuzaki et al., 2019). These globoid cells are associated with severe demyelination, axonopathy, and neuronal death (Wenger and Luzi 2020). On the other hand, psychosine accumulation is cytotoxic to oligodendrocytes (Komatsuzaki et al., 2019) and therefore may be the pathological driver of Krabbe disease symptoms. Indeed, in a human oligodendroglioma cell line, treatment with psychosine caused the mislocalization of fatty acid binding protein 5 (FABP5) to the mitochondria, the formation of mitochondrial pores, and reduced levels of mitochondrial DNA. The authors postulated that this could lead to oxidative stress and apoptosis (Cheng, Kawahata, and Fukunaga 2020). Thus, loss of GALC activity results in aberrant lipid accumulation and various downstream effects.
Surprisingly, of the >70 disease-causing mutations in GALC, most are not located in the enzymatic domain. For example, in the D528N mutation, the aberrant asparagine residue leads to hyperglycosylation and misfolding of GALC (Lee et al., 2010). This glycosylation defect is reminiscent of the D32N MPZ (myelin protein zero) mutation in Charcot-Marie-Tooth neuropathy. In the peripheral nervous system (PNS), this mutation leads to hyperglycosylation of MPZ and defects in MPZ trafficking to the plasma membrane (Prada et al., 2012). This is consistent with experiments in cell lines in which mutating the MPZ glycosylation site can lead to a membrane adhesion defect (Filbin and Tennekoon 1993), which is hypothesized to impact myelin compaction. Thus, Krabbe disease mutations can affect both enzymatic activity as well as post-translational modifications (PTMs).
Currently, multiple potential therapeutic strategies for Krabbe disease exist. HSC transplant has been performed and is most successful when it precedes symptom onset. Indeed, to facilitate early diagnosis and treatment, several states (e.g., New York, Ohio, Pennsylvania, Kentucky, Indiana, etc.) require all newborns to be screened within 48 h of birth (Orsini et al., 2016). Unfortunately, long-term outcomes of HSC transplant (regardless of whether the treatment was initiated prior to symptom onset) are poor and high-risk with elevated morbidity rates (Krivit et al., 1998; Escolar et al., 2005; Aldenhoven and Kurtzberg 2015; Wasserstein et al., 2016; Wright et al., 2017).
Ongoing research in mice has focused on another approach: viral delivery of the Galc gene. The twitcher mouse, which has a Galc mutation, is often used to model Krabbe disease. By postnatal day 30, these mice exhibit severe myelin loss, astrocytic gliosis, and the presence of globoid cells (Kobayashi et al., 1980; Li and Sands 2014). Several studies have looked at viral vector delivery of Galc to presymptomatic twitcher mice. Two studies administered AAV9 encoding mouse Galc via intrathecal (Karumuthil-Melethil et al., 2016) and intracisternal injections (Bradbury et al., 2020). Both studies found reduced galactosylsphingosine levels, ameliorated myelin abnormalities, and ultimately prolonged survival of the twitcher mice. Another study used a virus with a different serotype (AAVrh10) and a different delivery method (intravenous administration on postnatal day 10). This virus crossed the blood–brain barrier, and prevented both myelin loss and galactosylsphingosine accumulation (Rafi, Luzi, and Wenger 2021). Thus, these promising results in rodents may open the door for future pre-clinical studies on viral delivery of the Galc gene.
Metachromatic leukodystrophy (MLD)
MLD is an autosomal recessive demyelinating disorder that can be divided into four subtypes according to age of onset: late infantile, early-juvenile, late-juvenile, and adult-onset (Von Figura, Gieselmann, and Jaeken 2001; Sevin, Aubourg, and Cartier 2007). The most common form of MLD, which comprises ∼50% of all cases, is the late infantile subtype, which has symptom onset by ∼2 years of age (Cesani et al., 2016). Clinical presentations include muscle wasting, weakness and rigidity, developmental delays, progressive vision loss, seizures, paralysis, and dementia (Gieselmann and Krägeloh-Mann 2010). In the majority of cases, the prognosis is severe and often leads to a persistent vegetative state or death within a few years of symptom onset (Von Figura, Gieselmann, and Jaeken 2001; Gieselmann and Krägeloh-Mann 2010; Aubourg et al., 2011).
Most MLD subtypes can be traced back to >150 mutations in the gene encoding the enzyme arylsulfatase A enzyme (ARSA or ASA) (Von Figura, Gieselmann, and Jaeken 2001; Aubourg et al., 2011; Batzios and Zafeiriou 2012). In rare instances, MLD can also be caused by a mutation in the PSAP gene encoding presaposin, the precursor to saposin B (SAP-B), an activator of ARSA (Holtschmidt et al., 1991). Because ARSA is a lysosomal enzyme, MLD is often referred to as a lysosomal storage disorder. Mutations in ARSA lead to an enzymatic deficiency, ultimately resulting in the impaired metabolism of sulfatide, a major lipid component of the myelin sheath. Sulfatide can accumulate in microglia, neurons, and oligodendrocytes along with peripheral tissues (Gieselmann 2003; Molander-Melin et al., 2004; Patil and Maegawa 2013). Excess sulfatide triggers an inflammatory response which includes microglial activation, astrogliosis, and recruitment of peripheral macrophages. Together, these events are thought to cause apoptosis of glia and neurons as well as demyelination in the CNS (Sevin, Aubourg, and Cartier 2007; Patil and Maegawa 2013). Loss of function in MLD is also supported by a global Arsa knockout mouse that displays many clinical hallmarks of MLD, such as sulfatide accumulation, motor dysfunction, and cognitive impairment. However, these mice lacked shortened lifespans and white matter defects (Hess et al., 1996). Thus, additional research should clarify the mechanistic link between sulfatide accumulation and demyelination.
One therapeutic approach for MLD is enzyme replacement. Recent Phase 1 and 2 clinical trials evaluated the safety and efficacy of intravenous delivery of recombinant human ARSA. 13 children with MLD received ARSA at 50, 100 or 200 U/kg every 2 weeks for 1 year. The results revealed that cerebrospinal fluid (CSF) sulfatide levels significantly decreased in the 100 and 200 U/kg groups, but perplexingly increased in the 50 U/kg group. Nevertheless, despite improvements in sulfatide levels, the other efficacy variables, including MRI, and motor and cognitive function, showed either no change or worsening measurements (Í Dali et al., 2021). It is unclear whether intravenously administered ARSA can cross the blood–brain barrier and this could explain the lack of efficacy in the CNS. This is further confounded by a study indicating that different cell types have varying levels of ARSA uptake. In primary cultures of neurons, microglia, astrocytes, and oligodendrocytes treated with ARSA for 24 h, microglia had the highest uptake, whereas oligodendrocytes had less uptake (Kaminski et al., 2021). Thus, peripheral administration of recombinant ARSA has yet to yield promising results.
Another therapeutic approach for MLD is HSC transplant with the goal for donor cells to secrete functional ARSA (Rosenberg et al., 2016). Early work demonstrated that transplanted bone marrow cells expressing green fluorescent protein (GFP) from transgenic mice could be detected in the brains of wildtype recipients (Brazelton et al., 2000; Mezey et al., 2000) and these engrafted cells expressed microglia markers (Priller et al., 2001). In a clinical trial with three MLD patients, patient-derived HSCs were transduced with a lentivirus expressing a functional copy of ARSA, then reinfusion into patients. This led to functional ARSA expression and halting of disease progression (Biffi et al., 2013). Additional HSC transplant studies also report delays in disease progression (van Egmond et al., 2013; Groeschel et al., 2016; van Rappard et al., 2016). Thus, HSC transplantation is a promising approach for MLD treatment.
Peroxisome defects and fatty acid accumulation
Peroxisomes are organelles that play multiple important roles in lipid metabolism and the building of the myelin sheath. First, peroxisomes perform β-oxidation of fatty acids, such as very-long-chain fatty acids (VLCFAs), pristanic acid, long-chain dicarboxylic acids, and some eicosanoids and polyunsaturated fatty acids (PUFAs). Though mitochondria also perform β-oxidation of fatty acids, each organelle displays substrate exclusivity. Second, peroxisomes synthesize plasmalogens, a type of ether-phospholipid found in myelin (Kassmann 2014). Thus, it is not surprising that multiple leukodystrophies can be attributed to mutations in genes encoding peroxisomal proteins.
D-Bifunctional Protein (DBP) deficiency
DBP deficiency is an autosomal recessive disorder caused by mutations to the HSD17B4 (hydroxysteroid 17-β dehydrogenase 4) gene that encodes DBP. DBP enzyme catalyzes steps in the peroxisomal β-oxidation of fatty acids. Subtypes of this leukodystrophy are classified according to which DBP domains are affected. Type I, the most severe, is caused by mutations in both the hydratase and dehydrogenase domains; Type II is caused by mutations in the hydratase domain; Type III caused by mutations in the dehydrogenase domain (Ferdinandusse et al., 2006b). The patient phenotype includes abnormal facial features, reduced muscle tone, seizures within the first month of life, intellectual and sensory disability, increased plasma VLCFAs, as well as cerebellar atrophy, progressive white matter dystrophy, and demyelination visualized on MRI (van Grunsven et al., 1999; Ferdinandusse et al., 2006a; Yamamoto et al., 2021).
Several DBP deficient animal models have been generated. Zebrafish embryos lacking DBP display lipid accumulation, general growth delays, aberrant axon development, and reduced myelination (Kim et al., 2014). Interestingly, a neuronal-specific knockout mouse developed motor symptoms and cerebellar atrophy, but an oligodendrocyte-specific knockout mouse had no clinical features (Verheijden et al., 2013). These results hint at intricate neuron-glia interactions and highlight the need to untangle the precise mechanisms by which DBP deficiency causes a range of symptoms.
X-linked adrenoleukodystrophy (X-ALD)
X-ALD encompasses four distinct presentations: a severe childhood type (ALD), the milder adrenomyeloneuropathy (AMN), adrenal insufficiency (Addison’s Disease) and an asymptomatic form. The childhood form (ALD) typically begins between ages 4–10 with neurological symptoms that include worsening vision, difficulty swallowing, poor coordination, and seizures. Patient death from ALD typically occurs within a few years. Adult-onset AMN is the most common X-ALD and presents with weakness in the legs (paraparesis), changes in cognitive ability, and urogenital tract disorders. Adrenal insufficiency rarely presents with neurological symptoms (Berger, Forss-Petter, and Eichler 2014; Turk et al., 2020).
X-ALD is caused by mutations on the X chromosome in the ABCD1 gene that encodes adrenoleukodystrophy protein (ALDP). ALDP is a peroxisomal transmembrane protein involved in the transport of VLCFAs into peroxisomes for their subsequent β-oxidation (Wiesinger et al., 2013). ALDP deficiency causes increased plasma VLCFA levels and VLCFA accumulation in the adrenal cortex, testes, and brain regions preceding demyelination (Theda et al., 1992). However, it is unclear whether increasing levels of VLCFA contribute to disease severity. On the one hand, VLCFAs are toxic to cultured rat neurons, oligodendrocytes, and astrocytes, perhaps by altering inner mitochondrial membrane permeability (Hein et al., 2008). On the other hand, plasma VLCFA levels in ALD and AMN are similar, even though the AMN phenotype is far less severe (Moser et al., 1999; Stradomska and Tylki-Szymańska 2009; Stradomska et al., 2020). Moreover, mouse models expressing truncated deficient ALDP exhibit increased VLCFA levels and impaired β-oxidation, but no demyelination in adulthood (Forss-Petter et al., 1997; Kobayashi et al., 1997; Lu et al., 1997). By contrast, Abcd1-null mice display late-onset locomotion defects, and axonal degeneration in the spinal cord and sciatic nerve beginning ∼15 months of age (Pujol et al., 2002); therefore, this mouse more appropriately models AMN than ALD.
A number of approaches to treating ALD have been investigated. First, Lorenzo’s oil, a mixture of two long-chain fatty acids, can reduce plasma levels of VLCFA by competitively inhibiting their synthesis (Sassa et al., 2014). Second, bone marrow transplantation during the early stage of the disease led to reduced plasma VLCFA levels and stabilization or improvement of demyelinating lesions on MRI scans (Shapiro et al., 2000). Finally, patients transfused with HSCs that were transduced ex vivo with ABCD1 cDNA showed a halt in disease progression and even began to express functional ALDP (Eichler et al., 2017). Thus, basic understanding of peroxisome function and disease mechanism has contributed to promising steps towards the treatment of X-ALDs.
Zellweger syndrome spectrum disorders (ZSDs)
ZSDs are a set of neonatal-onset progressive leukodystrophies that include neonatal adrenoleukodystrophy (NALD) and Infantile Refsum disease. In newborns, symptoms include poor muscle tone, trouble with feeding, seizures, renal cysts, liver dysfunction, skeletal abnormalities, and hearing and vision loss. Children with ZSD have varying levels of symptom severity. Infants with severe ZSD rarely survive past the first year of life. Intermediate cases may show progressive hearing and vision loss as well as fatal myelin degeneration during childhood (Steinberg et al., 2003). Non-progressive cases often result in children surviving until school age with the possibility of normal intellect (Poll-The et al., 2004).
ZSDs are classified as peroxisome biogenesis disorders, because they are caused by autosomal recessive mutations in any of at least 12 peroxin (PEX) genes that are responsible for peroxisome assembly. The most commonly mutated genes are PEX1 and PEX6. The most severe cases usually result from loss-of-function mutations, such as large deletions and nonsense mutations (Steinberg et al., 2003). The prevailing hypothesis for the cause of ZSD posits that deficiencies in peroxisome assembly can cause dysregulation of fatty acid metabolism and subsequent accumulation of long-chain fatty acids, which may lead to membrane disruption and cellular toxicity. Indeed, when compared to unaffected brains, ZSD brains have higher levels of certain species of lipids (ceramide monohexosides, cholesterol ester, dipalmitoyl phosphatidylcholine and dipalmitoyl phosphatidylserine) (Steinberg et al., 2006; Saitoh et al., 2007). Interestingly, mouse studies indicate that cholesterol homeostasis and peroxisome assembly may be intricately connected. Lrp1 knockout mice with dysregulation of cholesterol homeostasis display aberrant peroxisome assembly and impaired oligodendrocyte maturation (Lin et al., 2017).
In mouse models, cell-specific knockout of Pex5 indicates that many cell types contribute to ZSD pathology. Knockout of Pex5 in both neurons and glia results in demyelination, axonal degeneration, lack of muscle control (ataxia), tremors, and premature death ∼6 months of age (Hulshagen et al., 2008). Knockout of Pex5 only in oligodendrocytes recapitulates these phenotypes, but mice die later, at ∼12 months of age (Kassmann et al., 2007). However, knockout of Pex5 in either neurons or astrocytes results in high VLCFA and reduced plasmalogens levels, but no behavioral phenotypes and unperturbed axonal integrity (Bottelbergs et al., 2010). Together, these studies show that while peroxisome dysfunction in oligodendrocytes can lead to ZSD phenotypes, contributions by peroxisomes in neurons and astrocytes likely play a role in ZSD pathology as well.
Currently, no cure exists for ZSD and treatments focus on managing symptoms (e.g., gastrostomy to provide nutrition, hearing aids for hearing loss). Unfortunately, dietary supplementation of lipids has not been able to affect disease course. An early study found that ZSD patients had very low levels of docosahexaenoic acid (DHA) in the brain, retina and other tissues (Martinez 2001). However, in a clinical trial, DHA supplementation for 1 year did not improve the visual function of individuals with peroxisome assembly disorders (Paker et al., 2010). Thus, additional therapeutic approaches should be considered for ZSD.
Mitochondrial protein defects and other metabolic deficiencies
Though mitochondria are commonly known as the “powerhouse of the cell” due to their role in the efficient production of adenosine triphosphate (ATP), they also have additional functions, such as regulation of cell death and metabolism of cholesterol. Several leukodystrophies can be attributed to mutations in genes encoding mitochondrial-specific proteins with diverse functions as well as other genes involved in cellular metabolism.
Cerebrotendinous xanthomatosis (CTX)
CTX is an autosomal recessive disorder caused by mutations in the gene encoding the mitochondrial enzyme sterol 27-hydroxylase (CYP27A1). These mutations result in defective versions of this enzyme with low or undetectable enzyme activity (Cali et al., 1991; Carson and De Jesus 2021). CTX is a lipid storage disease characterized by disruptions in bile acid synthesis. Normally, cholesterol is metabolized into a bile acid called chenodeoxycholic acid (CDCA) by CYP27A1. As a result of reduced feedback (i.e., lack of CDCA), cholesterol synthesis is upregulated, resulting in aberrant accumulation of the byproduct cholestanol in various tissues and organs (Serizawa et al., 1982; Carson and De Jesus 2021).
Sites of localized cholestanol accumulation, such as the brain, eyes, arteries, and tendons, reflect the clinical manifestation of CTX. Around puberty, CTX patients begin to show neurological signs and symptoms, such as cerebellar ataxia, pseudobulbar affect, and brain atrophy (Carson and De Jesus 2021). Furthermore, MRI scans revealed white matter atrophy in the dentate nuclei (Vanrietvelde et al., 2000; Chang et al., 2010). Other clinical features of CTX include cataracts, atherosclerosis, and fatty tumors on tendons and muscles. As the disease progresses, patients can develop paralysis and dementia. Eventually cholestanol deposits affect the brainstem and patient death occurs during mid-to-late adulthood from cardiac arrest (Carson and De Jesus 2021).
Many studies support the mechanistic link between CYP27A1 deficiency and cholestanol accumulation in the brain. An early study of postmortem CTX brains revealed that cholestanol preferentially accumulates in myelin fractions while healthy brain fractions had no detectable cholestanol (Stahl, Sumi, and Swanson 1971). Another study found that feeding mice 1% cholestanol led to significant accumulation of cholestanol in the cerebellum, suggesting that peripheral cholestanol can cross the blood–brain barrier (Byun et al., 1988). Cyp27a1 knockout mice, which have increased cholesterol synthesis and a two-fold increase in cholestanol levels in peripheral tissues, display a striking 12-fold increase in cholestanol levels in the cerebellum, indicating that cholestanol preferentially accumulates in the brain (Båvner et al., 2010). Indeed, a recent RNA-seq database from mouse brains indicates that Cyp27a1 mRNA is more highly enriched in oligodendrocytes than other brain cell types (e.g., neurons, astrocytes, microglia/macrophages) (Zhang et al., 2014). Thus, these studies suggest that oligodendrocyte-specific CYP27A1 activity and tissue-specific cholestanol accumulation in the brain play important roles in CTX.
CTX is currently treated through dietary supplementation therapy. CDCA supplementation lowers the accumulation of cholestanol in both plasma and CSF, likely by exerting negative feedback on cholesterol synthesis, and has led to improvements in many clinical symptoms, including cataracts, dementia, neuropathy, and other neurological symptoms (Salen, Meriwether, and Nicolau 1975; Berginer, Salen, and Shefer 1984; Duell et al., 2018). This treatment has shown success when implemented before the age of 25 (Yahalom et al., 2013). However, CDCA is hepatotoxic, prompting some studies to suggest cholic acid as an alternative treatment for patients who tolerate CDCA poorly (Pierre et al., 2008; Koyama et al., 2021). In addition, a recent study in a mouse model of CTX used an AAV to express CYP27A1 in the liver and this reestablished bile acid metabolism and restored normal plasma bile acid levels (Lumbreras et al., 2021). Thus, promising steps have been made in the management and treatment of this leukodystrophy, though more translational and clinical studies are needed.
Hypomyelination with spondylometaphyseal dysplasia (H-SMD)
H-SMD is a disorder characterized by abnormal growth and development of the vertebrae and bone growth plates (metaphyses), as well as significant lack of myelin on MRI. Beginning between 1 and 2 years of age, patients may start to lose the ability to walk, which may slowly progress into motor deterioration, spasticity and tremors. Other symptoms include mild cognitive impairment, bone and joint issues such as scoliosis, and visual symptoms such as involuntary eye movement (nystagmus) (Kettwig et al., 2015; Miyake et al., 2017).
Multiple studies have found mutations in the AIFM1 (apoptosis-inducing factor, mitochondria-associated 1) gene in H-SMD. AIFM1 has dual functions: 1) to regulate the assembly of the mitochondrial machinery that participate in oxidative phosphorylation (Vahsen et al., 2004), and 2) to act as a signal for cell death when it translocates to the nucleus (Susin et al., 1999). An early study discovered mutations in affected sons born to asymptomatic mothers, indicative of X-linked recessive inheritance (Bieganski, Dawydzik, and Kozlowski 1999). Most H-SMD mutations are missense mutations localized to a 70-base-pair region in exon seven of AIFM1. However, some cases involve intronic mutations and synonymous variant mutation (a codon substitution that doesn’t change the encoded amino acid) (Mierzewska et al., 2017; Edgerley et al., 2021). Conflicting evidence exists on whether AIFM1 mutations in H-SMD cause loss of function. In one study, patient osteoblast cells displayed significantly reduced Aifm1 mRNA and protein levels, which the authors suggest could be due to bioinformatically predicted mRNA splicing disruptions (Miyake et al., 2017). However, comparisons of four mutant AIFM1 proteins showed that the extent of structural alterations and changes in redox activity vary by mutation (Sevrioukova 2016), which indicates that only some AIFM1 mutations lead to loss of function. Hence, the molecular mechanisms underlying hypomyelination in H-SMD are still unclear.
2-Hydroxyglutaric aciduria (2HGA)
2HGA is an autosomal recessive neurometabolic disorder characterized by the accumulation of 2-hydroxyglutarate (2-HG). 2-HG is a side product of nonspecific Krebs Cycle enzyme activity and is closely related to the Krebs Cycle intermediate ɑ-ketoglutarate (Rzem et al., 2007). There are three forms of 2HGA; each form is associated with mutations in specific mitochondrial genes and named according to which 2-HG enantiomer (L or D) is affected.
First, L-2-HGA (LHGA) primarily affects the cerebellum, presenting with ataxia, seizures, and an enlarged head (macrocephaly). LHGA is associated with mutations in the L2HGDH gene, which codes for a mitochondrial enzyme that converts L-2-HG into α-ketoglutarate. A deficiency in this enzyme causes increased levels of L-2-HG in urine, plasma and CSF (Barth et al., 1993; Rzem et al., 2004).
Second, D-2-HGA (DHGA) falls into two categories. Type I is less severe with later onset and associated with mutations in the D2HGDH gene. Type II is more severe with earlier onset and can present with heart enlargement (cardiomyopathy). Unlike other 2HGA’s, Type II D-2-HGA is uniquely inherited in an autosomal dominant pattern and associated with mutations in the IHD2 gene (Struys et al., 2005; Kranendijk et al., 2010).
Finally, D,L-2-HGA is caused by mutations in the SLC25A1 gene, which encodes a protein that transports citrate across the inner mitochondrial membrane. In cultured patient cells, lack of SLC25A1 activity results in the increased presence of both D-2-HG and L-2-HG (Nota et al., 2013).
The mechanism for 2-HG accumulation leading to symptoms is likely due to two types of toxicity–oxidative damage and excitotoxicity. In primary neuron cultures, both L-2-HG and D-2-HG induce a number of metabolic effects, such as inhibiting mitochondrial creatine kinase and ATP synthase (Kölker et al., 2002; da Silva et al., 2003). In rats, intracerebroventricular administration of D-2-HG resulted in increased oxidative stress and upregulation of markers for reactive astrocytes and microglia (Ribeiro et al., 2021). Indeed, in L2HGDH knockout mice, vacuolar lesions appeared in oligodendrocyte and astrocyte cytoplasm as well as in myelin tracts in the cortex and corpus callosum (Rzem et al., 2015). In addition, 2-HG shares chemical structure similarity with the excitatory transmitter glutamate. D-2-HG treatment to cultured neurons led to excitotoxic cell damage (Kölker et al., 2002). In rats, a NMDA glutamate receptor antagonist administration prevented the formation of some oxidative species (Ribeiro et al., 2021). Thus, signs of both excitotoxicity and oxidative damage are observed in cell and animal models of 2HGA and these two types of damage might synergistically exacerbate symptoms in 2HGA.
Treatment of 2HGA primarily focuses on managing symptoms, especially seizures when they are present. One case study successfully managed patient tremors and reduced urinary 2-HG by supplementing with FAD and carnitine (Samuraki et al., 2008). Another found similar effects with riboflavin (Vitamin B2) supplementation (Yilmaz 2009). However, the mechanism of action for these supplements is unclear. Thus, further research is necessary to uncover effective treatments for 2HGA.
Canavan disease
Canavan disease is a progressive, autosomal recessive leukodystrophy. It typically affects infants ∼3 months of age, who begin to develop symptoms that include lethargy, poor vision, little-to-no motor development, seizures, and other progressive neurological defects. Symptoms continue to worsen until the child dies ∼10 years of age (Bokhari et al., 2022).
Canavan disease results from a mutation in the gene encoding aspartoacylase (ASPA) (Bokhari et al., 2022). ASPA is an oligodendrocyte-enriched enzyme that hydrolyzes N-acetyl L-aspartic acid (NAA), which is primarily synthesized in neurons, into acetate and aspartate. Canavan disease patients are unable to catabolize NAA, which progressively accumulates with age in oligodendrocytes. Studies in rodent oligodendrocytes indicate that the products of NAA hydrolysis are important for oligodendrocyte survival and differentiation (Kumar et al., 2009). Indeed, acetate, one of the products of ASPA, is converted to acetyl-CoA, which is an important building block in lipid synthesis. The other product of ASPA, aspartate, can enter cell metabolic pathways (I Amaral et al., 2017), promote oligodendrocyte precursor cell (OPC) differentiation and transcription of myelin basic protein (MBP) (Chakraborty et al., 2001), and may stimulate myelination and post-injury remyelination through glutamate receptor signaling (de Rosa et al., 2019). In a mouse model for Canavan disease, impaired NAA catabolism results in reduced myelin formation and spongy white matter degeneration (Hoshino and Kubota 2014). Therefore, the function of ASPA in NAA catabolism is important for oligodendrocyte health.
Currently, no treatments exist for Canavan disease, making this a fatal disorder, but recent research has targeted dietary supplementation as well as viral gene delivery. Standard of care is largely palliative and includes gastrostomy tubes that deliver nutrients directly to the stomach, anti-seizure medications, and physical therapy to improve posture and to reduce pressure ulcers (Bokhari et al., 2022). Dietary supplementation with glyceryl triacetate has been proposed to address low acetate levels in Canavan disease. Though this was successful in a mouse model, it has yet to cause improvement in human patients (Segel et al., 2011). Recently, researchers engineered a modified AAV with a capsid that has preferential tropism toward oligodendrocytes to express ASPA. Intracerebroventricular injections of this virus in a mouse model of Canavan disease rescued ASPA activity and motor function (Francis et al., 2021). Thus, ASPA replacement therapy may be a feasible therapeutic approach for human patients.
Cytoskeletal proteins
Cytoskeletal proteins, including microtubules, actin, septin, and intermediate filaments, form the cellular structure of glia. In oligodendrocytes, microtubules mediate long-distance transport and are found in processes that contact axons, as well as inside myelin sheaths (Weigel et al., 2021). Actin dynamics are important for the wrapping of myelin sheaths around axons (Nawaz et al., 2009; Zuchero et al., 2015). Septins also play an important role in oligodendrocyte development (Patzig et al., 2016). In astrocytes, the intermediate filament GFAP (glial fibrillary acidic protein) can play important roles in injury response and can be affected in leukodystrophy.
Alexander disease (AxD)
AxD is a progressive demyelinating leukodystrophy. Children often present with symptoms (e.g., megalencephaly, limb stiffness or rigidity, seizures, and developmental delays) by 2 years of age and have an average life expectancy of 14–25 years (Prust et al., 2011). AxD is a rare disease with an estimated 5-years prevalence of one in 2.7 million in a study in Japan (Yoshida et al., 2011). AxD is caused by autosomal dominant mutations in the GFAP gene. To date, over 100 missense variants in coding regions are associated with AxD (Saito et al., 2018). However, most AxD mutations are not inherited, but, instead, are de novo mutations (Li et al., 2006).
Many lines of evidence indicate that protein aggregation is a profound component in the AxD pathology due to the striking accumulation of Rosenthal fibers consisting of GFAP. GFAP, an intermediate filament protein, is a major component of astrocytic processes and becomes upregulated in reactive astrocytes responding to injury (Wilhelmsson et al., 2004). Overexpression of human wildtype GFAP in mice recapitulated Rosenthal fiber pathology and was lethal (Messing et al., 1998), indicating that GFAP protein levels are important in AxD pathology. Recent research suggests that certain GFAP isoforms or GFAP containing PTMs may be more prone to aggregation. For example, minor isoforms of GFAP (e.g., delta and kappa) preferentially aggregate in Rosenthal fibers (Lin et al., 2021). In addition, severe AxD patients selectively contain GFAP phosphorylated at Ser13, which facilitates GFAP aggregation in patient-derived iPSCs (Battaglia et al., 2019). Thus, many considerations, such as mutations, isoform specificity, and PTMs, may affect GFAP aggregation in AxD.
Though most treatment options for AxD are based on symptom management, researchers are exploring new approaches, such as ASOs to decrease GFAP expression. This approach is supported by results showing that mice lacking GFAP have very mild defects and therefore decreasing GFAP protein levels is unlikely to produce a toxic loss of function phenotype. In experiments in a mutant GFAP (R236H) mouse line, ASOs targeted against the 3′UTR of Gfap were injected intracerebroventricularly. By 2 weeks after injection, this resulted in less Gfap mRNA, near elimination of GFAP protein, fewer Rosenthal fibers, and improved body condition scores (Hagemann et al., 2018). Thus, GFAP-targeting ASOs are a viable therapeutic approach for AxD and additional follow-up studies should be performed.
Autosomal dominant leukodystrophy (ADLD) with autonomic disease
ADLD is an adult-onset leukodystrophy characterized by spasticity, ataxia, and autonomic issues, such as dysregulation of blood pressure and body temperature, and loss of bladder/bowel control (Zerbin-Rudin and Peiffer 1964; Eldridge et al., 1984). Patients display white matter lesions before the onset of symptoms, typically in the fifth decade, which progress over the course of 10–20 years (Finnsson et al., 2015). Most cases are autosomal dominant (Schuster et al., 2011) and caused by duplication of the LMNB1 gene that encodes lamin B1 (Padiath et al., 2006; Brussino et al., 2009).
Lamins are intermediate filament proteins that form the mesh-like network of the nuclear lamina, which lines the inner nucleoplasmic side of the nuclear envelope. Mutations in lamin A can cause premature aging (progeria), muscular dystrophy, cardiomyopathy, and peripheral neuropathy (Shin and Worman 2022). Patient fibroblasts overexpress lamin B1, which leads to increased nuclear rigidity, misshapen nuclei, and possible changes in nuclear signaling (Ferrera et al., 2014). Furthermore, a glioma cell line overexpressing LMNB1 also has misshapen nuclei with many aberrant layers of nuclear membranes (Ratti et al., 2021). Mouse models have shown that both cell-autonomous and non-cell-autonomous effects occur. Mice globally overexpressing lamin B1 show downregulation of PLP1 (Heng et al., 2013). Mice with oligodendrocyte-specific overexpression of lamin B1 show astrogliosis, microglia infiltration, decreased expression of genes that regulate lipid synthesis in oligodendrocytes, age-dependent demyelination (Rolyan et al., 2015), and motor dysfunction, but no autonomic phenotype (Lo Martire et al., 2018).
Recently, a drug screen in ADLD fibroblasts identified an inhibitor of heat shock protein 90 as a possible modulator of lamin B1 expression (Giorgio et al., 2021). Thus, therapeutic strategies for ADLD treatment are beginning to be identified.
Hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC)
H-ABC is a leukodystrophy inherited in an autosomal dominant manner. Beginning in infancy to early childhood, patients present with movement and speech abnormalities, including ataxia and spasticity, and may never develop fine motor skills. MRI studies have revealed atrophy of the basal ganglia, putamen, and cerebellum, and histology studies have confirmed hypomyelination (van der Knaap et al., 2002; van der Knaap et al., 2007).
H-ABC is caused by mutations in the TUBB4A gene which codes for β-tubulin, a major structural protein that dimerizes with ⍺-tubulin to form heterodimers that are the building blocks of the microtubule cytoskeleton. Compared to other β-tubulin genes, TUBB4A is highly expressed by oligodendrocytes and its expression increases postnatally, consistent with a role in myelination (Zhang et al., 2014; Park et al., 2021). In oligodendrocytes, microtubules play important roles in building the structure of oligodendrocyte branches and myelin sheaths (Fu et al., 2019) as well as in transporting mRNA and other cargos (Carson et al., 1997; Herbert et al., 2017). Loss of the microtubule nucleation protein TPPP (tubulin polymerization promoting protein) results in shorter and thinner myelin sheaths as well as behavioral defects in motor coordination (Fu et al., 2019) and in innate and memory-dependent fear responses (Nguyen et al., 2020).
H-ABC can be caused by many different point mutations in TUBB4A. TUBB4A mutations can affect microtubule stability and growth, and interfere with motor protein binding to microtubules (Vulinovic et al., 2018). One example is a de novo point mutation (D249N) at a highly conserved aspartic acid residue that is located on the dimer interface between α-tubulin and β-tubulin and is important for heterodimer formation (Simons et al., 2013). Indeed, a mouse model of the D249N mutation recapitulates disease-like motor dysfunction and hypomyelination and indicates that both neurons and oligodendrocytes are affected. Cultured cerebellar granule neurons expressing this mutation displayed a variety of morphological defects, including shorter axons, fewer dendrites, and less dendritic branching. Similarly, in the differentiated oligodendrocyte-like cell line Oli-neu, expression of this mutation resulted in lack of complex branching morphology (Curiel et al., 2017). Furthermore, this mouse model also displays fewer mature oligodendrocytes, striatal neurons, and cerebellar granular neurons, as well as more caspase-3-positive apoptotic OPCs and cerebellar granular neurons (Sase et al., 2020).
There are currently no treatments for H-ABC, though rehabilitation and other management strategies are often utilized (Rossi et al., 2018). However, Tubb4a knockout mice do not exhibit any deleterious effects (Sase et al., 2020), indicating that H-ABC may be an ideal target for ASO therapy that aims to decrease expression of mutant TUBB4A mRNAs.
Transcription defects
4H leukodystrophy
4H leukodystrophy is so named due to its alliterative primary clinical manifestations: hypomyelination, hypodontia, and hypogonadotropic hypogonadism (Bernard and Vanderver 1993). This leukodystrophy is also known as RNA polymerase III (POLR3)-related leukodystrophy due to mutations in POLR3A, POLR3B, and POLR3K genes (Bernard et al., 2011; Tétreault et al., 2011; Dorboz et al., 2018). The autosomal recessive inheritance pattern of 4H leukodystrophy requires biallelic expression of the pathogenic variant (e.g., homozygous or compound heterozygous mutations). Consistent with this inheritance pattern, a homozygous conditional knock-in mice expressing mutant Polr3a under the Olig2 promoter displays hypomyelination as well as a range of cognitive, sensory, and sensorimotor defects (Merheb et al., 2021).
Despite the existence of a mouse model for 4H leukodystrophy, the mechanism for how POLR3 mutations cause hypomyelination remains unclear. Multiple hypotheses have emerged in the literature. First, POLR3 mutations may lead to tRNA deficits that impact global translation. Indeed, POLR3 transcribes more than 100 tRNA genes (Goffeau et al., 1996), and mutations in POLR3 result in decreased tRNA transcription and increased tRNA post-translational modifications (Arimbasseri et al., 2015). In addition, oligodendrocytes may be more vulnerable to tRNA deficits due to their heavy reliance on local translation. For example, MBP is locally translated within the myelin sheath and many mRNAs are found locally in myelin fractions by RNA-seq (Thakurela et al., 2016; Herbert et al., 2017; Meservey, Topkar, and Fu 2021). Second, in a human oligodendrocyte-like cell line, POLR3 mutation can impair production of a non-coding RNA (ncRNA) that may be important for myelin formation and expression of myelin transcripts like MBP mRNA (Choquet et al., 2019). However, it is not clear which downstream RNAs could be feasible therapeutic targets for restoring myelination. Thus, future research elucidating the cellular pathology of oligodendrocytes in 4H leukodystrophy models will be important for uncovering logical therapeutic approaches.
NKX6-2-related spastic ataxia with hypomyelination (SPAX8)
De novo mutations in the NKX6-2 gene are associated with spastic cerebellar ataxia that presents with hypomyelination on MRI. While neonatal-onset SPAX8 is associated with global delays, childhood-onset SPAX8 typically results in only motor delays (Chelban et al., 2017; 2020). NKX6-2 is a homeobox transcription factor that is important for oligodendrocyte differentiation and myelination in the hindbrain and spinal cord (Vallstedt, Klos, and Ericson 2005; Cai et al., 2010). Indeed, transcription factors NKX6-2, SOX10, and OLIG2 can be used to reprogram human fibroblasts into O4-positive OPC-like cells that can differentiate into MBP-positive oligodendrocyte-like cells (Ehrlich et al., 2017). Specifically, NKX6-2 can regulate expression of the microtubule severing protein stathmin and paranodal cell adhesion molecules neurofascin and contactin (Southwood et al., 2004), which highlight a role for NKX6-2 in regulating oligodendrocyte cytoskeleton and paranodal structures. Currently, no animal model for this disease exists, though one may prove useful to determine disease mechanisms and investigate treatments.
Waardenburg-hirschsprung disease (WS4)
Mutations in the gene encoding the transcription factor SOX10 are associated with WS4 (Pingault et al., 1998), which is a subtype of a family of Waardenburg syndromes. WS4 is characterized by deafness, reduced pigmentation, skull and intestine abnormalities, and reduced myelin production by both oligodendrocytes and Schwann cells (Inoue et al., 1999; Inoue et al., 2002). In addition to its roles in the development of the colon and of melanocytes, SOX10 is important for the development of both CNS and PNS glia. In vivo, SOX10-null mice show impaired development of Schwann cells and other PNS glia (Britsch et al., 2001) and diminished oligodendrocyte differentiation in the CNS (Stolt et al., 2002).
Biochemical studies indicate that mutant SOX10 acquires a toxic gain of function. Though many SOX10 mutations result in truncated mRNA transcripts, they are not downregulated by the nonsense-mediated decay pathway. In fact, mutant SOX10 proteins surprisingly outcompete wildtype SOX10 for DNA binding when co-expressed in glioblastoma cells (Inoue et al., 2004). Thus, this dominant-negative mechanism indicates that WS4 may be best therapeutically targeted by decreasing mutant SOX10 mRNA expression or decreasing mutant SOX10 protein activity.
Translation machinery defects
tRNA synthetase-related leukodystrophies
Mutations in genes that encode components of the tRNA multisynthetase complex (DARS1, RARS1, EPRS1) are implicated in hypomyelination. These mutations cause a spectrum of clinical phenotypes, from severe seizures and brain atrophy within the first 3 months of age, to mild ataxia and cognitive deficits beginning at 1 year old or later (Taft et al., 2013; Mendes et al., 2020).
tRNA synthetases function to join the proper amino acid to each tRNA molecule. Though it is unclear how mutations in this mechanism cause hypomyelination, studies have shown reduced EPRS protein levels and activity in patients with EPRS mutations (Mendes et al., 2018). For RARS, the most common mutation is associated with mild disease, while truncation mutations and mutations located in the enzymatic domain are associated with severe disease (Mendes et al., 2020). These findings suggest that disruption in tRNA complex assembly, reduced tRNA synthetase activity, and translation disruption result in insufficient myelination in the developing brain.
Researchers have questioned whether tRNA synthetase mutations cause hypomyelination via oligodendrocyte dysfunction or neuron dysfunction (Mendes et al., 2020; Wolf, ffrench-Constant et al., 2018). Indeed, mutations in tRNA synthetase genes VARS (Friedman et al., 2019; Siekierska et al., 2019) and AIMP2 (Shukla et al., 2018) have been implicated in neuronal disorders with shared phenotypes in severely affected patients, such as cerebral atrophy and seizures. Thus, more research to dissect the cell-specific effects of mutations in tRNA synthetase machinery is important for better mechanistic understanding of these leukodystrophies.
Vanishing white matter (VWM) disease or childhood ataxia and cerebral hypomyelination (CACH)
VWM disease, also known as CACH, is one of the more prevalent leukodystrophies, with an estimated incidence of one in 80,000–100,000 live births (van der Knaap et al., 2022). VWM is highly variable in age of onset, severity, and progression. In some cases, children exhibit normal early development, whereas others experience ataxia, and delayed speech and cognitive milestones (Schiffmann et al., 1994; van der Knaap et al., 2003). MRI imaging of VWM patients shows distinct patterns of diffuse white matter signal (Schiffmann et al., 1994; van der Knaap et al., 1997). These signs of hypomyelination are present even prior to symptom onset (Schiffmann et al., 2003). As white matter MRI signals decrease and white matter tract deterioration progresses, neurological symptoms worsen. Unfortunately, this hypomyelination can become so severe that it can result in death around the first or second decade of life (van der Knaap et al., 2022).
A number of studies in postmortem brains have described the histopathological characteristics of VWM. First, one study observed oligodendrocytes with “foamy” cytoplasm, which was hypothesized to indicate abnormal glycosylation (Fogli et al., 2002). Second, a thorough literature review of VWM brain pathology studies highlighted a pattern of disproportionately high number of OPCs compared to mature oligodendrocytes. Lastly, oligodendrocytes in VWM brains have abnormal mitochondrial morphology (Wong et al., 2000), and increased rates of apoptosis (Brück et al., 2001; Van Haren et al., 2004). Thus, these histology results indicate that oligodendrocytes in VWM have maturation defects and cell health issues.
Genetics studies of VWM have revealed mutations in the genes encoding the five subunits of the eukaryotic initiation factor 2B (EIF2B1, EIF2B2, EIF2B3, EIF2B4, and EIF2B5). EIF2B regulates the first stage of translation initiation: the recruitment of ribosomes and proper identification of the start codon (Bogorad et al., 2018). While mutations can occur in any of the five subunits, 80% are missense mutations occuring in gene encoding the ε-subunit (EIF2B5) (van der Knaap et al., 2002). Since eIF2B is a guanine nucleotide exchange factor, EIF2B mutants display reduced GTP hydrolysis due to conformational changes, and therefore may adversely impact global mRNA translation (Fogli et al., 2004). These changes can trigger downstream activation of the integrated stress response, a system that regulates protein translation and folding in response to both intracellular and extracellular stress (van der Knaap et al., 2022).
Studies to elucidate the cellular mechanisms of VWM have uncovered both cell-autonomous and non-cell-autonomous mechanisms. Mutant-Eif2b5 mice exhibit delayed white matter development (Geva et al., 2010), and purified OPCs cultured from these mice had mitochondrial defects, as well as impaired differentiation and morphology (Herrero, Mandelboum, and Elroy-Stein 2019). However, in a co-culture system using glial cells isolated from wildtype and mutant-Eif2b5 mice, mutant astrocytes secreted factors that inhibited wildtype OPCs from differentiating into mature oligodendrocytes (Dooves et al., 2016). Thus hypomyelination in VWM likely involves defects in both OPCs and astrocytes.
Although there are no treatment options available for VWM, a recent study performed an ambitious small molecule drug screen. The authors conducted a screen of 2400 FDA-approved drugs using iPSC-derived astrocytes from a patient harboring mutations in two subunits of eIF2B. They identified 113 anti-inflammatory drugs with cytoprotective effects, including ursodiol, berberine, deflazacort, and zileuton. Of these, ursodiol rescued oxidative stress and mitochondrial dysfunction in VWM patient iPSC-derived astrocytes (Ng et al., 2020). However, for all the drugs identified, further in vivo animal testing is needed prior to moving to any clinical studies.
Cell junction defects
Megalencephalic leukoencephalopathy with subcortical cysts (MLC)
MLC is an autosomal-recessive vacuolating leukodystrophy that is also known as van der Knaap disease, after the pediatric neurologist who first described the condition (Bosch and Estévez 2021). MLC is characterized by prominent macrocephaly that progressively develops within the first year of life (van der Knaap et al., 2012). Due to the macrocephaly, children exhibit difficulty walking, while gradual onset of ataxia and spasticity further contribute to gross motor delays and difficulties. Typically, children have normal cognition, though mild mental deterioration can occur later during development. Computed tomography (CT) and MRI scans revealed that MLC patients have mild-to-severe swelling or edema, vacuolization of white matter, and subcortical cysts (van der Knaap et al., 1995; van der Knaap et al., 2002; Dash, Raj, and Sahu 2015).
MLC can be categorized into different subtypes based on the specific genes mutated. MLC1 subtype results from a missense mutation in the MLC1 gene. Mlc1 mRNA is expressed exclusively by astrocytes, but not by oligodendrocytes (Schmitt et al., 2003; Dubey et al., 2015). MLC1 encodes a transmembrane protein (Leegwater et al., 2001) that participates in osmoregulation by astrocytes. In astrocytoma cells, wildtype MLC1 localizes to the plasma membrane in a complex with aquaporin, the potassium channel Kir4.1, and the TRPV4 channel. By contrast, mutant MLC1 localizes intracellularly and this disrupts osmolarity homeostasis (Lanciotti et al., 2012). Consistent with this, a study in HeLa cells demonstrated that mutant MLC1 protein was retained in the ER (Duarri et al., 2008), likely due to protein misfolding. Both mouse and zebrafish models recapitulate human phenotypes, including macrocephaly and edema (Sirisi et al., 2014). A Mlc1-null mouse model unveiled morphologically abnormal astrocytes present in the swollen white matter. These astrocytes express lower levels of the adhesion molecule GLIALCAM, which is also known as HEPACAM (hepatocyte cell adhesion molecule). No neuronal or oligodendrocyte defects were observed, because MLC-null axons were fully myelinated in EMs and oligodendrocyte-specific transcripts were expressed at normal levels (Dubey et al., 2015).
MLC2a subtype is caused by mutations in the HEPACAM gene, which encodes a cell adhesion molecule in the immunoglobulin family (Sirisi et al., 2014). Hepacam mRNA is present in both astrocytes and oligodendrocytes (Hoegg-Beiler et al., 2014; Bugiani et al., 2017). Recent work indicates that HEPACAM plays a crucial role in gap junction coupling between adjacent astrocytes (Baldwin et al., 2021). Mutations in this gene have been shown to cause protein mislocalization (Elorza-Vidal et al., 2020).
MLC1 and HEPACAM likely function in a complex to mediate cell-cell interactions. In mouse models, MLC1 and HEPACAM mutually affect each other’s localization. Knockout of Hepacam affects Mlc1 expression and MLC protein localization (Hoegg-Beiler et al., 2014; Bugiani et al., 2017), while knockout of Mlc1 decreases HEPACAM levels and leads to its mislocalization (Dubey et al., 2015). A recent study showed that astrocytes in Mlc1 knockout mice are more branched, but their endfeet make fewer contacts with vasculature. In addition, Mlc1 knockout mice have high incidence of neurons aberrantly contacting vasculature (Gilbert et al., 2021). These defects in gliovascular coupling may underlie the fluid buildup and macrocephaly observed in MLC patients (van der Knaap et al., 2012; Dubey et al., 2015).
Although there are no treatments for MLC, recent work has provided some preclinical insight into viral replacement therapy. Using Mlc1 knockout mice, researchers demonstrated that cerebellar subarachnoid administration of an AAV expressing human MLC1 under a GFAP promoter increased MLC1 protein expression in the cerebellum and decreased white matter vacuolation in a dose-dependent manner (Bosch and Estévez 2021). This study indicates that replacement therapy is a feasible therapeutic approach for MLC.
Oculodentodigital dysplasia (ODDD)
ODDD is a multisystem syndrome whose symptoms present in three major categories: ocular, dental, and digital. Ocular symptoms include very small corneas (microcornea), eyes that do not look in the same direction (strabismus), and fluid buildup in the eye (glaucoma). Dental symptoms include abnormally small teeth, defective enamel, and a thickened lower jaw. Digital symptoms usually present as webbing between the fourth and fifth fingers and third, fourth, and fifth toes (syndactyly) (Traboulsi, Faris, and Der Kaloustian 1986). The neurological symptoms of ODDD are wide-ranging and include lower limb spasticity or paralysis (paraparesis), ataxia, bladder disturbances, and seizures. MRI imaging has shown decreased white matter intensity in the CNS, which likely contributes to motor symptoms in ODDD (Loddenkemper et al., 2002).
ODDD is caused by mutations in GJA1 (also known as Cx43), which encodes the gap junction α-1 protein (connexin 43). Mutations causing ODDD are typically inherited in an autosomal-dominant fashion (Paznekas et al., 2003). One screen of 17 families found 16 unique missense mutations and one codon duplication; these mutations were present only in affected individuals, indicating an autosomal-dominant pattern of inheritance (Paznekas et al., 2003). However, there are also documented cases of autosomal-recessive inheritance (Joss et al., 2008) and novel de novo mutations (Choi et al., 2018; Pace et al., 2019). Thus, ODDD displays a variety of inheritance patterns.
Cx43 is one protein component comprising gap junctions between astrocytes and other glial cells (Orthmann-Murphy et al., 2007). A mouse model deficient in the Cx43 and Cx30 genes had a complete loss of astrocyte–astrocyte gap junctions, which reduced the rate of potassium clearance and increased the probability of seizure-like activity in hippocampal neurons in vitro (Wallraff et al., 2006). Cx43 has also been shown to be involved in astrocyte–oligodendrocyte gap junctions (Nagy et al., 2003). Double knockout mice with astrocyte-specific deletion of Cx43 and global deletion of Cx30 displayed demyelination in multiple regions (corpus callosum, cerebellum, corticospinal and other tracts) and edema within myelin. These mice also performed worse on the rotarod motor coordination assay, which is consistent with ODDD neurological motor symptoms (Lutz et al., 2009).
Treatments for ODDD are symptomatic and supportive. For instance, an eye patch over the stronger eye can correct strabismus, corrective surgery can address defects in the fingers and toes, and antispastic medication can ameliorate neurological symptoms (Shinya et al., 2021). Thus, additional basic research is needed to elucidate the role of gap junctions in astrocytes and oligodendrocytes.
Connexin-47-related Pelizaeus-Merzbacher-like disease (PMLD)
PMLD presents with similar symptoms as Pelizaeus–Merzbacher Disease (PMD), including nystagmus, spasticity, head tremors, poor muscle tone, and progressive spasticity, but PMLD symptoms are less severe than PMD symptoms. PMLD is autosomal recessive and associated with mutations in connexin 47 (Cx47), which also known as connexin 46.6, gap junction protein γ2 (GJC2), and gap junction protein α12 (GJA12) (Uhlenberg et al., 2004; Bilir et al., 2013; Abrams et al., 2014; Owczarek-Lipska et al., 2019). Many patients present with biallelic (compound heterozygous) mutations in combination (Owczarek-Lipska et al., 2019).
Whether mutant Cx47 causes leukodystrophy via loss or gain of function remains unclear. In one study that treated HeLa cells with mutant Cx47, gap junction formation was normal, but electrical coupling was disrupted (Abrams et al., 2014). In a bulk RNA-seq transcriptome, Cx47 is expressed very highly and specifically in oligodendrocytes (Zhang et al., 2014). However, a Cx47 knockout mouse did not display any clinical symptoms (Odermatt et al., 2003), which hints that Cx47 mutations in PMLD patients may exert a toxic gain-of-function effect. Thus, elucidation of PMLD disease mechanisms is needed to lay the important groundwork for the road toward translational studies.
Myelin structural protein defects
Proteomic analysis of human myelin shows that the protein composition of myelin largely consists of two proteins - PLP1 (45%) and MBP (28%). Additional myelin proteins include CNP (4.5%), MOG (0.8%), MAG (0.3%), MOBP (0.1%), and others (Gargareta et al., 2022). Many of these proteins play a role in compaction, the formation of efficient myelin insulation by bringing together adjacent layers of membrane. Compaction happens both intracellularly (between membranes from the same wrap) and extracellularly (between membranes from different but juxtaposed wraps). While intracellular compaction largely relies on MBP, extracellular compaction relies on PLP. Additional transmembrane proteins, like MAG and MOG, are found in non-compact regions of the myelin sheath (Raasakka and Kursula 2020).
CNP-related hypomyelinating leukodystrophy
A recent study of multiple members of a consanguineous family found a homozygous mutation in the CNP (2′,3′-cyclic nucleotide-3′-phosphodiesterase) gene. Symptoms began ∼1 year of age and include arrest then regression in motor skill development, dystonia, irritability, microcephaly, scoliosis and hirsutism. MRI imaging showed hypomyelination, especially of periventricular and subcortical white matter. Ultimately these children passed away ∼five to eight years of age. This mutant CNP is unstable and has barely detectable protein levels by Western blot of patient fibroblast lysates (Al-Abdi et al., 2020). CNP plays an important role in establishment of the cytoplasmic channels in myelin sheaths that allow transport between outer to inner layers of myelin (Snaidero et al., 2017).
MAG-related PMLD
Another gene associated with PMLD is MAG (myelin-associated glycoprotein). Three siblings from a consanguineous family presented with infantile-onset PMLD that later in adulthood developed into additional symptoms of mental retardation, dysarthria, optic atrophy, and peripheral neuropathy. They share a homozygous missense mutation in MAG that results in mutant MAG retention in the ER, misshapen paranodal junctions, and thin myelin sheaths (Lossos et al., 2015).
MAL-related leukodystrophy
A recent report found that a patient presenting with PMLD had a novel missense mutation (A109D) in the gene encoding myelin and lymphocyte protein (MAL) (Elpidorou et al., 2022). MAL is a four-pass transmembrane protein that is important for the structure of paranodal loops, which are located at the ends of myelin sheaths and flank the nodes of Ranvier. In Mal knockout mice, paranodal loops are aberrantly oriented facing away from the axon (Schaeren-Wimers et al., 2004). Experiments in cultured kidney cells demonstrated that mutant MAL aggregates in the ER, likely due to a protein misfolding defect. Treatment with 4-phenylbutyrate, a chemical chaperone, appeared to restore MAL trafficking out of the ER (Elpidorou et al., 2022), which supports targeting protein misfolding as a putative therapeutic strategy for this leukodystrophy.
Pelizaeus–Merzbacher disease (PMD)
PMD is an X-linked, dysmyelinating leukodystrophy characterized by involuntary eye movement, head tremors, and systematically poor muscle tone. Symptom onset usually occurs within the first year of age (Laukka et al., 2016). PMD is caused by mutations in the gene encoding PLP1, one of the most abundant membrane-associated myelin proteins in the CNS. Many types of PLP1 mutations can cause PMD. These include duplications (Inoue et al., 1996), deletions (Raskind et al., 1991), and missense mutations. Of these, PLP1 gene duplication is the most common cause of PMD (Sistermans et al., 1998; Mimault et al., 1999), while missense point mutations in PLP1 also result in axonal degeneration in the CNS (Garbern et al., 2002).
Both loss-of-function and gain-of-function mechanisms have been suggested for how PLP1 mutations cause PMD. Early neuropathology studies of PMD brains revealed significant deficiencies in myelination as well as decreased levels of myelin proteins (MBP, MAG, and CNP) and lipids (cerebroside, sulfatides, and sphingomyelin) (Bourre et al., 1978). As a four-pass transmembrane protein, PLP1 functions in myelin compaction, bringing together adjacent extracellular layers of myelin. Indeed, PLP1-null mice exhibit myelin structural defects, such as incomplete compaction, and axonal accumulation of organelles (Klugmann et al., 1997; Edgar et al., 2004), which is indicative of axonal transport defects. Additional studies suggest that PMD mutations may lead to toxic gain of function. In patient-derived iPSCs differentiated into oligodendrocytes, mutant PLP1 proteins misfold and mislocalize to the ER (Numasawa-Kuroiwa et al., 2014) and can contribute to iron toxicity. These mechanisms may contribute to oligodendrocyte cell death and iron chelation therapy has even been suggested to prevent cell death (Nobuta et al., 2019). Thus, determining the disease-causing mechanisms for different PLP1 mutations will be crucial for assessing therapeutic rationales for different patient cohorts.
Recently, promising experiments in mice indicate that ASOs may be a feasible therapeutic approach for PMD. First, researchers ensured that decreasing PLP1 levels does not have detrimental effects. The jimpy mouse model expresses a truncated defective copy of PLP1 and shares similar cellular and neurological phenotypes with severe PMD patients. Interestingly, targeted germline deletion of the PLP1 gene improved the lifespan of this mouse and eliminated tremors, ataxia, and seizures. Thus, to recapitulate this positive effect, ASOs targeting Plp1 mRNA were administered to jimpy mice. This resulted in silencing of Plp1, restored white matter volume, and astonishingly improved survival rate (Elitt et al., 2020). However, it should be noted that jimpy is a spontaneously occurring PLP1 mutation in mice and does not directly model PMD mutations. Moreover, PLP1-null adult mice have myelin and axon defects (Klugmann et al., 1997; Edgar et al., 2004), indicating that complete loss of PLP1 can also be deleterious. Thus, it will be important for future research to focus on long-term effects of administering Plp1-targeted ASOs.
Another possible cause of PMD pathology is mutations causing aberrant splicing. Plp mRNA can be spliced into two isoforms: Plp1 and the shorter Dm20 (Nave et al., 1987). Some PMD patients have mutations in PLP1 Intron 3, which contains an alternative splicing site, (Taube et al., 2014; Kevelam et al., 2015). These mutations alter alternative splicing and disrupt the ratio of PLP1/DM20 protein in oligodendrocyte cultures (Hobson et al., 2006; Taube et al., 2014). In an attempt to target disease-causing mutations, morpholino oligomers blocking the DM20 splice site successfully shifted alternative splicing towards PLP1 in oligodendrocyte culture. In addition, intracerebroventricular delivery of the same oligomer to neonatal mice modeling this mutation rescued both Plp mRNA and PLP protein levels (Tantzer et al., 2018). Thus, approaches targeting both PLP overexpression and splicing defects are being developed and show promising results in animal models.
Other transmembrane protein defects
CNTNAP1-related arthrogryposis and leukodystrophy
Two children presenting with arthrogryposis (joint stiffness and muscle weakness) and hypomyelination by MRI were found to have either homozygous or compound heterozygous mutations in the CNTNAP1 gene, which encodes contactin-associated protein 1 (Caspr1). Caspr1 is a paranodal cell-adhesion protein in the axon that interacts with neurofascin 155 to form paranodal junctions. EM of sural nerve biopsies showed a striking absence of paranodal loop structures (Conant et al., 2018).
Hypomyelination with congenital cataracts (HCC)
HCC is characterized by the appearance of cataracts within the first year of life, followed by progressive neurological and cognitive impairment, and hypomyelination in the CNS and PNS (Biancheri et al., 2011; Wolf et al., 2021). HCC is caused by mutations in FAM126A gene that encodes the membrane protein hyccin, which is expressed ubiquitously in the adult brain and also in the lens of the eye (Zara et al., 2006).
Hyccin function may be important for myelin sheath development. Hyccin is a component of the PI4K (phosphatidylinositol 4-kinase) complex that synthesizes phosphatidylinositol 4-phosphate (PI4P). Absence of hyccin destabilizes the PI4K complex (Baskin et al., 2016), which may result in fewer PI4P and other phosphoinositides intermediates that are important for myelin formation. Indeed, phosphoinositides are important both for the association of MBP with adjacent myelin sheath plasma membranes (Nawaz et al., 2009; Zuchero et al., 2015), and for opening cytoplasmic channels that are found from outer to inner myelin sheath layers as they grow and wrap around the axon (Snaidero et al., 2014). More research on how hyccin mutations affect myelination are needed in order to better design novel therapeutic treatments for this leukodystrophy.
SLC35B2-related chondrodysplasia with hypomyelinating leukodystrophy
Homozygous mutations in SLC35B2 were found in two patients presenting with chondrodysplasia (disrupted growth of cartilage), intellectual disability, and hypomyelination on brain MRI (Guasto et al., 2022). The SLC35B2 gene (also known as PAPST1) encodes a 3′-phosphoadenosine 5′-phosphosulfate (PAPS) transporter that moves PAPS from the cytoplasm into the Golgi apparatus. In the Golgi, sulfotransferases add PAPS to proteins already modified with GAGs (glycosaminoglycan chains). This process generates sulfated proteoglycans (PGs), which are important in the extracellular matrix for building and maintaining connective tissues in bones, cartilage, and even brains (Schwartz and Domowicz, 2018). Biochemical analyses of sulfated proteins in both patient fibroblasts and serum indicate reductions in protein sulfation (Guasto et al., 2022). Moreover, in a transfected kidney cell line, mutant SLC35B2 lost its Golgi-specific localization and instead appeared throughout the cell. These results suggest that mutant SLC35B2 may act through a loss-of-function mechanism. Though the role of PAPS transporters in myelination is still unclear, the discovery of these mutations open the door toward exciting future research.
Transient infantile hypomyelinating leukodystrophy-19 (HLD19)
Recently, heterozygous missense mutations in the transmembrane protein 63A (TMEM63A) gene were identified in a hypomyelinating leukodystrophy that usually presents in the first weeks of life with nystagmus, neurological abnormalities, and hypomyelination by MRI. Surprisingly, all patients showed neurological improvement and nearly recovered hypomyelination by 4 years of age. TMEM63A is a mechanosensitive ion channel (Yan et al., 2019) that is preferentially expressed in oligodendrocytes and microglia (Zhang et al., 2014). While cultured cells expressing wildtype TMEM63A exhibited stretch-activated currents upon mechanical stimulation, cells expressing mutant TMEM63A did not (Yan et al., 2019). Though the mechanism leading to leukodystrophy is yet unknown, this discovery highlights an interesting finding that mechanosensitive ion channels play a role in myelination.
TMEM106B-related hypomyelinating leukodystrophy
Recently, a hypomyelinating leukodystrophy with relatively mild phenotypes was attributed to a single de novo mutation in the gene encoding transmembrane protein 106B (TMEM106B) leading to a D252N residue change. In 4 unrelated patients, symptoms include nystagmus, delays in motor skills development, and intellectual impairment (Simons et al., 2017).
Multiple studies support a role for TMEM106B in regulating both lysosomal function and acidification as well as myelin sheath structure. First, TMEM106B binds to the lysosomal protein vacuolar-ATPase accessory protein 1 (AP1). Loss of TMEM106B leads to fewer lysosomes in cultured neurons and decreased levels of lysosomal enzymes in mice (Klein et al., 2017). Second, early studies indicate that late endosomes and lysosomes transport the transmembrane protein PLP to the myelin sheath (Trajkovic et al., 2006). Indeed, Tmem106b knockout mice have reduced PLP and MOG levels by Western blot and their myelin sheaths contain vacuoles in between myelin layers visible by EM (Feng et al., 2020). Furthermore, Tmem106b knockout mice are more susceptible to cuprizone-induced demyelination damage and have poorer ability to remyelinate. Primary oligodendrocytes cultured from Tmem106b knockout mice contain clusters of PLP-positive lysosomes inside the cell body and decreased distribution of PLP outside of the cell body (Zhou et al., 2020). Together, these experiments indicate that TMEM106B dysfunction can lead to lysosomal defects in neurons as well as aberrant vacuoles and PLP trafficking defects in the myelin sheath.
Interestingly, TMEM106B dysfunction is also implicated in neurodegenerative diseases in elderly patients. Polymorphisms of TMEM106B were identified as a risk factor for frontotemporal degeneration (FTD) (Van Deerlin et al., 2010). Very recently, TMEM106B amyloid filaments were discovered in the brains of older patients with various neurodegenerative diseases, including tauopathies, synucleinopathies, Aβ-amyloidoses and TDP-43 proteinopathies. Similar TMEM106B filaments were also found in older but neurologically normal patients, indicating that TMEM106B is prone to forming filaments in aging brains. The D252N leukodystrophy mutation (Simons et al., 2017) resides within the amyloidogenic region in TMEM106B (residues 120–254) (Schweighauser et al., 2022), but it is unclear whether the D252N mutation could be more amyloidogenic than wildtype TMEM106B. Surprisingly, polymorphisms in other leukodystrophy genes, including ARSA and CYP27A1, are also linked to FTD (Lok and Kwok 2021). Thus, these connections between FTD and leukodystrophies hint at common mechanisms and an interesting topic for future research.
TMEM163-related hypomyelinating leukodystrophy
Recently, two patients with hypomyelinating leukodystrophy were found to carry two unique de novo variants of TMEM163 (Yan et al., 2022). Both patients presented initially with symptoms consistent with PMLD, such as nystagmus, motor deficits, and hypomyelination on MRI. Similarly to transient infantile HLD19, hypomyelination on MRI improved with age.
TMEM163 is a small protein with six predicted transmembrane domains. It is a member of the ZNT/SLC30 zinc efflux transporter family (Sanchez et al., 2019). In cultured cells exposed to the zinc indicator Newport Green (NPG), expression of wildtype TMEM163 leads to decreased NPG fluorescence, but expression of mutant L76P TMEM163 leads to even lower NPG signal, indicating that mutant TMEM163 has hyperactive zinc efflux activity. Zebrafish in which tmem163a and b were knocked down and replaced with mutant human TMEM163 had locomotion defects and decreased myelin protein expression (Yan et al., 2022), which recapitulates the motor deficits and hypomyelination seen in patients. These mutations raise the interesting hypothesis that dysregulation of zinc homeostasis can preferentially disrupt myelination. Consequently, further research should investigate how zinc efflux affects myelin growth and how this process is affected by these novel mutations.
Immune system activation
Aicardi-Goutieres syndrome (AGS)
AGS is an autosomal-recessive disorder that mainly affects the brain and the immune system. In the brain, there is a loss of white matter and the appearance of abnormal calcium deposits. Together, hypomyelination and calcification lead to severe disabilities, including joint and muscle rigidity, involuntary muscle contractions, and poor muscle tone. In the immune system, one case report found lymphocytes in the CSF, a 25-fold increase in CSF interferon α (IFN⍺) levels, and autoantibodies against phospholipids (Olivieri et al., 2013).
AGS is linked to mutations in genes encoding intracellular nucleic acid–sensor machinery, including ADAR1, IFIH1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and TREX1 (Crow 2013; Livingston and Crow 2016). Nucleic-acid sensors play an integral role in the innate immune response by inducing apoptosis, necroptosis, and pyroptosis when foreign or defective DNA and RNA are detected (Maelfait, Liverpool, and Rehwinkel 2020). Once these sensors are activated, they initiate induction of cytokines, leading to the transcriptional upregulation of type I interferons (McNab et al., 2015). AGS mutations lead to the accumulation of unusual double-stranded DNA species, which triggers chronic type I interferon release by astrocytes, and ultimately leads to autoimmunity. Indeed, elevated IFN⍺ level is a clinical marker for AGS (Crow et al., 2006).
Astrocytic release of type I interferon, specifically IFN⍺, may cause hypomyelination via multiple mechanisms. High levels of IFN⍺ adversely affect astrocytes, which may ultimately lead to insufficient trophic support for oligodendrocytes. Indeed, AGS patients have low astrocyte cell counts and chronic IFN⍺ exposure has deleterious effects on astrocytes, including reduced proliferation, increased reactivity, and gene dysregulation (Sase et al., 2018). Chronic IFN⍺ levels in AGS may also lead to hypomyelination by directly causing oligodendrocyte death. The type II interferon, interferon γ (IFNγ), harms cultured primary (in vitro) and in vivo oligodendrocytes by disrupting cellular bioenergetics pathways, such as glycolysis and mitochondrial respiration (Minchenberg and Massa 2019). However, currently there are no studies investigating the direct link between IFN⍺ and oligodendrocyte death (de Waard and Bugiani 2020).
The rarity of AGS presents difficulties in recruitment for clinical studies; however, anecdotal reports suggest that the kinase inhibitor Ruxolitinib may result in clinical improvements (Crow, Shetty, and Livingston 2020). Clearly, more basic research on cellular nucleic acid–sensor machinery and translation studies are needed in order to identify therapeutic targets in AGS.
Hereditary diffuse leukoencephalopathy with spheroids (HDLS)
HDLS is an adult-onset disorder typically beginning in the fourth decade of life; therefore, it likely represents a demyelinating disease rather than a developmental disease. HDLS is characterized by various neurological (seizures and parkinsonism), psychiatric (personality changes and dementia), and somatic symptoms (arthritis and gastrointestinal pain). Histopathological studies have found demyelination in the cerebral cortex, axonal spheroids containing aggregates of phosphorylated neurofilament and amyloid precursor protein, and oxidative damage in glia (Axelsson et al., 1984; Baba et al., 2006; Ali et al., 2007). HDLS is an autosomal-dominant disorder and whole-exome sequencing revealed that it is caused by mutations in the gene encoding colony stimulating factor 1 receptor (CSF1R) (Rademakers et al., 2011).
Multiple sources of evidence indicate that microglia defects cause HDLS. CSF1R is the receptor for the cytokine CSF-1, which regulates the proliferation and differentiation of microglia in the CNS (Stanley et al., 1997). Upon ligand binding, wildtype CSF1R homodimerizes and auto-phosphorylates tyrosines in its cytoplasmic domain, but mutant HDLS CSF1R does not (Rademakers et al., 2011). Moreover, a mouse model with reduced CSF1R expression in microglia displays memory deficits, microglial activation, and demyelination (Biundo et al., 2021). Thus, CSF1R inactivating mutations likely contribute to HDLS myelin pathology through microglia.
Though no treatment currently exists, microglia replacement therapy has been proposed for HDLS. This would involve microglia depletion followed by replacement with either HSCs or engineered microglia expressing wildtype CSF1R (Han et al., 2020). Indeed Csf1r knockout mice can be partially rescued by bone marrow transplants and donor-derived cells engraft into the brain and adopt signature microglial genes (Bennett et al., 2018). Though more validation of this approach is needed, it presents a promising avenue for treatment of this leukodystrophy.
Conclusion
The field of leukodystrophies is exemplary in identifying underlying genetic mutations. Perhaps due to decreased sequencing costs, many novel mutations are being discovered each year. This wealth of genetic information has provided not only a rich foundation for understanding the mechanistic underpinnings of leukodystrophies, but also insights into normal oligodendrocyte and myelin development, astrocyte cell biology, as well as glia–glia and glia–neuron interactions. Cellular mechanisms attributed to hypomyelination in leukodystrophies include contributions to lipid metabolism and myelin biogenesis by various organelles, such as lysosomes, peroxisomes, and mitochondria, as well as enzymes like ASPA. In addition, astrocytes may contribute to etiologies in leukodystrophies such as AxD, VWM, and AGS.
Selection of therapeutic strategies in translational studies of leukodystrophy have relied on basic research results that determined mode of genetic inheritance as well as cellular mechanisms. For autosomal recessive and X-linked leukodystrophies, replacement therapies and other therapies targeted at restoring loss of function have been validated for certain disorders. For autosomal dominant leukodystrophies, therapies aim to decrease expression of mutant or overexpressed proteins. ASOs, a new generation of neurotherapeutic that binds to RNA targeting sequences in order to achieve gene silencing, hold great promise. ASOs have been FDA-approved and are successful in targeting other genetic neurological diseases like SMA. Such applications would require careful validation in cell and animal models, but having a strong base of cellular targets and mechanisms is an important first step.
Future clinical studies in humans may face multiple challenges, including adverse or toxic effects, reproducibility in humans, and small sample sizes in rarer leukodystrophies. However, a solid foundation of many translational studies will hopefully lead to promising therapeutics approaches that may 1 day help patients affected by leukodystrophies.
Author contributions
JCN, AMF, and MMF wrote the manuscript.
Funding
This work was supported by the Division of Intramural Research of the NIH, NINDS (ZIA NS009432).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abrams, C. K., Scherer, S. S., Flores-Obando, R., Freidin, M. M., Wong, S., Lamantea, E., et al. (2014). A new mutation in GJC2 associated with subclinical leukodystrophy. J. Neurol. 261 (10), 1929–1938. doi:10.1007/s00415-014-7429-1
Aerts, J. M., Groener, J. E., Kuiper, S., Donker-Koopman, W. E., Strijland, A., Ottenhoff, R., et al. (2008). Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc. Natl. Acad. Sci. U. S. A. 105 (8), 2812–2817. doi:10.1073/pnas.0712309105
Al-Abdi, L., Al Murshedi, F., Elmanzalawy, A., Al Habsi, A., Helaby, R., Ganesh, A., et al. (2020). CNP deficiency causes severe hypomyelinating leukodystrophy in humans. Hum. Genet. 139 (5), 615–622. doi:10.1007/s00439-020-02144-4
Aldenhoven, M., and Kurtzberg, J. (2015). Cord blood is the optimal graft source for the treatment of pediatric patients with lysosomal storage diseases: Clinical outcomes and future directions. Cytotherapy 17 (6), 765–774. doi:10.1016/j.jcyt.2015.03.609
Ali, Z. S., Van Der Voorn, J. P., and Powers, J. M. (2007). A comparative morphologic analysis of adult onset leukodystrophy with neuroaxonal spheroids and pigmented glia-a role for oxidative damage. J. Neuropathol. Exp. Neurol. 66 (7), 660–672. doi:10.1097/nen.0b013e3180986247
Amado, D. A., and Davidson, B. L. (2021). Gene therapy for ALS: A review. Mol. Ther. 29 (12), 3345–3358. doi:10.1016/j.ymthe.2021.04.008
Arimbasseri, A. G., Blewett, N. H., Iben, J. R., Lamichhane, T. N., Cherkasova, V., Hafner, M., et al. (2015). RNA polymerase III output is functionally linked to tRNA dimethyl-G26 modification. PLoS Genet. 11 (12), e1005671. doi:10.1371/journal.pgen.1005671
Aubourg, P., Sevin, C., and Cartier, N. (2011). “Mouse models of metachromatic leukodystrophy and adrenoleukodystrophy,” in Animal Models of Dementia. Neuromethods ed. P. De Deyn, and D. Van Dam (Totowa, NJ: Humana Press), Vol. 48. doi:10.1007/978-1-60761-898-0_26
Axelsson, R., Röyttä, M., Sourander, P., Akesson, H. O., and Andersen, O. (1984). Hereditary diffuse leucoencephalopathy with spheroids. Acta Psychiatr. Scand. Suppl. 314, 1–65.
Baba, Y., Ghetti, B., Baker, M. C., Uitti, R. J., Hutton, M. L., Yamaguchi, K., et al. (2006). Hereditary diffuse leukoencephalopathy with spheroids: Clinical, pathologic and genetic studies of a new kindred. Acta Neuropathol. 111 (4), 300–311. doi:10.1007/s00401-006-0046-z
Back, S. A., Luo, N. L., Borenstein, N. S., Levine, J. M., Volpe, J. J., Kinney, H. C., et al. (2001). Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J. Neurosci. 21 (4), 1302–1312. doi:10.1523/JNEUROSCI.21-04-01302.2001
Baldwin, K. T., Tan, C. X., Strader, S. T., Jiang, C., Savage, J. T., Elorza-Vidal, X., et al. (2021). HepaCAM controls astrocyte self-organization and coupling. Neuron 109 (15), 2427–2442. doi:10.1016/j.neuron.2021.05.025
Barkovich, A. J., Kjos, B. O., Jackson, D. E., and Norman, D. (1988). Normal maturation of the neonatal and infant brain: MR imaging at 1.5 T. Radiology 166 (1 Pt 1), 173–180. doi:10.1148/radiology.166.1.3336675
Barth, P. G., Hoffmann, G. F., Jaeken, J., Wanders, R. J., Duran, M., Jansen, G. A., et al. (1993). L-2-hydroxyglutaric acidaemia: Clinical and biochemical findings in 12 patients and preliminary report on L-2-hydroxyacid dehydrogenase. J. Inherit. Metab. Dis. 16 (4), 753–761. doi:10.1007/BF00711907
Baskin, J. M., Wu, X., Christiano, R., Oh, M. S., Schauder, C. M., Gazzerro, E., et al. (2016). The leukodystrophy protein FAM126A (hyccin) regulates PtdIns(4)P synthesis at the plasma membrane. Nat. Cell. Biol. 18 (1), 132–138. doi:10.1038/ncb3271
Battaglia, R. A., Beltran, A. S., Delic, S., Dumitru, R., Robinson, J. A., Kabiraj, P., et al. (2019). Site-specific phosphorylation and caspase cleavage of GFAP are new markers of Alexander disease severity. Elife 8, e47789. doi:10.7554/eLife.47789
Batzios, S. P., and Zafeiriou, D. I. (2012). Developing treatment options for metachromatic leukodystrophy. Mol. Genet. Metab. 105 (1), 56–63. doi:10.1016/j.ymgme.2011.10.002
Båvner, A., Shafaati, M., Hansson, M., Olin, M., Shpitzen, S., Meiner, V., et al. (2010). On the mechanism of accumulation of cholestanol in the brain of mice with a disruption of sterol 27-hydroxylase. J. Lipid Res. 51 (9), 2722–2730. doi:10.1194/jlr.M008326
Beltran-Quintero, M. L., Bascou, N. A., Poe, M. D., Wenger, D. A., Saavedra-Matiz, C. A., Nichols, M. J., et al. (2019). Early progression of Krabbe disease in patients with symptom onset between 0 and 5 months. Orphanet J. Rare Dis. 14 (1), 46. doi:10.1186/s13023-019-1018-4
Benjamin, E. R., Flanagan, J. J., Schilling, A., Chang, H. H., Agarwal, L., Katz, E., et al. (2009). The pharmacological chaperone 1-deoxygalactonojirimycin increases alpha-galactosidase A levels in Fabry patient cell lines. J. Inherit. Metab. Dis. 32 (3), 424–440. doi:10.1007/s10545-009-1077-0
Bennett, F. C., Bennett, M. L., Yaqoob, F., Mulinyawe, S. B., Grant, G. A., Hayden Gephart, M., et al. (2018). A combination of ontogeny and CNS environment establishes microglial identity. Neuron 98 (6), 1170–1183. doi:10.1016/j.neuron.2018.05.014
Berger, J., Forss-Petter, S., and Eichler, F. S. (2014). Pathophysiology of X-linked adrenoleukodystrophy. Biochimie 98 (100), 135–142. doi:10.1016/j.biochi.2013.11.023
Berginer, V. M., Salen, G., and Shefer, S. (1984). Long-term treatment of cerebrotendinous xanthomatosis with chenodeoxycholic acid. N. Engl. J. Med. 311 (26), 1649–1652. doi:10.1056/NEJM198412273112601
Bernard, G., Chouery, E., Putorti, M. L., Tétreault, M., Takanohashi, A., Carosso, G., et al. (2011). Mutations of POLR3A encoding a catalytic subunit of RNA polymerase Pol III cause a recessive hypomyelinating leukodystrophy. Am. J. Hum. Genet. 89 (3), 415–423. doi:10.1016/j.ajhg.2011.07.014
Bernard, G., and Vanderver, A. (2017). “POLR3-Related leukodystrophy,” in GeneReviews® [internet], ed. M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. W. Grippet al. (Seattle, WA: University of Washington, Seattle).
Biancheri, R., Zara, F., Rossi, A., Mathot, M., Nassogne, M. C., Yalcinkaya, C., et al. (2011). Hypomyelination and congenital cataract: Broadening the clinical phenotype. Arch. Neurol. 68 (9), 1191–1194. doi:10.1001/archneurol.2011.201
Bidchol, A. M., Dalal, A., Trivedi, R., Shukla, A., Nampoothiri, S., Sankar, V. H., et al. (2015). Recurrent and novel GLB1 mutations in India. Gene 567 (2), 173–181. doi:10.1016/j.gene.2015.04.078
Bieganski, T., Dawydzik, B., and Kozlowski, K. (1999). Spondylo-epimetaphyseal dysplasia: A new X-linked variant with mental retardation. Eur. J. Pediatr. 158 (10), 809–814. doi:10.1007/s004310051211
Biffi, A., Montini, E., Lorioli, L., Cesani, M., Fumagalli, F., Plati, T., et al. (2013). Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 341 (6148), 1233158. doi:10.1126/science.1233158
Bilir, B., Yapici, Z., Yalcinkaya, C., Baris, I., Carvalho, C. M., Bartnik, M., et al. (2013). High frequency of GJA12/GJC2 mutations in Turkish patients with Pelizaeus-Merzbacher disease. Clin. Genet. 83 (1), 66–72. doi:10.1111/j.1399-0004.2012.01846.x
Biundo, F., Chitu, V., Shlager, G. G. L., Park, E. S., Gulinello, M. E., Saha, K., et al. (2021). Microglial reduction of colony stimulating factor-1 receptor expression is sufficient to confer adult onset leukodystrophy. Glia 69 (3), 779–791. doi:10.1002/glia.23929
Bogorad, A. M., Lin, K. Y., and Marintchev, A. (2018). eIF2B mechanisms of action and regulation: A thermodynamic view. Biochemistry 57 (9), 1426–1435. doi:10.1021/acs.biochem.7b00957
Bokhari, M. R., Samanta, D., and Bokhari, S. R. A. (2022). in Canavan Disease. StatPearls [internet] (Treasure Island, FL: StatPearls Publishing).
Bosch, A., and Estévez, R. (2021). Megalencephalic leukoencephalopathy: Insights into pathophysiology and perspectives for therapy. Front. Cell. Neurosci. 14, 627887. doi:10.3389/fncel.2020.627887
Bottelbergs, A., Verheijden, S., Hulshagen, L., Gutmann, D. H., Goebbels, S., Nave, K. A., et al. (2010). Axonal integrity in the absence of functional peroxisomes from projection neurons and astrocytes. Glia 58 (13), 1532–1543. doi:10.1002/glia.21027
Bourre, J. M., Jacque, C., Nguyen-Legros, J., Bornhofen, J. H., Araoz, C. A., Daudu, O., et al. (1978). Pelizaeus-merzbacher disease: Biochemical analysis of isolated myelin (electron-microscopy; protein, lipid and unsubstituted fatty acids analysis). Eur. Neurol. 17 (6), 317–326. doi:10.1159/000114969
Bradbury, A. M., Bagel, J. H., Nguyen, D., Lykken, E. A., Pesayco Salvador, J., Jiang, X., et al. (2020). Krabbe disease successfully treated via monotherapy of intrathecal gene therapy. J. Clin. Investig. 130 (9), 4906–4920. doi:10.1172/JCI133953
Brady, R. O., Gal, A. E., Bradley, R. M., Martensson, E., Warshaw, A. L., Laster, L., et al. (1967). Enzymatic defect in Fabry's disease. Ceramidetrihexosidase deficiency. N. Engl. J. Med. 276 (21), 1163–1167. doi:10.1056/NEJM196705252762101
Brazelton, T. R., Rossi, F. M., Keshet, G. I., and Blau, H. M. (2000). From marrow to brain: Expression of neuronal phenotypes in adult mice. Science 290 (5497), 1775-1779. doi:10.1126/science.290.5497.1775
Britsch, S., Goerich, D. E., Riethmacher, D., Peirano, R. I., Rossner, M., Nave, K. A., et al. (2001). The transcription factor Sox10 is a key regulator of peripheral glial development. Genes. Dev. 15 (1), 66–78. doi:10.1101/gad.186601
Brodsky, M. C., and Hunter, J. S. (2011). Positional ocular flutter and thickened optic nerves as sentinel signs of Krabbe disease. J. AAPOS 15 (6), 595–597. doi:10.1016/j.jaapos.2011.05.024
Brück, W., Herms, J., Brockmann, K., Schulz-Schaeffer, W., and Hanefeld, F. (2001). Myelinopathia centralis diffusa (vanishing white matter disease): Evidence of apoptotic oligodendrocyte degeneration in early lesion development. Ann. Neurol. 50 (4), 532–536. doi:10.1002/ana.1227
Brussino, A., Vaula, G., Cagnoli, C., Mauro, A., Pradotto, L., Daniele, D., et al. (2009). A novel family with Lamin B1 duplication associated with adult-onset leucoencephalopathy. J. Neurol. Neurosurg. Psychiatry 80 (2), 237–240. doi:10.1136/jnnp.2008.147330
Bugiani, M., Dubey, M., Breur, M., Postma, N. L., Dekker, M. P., Ter Braak, T., et al. (2017). Megalencephalic leukoencephalopathy with cysts: The glialcam-null mouse model. Ann. Clin. Transl. Neurol. 4 (7), 450–465. doi:10.1002/acn3.405
Byun, D. S., Kasama, T., Shimizu, T., Yorifuji, H., and Seyama, Y. (1988). Effect of cholestanol feeding on sterol concentrations in the serum, liver, and cerebellum of mice. J. Biochem. 103 (2), 375–379. doi:10.1093/oxfordjournals.jbchem.a122277
Cai, J., Zhu, Q., Zheng, K., Li, H., Qi, Y., Cao, Q., et al. (2010). Co-localization of Nkx6.2 and Nkx2.2 homeodomain proteins in differentiated myelinating oligodendrocytes. Glia58 (4), 458–68.
Cali, J. J., Hsieh, C. L., Francke, U., and Russell, D. W. (1991). Mutations in the bile acid biosynthetic enzyme sterol 27-hydroxylase underlie cerebrotendinous xanthomatosis. J. Biol. Chem. 266 (12), 7779–7783. doi:10.1016/s0021-9258(20)89518-0
Carson, B. E., and De Jesus, O. (20215202). in StatPearls [internet]. Treasure island (FL) (Tampa, Florida: StatPearls Publishing).Cerebrotendinous Xanthomatosis
Carson, J. H., Worboys, K., Ainger, K., and Barbarese, E. (1997). Translocation of myelin basic protein mRNA in oligodendrocytes requires microtubules and kinesin. Cell. Motil. Cytoskelet. 38 (4), 318–328. doi:10.1002/(SICI)1097-0169(1997)38:4<318::AID-CM2>3.0.CO;2-#
Cesani, M., Lorioli, L., Grossi, S., Amico, G., Fumagalli, F., Spiga, I., et al. (2016). Mutation update of ARSA and PSAP genes causing metachromatic leukodystrophy. Hum. Mutat. 37 (1), 16–27. doi:10.1002/humu.22919
Chakraborty, G., Mekala, P., Yahya, D., Wu, G., and Ledeen, R. W. (2001). Intraneuronal N-acetylaspartate supplies acetyl groups for myelin lipid synthesis: Evidence for myelin-associated aspartoacylase. J. Neurochem. 78 (4), 736–745. doi:10.1046/j.1471-4159.2001.00456.x
Chang, C. C., Lui, C. C., Wang, J. J., Huang, S. H., Lu, C. H., Chen, C., et al. (2010). Multi-parametric neuroimaging evaluation of cerebrotendinous xanthomatosis and its correlation with neuropsychological presentations. BMC Neurol. 10, 59. doi:10.1186/1471-2377-10-59
Chanoumidou, K., Mozafari, S., Baron-Van Evercooren, A., and Kuhlmann, T. (2020). Stem cell derived oligodendrocytes to study myelin diseases. Glia 68 (4), 705–720. doi:10.1002/glia.23733
Chelban, V., Alsagob, M., Kloth, K., Chirita-Emandi, A., Vandrovcova, J., Maroofian, R., et al. (2020). Genetic and phenotypic characterization of NKX6-2-related spastic ataxia and hypomyelination. Eur. J. Neurol. 27 (2), 334–342. doi:10.1111/ene.14082
Chelban, V., Patel, N., Vandrovcova, J., Zanetti, M. N., Lynch, D. S., Ryten, M., et al. (2017). Mutations in NKX6-2 cause progressive spastic ataxia and hypomyelination. Am. J. Hum. Genet. 100 (6), 969–977. doi:10.1016/j.ajhg.2017.05.009
Cheng, A., Kawahata, I., and Fukunaga, K. (2020). Fatty acid binding protein 5 mediates cell death by psychosine exposure through mitochondrial macropores formation in oligodendrocytes. Biomedicines 8 (12), 635.
Choi, J., Yang, A., Song, A., Lim, M., Kim, J., Jang, J. H., et al. (2018). Oculodentodigital dysplasia with a novel mutation in GJA1 diagnosed by targeted gene panel sequencing: A case report and literature review. Ann. Clin. Lab. Sci. 48 (6), 776–781.
Choquet, K., Forget, D., Meloche, E., Dicaire, M. J., Bernard, G., Vanderver, A., et al. (2019). Leukodystrophy-associated POLR3A mutations down-regulate the RNA polymerase III transcript and important regulatory RNA BC200. J. Biol. Chem. 294 (18), 7445–7459. doi:10.1074/jbc.RA118.006271
Chou, W. C., Guo, Z., Guo, H., Chen, L., Zhang, G., Liang, K., et al. (2021). AIM2 in regulatory T cells restrains autoimmune diseases. Nature 591 (7849), 300–305. doi:10.1038/s41586-021-03231-w
Conant, A., Curiel, J., Pizzino, A., Sabetrasekh, P., Murphy, J., Bloom, M., et al. (2018). Absence of axoglial paranodal junctions in a child with CNTNAP1 mutations, hypomyelination, and arthrogryposis. J. Child. Neurol. 33 (10), 642–650. doi:10.1177/0883073818776157
Crow, Y. J. (2013). Aicardi-Goutières syndrome. Handb. Clin. Neurol. 113, 1629–1635. doi:10.1016/B978-0-444-59565-2.00031-9
Crow, Y. J., Leitch, A., Hayward, B. E., Garner, A., Parmar, R., Griffith, E., et al. (2006). Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutières syndrome and mimic congenital viral brain infection. Nat. Genet. 38 (8), 910–916. doi:10.1038/ng1842
Crow, Y. J., Shetty, J., and Livingston, J. H. (2020). Treatments in aicardi-goutières syndrome. Dev. Med. Child. Neurol. 62 (1), 42–47. doi:10.1111/dmcn.14268
Curiel, J., Rodríguez Bey, G., Takanohashi, A., Bugiani, M., Fu, X., Wolf, N. I., et al. (2017). TUBB4A mutations result in specific neuronal and oligodendrocytic defects that closely match clinically distinct phenotypes. Hum. Mol. Genet. 26 (22), 4506–4518. doi:10.1093/hmg/ddx338
da Silva, C. G., Bueno, A. R., Schuck, P. F., Leipnitz, G., Ribeiro, C. A., Wannmacher, C. M., et al. (2003). L-2-hydroxyglutaric acid inhibits mitochondrial creatine kinase activity from cerebellum of developing rats. Int. J. Dev. Neurosci. 21 (4), 217–224. doi:10.1016/s0736-5748(03)00035-2
Dash, P. K., Raj, D. H., and Sahu, H. (2015). Megalencephalic leucoencephalopathy with subcortical cysts: Subcortical diffuse leucoencephalopathy associated with white matter cystic degeneration. BMJ Case Rep. 21, 2015, bcr2015211921. doi:10.1136/bcr-2015-211921
de Rosa, V., Secondo, A., Pannaccione, A., Ciccone, R., Formisano, L., Guida, N., et al. (2019). D-Aspartate treatment attenuates myelin damage and stimulates myelin repair. EMBO Mol. Med. 11 (1), e9278. doi:10.15252/emmm.201809278
de Waard, D. M., and Bugiani, M. (2020). Astrocyte-oligodendrocyte-microglia crosstalk in astrocytopathies. Front. Cell. Neurosci. 14, 608073. doi:10.3389/fncel.2020.608073
Dooves, S., Bugiani, M., Postma, N. L., Polder, E., Land, N., Horan, S. T., et al. (2016). Astrocytes are central in the pathomechanisms of vanishing white matter. J. Clin. Investig. 126 (4), 1512–1524. doi:10.1172/JCI83908
Dorboz, I., Dumay-Odelot, H., Boussaid, K., Bouyacoub, Y., Barreau, P., Samaan, S., et al. (2018). Mutation in POLR3K causes hypomyelinating leukodystrophy and abnormal ribosomal RNA regulation. Neurol. Genet. 4 (6), e289. doi:10.1212/NXG.0000000000000289
Du, X., and Hu, H. (2021). The roles of 2-hydroxyglutarate. Front. Cell. Dev. Biol. 9, 651317. doi:10.3389/fcell.2021.651317
Duarri, A., Teijido, O., López-Hernández, T., Scheper, G. C., Barriere, H., Boor, I., et al. (2008). Molecular pathogenesis of megalencephalic leukoencephalopathy with subcortical cysts: Mutations in MLC1 cause folding defects. Hum. Mol. Genet. 17 (23), 3728–3739. doi:10.1093/hmg/ddn269
Dubey, M., Bugiani, M., Ridder, M. C., Postma, N. L., Brouwers, E., Polder, E., et al. (2015). Mice with megalencephalic leukoencephalopathy with cysts: A developmental angle. Ann. Neurol. 77 (1), 114–131. doi:10.1002/ana.24307
Duell, P. B., Salen, G., Eichler, F. S., DeBarber, A. E., Connor, S. L., Casaday, L., et al. (2018). Diagnosis, treatment, and clinical outcomes in 43 cases with cerebrotendinous xanthomatosis. J. Clin. Lipidol. 12 (5), 1169–1178. doi:10.1016/j.jacl.2018.06.008
Dugas, J. C., Tai, Y. C., Speed, T. P., Ngai, J., and Barres, B. A. (2006). Functional genomic analysis of oligodendrocyte differentiation. J. Neurosci. 26 (43), 10967–10983. doi:10.1523/JNEUROSCI.2572-06.2006
Edgar, J. M., McLaughlin, M., Barrie, J. A., McCulloch, M. C., Garbern, J., Griffiths, I. R., et al. (2004). Age-related axonal and myelin changes in the rumpshaker mutation of the Plp gene. Acta Neuropathol. 107 (4), 331–335. doi:10.1007/s00401-003-0808-9
Edgerley, K., Barnicoat, A., Offiah, A. C., Calder, A. D., Mankad, K., Thomas, N. S., et al. (2021). AIFM1-associated X-linked spondylometaphyseal dysplasia with cerebral hypomyelination. Am. J. Med. Genet. A 185 (4), 1228–1235. doi:10.1002/ajmg.a.62072
Ehrlich, M., Mozafari, S., Glatza, M., Starost, L., Velychko, S., Hallmann, A. L., et al. (2017). Rapid and efficient generation of oligodendrocytes from human induced pluripotent stem cells using transcription factors. Proc. Natl. Acad. Sci. U. S. A. 114 (11), E2243-E2252. doi:10.1073/pnas.1614412114
Eichler, F., Duncan, C., Musolino, P. L., Orchard, P. J., De Oliveira, S., Thrasher, A. J., et al. (2017). Hematopoietic stem-cell gene therapy for cerebral adrenoleukodystrophy. N. Engl. J. Med. 377 (17), 1630–1638. doi:10.1056/NEJMoa1700554
Eldridge, R., Anayiotos, C. P., Schlesinger, S., Cowen, D., Bever, C., Patronas, N., et al. (1984). Hereditary adult-onset leukodystrophy simulating chronic progressive multiple sclerosis. N. Engl. J. Med. 311 (15), 948–953. doi:10.1056/NEJM198410113111504
Elitt, M. S., Barbar, L., Shick, H. E., Powers, B. E., Maeno-Hikichi, Y., Madhavan, M., et al. (2020). Suppression of proteolipid protein rescues Pelizaeus-Merzbacher disease. Nature 585 (7825), 397–403. doi:10.1038/s41586-020-2494-3
Elorza-Vidal, X., Xicoy-Espaulella, E., Pla-Casillanis, A., Alonso-Gardón, M., Gaitán-Peñas, H., Engel-Pizcueta, C., et al. (2020). Structural basis for the dominant or recessive character of GLIALCAM mutations found in leukodystrophies. Hum. Mol. Genet. 29, 1107–1120. doi:10.1093/hmg/ddaa009
Elpidorou, M., Poulter, J. A., Szymanska, K., Baron, W., Junger, K., Boldt, K., et al. (2022). Missense mutation of MAL causes a rare leukodystrophy similar to Pelizaeus-Merzbacher disease. Eur. J. Hum. Genet. 25. doi:10.1038/s41431-022-01050-9
Eng, C. M., Guffon, N., Wilcox, W. R., Germain, D. P., Lee, P., Waldek, S., et al. International Collaborative Fabry Disease Study Group (2001). Safety and efficacy of recombinant human alpha-galactosidase A replacement therapy in Fabry's disease. N. Engl. J. Med. 345 (1), 9–16. doi:10.1056/NEJM200107053450102
Escolar, M. L., Poe, M. D., Provenzale, J. M., Richards, K. C., Allison, J., Wood, S., et al. (2005). Transplantation of umbilical-cord blood in babies with infantile Krabbe's disease. N. Engl. J. Med. 352 (20), 2069–2081. doi:10.1056/NEJMoa042604
Fancy, S. P., Harrington, E. P., Yuen, T. J., Silbereis, J. C., Zhao, C., Baranzini, S. E., et al. (2011). Axin2 as regulatory and therapeutic target in newborn brain injury and remyelination. Nat. Neurosci. 14 (8), 1009–1016. doi:10.1038/nn.2855
Favrais, G., van de Looij, Y., Fleiss, B., Ramanantsoa, N., Bonnin, P., Stoltenburg-Didinger, G., et al. (2011). Systemic inflammation disrupts the developmental program of white matter. Ann. Neurol. 70 (4), 550–565. doi:10.1002/ana.22489
Fellgiebel, A., Müller, M. J., Mazanek, M., Baron, K., Beck, M., and Stoeter, P. (2005). White matter lesion severity in male and female patients with Fabry disease. Neurology 65 (4), 600–602. doi:10.1212/01.wnl.0000173030.70057.eb
Feng, T., Sheng, R. R., Solé-Domènech, S., Ullah, M., Zhou, X., Mendoza, C. S., et al. (2020). A role of the frontotemporal lobar degeneration risk factor TMEM106B in myelination. Brain 143 (7), 2255–2271. doi:10.1093/brain/awaa154
Ferdinandusse, S., Denis, S., Mooyer, P. A., Dekker, C., Duran, M., Soorani-Lunsing, R. J., et al. (2006a). Clinical and biochemical spectrum of D-bifunctional protein deficiency. Ann. Neurol. 59 (1), 92–104. doi:10.1002/ana.20702
Ferdinandusse, S., Ylianttila, M. S., Gloerich, J., Koski, M. K., Oostheim, W., Waterham, H. R., et al. (2006b). Mutational spectrum of D-bifunctional protein deficiency and structure-based genotype-phenotype analysis. Am. J. Hum. Genet. 78 (1), 112–124. doi:10.1086/498880
Ferrera, D., Canale, C., Marotta, R., Mazzaro, N., Gritti, M., Mazzanti, M., et al. (2014). Lamin B1 overexpression increases nuclear rigidity in autosomal dominant leukodystrophy fibroblasts. FASEB J. 28 (9), 3906–3918. doi:10.1096/fj.13-247635
Fields, R. D. (2010). Neuroscience. Change in the brain's white matter. Science 330 (6005), 768–769. doi:10.1126/science.1199139
Filbin, M. T., and Tennekoon, G. I. (1993). Homophilic adhesion of the myelin P0 protein requires glycosylation of both molecules in the homophilic pair. J. Cell. Biol. 122 (2), 451–459. doi:10.1083/jcb.122.2.451
Finnsson, J., Sundblom, J., Dahl, N., Melberg, A., and Raininko, R. (2015). LMNB1-related autosomal-dominant leukodystrophy: Clinical and radiological course. Ann. Neurol. 78 (3), 412–425. doi:10.1002/ana.24452
Flechsig, P. E. (1920). Anatomie des menschlichen gehirns und rückenmarks auf myelogenetischer grundlage. V. 1. Vol. 1. G. Thieme.
Fletcher, J. L., Kondagari, G. S., Vite, C. H., Williamson, P., and Taylor, R. M.. Oligodendrocyte loss during the disease course in a canine model of the lysosomal storage disease fucosidosis. J. Neuropathol. Exp. Neurol. 73 (6), 536–547. doi:10.1097/NEN.0000000000000075
Fletcher, J. L., Kondagari, G. S., Wright, A. L., Thomson, P. C., Williamson, P., Taylor, R. M., et al. (2011). Myelin genes are downregulated in canine fucosidosis. Biochim. Biophys. Acta 1812 (11), 1418–1426. doi:10.1016/j.bbadis.2011.06.001
Fogli, A., Schiffmann, R., Bertini, E., Ughetto, S., Combes, P., Eymard-Pierre, E., et al. (2004). The effect of genotype on the natural history of eIF2B-related leukodystrophies. Neurology 62 (9), 1509–1517. doi:10.1212/01.wnl.0000123259.67815.db
Fogli, A., Wong, K., Eymard-Pierre, E., Wenger, J., Bouffard, J. P., Goldin, E., et al. (2002). Cree leukoencephalopathy and CACH/VWM disease are allelic at the EIF2B5 locus. Ann. Neurol. 52 (4), 506–510. doi:10.1002/ana.10339
Forss-Petter, S., Werner, H., Berger, J., Lassmann, H., Molzer, B., Schwab, M. H., et al. (1997). Targeted inactivation of the X-linked adrenoleukodystrophy gene in mice. J. Neurosci. Res.50 (5), 829–843.
Francis, J. S., Markov, V., Wojtas, I. D., Gray, S., McCown, T., Samulski, R. J., et al. (2021). Preclinical biodistribution, tropism, and efficacy of oligotropic AAV/Olig001 in a mouse model of congenital white matter disease. Mol. Ther. Methods Clin. Dev. 20, 520–534. doi:10.1016/j.omtm.2021.01.009
Friedman, J., Smith, D. E., Issa, M. Y., Stanley, V., Wang, R., Mendes, M. I., et al. (2019). Biallelic mutations in valyl-tRNA synthetase gene VARS are associated with a progressive neurodevelopmental epileptic encephalopathy. Nat. Commun. 10 (1), 707. doi:10.1038/s41467-018-07067-3
Fu, M. M., McAlear, T. S., Nguyen, H., Oses-Prieto, J. A., Valenzuela, A., Shi, R. D., et al. (2019). The Golgi outpost protein TPPP nucleates microtubules and is critical for myelination. Cell. 179 (1), 132–146. doi:10.1016/j.cell.2019.08.025
Gal, A. (2010). “Molecular genetics of fabry disease and genotype–phenotype correlation,” in Fabry disease, ed. D. Elstein, G. Altarescu, and M. Beck (Dordrecht: Springer). doi:10.1007/978-90-481-9033-1_1
Garbern, J. Y., Yool, D. A., Moore, G. J., Wilds, I. B., Faulk, M. W., Klugmann, M., et al. (2002). Patients lacking the major CNS myelin protein, proteolipid protein 1, develop length-dependent axonal degeneration in the absence of demyelination and inflammation. Brain 125 (Pt 3), 551–561. doi:10.1093/brain/awf043
Gargareta, V. I., Reuschenbach, J., Siems, S. B., Sun, T., Piepkorn, L., Mangana, C., et al. (2022). Conservation and divergence of myelin proteome and oligodendrocyte transcriptome profiles between humans and mice. Elife 11, e77019. doi:10.7554/eLife.77019
Germain, D. P., Hughes, D. A., Nicholls, K., Bichet, D. G., Giugliani, R., Wilcox, W. R., et al. (2016). Treatment of fabry's disease with the pharmacologic chaperone migalastat. N. Engl. J. Med. 375 (6), 545–555. doi:10.1056/NEJMoa1510198
Geva, M., Cabilly, Y., Assaf, Y., Mindroul, N., Marom, L., Raini, G., et al. (2010). A mouse model for eukaryotic translation initiation factor 2B-leucodystrophy reveals abnormal development of brain white matter. Brain 133 (Pt 8), 2448–2461. doi:10.1093/brain/awq180
Gieselmann, V., and Krägeloh-Mann, I. (2010). Metachromatic leukodystrophy-an update. Neuropediatrics 41 (1), 1–6. doi:10.1055/s-0030-1253412
Gieselmann, V. (2003). Metachromatic leukodystrophy: Recent research developments. J. Child. Neurol. 18 (9), 591–594. doi:10.1177/08830738030180090301
Gilbert, A., Elorza-Vidal, X., Rancillac, A., Chagnot, A., Yetim, M., Hingot, V., et al. (2021). Megalencephalic leukoencephalopathy with subcortical cysts is a developmental disorder of the gliovascular unit. Elife 10, e71379. doi:10.7554/eLife.71379
Giorgio, E., Pesce, E., Pozzi, E., Sondo, E., Ferrero, M., Morerio, C., et al. (2021). A high-content drug screening strategy to identify protein level modulators for genetic diseases: A proof-of-principle in autosomal dominant leukodystrophy. Hum. Mutat. 42 (1), 102–116. doi:10.1002/humu.24147
Goffeau, A., Barrell, B. G., Bussey, H., Davis, R. W., Dujon, B., Feldmann, H., et al. (1996). Life with 6000 genes. Science 274 (5287), 563–567. doi:10.1126/science.274.5287.546
Groeschel, S., Kühl, J. S., Bley, A. E., Kehrer, C., Weschke, B., Döring, M., et al. (2016). Long-term outcome of allogeneic hematopoietic stem cell transplantation in patients with juvenile metachromatic leukodystrophy compared with nontransplanted control patients. JAMA Neurol. 73 (9), 1133–1140. doi:10.1001/jamaneurol.2016.2067
Groeschel, S., Vollmer, B., King, M. D., and Connelly, A. (2010). Developmental changes in cerebral grey and white matter volume from infancy to adulthood. Int. J. Dev. Neurosci. 28 (6), 481–489. doi:10.1016/j.ijdevneu.2010.06.004
Guasto, A., Dubail, J., Aguilera-Albesa, S., Paganini, C., Vanhulle, C., Haouari, W., et al. (2022). Biallelic variants in SLC35B2 cause a novel chondrodysplasia with hypomyelinating leukodystrophy. Brain. 24, awac110. doi:10.1093/brain/awac110
Hagberg, B., Sourander, P., and Svennerholm, L. (1963). Diagnosis of Krabbe's infantile leucodystrophy. J. Neurol. Neurosurg. Psychiatry 26 (3), 195–198. doi:10.1136/jnnp.26.3.195
Hagemann, T. L., Powers, B., Mazur, C., Kim, A., Wheeler, S., Hung, G., et al. (2018). Antisense suppression of glial fibrillary acidic protein as a treatment for Alexander disease. Ann. Neurol. 83 (1), 27–39. doi:10.1002/ana.25118
Han, J., Sarlus, H., Wszolek, Z. K., Karrenbauer, V. D., and Harris, R. A. (2020). Microglial replacement therapy: A potential therapeutic strategy for incurable CSF1R-related leukoencephalopathy. Acta Neuropathol. Commun. 8 (1), 217. doi:10.1186/s40478-020-01093-3
Hein, S., Schönfeld, P., Kahlert, S., and Reiser, G. (2008). Toxic effects of X-linked adrenoleukodystrophy-associated, very long chain fatty acids on glial cells and neurons from rat hippocampus in culture. Hum. Mol. Genet. 17 (12), 1750–1761. doi:10.1093/hmg/ddn066
Heng, M. Y., Lin, S. T., Verret, L., Huang, Y., Kamiya, S., Padiath, Q. S., et al. (2013). Lamin B1 mediates cell-autonomous neuropathology in a leukodystrophy mouse model. J. Clin. Investig. 123 (6), 2719–2729. doi:10.1172/JCI66737
Herbert, A. L., Fu, M. M., Drerup, C. M., Gray, R. S., Harty, B. L., Ackerman, S. D., et al. (2017). Dynein/dynactin is necessary for anterograde transport of Mbp mRNA in oligodendrocytes and for myelination in vivo. Proc. Natl. Acad. Sci. U. S. A. 114 (43), E9153-E9162. doi:10.1073/pnas.1711088114
Herrero, M., Mandelboum, S., and Elroy-Stein, O. (2019). eIF2B mutations cause mitochondrial malfunction in oligodendrocytes. Neuromolecular Med. 21 (3), 303–313. doi:10.1007/s12017-019-08551-9
Hess, B., Saftig, P., Hartmann, D., Coenen, R., Lüllmann-Rauch, R., Goebel, H. H., et al. (1996). Phenotype of arylsulfatase A-deficient mice: Relationship to human metachromatic leukodystrophy. Proc. Natl. Acad. Sci. U. S. A. 93 (25), 14821–14826. doi:10.1073/pnas.93.25.14821
Hilz, M. J., Kolodny, E. H., Brys, M., Stemper, B., Haendl, T., Marthol, H., et al. (2004). Reduced cerebral blood flow velocity and impaired cerebral autoregulation in patients with Fabry disease. J. Neurol. 251 (5), 564–570. doi:10.1007/s00415-004-0364-9
Hobson, G. M., Huang, Z., Sperle, K., Sistermans, E., Rogan, P. K., Garbern, J. Y., et al. (2006). Splice-site contribution in alternative splicing of PLP1 and DM20: Molecular studies in oligodendrocytes. Hum. Mutat. 27 (1), 69–77. doi:10.1002/humu.20276
Hoegg-Beiler, M. B., Sirisi, S., Orozco, I. J., Ferrer, I., Hohensee, S., Auberson, M., et al. (2014). Disrupting MLC1 and GlialCAM and ClC-2 interactions in leukodystrophy entails glial chloride channel dysfunction. Nat. Commun. 5, 3475. doi:10.1038/ncomms4475
Holtschmidt, H., Sandhoff, K., Kwon, H. Y., Harzer, K., Nakano, T., Suzuki, K., et al. (1991). Sulfatide activator protein. Alternative splicing that generates three mRNAs and a newly found mutation responsible for a clinical disease. J. Biol. Chem. 266 (12), 7556–7560. doi:10.1016/s0021-9258(20)89483-6
Hopkin, R. J., Bissler, J., Banikazemi, M., Clarke, L., Eng, C. M., Germain, D. P., et al. (2008). Characterization of fabry disease in 352 pediatric patients in the fabry registry. Pediatr. Res. 64 (5), 550–555. doi:10.1203/PDR.0b013e318183f132
Hoshino, H., and Kubota, M. (2014). Canavan disease: Clinical features and recent advances in research. Pediatr. Int. 56 (4), 477–483. doi:10.1111/ped.12422
Hughes, D. A., Nicholls, K., Shankar, S. P., Sunder-Plassmann, G., Koeller, D., Nedd, K., et al. (2017). Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in fabry disease: 18-month results from the randomised phase III ATTRACT study. J. Med. Genet. 54 (4), 288–296. doi:10.1136/jmedgenet-2016-104178
Hughes, E. G., Kang, S. H., Fukaya, M., and Bergles, D. E. (2013). Oligodendrocyte progenitors balance growth with self-repulsion to achieve homeostasis in the adult brain. Nat. Neurosci. 16 (6), 668–676. doi:10.1038/nn.3390
Hulshagen, L., Krysko, O., Bottelbergs, A., Huyghe, S., Klein, R., Van Veldhoven, P. P., et al. (2008). Absence of functional peroxisomes from mouse CNS causes dysmyelination and axon degeneration. J. Neurosci. 28 (15), 4015–4027. doi:10.1523/JNEUROSCI.4968-07.2008
I Amaral, A., Hadera, M. G., Kotter, M., and Sonnewald, U. (2017). Oligodendrocytes do not export NAA-derived aspartate in vitro. Neurochem. Res. 42 (3), 827–837. doi:10.1007/s11064-016-1985-y
Í Dali, C., Groeschel, S., Moldovan, M., Farah, M. H., Krägeloh-Mann, I., Wasilewski, M., et al. (2021). Intravenous arylsulfatase A in metachromatic leukodystrophy: A phase 1/2 study. Ann. Clin. Transl. Neurol. 8 (1), 66–80. doi:10.1002/acn3.51254
Inoue, K., Khajavi, M., Ohyama, T., Hirabayashi, S., Wilson, J., Reggin, J. D., et al. (2004). Molecular mechanism for distinct neurological phenotypes conveyed by allelic truncating mutations. Nat. Genet. 36 (4), 361–369. doi:10.1038/ng1322
Inoue, K., Osaka, H., Sugiyama, N., Kawanishi, C., Onishi, H., Nezu, A., et al. (1996). A duplicated PLP gene causing Pelizaeus-Merzbacher disease detected by comparative multiplex PCR. Am. J. Hum. Genet. 59 (1), 32–39.
Inoue, K., Shilo, K., Boerkoel, C. F., Crowe, C., Sawady, J., Lupski, J. R., et al. (2002). Congenital hypomyelinating neuropathy, central dysmyelination, and waardenburg-hirschsprung disease: Phenotypes linked by SOX10 mutation. Ann. Neurol. 52 (6), 836–842. doi:10.1002/ana.10404
Inoue, K., Tanabe, Y., and Lupski, J. R. (1999). Myelin deficiencies in both the central and the peripheral nervous systems associated with a SOX10 mutation. Ann. Neurol. 46 (3), 313–318. doi:10.1002/1531-8249(199909)46:3<313::aid-ana6>3.0.co;2-7
Itoh, Y., Esaki, T., Cook, M., Qasba, P., Shimoji, K., Alroy, J., et al. (2001). Local and global cerebral blood flow and glucose utilization in the alpha-galactosidase A knockout mouse model of Fabry disease. J. Neurochem. 79 (6), 1217–1224. doi:10.1046/j.1471-4159.2001.00669.x
Jablonska, B., Scafidi, J., Aguirre, A., Vaccarino, F., Nguyen, V., Borok, E., et al. (2012). Oligodendrocyte regeneration after neonatal hypoxia requires FoxO1-mediated p27Kip1 expression. J. Neurosci. 32 (42), 14775–14793. doi:10.1523/JNEUROSCI.2060-12.2012
Jain, P., Ramesh, K., Mohamed, A., Kumar, A., and Gulati, S. (2012). Teaching NeuroImages: Distinct neuroimaging features of fucosidosis. Neurology 78 (5), e33. doi:10.1212/WNL.0b013e3182452910
Jakovcevski, I., Filipovic, R., Mo, Z., Rakic, S., and Zecevic, N. (2009). Oligodendrocyte development and the onset of myelination in the human fetal brain. Front. Neuroanat. 3, 5. doi:10.3389/neuro.05.005.2009
Joss, S. K., Ghazawy, S., Tomkins, S., Ahmed, M., Bradbury, J., Sheridan, E., et al. (2008). Variable expression of neurological phenotype in autosomal recessive oculodentodigital dysplasia of two sibs and review of the literature. Eur. J. Pediatr. 167 (3), 341–345. doi:10.1007/s00431-007-0468-1
Kajihara, R., Numakawa, T., Odaka, H., Yaginuma, Y., Fusaki, N., Okumiya, T., et al. (2020). Novel drug candidates improve ganglioside accumulation and neural dysfunction in GM1 gangliosidosis models with autophagy activation. Stem Cell. Rep.14 (5): 909–923.
Kaminski, D., Yaghootfam, C., Matthes, F., Reßing, A., Gieselmann, V., Matzner, U., et al. (2021). Brain cell type-specific endocytosis of arylsulfatase A identifies limitations of enzyme-based therapies for metachromatic leukodystrophy. Hum. Mol. Genet. 29 (23), 3807–3817. doi:10.1093/hmg/ddaa277
Karumuthil-Melethil, S., Marshall, M. S., Heindel, C., Jakubauskas, B., Bongarzone, E. R., and Gray, S. J. (2016). Intrathecal administration of AAV/GALC vectors in 10-11-day-old twitcher mice improves survival and is enhanced by bone marrow transplant. J. Neurosci. Res. 94 (11), 1138–1151. doi:10.1002/jnr.23882
Kassmann, C. M., Lappe-Siefke, C., Baes, M., Brügger, B., Mildner, A., Werner, H. B., et al. (2007). Axonal loss and neuroinflammation caused by peroxisome-deficient oligodendrocytes. Nat. Genet. 39 (8), 969–976. doi:10.1038/ng2070
Kassmann, C. M. (2014). Myelin peroxisomes - essential organelles for the maintenance of white matter in the nervous system. Biochimie 98, 111–118. doi:10.1016/j.biochi.2013.09.020
Kettwig, M., Schubach, M., Zimmermann, F. A., Klinge, L., Mayr, J. A., Biskup, S., et al. (2015). From ventriculomegaly to severe muscular atrophy: Expansion of the clinical spectrum related to mutations in AIFM1. Mitochondrion 21, 12–18. doi:10.1016/j.mito.2015.01.001
Kevelam, S. H., Taube, J. R., van Spaendonk, R. M., Bertini, E., Sperle, K., Tarnopolsky, M., et al. (2015). Altered PLP1 splicing causes hypomyelination of early myelinating structures. Ann. Clin. Transl. Neurol. 2 (6), 648–661. doi:10.1002/acn3.203
Khan, A., Barber, D. L., Huang, J., Rupar, C. A., Rip, J. W., Auray-Blais, C., et al. (2021). Lentivirus-mediated gene therapy for Fabry disease. Nat. Commun. 12 (1), 1178. doi:10.1038/s41467-021-21371-5
Kim, D., Lee, Y. R., Choi, T. I., Kim, S. H., Kang, H. C., Kim, C. H., et al. (2021). Comparative proteome research in a zebrafish model for vanishing white matter disease. Int. J. Mol. Sci. 22 (5), 2707. doi:10.3390/ijms22052707
Kim, Y. I., Bhandari, S., Lee, J. N., Yoo, K. W., Kim, S. J., Oh, G. S., et al. (2014). Developmental roles of D-bifunctional protein-A zebrafish model of peroxisome dysfunction. Mol. Cells 37 (1), 74–80. doi:10.14348/molcells.2014.2300
King, K. E., Kim, S., Whitley, C. B., and Jarnes-Utz, J. R. (2020). The juvenile gangliosidoses: A timeline of clinical change. Mol. Genet. Metab. Rep. 25, 100676. doi:10.1016/j.ymgmr.2020.100676
Klein, Z. A., Takahashi, H., Ma, M., Stagi, M., Zhou, M., Lam, T. T., et al. (2017). Loss of TMEM106B ameliorates lysosomal and frontotemporal dementia-related phenotypes in progranulin-deficient mice. Neuron 95 (2), 281–296. doi:10.1016/j.neuron.2017.06.026
Klugmann, M., Schwab, M. H., Pühlhofer, A., Schneider, A., Zimmermann, F., Griffiths, I. R., et al. (1997). Assembly of CNS myelin in the absence of proteolipid protein. Neuron 18 (1), 59–70. doi:10.1016/s0896-6273(01)80046-5
Kobayashi, T., Shinnoh, N., Kondo, A., and Yamada, T. (1997). Adrenoleukodystrophy protein-deficient mice represent abnormality of very long chain fatty acid metabolism. Biochem. Biophys. Res. Commun. 232 (3), 631–636. doi:10.1006/bbrc.1997.6340
Kobayashi, T., Yamanaka, T., Jacobs, J. M., Teixeira, F., and Suzuki, K. (1980). The twitcher mouse: An enzymatically authentic model of human globoid cell leukodystrophy (Krabbe disease). Brain Res. 202 (2), 479–483. doi:10.1016/0006-8993(80)90159-6
Kölker, S., Pawlak, V., Ahlemeyer, B., Okun, J. G., Hörster, F., Mayatepek, E., et al. (2002). NMDA receptor activation and respiratory chain complex V inhibition contribute to neurodegeneration in d-2-hydroxyglutaric aciduria. Eur. J. Neurosci. 16 (1), 21–28. doi:10.1046/j.1460-9568.2002.02055.x
Komatsuzaki, S., Zielonka, M., Mountford, W. K., Kölker, S., Hoffmann, G. F., Garbade, S. F., et al. (2019). Clinical characteristics of 248 patients with Krabbe disease: Quantitative natural history modeling based on published cases. Genet. Med. 21 (10), 2208–2215. doi:10.1038/s41436-019-0480-7
Kondagari, G. S., Fletcher, J. L., Cruz, R., Williamson, P., Hopwood, J. J., and Taylor, R. M. (2015). The effects of intracisternal enzyme replacement versus sham treatment on central neuropathology in preclinical canine fucosidosis. Orphanet J. Rare Dis. 10, 143. doi:10.1186/s13023-015-0357-z
Kondagari, G. S., King, B. M., Thomson, P. C., Williamson, P., Clements, P. R., Fuller, M., et al. (2011). Treatment of canine fucosidosis by intracisternal enzyme infusion. Exp. Neurol. 230 (2), 218–226. doi:10.1016/j.expneurol.2011.04.019
Kousseff, B. G., Beratis, N. G., Strauss, L., Brill, P. W., Rosenfield, R. E., Kaplan, B., et al. (1976). Fucosidosis type 2. Pediatrics 57 (2), 205–213. doi:10.1542/peds.57.2.205
Koyama, S., Sekijima, Y., Ogura, M., Hori, M., Matsuki, K., Miida, T., et al. (2021). Cerebrotendinous xanthomatosis: Molecular pathogenesis, clinical spectrum, diagnosis, and disease-modifying treatments. J. Atheroscler. Thromb. 28 (9), 905–925. doi:10.5551/jat.RV17055
Kranendijk, M., Struys, E. A., van Schaftingen, E., Gibson, K. M., Kanhai, W. A., van der Knaap, M. S., et al. (2010). IDH2 mutations in patients with D-2-hydroxyglutaric aciduria. Science 330 ((6002), 336. doi:10.1126/science.1192632
Krivit, W., Shapiro, E. G., Peters, C., Wagner, J. E., Cornu, G., Kurtzberg, J., et al. (1998). Hematopoietic stem-cell transplantation in globoid-cell leukodystrophy. N. Engl. J. Med. 338 (16), 1119–1126. doi:10.1056/NEJM199804163381605
Kumar, S., Biancotti, J. C., Matalon, R., and de Vellis, J. (2009). Lack of aspartoacylase activity disrupts survival and differentiation of neural progenitors and oligodendrocytes in a mouse model of Canavan disease. J. Neurosci. Res. 87 (15), 3415–3427. doi:10.1002/jnr.22233
Lanciotti, A., Brignone, M. S., Molinari, P., Visentin, S., De Nuccio, C., Macchia, G., et al. (2012). Megalencephalic leukoencephalopathy with subcortical cysts protein 1 functionally cooperates with the TRPV4 cation channel to activate the response of astrocytes to osmotic stress: Dysregulation by pathological mutations. Hum. Mol. Genet. 21 (10), 2166–2180. doi:10.1093/hmg/dds032
Latour, Y. L., Yoon, R., Thomas, S. E., Grant, C., Li, C., Sena-Esteves, M., et al. (2019). Human GLB1 knockout cerebral organoids: A model system for testing AAV9-mediated GLB1 gene therapy for reducing GM1 ganglioside storage in GM1 gangliosidosis. Mol. Genet. Metab. Rep. 21, 100513. doi:10.1016/j.ymgmr.2019.100513
Laukka, J. J., Kamholz, J., Bessert, D., and Skoff, R. P. (2016). Novel pathologic findings in patients with Pelizaeus-Merzbacher disease. Neurosci. Lett. 627, 222–232. doi:10.1016/j.neulet.2016.05.028
Lee, W. C., Kang, D., Causevic, E., Herdt, A. R., Eckman, E. A., and Eckman, C. B. (2010). Molecular characterization of mutations that cause globoid cell leukodystrophy and pharmacological rescue using small molecule chemical chaperones. J. Neurosci. 30 (16), 5489–5497. doi:10.1523/JNEUROSCI.6383-09.2010
Leegwater, P. A., Yuan, B. Q., van der Steen, J., Mulders, J., Könst, A. A., Boor, P. K., et al. (2001). Mutations of MLC1 (KIAA0027), encoding a putative membrane protein, cause megalencephalic leukoencephalopathy with subcortical cysts. Am. J. Hum. Genet. 68 (4), 831–838. doi:10.1086/319519
Lenders, M., Stypmann, J., Duning, T., Schmitz, B., Brand, S. M., Brand, E., et al. (2016). Serum-mediated inhibition of enzyme replacement therapy in fabry disease. J. Am. Soc. Nephrol. 27 (1), 256–264. doi:10.1681/ASN.2014121226
Li, R., Johnson, A. B., Salomons, G. S., van der Knaap, M. S., Rodriguez, D., Boespflug-Tanguy, O., et al. (2006). Propensity for paternal inheritance of de novo mutations in Alexander disease. Hum. Genet. 119 (1-2), 137–144. doi:10.1007/s00439-005-0116-7
Li, Y., and Sands, M. S. (2014). Experimental therapies in the murine model of globoid cell leukodystrophy. Pediatr. Neurol. 51 (5), 600–606. doi:10.1016/j.pediatrneurol.2014.08.003
Lieblein-Boff, J. C., McKim, D. B., Shea, D. T., Wei, P., Deng, Z., Sawicki, C., et al. (2013). Neonatal E. coli infection causes neuro-behavioral deficits associated with hypomyelination and neuronal sequestration of iron. J. Neurosci. 33 (41), 16334–16345. doi:10.1523/JNEUROSCI.0708-13.2013
Lin, J. P., Mironova, Y. A., Shrager, P., and Giger, R. J. (2017). LRP1 regulates peroxisome biogenesis and cholesterol homeostasis in oligodendrocytes and is required for proper CNS myelin development and repair. Elife 6, e30498. doi:10.7554/eLife.30498
Lin, N. H., Yang, A. W., Chang, C. H., and Perng, M. D. (2021). Elevated GFAP isoform expression promotes protein aggregation and compromises astrocyte function. FASEB J. 35 (5), e21614. doi:10.1096/fj.202100087R
Livingston, J. H., and Crow, Y. J. (2016). Neurologic phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR1, and IFIH1: Aicardi-goutières syndrome and beyond. Neuropediatrics 47 (6), 355–360. doi:10.1055/s-0036-1592307
Lo Martire, V., Alvente, S., Bastianini, S., Berteotti, C., Bombardi, C., Calandra-Buonaura, G., et al. (2018). Mice overexpressing lamin B1 in oligodendrocytes recapitulate the age-dependent motor signs, but not the early autonomic cardiovascular dysfunction of autosomal-dominant leukodystrophy (ADLD). Exp. Neurol.301 (Pt A), 1–12.
Loddenkemper, T., Grote, K., Evers, S., Oelerich, M., and Stögbauer, F. (2002). Neurological manifestations of the oculodentodigital dysplasia syndrome. J. Neurol. 249 (5), 584–595. doi:10.1007/s004150200068
Loeb, H., Tondeur, M., Jonniaux, G., Mockel-Pohl, S., and Vamos-Hurwitz, E. (1969). Biochemical and ultrastructural studies in a case of mucopolysaccharidosis "F" (fucosidosis). Helv. Paediatr. Acta 24 (5), 519–537.
Lok, H. C., and Kwok, J. B. (2021). The role of white matter dysfunction and leukoencephalopathy/leukodystrophy genes in the aetiology of frontotemporal dementias: Implications for novel approaches to therapeutics. Int. J. Mol. Sci. 22 (5), 2541. doi:10.3390/ijms22052541
López-Hernández, T., Ridder, M. C., Montolio, M., Capdevila-Nortes, X., Polder, E., Sirisi, S., et al. (2011). Mutant GlialCAM causes megalencephalic leukoencephalopathy with subcortical cysts, benign familial macrocephaly, and macrocephaly with retardation and autism. Am. J. Hum. Genet. 88 (4), 422–432. doi:10.1016/j.ajhg.2011.02.009
Lossos, A., Elazar, N., Lerer, I., Schueler-Furman, O., Fellig, Y., Glick, B., et al. (2015). Myelin-associated glycoprotein gene mutation causes Pelizaeus-Merzbacher disease-like disorder. Brain 138 (Pt 9), 2521–2536. doi:10.1093/brain/awv204
Lu, J. F., Lawler, A. M., Watkins, P. A., Powers, J. M., Moser, A. B., Moser, H. W., et al. (1997). A mouse model for X-linked adrenoleukodystrophy. Proc. Natl. Acad. Sci. U. S. A. 94 (17), 9366–9371. doi:10.1073/pnas.94.17.9366
Lumbreras, S., Ricobaraza, A., Baila-Rueda, L., Gonzalez-Aparicio, M., Mora-Jimenez, L., Uriarte, I., et al. (2021). Gene supplementation of CYP27A1 in the liver restores bile acid metabolism in a mouse model of cerebrotendinous xanthomatosis. Mol. Ther. Methods Clin. Dev.22, 210–221.
Lutz, S. E., Zhao, Y., Gulinello, M., Lee, S. C., Raine, C. S., and Brosnan, C. F. (2009). Deletion of astrocyte connexins 43 and 30 leads to a dysmyelinating phenotype and hippocampal CA1 vacuolation. J. Neurosci. 29 (24), 7743–7752. doi:10.1523/JNEUROSCI.0341-09.2009
MacDermot, K. D., Holmes, A., and Miners, A. H. (2001). Anderson-Fabry disease: Clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J. Med. Genet. 38 (11), 769–775. doi:10.1136/jmg.38.11.769
MacDermot, K. D., Holmes, A., and Miners, A. H. (2001). Anderson-Fabry disease: Clinical manifestations and impact of disease in a cohort of 98 hemizygous males. J. Med. Genet. 38 (11), 750–760. doi:10.1136/jmg.38.11.750
Maelfait, J., Liverpool, L., and Rehwinkel, J. (2020). Nucleic acid sensors and programmed cell death. J. Mol. Biol. 432 (2), 552–568. doi:10.1016/j.jmb.2019.11.016
Marelli, A. J., Mackie, A. S., Ionescu-Ittu, R., Rahme, E., and Pilote, L. (2007). Congenital heart disease in the general population: Changing prevalence and age distribution. Circulation 115 (2), 163–172. doi:10.1161/CIRCULATIONAHA.106.627224
Martinez, M. (2001). Restoring the DHA levels in the brains of Zellweger patients. J. Mol. Neurosci. 16 (2-3), 309–316. doi:10.1385/JMN:16:2-3:309
McNab, F., Mayer-Barber, K., Sher, A., Wack, A., and O'Garra, A. (2015). Type I interferons in infectious disease. Nat. Rev. Immunol. 15 (2), 87–103. doi:10.1038/nri3787
Mendes, M. I., Green, L. M. C., Bertini, E., Tonduti, D., Aiello, C., Smith, D., et al. (2020). RARS1-related hypomyelinating leukodystrophy: Expanding the spectrum. Ann. Clin. Transl. Neurol. 7 (1), 83–93. doi:10.1002/acn3.50960
Mendes, M. I., Gutierrez Salazar, M., Guerrero, K., Thiffault, I., Salomons, G. S., Gauquelin, L., et al. (2018). Bi-Allelic mutations in EPRS, encoding the glutamyl-prolyl-aminoacyl-tRNA synthetase, cause a hypomyelinating leukodystrophy. Am. J. Hum. Genet. 102 (4), 676–684. doi:10.1016/j.ajhg.2018.02.011
Merheb, E., Cui, M. H., DuBois, J. C., Branch, C. A., Gulinello, M., Shafit-Zagardo, B., et al. (2021). Defective myelination in an RNA polymerase III mutant leukodystrophic mouse. Proc. Natl. Acad. Sci. U. S. A. 118 (40), e2024378118. doi:10.1073/pnas.2024378118
Meschkat, M., Steyer, A. M., Weil, M. T., Kusch, K., Jahn, O., Piepkorn, L., et al. (2022). White matter integrity in mice requires continuous myelin synthesis at the inner tongue. Nat. Commun. 13 (1), 1163. doi:10.1038/s41467-022-28720-y
Meservey, L. M., Topkar, V. V., and Fu, M. M. (2021). mRNA transport and local translation in glia. Trends Cell. Biol. 31 (6), 419–423. doi:10.1016/j.tcb.2021.03.006
Messing, A., Head, M. W., Galles, K., Galbreath, E. J., Goldman, J. E., Brenner, M., et al. (1998). Fatal encephalopathy with astrocyte inclusions in GFAP transgenic mice. Am. J. Pathol. 152 (2), 391–398.
Mezey, E., Chandross, K. J., Harta, G., Maki, R. A., and McKercher, S. R. (2000). Turning blood into brain: Cells bearing neuronal antigens generated in vivo from bone marrow. Science290 (5497), 1779–1782. doi:10.1126/science.290.5497.1779
Miano, M., Lanino, E., Gatti, R., Morreale, G., Fondelli, P., Celle, M. E., et al. (2001). Four year follow-up of a case of fucosidosis treated with unrelated donor bone marrow transplantation. Bone Marrow Transpl. 27 (7), 747–751. doi:10.1038/sj.bmt.1702994
Mierzewska, H., Rydzanicz, M., Biegański, T., Kosinska, J., Mierzewska-Schmidt, M., Ługowska, A., et al. (2017). Spondyloepimetaphyseal dysplasia with neurodegeneration associated with AIFM1 mutation - a novel phenotype of the mitochondrial disease. Clin. Genet. 91 (1), 30–37. doi:10.1111/cge.12792
Mimault, C., Giraud, G., Courtois, V., Cailloux, F., Boire, J. Y., Dastugue, B., et al. (1999). Proteolipoprotein gene analysis in 82 patients with sporadic pelizaeus-merzbacher disease: Duplications, the major cause of the disease, originate more frequently in male germ cells, but point mutations do not. The clinical European network on brain dysmyelinating disease. Am. J. Hum. Genet. 65 (2), 360–369. doi:10.1086/302483
Minchenberg, S. B., and Massa, P. T. (2019). The control of oligodendrocyte bioenergetics by interferon-gamma (IFN-γ) and Src homology region 2 domain-containing phosphatase-1 (SHP-1). J. Neuroimmunol. 331, 46–57. doi:10.1016/j.jneuroim.2017.10.015
Miyake, N., Wolf, N. I., Cayami, F. K., Crawford, J., Bley, A., Bulas, D., et al. (2017). X-linked hypomyelination with spondylometaphyseal dysplasia (H-SMD) associated with mutations in AIFM1. Neurogenetics 18 (4), 185–194. doi:10.1007/s10048-017-0520-x
Molander-Melin, M., Pernber, Z., Franken, S., Gieselmann, V., Månsson, J. E., Fredman, P., et al. (2004). Accumulation of sulfatide in neuronal and glial cells of arylsulfatase A deficient mice. J. Neurocytol. 33 (4), 417–427. doi:10.1023/B:NEUR.0000046572.53905.2c
Moore, D. F., Altarescu, G., Barker, W. C., Patronas, N. J., Herscovitch, P., Schiffmann, R., et al. (2003). White matter lesions in Fabry disease occur in 'prior' selectively hypometabolic and hyperperfused brain regions. Brain Res. Bull. 62 (3), 231–240. doi:10.1016/j.brainresbull.2003.09.021
Morrone, A., Bardelli, T., Donati, M. A., Giorgi, M., Di Rocco, M., Gatti, R., et al. (2000). beta-galactosidase gene mutations affecting the lysosomal enzyme and the elastin-binding protein in GM1-gangliosidosis patients with cardiac involvement. Hum. Mutat.15 (4), 354–366
Morse, L. E., and Rosman, N. P. (2006). Myoclonic seizures in Krabbe disease: A unique presentation in late-onset type. Pediatr. Neurol. 35 (2), 154–157. doi:10.1016/j.pediatrneurol.2006.02.004
Moser, A. B., Kreiter, N., Bezman, L., Lu, S., Raymond, G. V., Naidu, S., et al. (1999). Plasma very long chain fatty acids in 3, 000 peroxisome disease patients and 29, 000 controls. Ann. Neurol. 45 (1), 100–110. doi:10.1002/1531-8249(199901)45:1<100::aid-art16>3.0.co;2-u
Nagy, J. I., Ionescu, A. V., Lynn, B. D., and Rash, J. E. (2003). Coupling of astrocyte connexins Cx26, Cx30, Cx43 to oligodendrocyte Cx29, Cx32, Cx47: Implications from normal and connexin32 knockout mice. Glia 44 (3), 205–218. doi:10.1002/glia.10278
Nave, K. A., Lai, C., Bloom, F. E., and Milner, R. J. (1987). Splice site selection in the proteolipid protein (PLP) gene transcript and primary structure of the DM-20 protein of central nervous system myelin. Proc. Natl. Acad. Sci. U. S. A. 84 (16), 5665–5669. doi:10.1073/pnas.84.16.5665
Nawaz, S., Kippert, A., Saab, A. S., Werner, H. B., Lang, T., Nave, K. A., et al. (2009). Phosphatidylinositol 4,5-bisphosphate-dependent interaction of myelin basic protein with the plasma membrane in oligodendroglial cells and its rapid perturbation by elevated calcium. J. Neurosci. 29 (15), 4794–4807. doi:10.1523/JNEUROSCI.3955-08.2009
Nestrasil, I., Ahmed, A., Utz, J. M., Rudser, K., Whitley, C. B., Jarnes-Utz, J. R., et al. (2018). Distinct progression patterns of brain disease in infantile and juvenile gangliosidoses: Volumetric quantitative MRI study. Mol. Genet. Metab. 123 (2), 97–104. doi:10.1016/j.ymgme.2017.12.432
Ng, N., Cabral-da-Silva, M. C., Maksour, S., Berg, T., Engel, M., Silva, D. M., et al. (2020). Identification of repurposable cytoprotective drugs in vanishing white matter disease patient-derived cells. Transl. Med. Commun. 21, 18. doi:10.1186/s41231-020-00071-0
Nguyen, H., Meservey, L. M., Ishiko-Silveria, N., Zhou, M., Huang, T. T., Fu, M. M., et al. (2020). Fear deficits in hypomyelinated tppp knock-out mice. eNeuro 7 (5), ENEURO.0170–20.2020. doi:10.1523/ENEURO.0170-20.2020
Nobuta, H., Yang, N., Ng, Y. H., Marro, S. G., Sabeur, K., Chavali, M., et al. (2019). Oligodendrocyte death in pelizaeus-merzbacher disease is rescued by iron chelation. Cell. Stem Cell. 25 (4), 531–541. doi:10.1016/j.stem.2019.09.003
Norton, W. T., and Poduslo, S. E. (1973). Myelination in rat brain: Changes in myelin composition during brain maturation. J. Neurochem. 21 (4), 759–773. doi:10.1111/j.1471-4159.1973.tb07520.x
Nota, B., Struys, E. A., Pop, A., Jansen, E. E., Fernandez Ojeda, M. R., Kanhai, W. A., et al. (2013). Deficiency in SLC25A1, encoding the mitochondrial citrate carrier, causes combined D-2- and L-2-hydroxyglutaric aciduria. Am. J. Hum. Genet. 92 (4), 627–631. doi:10.1016/j.ajhg.2013.03.009
Numasawa-Kuroiwa, Y., Okada, Y., Shibata, S., Kishi, N., Akamatsu, W., Shoji, M., et al. (2014). Involvement of ER stress in dysmyelination of Pelizaeus-Merzbacher Disease with PLP1 missense mutations shown by iPSC-derived oligodendrocytes. Stem Cell. Rep. 24 (51), 648–661. doi:10.1016/j.stemcr.2014.03.007
O'Brien, J. S. (1965). Stability of the myelin membrane. Science 147 (3662), 1099–1107. doi:10.1126/science.147.3662.1099
Odermatt, B., Wellershaus, K., Wallraff, A., Seifert, G., Degen, J., Euwens, C., et al. (2003). Connexin 47 (Cx47)-deficient mice with enhanced green fluorescent protein reporter gene reveal predominant oligodendrocytic expression of Cx47 and display vacuolized myelin in the CNS. J. Neurosci. 23 (11), 4549–4559. doi:10.1523/JNEUROSCI.23-11-04549.2003
Olivieri, I., Cattalini, M., Tonduti, D., La Piana, R., Uggetti, C., Galli, J., et al. (2013). Dysregulation of the immune system in aicardi-goutières syndrome: Another example in a TREX1-mutated patient. Lupus 22 (10), 1064–1069. doi:10.1177/0961203313498800
Orsini, J. J., Saavedra-Matiz, C. A., Gelb, M. H., and Caggana, M. (2016). Newborn screening for Krabbe's disease. J. Neurosci. Res. 94 (11), 1063–1075. doi:10.1002/jnr.23781
Orthmann-Murphy, J. L., Freidin, M., Fischer, E., Scherer, S. S., and Abrams, C. K. (2007). Two distinct heterotypic channels mediate gap junction coupling between astrocyte and oligodendrocyte connexins. J. Neurosci. 27 (51), 13949–13957. doi:10.1523/JNEUROSCI.3395-07.2007
Owczarek-Lipska, M., Mulahasanovic, L., Obermaier, C. D., Hörtnagel, K., Neubauer, B. A., Korenke, G. C., et al. (2019). Novel mutations in the GJC2 gene associated with Pelizaeus-Merzbacher-like disease. Mol. Biol. Rep. 46 (4), 4507–4516. doi:10.1007/s11033-019-04906-4
Pace, N. P., Benoit, V., Agius, D., Grima, M. A., Parascandalo, R., Hilbert, P., et al. (2019). Two novel GJA1 variants in oculodentodigital dysplasia. Mol. Genet. Genomic Med. 7 (9), e882. doi:10.1002/mgg3.882
Padiath, Q. S., Saigoh, K., Schiffmann, R., Asahara, H., Yamada, T., Koeppen, A., et al. (2006). Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat Genet. 38 (10), 1114–1123. doi:10.1038/ng0207-276c
Paker, A. M., Sunness, J. S., Brereton, N. H., Speedie, L. J., Albanna, L., Dharmaraj, S., et al. (2010). Docosahexaenoic acid therapy in peroxisomal diseases: Results of a double-blind, randomized trial. Neurology 75 (9), 826–830. doi:10.1212/WNL.0b013e3181f07061
Park, K., Hoff, K. J., Wethekam, L., Stence, N., Saenz, M., and Moore, J. K. (2021). Kinetically stabilizing mutations in beta tubulins create isotype-specific brain malformations. Front. Cell. Dev. Biol. 9, 765992. doi:10.3389/fcell.2021.765992
Patil, S. A., and Maegawa, G. H. (2013). Developing therapeutic approaches for metachromatic leukodystrophy. Drug Des. devel. Ther. 7, 729–745. doi:10.2147/DDDT.S15467
Patzig, J., Erwig, M. S., Tenzer, S., Kusch, K., Dibaj, P., Möbius, W., et al. (2016). Septin/anillin filaments scaffold central nervous system myelin to accelerate nerve conduction. Elife 5, e17119. doi:10.7554/eLife.17119
Paznekas, W. A., Boyadjiev, S. A., Shapiro, R. E., Daniels, O., Wollnik, B., Keegan, C. E., et al. (2003). Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia. Am. J. Hum. Genet. 72 (2), 408–418. doi:10.1086/346090
Pierre, G., Setchell, K., Blyth, J., Preece, M. A., Chakrapani, A., McKiernan, P., et al. (2008). Prospective treatment of cerebrotendinous xanthomatosis with cholic acid therapy. J. Inherit. Metab. Dis. 31 (Suppl. 2), S241–S245. doi:10.1007/s10545-008-0815-z
Pingault, V., Bondurand, N., Kuhlbrodt, K., Goerich, D. E., Préhu, M. O., Puliti, A., et al. (1998). SOX10 mutations in patients with Waardenburg-Hirschsprung disease. Nat. Genet. 18 (2), 171–173. doi:10.1038/ng0298-171
Poitelon, Y., Kopec, A. M., and Belin, S. (2020). Myelin fat facts: An overview of lipids and fatty acid metabolism. Cells 9 (4), 812. doi:10.3390/cells9040812
Poll-The, B. T., Gootjes, J., Duran, M., De Klerk, J. B., Wenniger-Prick, L. J., Admiraal, R. J., et al. (2004). Peroxisome biogenesis disorders with prolonged survival: Phenotypic expression in a cohort of 31 patients. Am. J. Med. Genet. A 126A (4), 333–338. doi:10.1002/ajmg.a.20664
Prada, V., Passalacqua, M., Bono, M., Luzzi, P., Scazzola, S., Nobbio, L. A., et al. (2012). Gain of glycosylation: A new pathomechanism of myelin protein zero mutations. Ann. Neurol. 71 (3), 427–431. doi:10.1002/ana.22695
Priller, J., Flügel, A., Wehner, T., Boentert, M., Haas, C. A., Prinz, M., et al. (2001). Targeting gene-modified hematopoietic cells to the central nervous system: Use of green fluorescent protein uncovers microglial engraftment. Nat. Med. 7 (12), 1356–1361. doi:10.1038/nm1201-1356
Prust, M., Wang, J., Morizono, H., Messing, A., Brenner, M., Gordon, E., et al. (2011). GFAP mutations, age at onset, and clinical subtypes in Alexander disease. Neurology 77 (13), 1287–1294. doi:10.1212/WNL.0b013e3182309f72
Pujol, A., Hindelang, C., Callizot, N., Bartsch, U., Schachner, M., Mandel, J. L., et al. (2002). Late onset neurological phenotype of the X-ALD gene inactivation in mice: A mouse model for adrenomyeloneuropathy. Hum. Mol. Genet. 11 (5), 499–505. doi:10.1093/hmg/11.5.499
Raasakka, A., and Kursula, P. (20201820). Flexible players within the sheaths: The intrinsically disordered proteins of myelin in health and disease. Cells9 (2), 470.
Rademakers, R., Baker, M., Nicholson, A. M., Rutherford, N. J., Finch, N., Soto-Ortolaza, A., et al. (2011). Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat. Genet. 44 (2), 200–205. doi:10.1038/ng.1027
Rafi, M. A., Luzi, P., and Wenger, D. A. (2021). Can early treatment of twitcher mice with high dose AAVrh10-GALC eliminate the need for BMT? Bioimpacts 11 (2), 135–146. doi:10.34172/bi.2021.21
Raskind, W. H., Williams, C. A., Hudson, L. D., and Bird, T. D. (1991). Complete deletion of the proteolipid protein gene (PLP) in a family with X-linked Pelizaeus-Merzbacher disease. Am. J. Hum. Genet. 49 (6), 1355–1360.
Ratti, S., Rusciano, I., Mongiorgi, S., Owusu Obeng, E., Cappellini, A., Teti, G., et al. (2021). Cell signaling pathways in autosomal-dominant leukodystrophy (ADLD): The intriguing role of the astrocytes. Cell. Mol. Life Sci. 78 (6), 2781–2795. doi:10.1007/s00018-020-03661-1
Regier, D. S., Tifft, C. J., and Rothermel, C. E. (20131993–2022). “GLB1-Related disorders,” in GeneReviews® [internet], ed. M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. W. Grippet al. (Seattle, WA: University of Washington, Seattle).
Ribeiro, R. T., Seminotti, B., Zanatta, Â., de Oliveira, F. H., Amaral, A. U., Leipnitz, G., et al. (2021). Neuronal death, glial reactivity, microglia activation, oxidative stress and bioenergetics impairment caused by intracerebroventricular administration of D-2-hydroxyglutaric acid to neonatal rats. Neuroscience 471, 115–132. doi:10.1016/j.neuroscience.2021.07.024
Rolyan, H., Tyurina, Y. Y., Hernandez, M., Amoscato, A. A., Sparvero, L. J., Nmezi, B. C., et al. (2015). Defects of lipid synthesis are linked to the age-dependent demyelination caused by lamin B1 overexpression. J. Neurosci. 35 (34), 12002–12017. doi:10.1523/JNEUROSCI.1668-15.2015
Rombach, S. M., Smid, B. E., Bouwman, M. G., Linthorst, G. E., Dijkgraaf, M. G., and Hollak, C. E. (2013). Long term enzyme replacement therapy for fabry disease: Effectiveness on kidney, heart and brain. Orphanet J. Rare Dis. 8, 47. doi:10.1186/1750-1172-8-47
Rosenberg, J. B., Kaminsky, S. M., Aubourg, P., Crystal, R. G., and Sondhi, D. (2016). Gene therapy for metachromatic leukodystrophy. J. Neurosci. Res. 94 (11), 1169–1179. doi:10.1002/jnr.23792
Rossi, M., Balint, B., Millar Vernetti, P., Bhatia, K. P., and Merello, M. (2018). Genetic dystonia-ataxia syndromes: Clinical spectrum, diagnostic approach, and treatment options. Mov. Disord. Clin. Pract. 5 (4), 373–382. doi:10.1002/mdc3.12635
Rzem, R., Achouri, Y., Marbaix, E., Schakman, O., Wiame, E., Marie, S., et al. (2015). A mouse model of L-2-hydroxyglutaric aciduria, a disorder of metabolite repair. PLoS One 10 (3), e0119540. doi:10.1371/journal.pone.0119540
Rzem, R., Veiga-da-Cunha, M., Noël, G., Goffette, S., Nassogne, M. C., Tabarki, B., et al. (2004). A gene encoding a putative FAD-dependent L-2-hydroxyglutarate dehydrogenase is mutated in L-2-hydroxyglutaric aciduria. Proc. Natl. Acad. Sci. U. S. A. 101 (48), 16849–16854. doi:10.1073/pnas.0404840101
Rzem, R., Vincent, M. F., Van Schaftingen, E., and Veiga-da-Cunha, M. (2007). L-2-hydroxyglutaric aciduria, a defect of metabolite repair. J. Inherit. Metab. Dis. 30 (5), 681–689. doi:10.1007/s10545-007-0487-0
Saito, K., Shigetomi, E., Yasuda, R., Sato, R., Nakano, M., Tashiro, K., et al. (2018). Aberrant astrocyte Ca2+ signals "AxCa signals" exacerbate pathological alterations in an Alexander disease model. Glia 66 (5), 1053–1067. doi:10.1002/glia.23300
Saitoh, M., Sakakihara, Y., Mizuguchi, M., Itoh, M., Takashima, S., Iwamori, M., et al. (2007). Increase of ceramide monohexoside and dipalmitoyl glycerophospholipids in the brain of Zellweger syndrome. Neurosci. Lett. 417 (2), 165–170. doi:10.1016/j.neulet.2007.01.083
Sakai, N. (2009). Pathogenesis of leukodystrophy for Krabbe disease: Molecular mechanism and clinical treatment. Brain Dev. 31 (7), 485–487. doi:10.1016/j.braindev.2009.03.001
Salen, G., Meriwether, T. W., and Nicolau, G. (1975). Chenodeoxycholic acid inhibits increased cholesterol and cholestanol synthesis in patients with cerebrotendinous xanthomatosis. Biochem. Med. 14 (1), 57–74. doi:10.1016/0006-2944(75)90020-4
Samuraki, M., Komai, K., Hasegawa, Y., Kimura, M., Yamaguchi, S., Terada, N., et al. (2008). A successfully treated adult patient with L-2-hydroxyglutaric aciduria. Neurology 70 (13), 1051–1052. doi:10.1212/01.wnl.0000287141.90944.95
Sanchez, V. B., Ali, S., Escobar, A., and Cuajungco, M. P. (2019). Transmembrane 163 (TMEM163) protein effluxes zinc. Arch. Biochem. Biophys., 677, 108166. doi:10.1016/j.abb.2019.108166
Sano, R., Annunziata, I., Patterson, A., Moshiach, S., Gomero, E., Opferman, J., et al. (2009). GM1-ganglioside accumulation at the mitochondria-associated ER membranes links ER stress to Ca(2+)-dependent mitochondrial apoptosis. Mol. Cell. 36 (3), 500–511. doi:10.1016/j.molcel.2009.10.021
Sase, S., Almad, A. A., Boecker, C. A., Guedes-Dias, P., Li, J. J., Takanohashi, A., et al. (2020). TUBB4A mutations result in both glial and neuronal degeneration in an H-ABC leukodystrophy mouse model. Elife 9, e52986. doi:10.7554/eLife.52986
Sase, S., Takanohashi, A., Vanderver, A., and Almad, A. (2018). Astrocytes, an active player in Aicardi-Goutières syndrome. Brain Pathol.28 (3), 399–407
Sassa, T., Wakashima, T., Ohno, Y., and Kihara, A. (2014). Lorenzo's oil inhibits ELOVL1 and lowers the level of sphingomyelin with a saturated very long-chain fatty acid. J. Lipid Res. 55 (3), 524–530. doi:10.1194/jlr.M044586
Schaeren-Wiemers, N., Bonnet, A., Erb, M., Erne, B., Bartsch, U., Kern, F., et al. (2004). The raft-associated protein MAL is required for maintenance of proper axon-glia interactions in the central nervous system. J. Cell. Biol. 166 (5), 731–742. doi:10.1083/jcb.200406092
Schiffmann, R., Moller, J. R., Trapp, B. D., Shih, H. H., Farrer, R. G., Katz, D. A., et al. (1994). Childhood ataxia with diffuse central nervous system hypomyelination. Ann. Neurol. 35 (3), 331–340. doi:10.1002/ana.410350314
Schiffmann, R., van der Knaap, M. S., Fogli, A., and Boespflug-Tanguy, O. (2019). Abbink TEM, childhood ataxia with central nervous system hypomyelination/vanishing white matter. GeneReviews® [internet] ed. M. P. Adam, H. H. Ardinger, R. A. Pagonet al. (Seattle, WA: University of Washington, Seattle). https://www.ncbi.nlm.nih.gov/books/NBK1258/.
Schiffmann, R., Warnock, D. G., Banikazemi, M., Bultas, J., Linthorst, G. E., Packman, S., et al. (2009). Fabry disease: Progression of nephropathy, and prevalence of cardiac and cerebrovascular events before enzyme replacement therapy. Nephrol. Dial. Transpl. 24 (7), 2102–2111. doi:10.1093/ndt/gfp031
Schmitt, A., Gofferje, V., Weber, M., Meyer, J., Mössner, R., Lesch, K. P., et al. (2003). The brain-specific protein MLC1 implicated in megalencephalic leukoencephalopathy with subcortical cysts is expressed in glial cells in the murine brain. Glia 44 (3), 283–295. doi:10.1002/glia.10304
Schuster, J., Sundblom, J., Thuresson, A. C., Hassin-Baer, S., Klopstock, T., Dichgans, M., et al. (2011). Genomic duplications mediate overexpression of lamin B1 in adult-onset autosomal dominant leukodystrophy (ADLD) with autonomic symptoms. Neurogenetics 12 (1), 65–72. doi:10.1007/s10048-010-0269-y
Schwartz, N. B., and Domowicz, M. S. (2018). Proteoglycans in brain development and pathogenesis. FEBS Lett. 592 (23), 3791–3805. doi:10.1002/1873-3468.13026
Schweighauser, M., Arseni, D., Bacioglu, M., Huang, M., Lövestam, S., Shi, Y., et al. (2022). Age-dependent formation of TMEM106B amyloid filaments in human brains. Nature 28, 310–314. doi:10.1038/s41586-022-04650-z
Segel, R., Anikster, Y., Zevin, S., Steinberg, A., Gahl, W. A., Fisher, D., et al. (2011). A safety trial of high dose glyceryl triacetate for Canavan disease. Mol. Genet. Metab. 103 (3), 203–206. doi:10.1016/j.ymgme.2011.03.012
Serizawa, S., Otsuka, H., Seyama, Y., and Yamakawa, T. (1982). Studies on the biosynthesis of cholestanol in cultured cells. J. Biochem. 92 (5), 1547–1557. doi:10.1093/oxfordjournals.jbchem.a134079
Sevin, C., Aubourg, P., and Cartier, N. (2007). Enzyme, cell and gene-based therapies for metachromatic leukodystrophy. J. Inherit. Metab. Dis. 30 (2), 175–183. doi:10.1007/s10545-007-0540-z
Sevrioukova, I. F. (2016). Structure/function relations in AIFM1 variants associated with neurodegenerative disorders. J. Mol. Biol. 428 (18), 3650–3665. doi:10.1016/j.jmb.2016.05.004
Shapiro, E., Krivit, W., Lockman, L., Jambaqué, I., Peters, C., Cowan, M., et al. (2000). Long-term effect of bone-marrow transplantation for childhood-onset cerebral X-linked adrenoleukodystrophy. Lancet356 (9231), 713–718. doi:10.1016/S0140-6736(00)02629-5
Shin, J. Y., and Worman, H. J. (2022). Molecular pathology of laminopathies. Annu. Rev. Pathol. 17, 159–180. doi:10.1146/annurev-pathol-042220-034240
Shinya, A., Takahashi, M., Sato, N., Nishida, Y., Inaba, A., Inaji, M., et al. (2021). Oculo-dento-digital dysplasia presenting as spastic paraparesis which was successfully treated by intrathecal baclofen therapy. Intern Med. 60 (14), 2301–2305. doi:10.2169/internalmedicine.6145-20
Shukla, A., Das Bhowmik, A., Hebbar, M., Rajagopal, K. V., Girisha, K. M., Gupta, N., et al. (2018). Homozygosity for a nonsense variant in AIMP2 is associated with a progressive neurodevelopmental disorder with microcephaly, seizures, and spastic quadriparesis. J. Hum. Genet. 63 (1), 19–25. doi:10.1038/s10038-017-0363-1
Siekierska, A., Stamberger, H., Deconinck, T., Oprescu, S. N., Partoens, M., Zhang, Y., et al. C4RCD Research Group; AR working group of the EuroEPINOMICS RES Consortium (2019). Biallelic VARS variants cause developmental encephalopathy with microcephaly that is recapitulated in vars knockout zebrafish. Nat. Commun. 10 (1), 708. doi:10.1038/s41467-018-07953-w
Simons, C., Dyment, D., Bent, S. J., Crawford, J., D'Hooghe, M., Kohlschütter, A., Venkateswaran, S., et al. Care4Rare Consortium (2017). A recurrent de novo mutation in TMEM106B causes hypomyelinating leukodystrophy. Brain 140 (12), 3105–3111. doi:10.1093/brain/awx314
Simons, C., Wolf, N. I., McNeil, N., Caldovic, L., Devaney, J. M., Takanohashi, A., et al. (2013). A de novo mutation in the β-tubulin gene TUBB4A results in the leukoencephalopathy hypomyelination with atrophy of the basal ganglia and cerebellum. Am. J. Hum. Genet. 92 (5), 767–773. doi:10.1016/j.ajhg.2013.03.018
Sipione, S., Monyror, J., Galleguillos, D., Steinberg, N., and Kadam, V. (2020). Gangliosides in the brain: Physiology, pathophysiology and therapeutic applications. Front. Neurosci. 14, 572965. doi:10.3389/fnins.2020.572965
Sirisi, S., Folgueira, M., López-Hernández, T., Minieri, L., Pérez-Rius, C., Gaitán-Peñas, H., et al. (2014). Megalencephalic leukoencephalopathy with subcortical cysts protein 1 regulates glial surface localization of GLIALCAM from fish to humans. Hum. Mol. Genet. 23 (19), 5069–5086. doi:10.1093/hmg/ddu231
Sistermans, E. A., de Coo, R. F., De Wijs, I. J., and Van Oost, B. A. (1998). Duplication of the proteolipid protein gene is the major cause of Pelizaeus-Merzbacher disease. Neurology 50 (6), 1749–1754. doi:10.1212/wnl.50.6.1749
Snaidero, N., Möbius, W., Czopka, T., Hekking, L. H., Mathisen, C., Verkleij, D., et al. (2014). Myelin membrane wrapping of CNS axons by PI(3,4,5)P3-dependent polarized growth at the inner tongue. Cell. 156 (1-2), 277–290. doi:10.1016/j.cell.2013.11.044
Snaidero, N., Velte, C., Myllykoski, M., Raasakka, A., Ignatev, A., Werner, H. B., et al. (2017). Antagonistic functions of MBP and CNP establish cytosolic channels in CNS myelin. Cell. Rep. 18 (2), 314–323. doi:10.1016/j.celrep.2016.12.053
Son, M. Y., Kwak, J. E., Seol, B., Lee, D. Y., Jeon, H., Cho, Y. S., et al. (2015). A novel human model of the neurodegenerative disease GM1 gangliosidosis using induced pluripotent stem cells demonstrates inflammasome activation. J. Pathol. 237 (1), 98–110. doi:10.1002/path.4551
Southwood, C., He, C., Garbern, J., Kamholz, J., Arroyo, E., and Gow, A. (2004). CNS myelin paranodes require Nkx6-2 homeoprotein transcriptional activity for normal structure. J. Neurosci. 24 (50), 11215–11225. doi:10.1523/JNEUROSCI.3479-04.2004
Spada, M., Pagliardini, S., Yasuda, M., Tukel, T., Thiagarajan, G., Sakuraba, H., et al. (2006). High incidence of later-onset fabry disease revealed by newborn screening. Am. J. Hum. Genet. 79 (1), 31–40. doi:10.1086/504601
Stahl, W. L., Sumi, S. M., and Swanson, P. D. (1971). Subcellular distribution of cerebral cholestanol in cerebrotendinous xanthomatosis. J. Neurochem. 18 (3), 403–413. doi:10.1111/j.1471-4159.1971.tb11968.x
Stanley, E. R., Berg, K. L., Einstein, D. B., Lee, P. S., Pixley, F. J., Wang, Y., et al. (1997). Biology and action of colony-stimulating factor-1. Mol. Reprod. Dev.46 (1), 4–10
Steenweg, M. E., Vanderver, A., Blaser, S., Bizzi, A., de Koning, T. J., Mancini, G. M., et al. (2010). Magnetic resonance imaging pattern recognition in hypomyelinating disorders. Brain 133 (10), 2971–2982. doi:10.1093/brain/awq257
Steinberg, S. J., Dodt, G., Raymond, G. V., Braverman, N. E., Moser, A. B., Moser, H. W., et al. (2006). Peroxisome biogenesis disorders. Biochim. Biophys. Acta 1763 (12), 1733–1748. doi:10.1016/j.bbamcr.2006.09.010
Steinberg, S. J., Raymond, G. V., Braverman, N. E., and Moser, A. B. (2020). “Zellweger spectrum disorder,” in GeneReviews® [internet], ed. M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. W. Grippet al. (Seattle, WA: University of Washington, Seattle).
Stepien, K. M., Ciara, E., and Jezela-Stanek, A. (2020). Fucosidosis-clinical manifestation, long-term outcomes, and genetic profile-review and case series. Genes. (Basel) 11 (11), 1383. doi:10.3390/genes11111383
Stoll, B. J., Hansen, N. I., Adams-Chapman, I., Fanaroff, A. A., Hintz, S. R., and Vohr, B.National Institute of Child Health and Human Development Neonatal Research Networket al. (2004). Neurodevelopmental and growth impairment among extremely low-birth-weight infants with neonatal infection. JAMA 292 (19), 2357–2365. doi:10.1001/jama.292.19.2357
Stolt, C. C., Rehberg, S., Ader, M., Lommes, P., Riethmacher, D., Schachner, M., et al. (2002). Terminal differentiation of myelin-forming oligodendrocytes depends on the transcription factor Sox10. Genes. Dev. 16 (2), 165–170. doi:10.1101/gad.215802
Stradomska, T. J., Syczewska, M., Jamroz, E., Pleskaczyńska, A., Kruczek, P., Ciara, E., et al. (2020). Serum very long-chain fatty acids (VLCFA) levels as predictive biomarkers of diseases severity and probability of survival in peroxisomal disorders. PLoS One15 (9), e0238796.
Stradomska, T. J., and Tylki-Szymańska, A. (2009). Serum very-long-chain fatty acids levels determined by gas chromatography in the diagnosis of peroxisomal disorders in Poland. Folia Neuropathol. 47 (4), 306–313.
Stroobants, S., Wolf, H., Callaerts-Vegh, Z., Dierks, T., Lübke, T., and D'Hooge, R. (2018). Sensorimotor and neurocognitive dysfunctions parallel early telencephalic neuropathology in fucosidosis mice. Front. Behav. Neurosci. 12, 69. doi:10.3389/fnbeh.2018.00069
Struys, E. A., Salomons, G. S., Achouri, Y., Van Schaftingen, E., Grosso, S., Craigen, W. J., et al. (2005). Mutations in the D-2-hydroxyglutarate dehydrogenase gene cause D-2-hydroxyglutaric aciduria. Am. J. Hum. Genet. 76 (2), 358–360. doi:10.1086/427890
Susin, S. A., Lorenzo, H. K., Zamzami, N., Marzo, I., Snow, B. E., Brothers, G. M., et al. (1999). Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397 (6718), 441–446. doi:10.1038/17135
Suzuki, K., and Suzuki, Y. (1970). Globoid cell leucodystrophy (Krabbe's disease): deficiency of galactocerebroside beta-galactosidase. Proc. Natl. Acad. Sci. U. S. A. 66 (2), 302–309. doi:10.1073/pnas.66.2.302
Sweeley, C. C., and Fabry's Disease, Klionsky B. (1963). Fabry's Disease: Classification as a Sphingolipidosis and Partial Characterization of a Novel Glycolipid. J. Biol. Chem. 238, 3148–3150. doi:10.1016/s0021-9258(18)51888-3
Taft, R. J., Vanderver, A., Leventer, R. J., Damiani, S. A., Simons, C., Grimmond, S. M., et al. (2013). Mutations in DARS cause hypomyelination with brain stem and spinal cord involvement and leg spasticity. Am. J. Hum. Genet. 92 (5), 774–780. doi:10.1016/j.ajhg.2013.04.006
Takamura, A., Higaki, K., Kajimaki, K., Otsuka, S., Ninomiya, H., Matsuda, J., et al. (2008). Enhanced autophagy and mitochondrial aberrations in murine G(M1)-gangliosidosis. Biochem. Biophys. Res. Commun. 367 (3), 616–622. doi:10.1016/j.bbrc.2007.12.187
Tantzer, S., Sperle, K., Kenaley, K., Taube, J., and Hobson, G. M. (2018). Morpholino Antisense Oligomers as a Potential Therapeutic Option for the Correction of Alternative Splicing in PMD, SPG2, and HEMS. Mol. Ther. Nucleic Acids 12, 420–432. doi:10.1016/j.omtn.2018.05.019
Taube, J. R., Sperle, K., Banser, L., Seeman, P., Cavan, B. C., Garbern, J. Y., et al. (2014). PMD patient mutations reveal a long-distance intronic interaction that regulates PLP1/DM20 alternative splicing. Hum. Mol. Genet. 23 (20), 5464–5478. doi:10.1093/hmg/ddu271
Taylor, R. M., Farrow, B. R., and Stewart, G. J. (1992). Amelioration of clinical disease following bone marrow transplantation in fucosidase-deficient dogs. Am. J. Med. Genet. 42 (4), 628–632. doi:10.1002/ajmg.1320420439
Tessitore, A., del, P., Martin, M., Sano, R., Ma, Y., Mann, L., et al. (2004). GM1-ganglioside-mediated activation of the unfolded protein response causes neuronal death in a neurodegenerative gangliosidosis. Mol. Cell. 15 (5), 753–766. doi:10.1016/j.molcel.2004.08.029
Tétreault, M., Choquet, K., Orcesi, S., Tonduti, D., Balottin, U., Teichmann, M., et al. (2011). Recessive mutations in POLR3B, encoding the second largest subunit of Pol III, cause a rare hypomyelinating leukodystrophy. Am. J. Hum. Genet. 89 (5), 652–655. doi:10.1016/j.ajhg.2011.10.006
Thakurela, S., Garding, A., Jung, R. B., Müller, C., Goebbels, S., White, R., et al. (2016). The transcriptome of mouse central nervous system myelin. Sci. Rep. 6, 25828. doi:10.1038/srep25828
Theda, C., Moser, A. B., Powers, J. M., and Moser, H. W. (1992). Phospholipids in X-linked adrenoleukodystrophy white matter: fatty acid abnormalities before the onset of demyelination. J. Neurol. Sci. 110 (1-2), 195–204. doi:10.1016/0022-510x(92)90028-j
Tomassy, G. S., Dershowitz, L. B., and Arlotta, P. (2016). Diversity Matters: A Revised Guide to Myelination. Trends Cell. Biol. 26 (2), 135–147. doi:10.1016/j.tcb.2015.09.002
Traboulsi, E. I., Faris, B. M., and Der Kaloustian, V. M. (1986). Persistent hyperplastic primary vitreous and recessive oculo-dento-osseous dysplasia. Am. J. Med. Genet. 24 (1), 95–100. doi:10.1002/ajmg.1320240111
Trajkovic, K., Dhaunchak, A. S., Goncalves, J. T., Wenzel, D., Schneider, A., Bunt, G., et al. (2006). Neuron to glia signaling triggers myelin membrane exocytosis from endosomal storage sites. J. Cell. Biol. 172 (6), 937–948. doi:10.1083/jcb.200509022
Turk, B. R., Theda, C., Fatemi, A., and Moser, A. B. (2020). X-linked adrenoleukodystrophy: Pathology, pathophysiology, diagnostic testing, newborn screening and therapies. Int. J. Dev. Neurosci. 80 (1), 52–72. doi:10.1002/jdn.10003
Uchino, A., Nagai, M., Kanazawa, N., Ichinoe, M., Yanagisawa, N., Adachi, K., et al. (2020). An autopsy case of GM1 gangliosidosis type II in a patient who survived a long duration with artificial respiratory support. Neuropathology 40 (4), 379–388. doi:10.1111/neup.12651
Uhlenberg, B., Schuelke, M., Rüschendorf, F., Ruf, N., Kaindl, A. M., Henneke, M., et al. (2004). Mutations in the gene encoding gap junction protein alpha 12 (connexin 46.6) cause Pelizaeus-Merzbacher-like disease. Am. J. Hum. Genet. 75 (2), 251–260. doi:10.1086/422763
Vagli, C., Fisicaro, F., Vinciguerra, L., Puglisi, V., Rodolico, M. S., Giordano, A., et al. (2020). Cerebral Hemodynamic Changes to Transcranial Doppler in Asymptomatic Patients with Fabry's Disease. Brain Sci. 10 (8), 546. doi:10.3390/brainsci10080546
Vahsen, N., Candé, C., Brière, J. J., Bénit, P., Joza, N., Larochette, N., et al. (2004). AIF deficiency compromises oxidative phosphorylation. EMBO J. 23 (23), 4679–4689. doi:10.1038/sj.emboj.7600461
Vallstedt, A., Klos, J. M., and Ericson, J. (2005). Multiple dorsoventral origins of oligodendrocyte generation in the spinal cord and hindbrain. Neuron 45 (1), 55–67. doi:10.1016/j.neuron.2004.12.026
Van Deerlin, V. M., Sleiman, P. M., Martinez-Lage, M., Chen-Plotkin, A., Wang, L. S., Graff-Radford, N. R., et al. (2010). Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat. Genet. 42 (3), 234–239. doi:10.1038/ng.536
van der Knaap, M. S., Fogli, A., Boespflug-Tanguy, O., Abbink, T. E. M., and Schiffmann, R. (2003). “Childhood ataxia with central nervous system hypomyelination/vanishing white matter,” in GeneReviews® [internet], ed. M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. W. Grippet al. (Seattle, WA: University of Washington, Seattle).
van der Knaap, M. S., Abbink, T. E. M., and Min, R. (20031993). “Megalencephalic Leukoencephalopathy with Subcortical Cysts,” in GeneReviews® [internet]. Editors M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. W. Grippet al. (Seattle (WA): University of Washington, Seattle).
van der Knaap, M. S., Barth, P. G., Gabreëls, F. J., Franzoni, E., Begeer, J. H., Stroink, H., et al. (1997). A new leukoencephalopathy with vanishing white matter. Neurology 48 (4), 845–855. doi:10.1212/wnl.48.4.845
van der Knaap, M. S., Barth, P. G., Stroink, H., van Nieuwenhuizen, O., Arts, W. F., Hoogenraad, F., et al. (1995). Leukoencephalopathy with swelling and a discrepantly mild clinical course in eight children. Ann. Neurol. 37 (3), 324–334. doi:10.1002/ana.410370308
van der Knaap, M. S., Bonkowsky, J. L., Vanderver, A., Schiffmann, R., Krägeloh-Mann, I., Bertini, E., et al. (2022). Therapy trial design in vanishing white matter: an expert consortium opinion. Neurol. Genet. 8 (2), e657. doi:10.1212/NXG.0000000000000657
van der Knaap, M. S., Boor, I., and Estévez, R. (2012). Megalencephalic leukoencephalopathy with subcortical cysts: chronic white matter oedema due to a defect in brain ion and water homoeostasis. Lancet. Neurol. 11 (11), 973–985. doi:10.1016/S1474-4422(12)70192-8
van der Knaap, M. S., Leegwater, P. A., Könst, A. A., Visser, A., Naidu, S., Oudejans, C. B., et al. (2002). Mutations in each of the five subunits of translation initiation factor eIF2B can cause leukoencephalopathy with vanishing white matter. Ann. Neurol. 51 (2), 264–270. doi:10.1002/ana.10112
van der Knaap, M. S., Linnankivi, T., Paetau, A., Feigenbaum, A., Wakusawa, K., Haginoya, K., et al. (2007). Hypomyelination with atrophy of the basal ganglia and cerebellum: follow-up and pathology. Neurology 69 (2), 166–171. doi:10.1212/01.wnl.0000265592.74483.a6
van der Knaap, M. S., Naidu, S., Pouwels, P. J., Bonavita, S., van Coster, R., Lagae, L., et al. (2002). New syndrome characterized by hypomyelination with atrophy of the basal ganglia and cerebellum. AJNR. Am. J. Neuroradiol. 23 (9), 1466–1474.
van Egmond, M. E., Pouwels, P. J., Boelens, J. J., Lindemans, C. A., Barkhof, F., Steenwijk, M. D., et al. (2013). Improvement of white matter changes on neuroimaging modalities after stem cell transplant in metachromatic leukodystrophy. JAMA Neurol. 70 (6), 779–782. doi:10.1001/jamaneurol.2013.629
van Grunsven, E. G., Mooijer, P. A., Aubourg, P., and Wanders, R. J. (1999). Enoyl-CoA hydratase deficiency: identification of a new type of D-bifunctional protein deficiency. Hum. Mol. Genet. 8 (8), 1509–1516. doi:10.1093/hmg/8.8.1509
Van Haren, K., van der Voorn, J. P., Peterson, D. R., van der Knaap, M. S., and Powers, J. M. (2004). The life and death of oligodendrocytes in vanishing white matter disease. J. Neuropathol. Exp. Neurol. 63 (6), 618–630. doi:10.1093/jnen/63.6.618
Van Hoof, F., and Hers, H. G. (1968). Mucopolysaccharidosis by absence of alpha-fucosidase. Lancet 1 (7553), 1198. doi:10.1016/s0140-6736(68)91895-3
van Rappard, D. F., Boelens, J. J., van Egmond, M. E., Kuball, J., van Hasselt, P. M., Oostrom, K. J., et al. (2016). Efficacy of hematopoietic cell transplantation in metachromatic leukodystrophy: the Dutch experience. Blood 127 (24), 3098–3101. doi:10.1182/blood-2016-03-708479
Vanderver, A., Hussey, H., Schmidt, J. L., Pastor, W., and Hoffman, H. J. (2012). Relative incidence of inherited white matter disorders in childhood to acquired pediatric demyelinating disorders. Semin. Pediatr. Neurol. 19 (4), 219–223. doi:10.1016/j.spen.2012.10.001
Vanrietvelde, F., Lemmerling, M., Mespreuve, M., Crevits, L., De Reuck, J., Kunnen, M., et al. (2000). MRI of the brain in cerebrotendinous xanthomatosis (van Bogaert-Scherer-Epstein disease). Eur. Radiol. 10 (4), 576–578. doi:10.1007/s003300050964
Vellodi, A., Cragg, H., Winchester, B., Young, E., Young, J., Downie, C. J., et al. (1995). Allogeneic bone marrow transplantation for fucosidosis. Bone Marrow Transpl. 15 (1), 153–158.
Verheijden, S., Bottelbergs, A., Krysko, O., Krysko, D. V., Beckers, L., De Munter, S., et al. (2013). Peroxisomal multifunctional protein-2 deficiency causes neuroinflammation and degeneration of Purkinje cells independent of very long chain fatty acid accumulation. Neurobiol. Dis. 58, 258–269. doi:10.1016/j.nbd.2013.06.006
Von Figura, K., Gieselmann, V., and Jaeken, J. (2001). Metachromatic leukodystrophy. New York: McGraw-Hill, 3695–3724.The Metabolic and Molecular Bases of Inherited Disease
Vulinovic, F., Krajka, V., Hausrat, T. J., Seibler, P., Alvarez-Fischer, D., Madoev, H., et al. (2018). Motor protein binding and mitochondrial transport are altered by pathogenic TUBB4A variants. Hum. Mutat. 39 (12), 1901–1915. doi:10.1002/humu.23913
Waldek, S., Patel, M. R., Banikazemi, M., Lemay, R., and Lee, P. (2009). Life expectancy and cause of death in males and females with Fabry disease: findings from the Fabry Registry. Genet. Med. 11 (11), 790–796. doi:10.1097/GIM.0b013e3181bb05bb
Wallraff, A., Köhling, R., Heinemann, U., Theis, M., Willecke, K., Steinhäuser, C., et al. (2006). The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J. Neurosci. 26 (20), 5438–5447. doi:10.1523/JNEUROSCI.0037-06.2006
Wang, R. Y., Lelis, A., Mirocha, J., and Wilcox, W. R. (2007). Heterozygous Fabry women are not just carriers, but have a significant burden of disease and impaired quality of life. Genet. Med. 9 (1), 34–45. doi:10.1097/gim.0b013e31802d8321
Wasserstein, M. P., Andriola, M., Arnold, G., Aron, A., Duffner, P., Erbe, R. W., et al. (2016). Clinical outcomes of children with abnormal newborn screening results for Krabbe disease in New York State. Genet. Med. 18 (12), 1235–1243. doi:10.1038/gim.2016.35
Weigel, M., Wang, L., and Fu, M. M. (2021). Microtubule organization and dynamics in oligodendrocytes, astrocytes, and microglia. Dev. Neurobiol. 81 (3), 310–320. doi:10.1002/dneu.22753
Weismann, C. M., Ferreira, J., Keeler, A. M., Su, Q., Qui, L., Shaffer, S. A., et al. (2015). Systemic AAV9 gene transfer in adult GM1 gangliosidosis mice reduces lysosomal storage in CNS and extends lifespan. Hum. Mol. Genet. 24 (15), 4353–4364. doi:10.1093/hmg/ddv168
Wenger, D. A., and Luzi, P. (2020). “Krabbe Disease: Globoid Cell Leukodystrophy,” in Rosenberg’s molecular and genetic basis of neurological and psychiatric disease (Elsevier), 481–491.
Wiesinger, C., Kunze, M., Regelsberger, G., Forss-Petter, S., and Berger, J. (2013). Impaired very long-chain acyl-CoA β-oxidation in human X-linked adrenoleukodystrophy fibroblasts is a direct consequence of ABCD1 transporter dysfunction. J. Biol. Chem. 288 (26), 19269–19279. doi:10.1074/jbc.M112.445445
Wilcox, W. R., Linthorst, G. E., Germain, D. P., Feldt-Rasmussen, U., Waldek, S., Richards, S. M., et al. (2012). Anti-α-galactosidase A antibody response to agalsidase beta treatment: data from the Fabry Registry. Mol. Genet. Metab. 105 (3), 443–449. doi:10.1016/j.ymgme.2011.12.006
Wilhelmsson, U., Li, L., Pekna, M., Berthold, C. H., Blom, S., Eliasson, C., et al. (2004). Absence of glial fibrillary acidic protein and vimentin prevents hypertrophy of astrocytic processes and improves post-traumatic regeneration. J. Neurosci. 24 (21), 5016–5021. doi:10.1523/JNEUROSCI.0820-04.2004
Willems, P. J., Darby, J. K., DiCioccio, R. A., Nakashima, P., Eng, C., Kretz, K. A., et al. (1988). Identification of a mutation in the structural alpha-L-fucosidase gene in fucosidosis. Am. J. Hum. Genet. 43 (5), 756–763.
Willems, P. J., Gatti, R., Darby, J. K., Romeo, G., Durand, P., Dumon, J. E., et al. (1991). Fucosidosis revisited: a review of 77 patients. Am. J. Med. Genet. 38 (1), 111–131. doi:10.1002/ajmg.1320380125
Wolf, N. I., Biancheri, R., Zara, F., Bruno, C., Gazzerro, E., Rossi, A., et al. (2021). “Hypomyelination and Congenital Cataract,” in GeneReviews® [internet], ed. M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. W. Grippet al. (Seattle, WA: University of Washington, Seattle).
Wolf, N. I., ffrench-Constant, C., and van der Knaap, M. S. (2021). Hypomyelinating leukodystrophies - unravelling myelin biology. Nat. Rev. Neurol. 17 (2), 88–103. doi:10.1038/s41582-020-00432-1
Wong, K., Armstrong, R. C., Gyure, K. A., Morrison, A. L., Rodriguez, D., Matalon, R., et al. (2000). Foamy cells with oligodendroglial phenotype in childhood ataxia with diffuse central nervous system hypomyelination syndrome. Acta Neuropathol. 100 (6), 635–646. doi:10.1007/s004010000234
Woodward, L. J., Anderson, P. J., Austin, N. C., Howard, K., and Inder, T. E. (2006). Neonatal MRI to predict neurodevelopmental outcomes in preterm infants. N. Engl. J. Med. 355 (7), 685–694. doi:10.1056/NEJMoa053792
Wright, M. D., Poe, M. D., DeRenzo, A., Haldal, S., and Escolar, M. L. (2017). Developmental outcomes of cord blood transplantation for Krabbe disease: A 15-year study. Neurology 89 (13), 1365–1372. doi:10.1212/WNL.0000000000004418
Yahalom, G., Tsabari, R., Molshatzki, N., Ephraty, L., Cohen, H., Hassin-Baer, S., et al. (2013). Neurological outcome in cerebrotendinous xanthomatosis treated with chenodeoxycholic acid: early versus late diagnosis. Clin. Neuropharmacol. 36 (3), 78–83. doi:10.1097/WNF.0b013e318288076a
Yam, G. H., Zuber, C., and Roth, J. (2005). A synthetic chaperone corrects the trafficking defect and disease phenotype in a protein misfolding disorder. FASEB J. 19 (1), 12–18. doi:10.1096/fj.04-2375com
Yamamoto, A., Fukumura, S., Habata, Y., Miyamoto, S., Nakashima, M., Takashima, S., et al. (2021). Novel HSD17B4 Variants Cause Progressive Leukodystrophy in Childhood: Case Report and Literature Review. Child. Neurol. Open 8, 2329048X211048613. doi:10.1177/2329048X211048613
Yan, H., Helman, G., Murthy, S. E., Ji, H., Crawford, J., Kubisiak, T., et al. (2019). Heterozygous Variants in the Mechanosensitive Ion Channel TMEM63A Result in Transient Hypomyelination during Infancy. Am. J. Hum. Genet. 105 (5), 996–1004. doi:10.1016/j.ajhg.2019.09.011
Yan, H., Yang, S., Hou, Y., Ali, S., Escobar, A., Gao, K., et al. (2022). Functional Study of TMEM163 Gene Variants Associated with Hypomyelination Leukodystrophy. Cells 11 (8), 1285. doi:10.3390/cells11081285
Yilmaz, K. (2009). Riboflavin treatment in a case with l-2-hydroxyglutaric aciduria. Eur. J. Paediatr. Neurol. 13 (1), 57–60. doi:10.1016/j.ejpn.2008.01.003
Yoshida, T., Sasaki, M., Yoshida, M., Namekawa, M., Okamoto, Y., and Tsujino, S.Alexander Disease Study Group in Japanet al. (2011). Nationwide survey of Alexander disease in Japan and proposed new guidelines for diagnosis. J. Neurol. 258 (11), 1998–2008. doi:10.1007/s00415-011-6056-3
Zara, F., Biancheri, R., Bruno, C., Bordo, L., Assereto, S., Gazzerro, E., et al. (2006). Deficiency of hyccin, a newly identified membrane protein, causes hypomyelination and congenital cataract. Nat. Genet. 38 (10), 1111–1113. doi:10.1038/ng1870
Zerbin-Rüdin, E., and Peiffer, J. (1964). [Genetic contribution to the problem of the late form of Pelizaeus-Merzbacher disease]. Humangenetik 1 (2), 107–122. doi:10.1007/BF00389627
Zhang, Y., Chen, K., Sloan, S. A., Bennett, M. L., Scholze, A. R., O'Keeffe, S., et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 3435 (362), 11929–11947. doi:10.1523/JNEUROSCI.1860-14.2014
Zhou, X., Nicholson, A. M., Ren, Y., Brooks, M., Jiang, P., Zuberi, A., et al. (2020). Loss of TMEM106B leads to myelination deficits: implications for frontotemporal dementia treatment strategies. Brain 143 (6), 1905–1919. doi:10.1093/brain/awaa141
Keywords: leukodystrophy, oligodendrocyte, myelin, white matter (WM), astrocyte, microglia, glia
Citation: Nowacki JC, Fields AM and Fu MM (2022) Emerging cellular themes in leukodystrophies. Front. Cell Dev. Biol. 10:902261. doi: 10.3389/fcell.2022.902261
Received: 22 March 2022; Accepted: 30 June 2022;
Published: 08 August 2022.
Edited by:
Nobuharu Suzuki, Tokyo Medical and Dental University, JapanReviewed by:
Arne Raasakka, University of Bergen, NorwayQuasar Padiath, University of Pittsburgh, United States
Copyright © 2022 Nowacki, Fields and Fu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Meng Meng Fu, mengmengfu@gmail.com
†These authors share first authorship