Non-alcoholic Steatohepatitis Pathogenesis, Diagnosis, and Treatment
 Bo Zhu
Bo Zhu Siu-Lung Chan
Siu-Lung Chan Jack Li
Jack Li  Hao Wu
Hao Wu Kui Cui
Kui Cui Hong Chen
Hong Chen- Department of Surgery, Vascular Biology Program, Harvard Medical School, Boston Children's Hospital, Boston, MA, United States
There has been a rise in the prevalence of non-alcohol fatty liver disease (NAFLD) due to the popularity of western diets and sedentary lifestyles. One quarter of NAFLD patients is diagnosed with non-alcoholic steatohepatitis (NASH), with histological evidence not only of fat accumulation in hepatocytes but also of liver cell injury and death due to long-term inflammation. Severe NASH patients have increased risks of cirrhosis and liver cancer. In this review, we discuss the pathogenesis and current methods of diagnosis for NASH, and current status of drug development for this life-threatening liver disease.
Introduction
There has been an increased interest in non-alcoholic fatty liver disease (NAFLD), and its advanced stage, non-alcoholic steatohepatitis (NASH) because of their increasing impact on global health (1). In the United States, the number of NAFLD cases is rapidly expanding, and is expected to reach 100.9 million patients in 2030 (~1/3 of the population) (2). During the past three decades, the number of patients with NAFLD has increased from 20 to 32% of the United States population (3). Recently, there is an increasing tendency for young people to be diagnosed with NAFLD due to being overweight or obese (4). As an increased proportion (~25% of NAFLD cases) of NAFLD will lead to NASH, there will be increased number of NASH patients with cirrhosis, leading to elevated liver transplantation for end-stage cirrhosis (5). Worse still, the risk of hepatocellular carcinoma (HCC) increases significantly for NAFLD or NASH patients who have cirrhosis (6–8).
NASH is strongly associated with overweight or obesity and metabolic syndromes (9, 10). Recent studies have shown that more than 80% of patients with NASH are overweight or obese (11, 12). NASH is highly associated with type 2 diabetes mellitus (13–15). NAFLD is the general term that comprises hepatic steatosis and steatohepatitis. Unlike isolated hepatic steatosis, NASH is strongly associated with fibrosis found in liver biopsy (5). The level of fibrosis varies among different NASH patients and advanced fibrosis progresses into liver cirrhosis and the eventual scarring (16). Currently, fibrosis-induced liver cell death and further functional failure is a major cause of liver transplantation (17). In addition to liver functional failure, cirrhosis and HCC, associated non-liver adverse outcomes are primarily related to increased cardiovascular diseases (18–21) and type 2 diabetes mellitus (22). Recently, studies have suggested that NAFLD should be defined as a disease of global metabolic dysfunction (23), and not just limited to the liver. The metabolic dysfunctions in fat tissue and muscle, and microbiota variation in the gut contribute to fatty liver disease progression. Therefore, to more accurately describe NAFLD, scientists suggest renaming it as “metabolic associated fatty liver disease” or MAFLD (24).
At present, steady progress in clarifying the pathogenesis of NASH has been made, leading to the identification of therapeutic targets for drug development. However, there is currently no FDA-approved drug that can cure NASH. In addition, the lack of precise predictive biomarkers limits early diagnosis of NASH. Liver biopsy remains the gold standard for diagnosis of NASH. This review focuses on discussing the risks for NASH pathogenesis, current development of biomarkers, and therapeutic target identification for drug development.
Pathogenesis of NAFL/NASH
The “two-hit” theory for the development of NASH was first proposed more than two decades ago (25). The theory assumed that the setting in of steatosis is the first hit, and that a second hit from other factors is required for the development of NASH (26), such as oxidative stress. However, this theory is now considered outdated. Multiple-hit pathogenesis was proposed, suggesting that many different factors have been considered to contribute to NASH progression, such as inherited and environmental factors (27, 28). Diet-induced obesity is the most common inducer of NASH development, because the severe accumulation of fat in liver leads to dysfunction of lipid metabolism. At present, the accumulation of hepatic free cholesterol and free fatty acid is considered the primary source of stress to the liver (29–31). Particularly, hepatic free cholesterol is a major lipotoxic molecule critical for NASH progression (32). Its metabolites trigger hepatocellular stress (for example, oxidative stress) and induce hepatocyte injury and death, leading to fibrosis and further cirrhosis (Figure 1A).
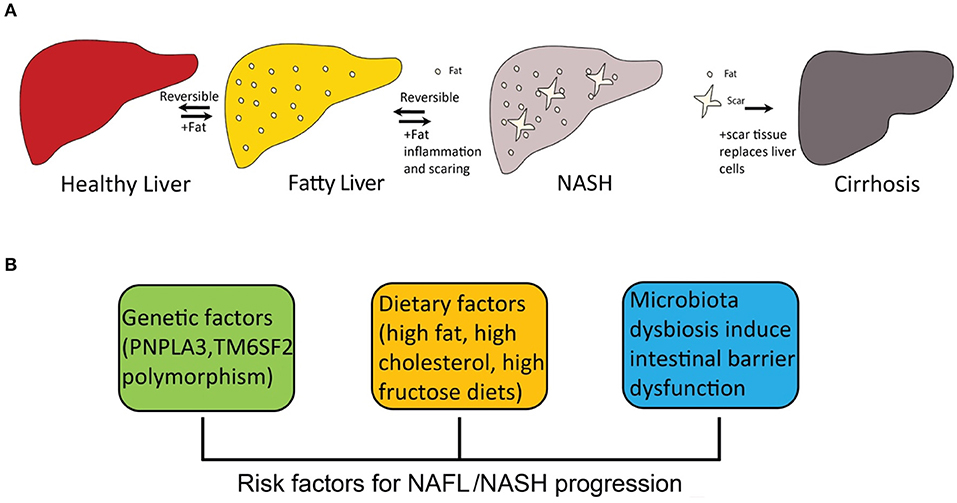
Figure 1. The progression of fatty liver disease and risk factors for NAFL/NASH progression. (A) the spectrum of fatty liver disease; (B) risk factors for NAFL/NASH progression.
Fibrosis in the liver and hepatocyte injury and death are the key features that distinguish NASH from isolated steatosis (5, 33). It is, however, debatable whether hepatocyte injury causes liver inflammation or if hepatocyte injury is the consequence of liver inflammation. Both hepatocyte injury and liver inflammation are relevant to the pathogenesis of NASH because injured hepatocytes release factors that promote inflammation, resulting in a downward spiral as inflammation further triggers hepatocyte injury (34, 35). This is because inflammation in the liver is caused by released proinflammatory chemokines and cytokines (such as TNF-α, IL-6, and CCL2), which further damage the injured hepatocytes (36). In the liver, Kupffer cells, a kind of resident macrophage, are localized in the lumen of the liver sinusoids and play a central role in liver inflammation (37). The initiation of liver injury stems from the activation of Kupffer cells resulting in cytokine and chemokine production (38). Kupffer cells can be divided into classically activated M1 Kupffer cells (proinflammatory M1) and alternatively activated M2 Kupffer cells (wound-healing M2) (39). The activation of pro-inflammatory M1 Kupffer cells is the critical step that contributes to the pathogenesis of fibrogenesis during NASH progression. In contrast, the polarization and activation of anti-inflammatory M2 Kupffer cells play a protective role against fibrogenesis in NASH (40). Importantly, M2 Kupffer cells promote the apoptosis of M1 Kupffer cells, which is protective against NAFL/NASH (41). Under high fat diet treatment, mice with a high M2:M1 Kupffer cell ratio are resistant to developing liver lesions, while mice with a high M1:M2 Kupffer cell ratio are more likely to develop liver lesions (42).
In addition to resident Kupffer cells in liver, monocyte-derived macrophages play an important role in the pathogenesis of NAFL/NASH (40). The infiltrating monocyte-derived macrophages can be divided into two major subtypes, Ly-6Chi macrophages and Ly-6Clo macrophages (43), both of which affect hepatic stellate cells (HSCs), though in differing ways. HSCs have been identified as the major extracellular matrix protein (ECM) producing cells in injured liver (44) that play the central role in the formation of hepatic fibrosis (45). HSCs have two different states, quiescent and activated states, and the transdifferentiation from the quiescent into the activated state is the major cause of fibrosis (44). The proinflammatory Ly-6Chi macrophages activate HSCs by secreting IL-1β and CCL2 that enhance the fibrotic process (46), while the pro-restorative Ly-6Clo macrophages promote apoptosis of HSCs and accelerate extracellular matrix degradation by upregulation of matrix metalloproteinase 9 (MMP9), MMP12 and MMP13 (47). The pro-restorative Ly-6Clo macrophages express chemokine (C-X3-C motif) receptor 1 (CX3CR1), and because its ligand1 (CX3CL1) is mainly expressed in HSCs, CX3CL1-CX3CR1 interaction negatively regulates inflammatory properties in macrophages within the liver (48). Chemokine CCL2 production, working through CCR2, is a major cause of monocyte-derived macrophage (inflammatory Ly-6Chi macrophages) recruitment induced by Toll-like receptor 4 (TLR4) signaling (49). The infiltration of monocytes from blood that rapidly differentiate into pro-inflammatory macrophages in the liver contributes to NAFL/NASH progression (46). Extensive studies have reported that genetic deficiency or pharmacological inhibition of CCR2 decreased monocyte recruitment to the liver and ameliorated NASH in mice (50–52). In addition to chemokine CCL2, cytokines like TNF-α and IL-1β released from macrophages are important drivers of steatosis, inflammation, and fibrosis in NAFL/NASH (40).
A number of risk factors promote the pathogenesis of NASH, including inherited and environmental factors (Figure 1B). Dietary factors are one of the most important environmental factors that lead to NASH.
Genetic and Epigenetic Factors
Genome-wide association studies (GWAS) suggest that polymorphisms in patatin-like phospholipase domain-containing 3 (PNPLA3) and transmembrane 6 superfamily, member 2 (TM6SF2) promote the development of NASH (53). Particularly, PNPLA3 polymorphisms (I148M) are strongly associated with hepatic steatosis caused by accumulation of PNPLA3 on hepatic lipid droplets (54–56). TM6SF2 polymorphism reduces very-low density lipoprotein (VLDL) secretion that is associated with NASH and fibrosis (57, 58). The polymorphism of several other genes, including glucokinase regulator (GCKR), membrane bound O-acyltransferase domain-containing 7 (MBOAT7) and hydroxysteroid 17β-dehydrogenase (HSD17B13), are closely associated with susceptibility to NAFL and progression of NASH (53).
Epigenetic mechanisms are special genetic regulations without any change in gene sequence in the genome but with different modifications, such as DNA methylation, histone modification, and non-coding RNAs (59). By analysis of liver samples from NAFLD patients, the heavy methylation of NAFLD associated genes, such as PNPLA3 (60) and PPARG (61), increases the severity of NAFLD. Recently studies have indicated that non-coding RNAs could regulate NAFLD progression, including miR-122 and miR-125b, which show significantly decreased expression in NAFLD patients (62, 63). Moreover, hepatic-specific deletion of miR-21 prevents steatosis, and thus may be a potential therapeutic target for NAFLD (64). In addition, changes in hepatic lncRNA expression patterns are associated with NAFLD. For example, a strong increase in hepatic lncRNA un.372 is detected in NAFLD patients (65), while hepatic lncRNA lnc18q22.2 is reported to correlate with the severity of NASH (66). A list of microRNAs and long non-coding RNAs that are involved in NAFL/NASH progression are summarized in Table 1. Furthermore, studies in rodents have demonstrated the existence of epigenetic factors that regulate fibrogenic liver cell development to cirrhosis (124). A similar phenomenon is observed in NASH patients. Therefore, epigenetic mechanisms may be related to susceptibility for NASH.
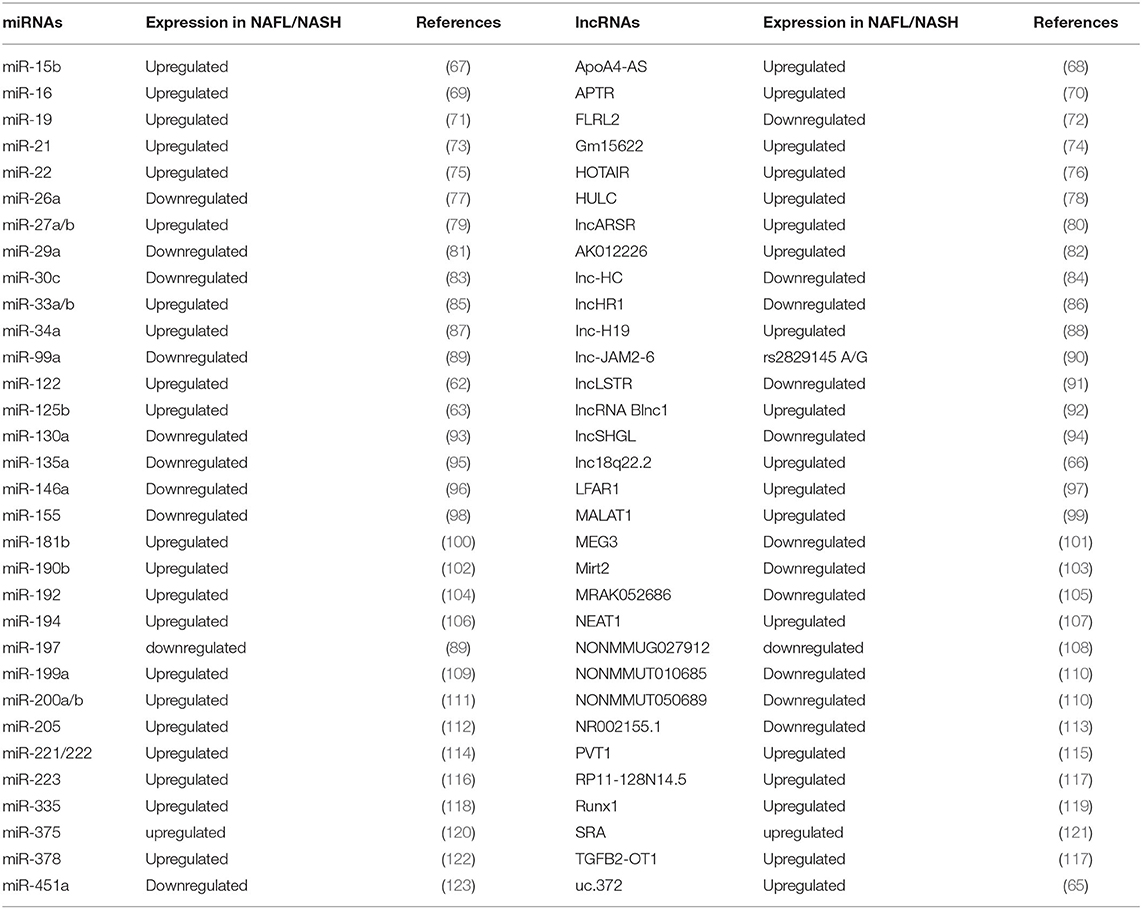
Table 1. microRNAs and long non-coding RNAs that are involved in NAFLD pathogenesis.
Dietary Factors
Consumption of a high-calorie diet with high fat and high sugar (western diet) results in weight gain and is the initial event for the development of fatty liver (125–127). Particular types of lipids and carbohydrates play important roles in the progression of NASH. For dietary lipids, polyunsaturated fatty acids (PUFAs) induce inflammation and fibrosis formation in NASH (128). For dietary carbohydrates, over-consumption of carbohydrates extensively promotes the development of NAFLD (129), especially fructose, a highly lipogenic sugar and a common component in almost all major sweet foods (130, 131). Extensive experimental studies support the association between the increasing rates of obesity and the progression of NAFLD (132–134). Obese individuals who have an excessive body mass index (BMI) and visceral obesity are at a high risk for developing NAFLD (135). Studies have reported that more than 95% people that had severe obesity have NAFLD (136). Type 2 diabetes mellitus (T2DM) patients have higher risks for NAFLD due to insulin resistance, and T2DM and NAFLD can be developed simultaneously (137). In addition, people who have dyslipidemia and hypertension are also at high risk for NAFLD (138).
Microbiota Dysbiosis
The relationship between intestinal microbiota and NAFL/NASH has been proposed for decades. Intestinal microbiota is altered in genetically obese mice that have metabolic syndrome and fatty liver (139). Studies indicate that microbiota dysbiosis is associated with inflammatory signaling, which promotes hepatic steatosis and NASH (140, 141). Recently, extensive studies on the gut-liver axis have suggested that intestinal microbiota influenced host susceptibility to obesity, hepatic steatosis, and NASH (142–144). The homeostasis of intestinal microbiota is essential to maintaining proper function of the intestinal barrier, and recent studies have shown that intestinal microbiota dysbiosis triggers intestinal inflammation and further impairs the intestinal barrier. Microbial products released can reach the liver and induce hepatic inflammation and further lead to NAFL/NASH (145). This intestinal barrier is hepatoprotective, and microbiota-driven gut-vascular barrier disruption is a prerequisite for NASH development (146). This barrier disruption is caused by diet-induced dysbiosis (147). Patients with NAFL and NASH usually show dysbiosis in the gut microbiota (148). Overall, patients with NAFL and NASH have lower microbiota diversity than healthy people and have different microbiota species abundance patterns (149). For example, patients with NASH have an increased abundance of Escherichia, where patients with advanced fibrotic NASH or cirrhosis have dramatically increased proportions of Bacteroides and Ruminococcus (150).
Diagnosis of NAFL/NASH
At present, liver biopsy remains the gold standard for diagnosis of (151), since invasive liver biopsy for assessing different fibrosis stages is the most accurate method for NASH diagnosis (Figure 2A) (152). However, liver biopsy is painful for patients, and has sampling error limitations (153). Therefore, new developments in non-invasive biomarkers for early diagnosis and treatment of NAFL and NASH are urgently needed. Current progression on developing non-invasive biomarkers is mainly based on the detection of hepatic steatosis (154). There are several indices and scores for the evaluation of hepatic steatosis. Testing of blood biomarkers, determining the fatty liver index (FLI) and determining the hepatic steatosis index (HSI) are the most popular methods to evaluate the risks of NAFLD (155). Fatty liver index, BMI, and serum level of triglycerides also have moderate accuracy for fatty liver diagnosis (156). Hepatic steatosis index can be evaluated by the ratio between serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) and has moderate accuracy for the detection of fatty liver (156). The limitation of both FLI and HSI is insensitive to mild steatosis since they are designed to target indirectly on blood fat rather than liver fat.
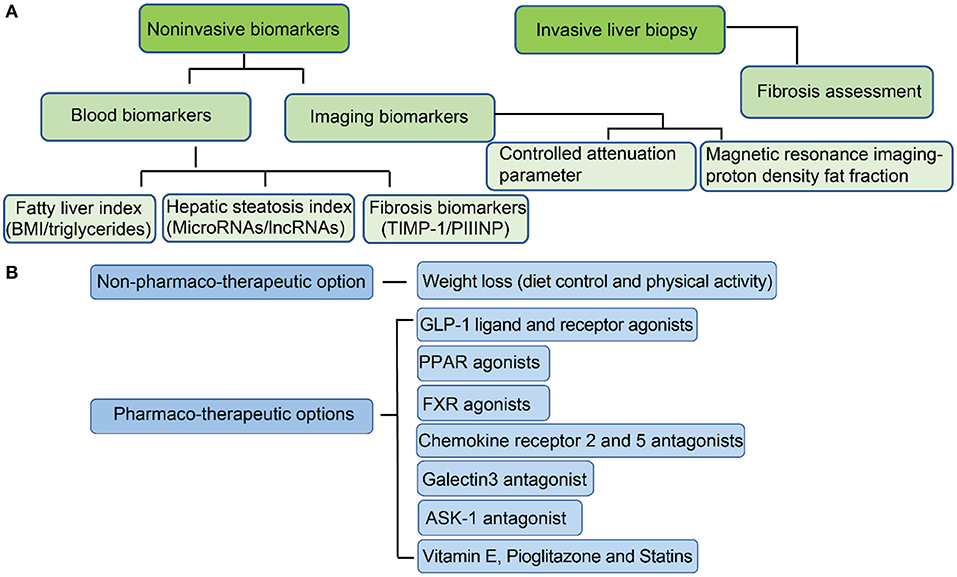
Figure 2. Current approaches for the diagnosis of NAFL/NASH and options for treatment. (A) Methods for NAFL/NASH diagnosis; (B) Options for NAFL/NASH treatment.
A more sensitive and accurate method is to measure liver fat directly. The development of imaging biomarkers has dramatically improved the progress of NAFLD diagnosis (157). Ultrasonography is the most common method for detection of hepatic steatosis and can accurately identify moderate to severe steatosis (158). However, it is less sensitive when detecting steatosis in NASH patients who have advanced fibrosis (159). Computed tomography (CT), although sensitive in detecting moderate and severe steatosis with mild histological fibrosis (159), has not been shown to have improved sensitivity in mild steatosis (160). Both controlled attenuation parameter (CAP) and magnetic resonance imaging-proton density fat fraction (MRI-PDFF) can address sensitivity issues in mild steatosis (161, 162). Controlled attenuation parameter is a method for grading steatosis that measures the degree of ultrasound attenuation by hepatic fat using a process based on simultaneous transient elastography, which is more sensitive and accurate than previous imaging markers (163). MRI-PDFF maps can be generated within seconds and detection of hepatic fat is more accurate than CAP for detecting all grades of steatosis in patients with NAFLD (164). Magnetic resonance imaging (MRI) approaches 100% sensitivity in the detection of hepatic steatosis, even at low steatosis levels (165). At present, MRI is the best method for detecting hepatic steatosis accurately and efficiently due to its high sensitivity.
The current progress on non-invasive biomarkers development for fibrosis diagnosis has advanced. Several potential non-invasive biomarkers have been reported for NASH diagnosis. Studies indicate that serum hyaluronic acid (HA) is a non-invasive marker of liver fibrosis (166), and serum YKL-40 can also be a marker of liver fibrosis in patients with NAFLD (167). In addition, the expression of tissue inhibitor of matrix metalloproteinase 1 (TIMP-1) is increased in human and rat models of liver fibrosis (168), and its degree of expression correlates with the extent of fibrosis in human liver (169). Although functional studies indicate TIMP-1 promotes hepatic fibrogenesis, TIMP-1 deficiency does not prevent carbon tetrachloride (CCl4)-induced hepatic fibrogenesis (170). Terminal peptide of procollagen III (PIIINP) is released during the synthesis and deposition of type III collagen (171). At present, PIIINP has been validated to be effective in the detection of fibrosis, and is particularly effective in the detection of severe fibrosis (172). PIIINP is elevated in patients with advanced fibrosis (172). Therefore, serum PIIINP is potentially a good non-invasive marker of liver fibrosis.
Treatment of NAFL/NASH
Most NASH patients are obese. The first and simplest management against NAFLD or NASH is to make lifestyle modifications (173). Through sustained weight loss (such as through a calorie-restricted diet) and increased physical activity (exercise), hepatic steatosis caused by western-style diet and sedentary lifestyle can be reduced significantly (174). A modest weight loss of about 3% may reduce hepatic steatosis, however, up to 10% or more is required for the resolution of NAFLD and the regression of fibrosis in NASH (175). Thus, resolution of steatosis in NAFLD and even fibrosis reversal in NASH can be achieved after significant weight loss through lifestyle modifications. Therefore, as a non-pharmaco-therapeutic option, transitioning from a high-calorie diet and a sedentary lifestyle into a restricted calorie diet with increased physical activity is the healthiest treatment for NAFL and early stage of NASH.
In addition to lifestyle modifications, there are several therapeutic targets that are under clinical trials (Figure 2B). The most promising pharmaco-therapeutic candidates will be discussed.
Vitamin E, Pioglitazone, and Statins
Oxidative stress plays an essential role in the development from isolated steatosis to NASH (176). Anti-oxidation treats NAFL/NASH by removing excessive reactive oxygen species (ROS) (177). Extensive studies have indicated that vitamin E reduced steatosis and ameliorated fibrosis (178–180). However, for NASH patients with diabetes, vitamin E alone does not significantly improve histological outcomes (181). Considering NAFL/NASH is a metabolic syndrome, and thus is usually accompanied by other metabolic diseases, such as diabetes and CVD, drugs for the treatment of diabetes and CVD are used for treating NAFL/NASH, particularly pioglitazone and statins (182, 183). Pioglitazone is used for treatment of type 2 diabetes mellitus, and has been reported to significantly reduce fibrosis in NASH patients (184). The safety and efficacy of low-dose pioglitazone (NCT04501406) has been tested (185). In addition, statins, a class of drugs used for reducing cardiovascular disease risk, has been tested on NAFL/NASH patients who then displayed substantial improvement on steatosis and fibrosis (186, 187). While piloglitazone and statins have already been in the market for a while for treating other conditions, they also represent new potential therapies for NAFL/NASH.
GLP-1 Ligand and Receptor Agonists
Glucagon-like peptide 1 (GLP-1) is essential for glucose homeostasis (188). GLP-1 and its receptor agonists (GLP-1 RAs) can treat type 2 diabetes mellitus (189), which is closely associated with NAFLD (13). Evidence supports that GLP-1 secretion is impaired in patients with NAFL and NASH (190), indicating that GLP-1 agonists may be potential treatment candidates for NAFLD. GLP-1 agonists and GLP-1 RAs, including liraglutide (NCT01237119), exenatide (NCT00650546) and semaglutide (NCT02970942), have been tested for improving liver histology in NASH patients (191–193), and are currently under phase II clinical trials for NASH.
PPAR Agonists
Peroxisome proliferator-activator receptors (PPARs) are a group of nuclear receptors that play a critical role in intracellular lipid metabolism (194). Extensive studies indicate the dual PPAR alpha and delta agonist, elafibranor, is effective against NASH (195). Taking elafibranor at a dose of 120 mg/day has been shown to cause significant regression of fibrosis in NASH patients. In addition, other metabolic parameters, such as liver enzymes, are also greatly improved by elafibranor. Elafibranor is now under phase III clinical trials (NCT02704403).
FXR Agonists
Farnesoid X receptor (FXR) is a master regulator of hepatic triglyceride and glucose homeostasis (196). Obeticholic acid (OCA), a FXR agonist, has been recently studied and shown to improve histology and fibrosis scores (197). OCA is highly promising for NASH treatment and is under phase III clinical trials (NCT02548351). In addition to OCA, other FXR agonists are also under clinical trials for treating NASH. Another FXR agonist, MET-409, significantly reduces liver fat after 12 weeks of treatment in NASH patients (198). MET-409 is under phase II clinical trials for NASH (NCT04702490). EDP-305, a potent FXR agonist, reduces liver fat and is a potent inhibitor of fibrosis (199). EDP-305 is under phase II clinical trials for NASH (NCT03421413).
CCR2/5 Antagonists
NASH is a disease with inflammation and fibrosis. Chemokine receptor 2/5 (CCR2 and CCR5) are commonly increased in the liver from NASH patients. Cenicriviroc (CVC), a C-C motif chemokine receptor 2/5 (CCR2/5) antagonist, was developed to target inflammation (200). In NASH patients, liver fibrosis is caused by the accumulation of extracellular matrix proteins, mainly composed of collagen (201). CCR5 antagonist can inhibit collagen production by HSCs by impairing the migration, activation, and proliferation of HSCs (202). CVC has been shown to have antifibrotic function and has significantly improved fibrosis in NASH patients after 1 year of treatment (203). CVC is currently under phase III clinical evaluation for NASH (NCT03028740).
Galectin-3 Antagonist
Galectin-3 protein expression is essential for the development of hepatic fibrosis, which is significantly increased in NASH liver (204). In mice models, GR-MD-02, a galectin-3 inhibitor, markedly ameliorates liver fibrosis through inhibition of collagen deposition (205). GR-MD-02 has been shown to be safe and efficient for patients with NASH cirrhosis with portal hypertension (165). The safety and efficacy of GR-MD-02 for the treatment of liver fibrosis is under phase II clinical trials (NCT02462967) (206).
ASK-1 Antagonist
To improve inflammation and fibrosis in NASH, many therapeutic targets have been tested. The inhibitors of apoptosis signal-regulating kinase 1 (ASK1), a serine/threonine kinase, have been shown to significantly improve fibrosis in NASH animal models (207). Selonsertib (aka GS-4997), a selective inhibitor of ASK1, can reduce hepatic steatosis and fibrosis in NASH animal models fed with high fat and sugar. In phase II clinical trials, patients treated with selonsertib showed huge improvements in fibrosis through reduction of hepatic collagen content (208). However, a recent study has indicated that selonsertib neither leads to fibrosis regression nor reduces NASH progression (209). At present, selonsertib is undergoing phase III clinical evaluation and being studied for its efficacy on NASH (NCT03053050).
Gut and Microbiome Related Therapies
Studies between microbiome and NAFL/NASH are relatively nascent, but the consensus is that dysbiosis leads to increased intestinal permeability, which may increase NAFL/NASH progression (210). Thus, disturbed gut-liver barrier integrity is essential in the pathogenesis of both NAFL and NASH since the release of bacterial products from the gut into blood circulation may cause a massive inflammatory response from the liver (211). Orlistat, an FDA-approved lipase inhibitor for treating obesity, reduces absorption of dietary fat (212) and may have beneficial effects on body weight through modification of the composition of gut microbiota (213), however, the efficacy of orlistat on NASH has not been clearly demonstrated. Other drugs targeting the microbiome include solithromycin. Solithromycin is an antibiotic in clinical trials for the treatment of bacterial infection. Studies in a mouse model of NASH have displayed that solithromycin had beneficial effects by reducing hepatocyte ballooning and inflammation (214). Solithromycin is currently in a phase II trial (NCT02510599) for NASH. All six patients with NASH it was tested on showed reduction in NASH parameters after 90 days of treatment (215). Notably, solithromycin may not impair the gut microenvironment since its mechanism of action against NASH may not be related to its antibacterial activity, as it is not active against gut Gram-negative bacteria (214).
Although many potential candidates for the treatment of NASH are under phase II and III clinical trials as discussed above, no drug has yet been approved by the FDA. In addition to the discovery of novel promising targets for clinical testing, existing drugs like piloglitazone and statins used for the treatment of diabetes and CVD diseases can also be viable therapeutic options for NAFL/NASH. Furthermore, antioxidant vitamin E can be a beneficial supplement for NAFL/NASH patients.
Future Perspectives
Considering NAFLD is closely associated with metabolic dysfunction, metabolic dysfunction-associated fatty liver disease (MAFLD) may better describe the disease than NAFLD (216). The lack of appropriate NASH animal models is the bottleneck for NASH investigation. Although a methionine-choline-deficient (MCD) diet is a widely used model for NASH study, the body weight loss and increased insulin sensitivity of this model are not features of NASH (217). Recently, high fat, high fructose, high cholesterol diets and use of chemical inducers (such as carbon tetrachloride, CCl4) have been extensively applied as NASH models, though the feeding or induction cycle is very long (218). Despite its long-term feeding cycle, this animal model may become more popular in the future since it can accurately mimic the features of NASH.
Non-invasive diagnosis approaches can primarily detect hepatic steatosis sensitively but struggle to detect fibrosis, meaning the diagnosis of NASH still requires invasive liver biopsy. Development of non-invasive methods that can sensitively and accurately diagnose NASH is currently ongoing and such methods may be available in the future. At present, because of the absence of any existing FDA-approved medication for the treatment of NASH, an effective clinical pharmacotherapy is urgently needed for advanced NASH. For mild NASH, the most healthy and effective treatment is through management of diet and lifestyle. NASH fibrosis regression can occur with 10% weight loss, but such weight loss is difficult to achieve. In addition, NASH is commonly associated with diabetes, meaning drugs that target diabetes may have potential for treating NASH. In addition, the development of drug targets on gut microbiota may be a novel direction in the coming decades, because accumulating evidence shows microbiota dysbiosis is one of the critical causes of NASH through the gut-liver axis. Alternatively, the development of pharmacological interventions targeting the polarization of M2 Kupffer cells during the early stages of NASH may become an attractive strategy for reducing inflammation and hepatocyte injury. Similarly, in monocyte-derived macrophages, it is attractive to develop pharmacological interventions that target the polarization of pro-restorative Ly-6Clo macrophages to treat NASH.
Conclusion
Great developments in NASH pathogenesis, diagnosis and treatment have been achieved in the past decades. This review discusses factors that may induce NASH, non-invasive methods for NASH diagnosis, and potential pharmaco-therapeutic options to resolve NASH. Dietary factors are a major cause for NAFLD that may further develop into NASH, particularly for people under long-term high fat and high sugar consumption who are obese or overweight. Body weight loss is beneficial for early-stage NASH. Accurate and sensitive non-invasive diagnosis methods for NASH are needed to replace invasive liver biopsy. Although no FDA-approved drug is available for NASH, many clinical pharmacotherapies are under phase II or phase III clinical trials. Moving forward, more potential NASH targets will likely be identified, offering more opportunities to discover effective and specific drugs to treat and resolve NASH.
Author Contributions
BZ, S-LC, and HC wrote the manuscript. JL and KL edited the manuscript. HW and KC critically read the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported in part by NIH Grants Nos. R01HL093242, R01HL137229, R01HL133216, and R01HL156362 to HC.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. (2018) 15:11–20. doi: 10.1038/nrgastro.2017.109
2. Estes C, Razavi H, Loomba R, Younossi Z, Sanyal AJ. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology. (2018) 67:123–33. doi: 10.1002/hep.29466
3. Loomba R, Friedman SL, Shulman GI. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell. (2021) 184:2537–64. doi: 10.1016/j.cell.2021.04.015
4. Scapaticci S, D'Adamo E, Mohn A, Chiarelli F, Giannini C. Non-alcoholic fatty liver disease in obese youth with insulin resistance and type 2 diabetes. Front Endocrinol. (2021) 12:639548. doi: 10.3389/fendo.2021.639548
5. Diehl AM, Day C. Cause pathogenesis, and treatment of nonalcoholic steatohepatitis. N Engl J Med. (2017) 377:2063–72. doi: 10.1056/NEJMra1503519
6. Ascha MS, Hanouneh IA, Lopez R, Tamimi TA, Feldstein AF, Zein NN. The incidence and risk factors of hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. Hepatology. (2010) 51:1972–8. doi: 10.1002/hep.23527
7. Starley BQ, Calcagno CJ, Harrison SA. Nonalcoholic fatty liver disease and hepatocellular carcinoma: a weighty connection. Hepatology. (2010) 51:1820–32. doi: 10.1002/hep.23594
8. Kogiso T, Tokushige K. The current view of nonalcoholic fatty liver disease-related hepatocellular Carcinoma. Cancers. (2021) 13:516. doi: 10.3390/cancers13030516
9. Godoy-Matos AF, Silva Junior WS, Valerio CM. NAFLD as a continuum: from obesity to metabolic syndrome and diabetes. Diabetol Metab Syndr. (2020) 12:60. doi: 10.1186/s13098-020-00570-y
10. Sarwar R, Pierce N, Koppe S. Obesity and nonalcoholic fatty liver disease: current perspectives. Diabetes Metab Syndr Obes. (2018) 11:533–42. doi: 10.2147/DMSO.S146339
11. Milic S, Lulic D, Stimac D. Non-alcoholic fatty liver disease and obesity: biochemical, metabolic and clinical presentations. World J Gastroenterol. (2014) 20:9330–7. doi: 10.3748/wjg.v20.i28.9330
12. Younossi ZM. The epidemiology of nonalcoholic steatohepatitis. Clin Liver Dis. (2018) 11:92–4. doi: 10.1002/cld.710
13. Gastaldelli A, Cusi K. From NASH to diabetes and from diabetes to NASH: mechanisms and treatment options. JHEP Rep. (2019) 1:312–28. doi: 10.1016/j.jhepr.2019.07.002
14. Amiri Dash Atan N, Koushki M, Motedayen M, Dousti M, Sayehmiri F, Vafaee R, et al. Type 2 diabetes mellitus and non-alcoholic fatty liver disease: a systematic review and meta-analysis. Gastroenterol Hepatol Bed Bench. (2017) 10:S1–7.
15. Dharmalingam M, Yamasandhi PG. Nonalcoholic fatty liver disease and type 2 diabetes mellitus. Indian J Endocrinol Metab. (2018) 22:421–8. doi: 10.4103/ijem.IJEM_585_17
17. Pais R, Barritt A, St Calmus Y, Scatton O, Runge T, Lebray P, et al. NAFLD and liver transplantation: Current burden and expected challenges. J Hepatol. (2016) 65:1245–57. doi: 10.1016/j.jhep.2016.07.033
18. Francque SM, van der Graaff D, Kwanten WJ. Non-alcoholic fatty liver disease and cardiovascular risk: Pathophysiological mechanisms and implications. J Hepatol. (2016) 65:425–43. doi: 10.1016/j.jhep.2016.04.005
19. Patil R, Sood GK. Non-alcoholic fatty liver disease and cardiovascular risk. World J Gastrointest Pathophysiol. (2017) 8:51–8. doi: 10.4291/wjgp.v8.i2.51
20. Kasper P, Martin A, Lang S, Kutting F, Goeser T, Demir M, et al. NAFLD and cardiovascular diseases: a clinical review. Clin Res Cardiol. (2021) 110:921–37. doi: 10.1007/s00392-020-01709-7
21. Ismaiel A, Dumitrascu DL. Genetic predisposition in metabolic-dysfunction-associated fatty liver disease and cardiovascular outcomes-Systematic review. Eur J Clin Invest. (2020) 50:e13331. doi: 10.1111/eci.13331
22. Younossi ZM, Golabi P, de Avila L, Paik JM, Srishord M, Fukui N, et al. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic review and meta-analysis. J Hepatol. (2019) 71:793–801. doi: 10.1016/j.jhep.2019.06.021
23. Fouad Y, Waked I, Bollipo S, Gomaa A, Ajlouni Y, Attia D. What's in a name? Renaming 'NAFLD' to 'MAFLD'. Liver Int. (2020) 40:1254–61. doi: 10.1111/liv.14478
24. Eslam M, Sanyal AJ, George J. International consensus, MAFLD: A consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology. (2020) 158:1999–2014.e1. doi: 10.1053/j.gastro.2019.11.312
25. Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. (1998) 114:842–5. doi: 10.1016/S0016-5085(98)70599-2
26. Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. (2018) 24:908–22. doi: 10.1038/s41591-018-0104-9
27. Buzzetti E, Pinzani M, Tsochatzis EA. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism. (2016) 65:1038–48. doi: 10.1016/j.metabol.2015.12.012
28. Peng C, Stewart AG, Woodman OL, Ritchie RH, Qin CX. Non-alcoholic steatohepatitis: a review of its mechanism, models and medical treatments. Front Pharmacol. (2020) 11:603926. doi: 10.3389/fphar.2020.603926
29. Neuschwander-Tetri BA. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology. (2010) 52:774–88. doi: 10.1002/hep.23719
30. Arguello G, Balboa E, Arrese M, Zanlungo S. Recent insights on the role of cholesterol in non-alcoholic fatty liver disease. Biochim Biophys Acta. (2015) 1852:1765–78. doi: 10.1016/j.bbadis.2015.05.015
31. Ho CM, Ho SL, Jeng YM, Lai YS, Chen YH, Lu SC, et al. Accumulation of free cholesterol and oxidized low-density lipoprotein is associated with portal inflammation and fibrosis in nonalcoholic fatty liver disease. J Inflamm (Lond). (2019) 16:7. doi: 10.1186/s12950-019-0211-5
32. Ioannou GN. The role of cholesterol in the pathogenesis of NASH. Trends Endocrinol Metab. (2016) 27:84–95. doi: 10.1016/j.tem.2015.11.008
33. Parthasarathy G, Revelo X, Malhi H. Pathogenesis of nonalcoholic steatohepatitis: an overview. Hepatol Commun. (2020) 4:478–92. doi: 10.1002/hep4.1479
34. Ibrahim SH, Hirsova P, Gores GJ. Non-alcoholic steatohepatitis pathogenesis: sublethal hepatocyte injury as a driver of liver inflammation. Gut. (2018) 67:963–72. doi: 10.1136/gutjnl-2017-315691
35. Liu W, Baker RD, Bhatia T, Zhu L, Baker SS. Pathogenesis of nonalcoholic steatohepatitis. Cell Mol Life Sci. (2016) 73:1969–87. doi: 10.1007/s00018-016-2161-x
36. Koyama Y, Brenner DA. Liver inflammation and fibrosis. J Clin Invest. (2017) 127:55–64. doi: 10.1172/JCI88881
37. Bouwens L, Baekeland M, De Zanger R, Wisse E. Quantitation, tissue distribution and proliferation kinetics of Kupffer cells in normal rat liver. Hepatology. (1986) 6:718–22. doi: 10.1002/hep.1840060430
38. Kolios G, Valatas V, Kouroumalis E. Role of Kupffer cells in the pathogenesis of liver disease. World J Gastroenterol. (2006) 12:7413–20. doi: 10.3748/wjg.v12.i46.7413
39. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. (2008) 8:958–69. doi: 10.1038/nri2448
40. Kazankov K, Jorgensen SMD, Thomsen KL, Moller HJ, Vilstrup H, George J, et al. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat Rev Gastroenterol Hepatol. (2019) 16:145–59. doi: 10.1038/s41575-018-0082-x
41. Wan J, Benkdane M, Teixeira-Clerc F, Bonnafous S, Louvet A, Lafdil F, et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: a protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology. (2014) 59:130–42. doi: 10.1002/hep.26607
42. Smith K. Liver disease: Kupffer cells regulate the progression of ALD and NAFLD. Nat Rev Gastroenterol Hepatol. (2013) 10:503. doi: 10.1038/nrgastro.2013.140
43. Li H, Zhou Y, Wang H, Zhang M, Qiu P, Zhang M, et al. Crosstalk between liver macrophages and surrounding cells in nonalcoholic steatohepatitis. Front Immunol. (2020) 11:1169. doi: 10.3389/fimmu.2020.01169
44. Lee UE, Friedman SL. Mechanisms of hepatic fibrogenesis. Best Pract Res Clin Gastroenterol. (2011) 25:195–206. doi: 10.1016/j.bpg.2011.02.005
45. Zhang CY, Yuan WG, He P, Lei JH, Wang CX. Liver fibrosis and hepatic stellate cells: Etiology, pathological hallmarks and therapeutic targets. World J Gastroenterol. (2016) 22:10512–22. doi: 10.3748/wjg.v22.i48.10512
46. Cha JY, Kim DH, Chun KH. The role of hepatic macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Lab Anim Res. (2018) 34:133–9. doi: 10.5625/lar.2018.34.4.133
47. Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A, et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc Natl Acad Sci USA. (2012) 109:E3186–95. doi: 10.1073/pnas.1119964109
48. Seki E, Schwabe RF. Hepatic inflammation and fibrosis: functional links and key pathways. Hepatology. (2015) 61:1066–79. doi: 10.1002/hep.27332
49. Miura K, Yang L, van Rooijen N, Ohnishi H, Seki E. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am J Physiol Gastrointest Liver Physiol. (2012) 302:G1310–21. doi: 10.1152/ajpgi.00365.2011
50. Yang SJ, IglayReger HB, Kadouh HC, Bodary PF. Inhibition of the chemokine (C-C motif) ligand 2/chemokine (C-C motif) receptor 2 pathway attenuates hyperglycaemia and inflammation in a mouse model of hepatic steatosis and lipoatrophy. Diabetologia. (2009) 52:972–81. doi: 10.1007/s00125-009-1309-8
51. Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, et al. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest. (2006) 116:115–24. doi: 10.1172/JCI24335
52. Baeck C, Wehr A, Karlmark KR, Heymann F, Vucur M, Gassler N, et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut. (2012) 61:416–26. doi: 10.1136/gutjnl-2011-300304
53. Eslam M, George J. Genetic contributions to NAFLD: leveraging shared genetics to uncover systems biology. Nat Rev Gastroenterol Hepatol. (2020) 17:40–52. doi: 10.1038/s41575-019-0212-0
54. Smagris E, BasuRay S, Li J, Huang Y, Lai KM, Gromada J, et al. Pnpla3I148M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology. (2015) 61:108–18. doi: 10.1002/hep.27242
55. BasuRay S, Wang Y, Smagris E, Cohen JC, Hobbs HH. Accumulation of PNPLA3 on lipid droplets is the basis of associated hepatic steatosis. Proc Natl Acad Sci USA. (2019) 116:9521–6. doi: 10.1073/pnas.1901974116
56. Dong XC. PNPLA3-A potential therapeutic target for personalized treatment of chronic liver disease. Front Med. (2019) 6:304. doi: 10.3389/fmed.2019.00304
57. Liu YL, Reeves HL, Burt AD, Tiniakos D, McPherson S, Leathart JB, et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat Commun. (2014) 5:4309. doi: 10.1038/ncomms5309
58. Jiang ZG, Tapper EB, Kim M, Connelly MA, Krawczyk SA, Yee EU, et al. Genetic determinants of circulating lipoproteins in nonalcoholic fatty liver disease. J Clin Gastroenterol. (2018) 52:444–51. doi: 10.1097/MCG.0000000000000816
59. Gibney ER, Nolan CM. Epigenetics and gene expression. Heredity (Edinb). (2010) 105:4–13. doi: 10.1038/hdy.2010.54
60. Kitamoto T, Kitamoto A, Ogawa Y, Honda Y, Imajo K, Saito S, et al. Targeted-bisulfite sequence analysis of the methylation of CpG islands in genes encoding PNPLA3, SAMM50, and PARVB of patients with non-alcoholic fatty liver disease. J Hepatol. (2015) 63:494–502. doi: 10.1016/j.jhep.2015.02.049
61. Hardy T, Zeybel M, Day CP, Dipper C, Masson S, McPherson S, et al. Plasma DNA methylation: a potential biomarker for stratification of liver fibrosis in non-alcoholic fatty liver disease. Gut. (2017) 66:1321–8. doi: 10.1136/gutjnl-2016-311526
62. Cheung O, Puri P, Eicken C, Contos MJ, Mirshahi F, Maher JW, et al. Nonalcoholic steatohepatitis is associated with altered hepatic MicroRNA expression. Hepatology. (2008) 48:1810–20. doi: 10.1002/hep.22569
63. Zhang ZC, Liu Y, Xiao LL, Li SF, Jiang JH, Zhao Y, et al. Upregulation of miR-125b by estrogen protects against non-alcoholic fatty liver in female mice. J Hepatol. (2015) 63:1466–75. doi: 10.1016/j.jhep.2015.07.037
64. Calo N, Ramadori P, Sobolewski C, Romero Y, Maeder C, Fournier M, et al. Stress-activated miR-21/miR-21* in hepatocytes promotes lipid and glucose metabolic disorders associated with high-fat diet consumption. Gut. (2016) 65:1871–81. doi: 10.1136/gutjnl-2015-310822
65. Guo J, Fang W, Sun L, Lu Y, Dou L, Huang X, et al. Ultraconserved element uc.372 drives hepatic lipid accumulation by suppressing miR-195/miR4668 maturation. Nat Commun. (2018) 9:612. doi: 10.1038/s41467-018-03072-8
66. Atanasovska B, Rensen SS, van der Sijde MR, Marsman G, Kumar V, Jonkers I, et al. A liver-specific long noncoding RNA with a role in cell viability is elevated in human nonalcoholic steatohepatitis. Hepatology. (2017) 66:794–808. doi: 10.1002/hep.29034
67. Zhang Y, Cheng X, Lu Z, Wang J, Chen H, Fan W, et al. Upregulation of miR-15b in NAFLD models and in the serum of patients with fatty liver disease. Diabetes Res Clin Pract. (2013) 99:327–34. doi: 10.1016/j.diabres.2012.11.025
68. Qin W, Li X, Xie L, Li S, Liu J, Jia L, et al. A long non-coding RNA, APOA4-AS, regulates APOA4 expression depending on HuR in mice. Nucleic Acids Res. (2016) 44:6423–33. doi: 10.1093/nar/gkw341
69. Cermelli S, Ruggieri A, Marrero JA, Ioannou GN, Beretta L. Circulating microRNAs in patients with chronic hepatitis C and non-alcoholic fatty liver disease. PLoS ONE. (2011) 6:e23937. doi: 10.1371/journal.pone.0023937
70. Yu F, Zheng J, Mao Y, Dong P, Li G, Lu Z, et al. Long non-coding RNA APTR promotes the activation of hepatic stellate cells and the progression of liver fibrosis. Biochem Biophys Res Commun. (2015) 463:679–85. doi: 10.1016/j.bbrc.2015.05.124
71. Gerhard GS, DiStefano JK. Micro RNAs in the development of non-alcoholic fatty liver disease. World J Hepatol. (2015) 7:226–34. doi: 10.4254/wjh.v7.i2.226
72. Chen Y, Chen X, Gao J, Xu C, Xu P, Li Y, et al. Long noncoding RNA FLRL2 alleviated nonalcoholic fatty liver disease through Arntl-Sirt1 pathway. FASEB J. (2019) 33:11411–9. doi: 10.1096/fj.201900643RRR
73. Zhang T, Yang Z, Kusumanchi P, Han S, Liangpunsakul S. Critical role of microRNA-21 in the pathogenesis of liver diseases. Front Med. (2020) 7:7. doi: 10.3389/fmed.2020.00007
74. Ma M, Duan R, Shen L, Liu M, Ji Y, Zhou H, et al. The lncRNA Gm15622 stimulates SREBP-1c expression and hepatic lipid accumulation by sponging the miR-742-3p in mice. J Lipid Res. (2020) 61:1052–64. doi: 10.1194/jlr.RA120000664
75. Hu Y, Liu HX, Jena PK, Sheng L, Ali MR, Wan YY. miR-22 inhibition reduces hepatic steatosis via FGF21 and FGFR1 induction. JHEP Rep. (2020) 2:100093. doi: 10.1016/j.jhepr.2020.100093
76. Yu F, Chen B, Dong P, Zheng J. HOTAIR epigenetically modulates PTEN expression via MicroRNA-29b: a novel mechanism in regulation of liver fibrosis. Mol Ther. (2017) 25:205–17. doi: 10.1016/j.ymthe.2016.10.015
77. Ali O, Darwish HA, Eldeib KM, Abdel Azim SA. miR-26a potentially contributes to the regulation of fatty acid and sterol metabolism in vitro human HepG2 cell model of nonalcoholic fatty liver disease. Oxid Med Cell Longev. (2018) 2018:8515343. doi: 10.1155/2018/8515343
78. Shen X, Guo H, Xu J, Wang J. Inhibition of lncRNA HULC improves hepatic fibrosis and hepatocyte apoptosis by inhibiting the MAPK signaling pathway in rats with nonalcoholic fatty liver disease. J Cell Physiol. (2019) 234:18169–79. doi: 10.1002/jcp.28450
79. Zhang M, Sun W, Zhou M, Tang Y. MicroRNA-27a regulates hepatic lipid metabolism and alleviates NAFLD via repressing FAS and SCD1. Sci Rep. (2017) 7:14493. doi: 10.1038/s41598-017-15141-x
80. Zhang M, Chi X, Qu N, Wang C. Long noncoding RNA lncARSR promotes hepatic lipogenesis via Akt/SREBP-1c pathway and contributes to the pathogenesis of nonalcoholic steatohepatitis. Biochem Biophys Res Commun. (2018) 499:66–70. doi: 10.1016/j.bbrc.2018.03.127
81. Jampoka K, Muangpaisarn P, Khongnomnan K, Treeprasertsuk S, Tangkijvanich P, Payungporn S. Serum miR-29a and miR-122 as potential biomarkers for non-alcoholic fatty liver disease (NAFLD). Microrna. (2018) 7:215–22. doi: 10.2174/2211536607666180531093302
82. Chen X, Xu Y, Zhao D, Chen T, Gu C, Yu G, et al. LncRNA-AK012226 is involved in fat accumulation in db/db mice fatty liver and non-alcoholic fatty liver disease cell model. Front Pharmacol. (2018) 9:888. doi: 10.3389/fphar.2018.00888
83. Fan J, Li H, Nie X, Yin Z, Zhao Y, Chen C, et al. MiR-30c-5p ameliorates hepatic steatosis in leptin receptor-deficient (db/db) mice via down-regulating FASN. Oncotarget. (2017) 8:13450–63. doi: 10.18632/oncotarget.14561
84. Lan X, Wu L, Wu N, Chen Q, Li Y, Du X, et al. Long noncoding RNA lnc-HC regulates PPARgamma-mediated hepatic lipid metabolism through miR-130b-3p. Mol Ther Nucleic Acids. (2019) 18:954–65. doi: 10.1016/j.omtn.2019.10.018
85. Auguet T, Aragones G, Berlanga A, Guiu-Jurado E, Marti A, Martinez S, et al. miR33a/miR33b* and miR122 as possible contributors to hepatic lipid metabolism in obese women with nonalcoholic fatty liver disease. Int J Mol Sci. (2016) 17:1620. doi: 10.3390/ijms17101620
86. Li D, Cheng M, Niu Y, Chi X, Liu X, Fan J, et al. Identification of a novel human long non-coding RNA that regulates hepatic lipid metabolism by inhibiting SREBP-1c. Int J Biol Sci. (2017) 13:349–57. doi: 10.7150/ijbs.16635
87. Ding J, Li M, Wan X, Jin X, Chen S, Yu C, et al. Effect of miR-34a in regulating steatosis by targeting PPARalpha expression in nonalcoholic fatty liver disease. Sci Rep. (2015) 5:13729. doi: 10.1038/srep13729
88. Yang Z, Zhang T, Han S, Kusumanchi P, Huda N, Jiang Y, et al. Long noncoding RNA H19 - a new player in the pathogenesis of liver diseases. Transl Res. (2021) 230:139–50. doi: 10.1016/j.trsl.2020.11.010
89. Celikbilek M, Baskol M, Taheri S, Deniz K, Dogan S, Zararsiz G, et al. Circulating microRNAs in patients with non-alcoholic fatty liver disease. World J Hepatol. (2014) 6:613–20. doi: 10.4254/wjh.v6.i8.613
90. Sookoian S, Rohr C, Salatino A, Dopazo H, Fernandez Gianotti T, Castano GO, et al. Genetic variation in long noncoding RNAs and the risk of nonalcoholic fatty liver disease. Oncotarget. (2017) 8:22917–26. doi: 10.18632/oncotarget.15286
91. Li P, Ruan X, Yang L, Kiesewetter K, Zhao Y, Luo H, et al. A liver-enriched long non-coding RNA, lncLSTR, regulates systemic lipid metabolism in mice. Cell Metab. (2015) 21:455–67. doi: 10.1016/j.cmet.2015.02.004
92. Zhao XY, Xiong X, Liu T, Mi L, Peng X, Rui C, et al. Long noncoding RNA licensing of obesity-linked hepatic lipogenesis and NAFLD pathogenesis. Nat Commun. (2018) 9:2986. doi: 10.1038/s41467-018-05383-2
93. Wang Y, Du J, Niu X, Fu N, Wang R, Zhang Y, et al. MiR-130a-3p attenuates activation and induces apoptosis of hepatic stellate cells in nonalcoholic fibrosing steatohepatitis by directly targeting TGFBR1 and TGFBR2. Cell Death Dis. (2017) 8:e2792. doi: 10.1038/cddis.2017.10
94. Wang J, Yang W, Chen Z, Chen J, Meng Y, Feng B, et al. Long noncoding RNA lncSHGL recruits hnRNPA1 to suppress hepatic gluconeogenesis and lipogenesis. Diabetes. (2018) 67:581–93. doi: 10.2337/db17-0799
95. Jiang H, Qian Y, Shen Z, Liu Y, He Y, Gao R, et al. Circulating microRNA135a3p in serum extracellular vesicles as a potential biological marker of nonalcoholic fatty liver disease. Mol Med Rep. (2021) 24:12137. doi: 10.3892/mmr.2021.12137
96. Du J, Niu X, Wang Y, Kong L, Wang R, Zhang Y, et al. MiR-146a-5p suppresses activation and proliferation of hepatic stellate cells in nonalcoholic fibrosing steatohepatitis through directly targeting Wnt1 and Wnt5a. Sci Rep. (2015) 5:16163. doi: 10.1038/srep16163
97. Zhang K, Han X, Zhang Z, Zheng L, Hu Z, Yao Q, et al. The liver-enriched lnc-LFAR1 promotes liver fibrosis by activating TGFbeta and Notch pathways. Nat Commun. (2017) 8:144. doi: 10.1038/s41467-017-00204-4
98. Miller AM, Gilchrist DS, Nijjar J, Araldi E, Ramirez CM, Lavery CA, et al. MiR-155 has a protective role in the development of non-alcoholic hepatosteatosis in mice. PLoS ONE. (2013) 8:e72324. doi: 10.1371/journal.pone.0072324
99. Leti F, Legendre C, Still CD, Chu X, Petrick A, Gerhard GS, et al. Altered expression of MALAT1 lncRNA in nonalcoholic steatohepatitis fibrosis regulates CXCL5 in hepatic stellate cells. Transl Res. (2017) 190:25–39.e21. doi: 10.1016/j.trsl.2017.09.001
100. Wang Y, Zhu K, Yu W, Wang H, Liu L, Wu Q, et al. MiR-181b regulates steatosis in nonalcoholic fatty liver disease via targeting SIRT1. Biochem Biophys Res Commun. (2017) 493:227–32. doi: 10.1016/j.bbrc.2017.09.042
101. Huang P, Huang FZ, Liu HZ, Zhang TY, Yang MS, Sun CZ. LncRNA MEG3 functions as a ceRNA in regulating hepatic lipogenesis by competitively binding to miR-21 with LRP6. Metabolism. (2019) 94:1–8. doi: 10.1016/j.metabol.2019.01.018
102. Xu M, Zheng XM, Jiang F, Qiu WQ. MicroRNA-190b regulates lipid metabolism and insulin sensitivity by targeting IGF-1 and ADAMTS9 in non-alcoholic fatty liver disease. J Cell Biochem. (2018) 119:5864–74. doi: 10.1002/jcb.26776
103. Zhang B, Li H, Li D, Sun H, Li M, Hu H. Long noncoding RNA Mirt2 upregulates USP10 expression to suppress hepatic steatosis by sponging miR-34a-5p. Gene. (2019) 700:139–48. doi: 10.1016/j.gene.2019.02.096
104. Liu XL, Cao HX, Wang BC, Xin FZ, Zhang RN, Zhou D, et al. miR-192-5p regulates lipid synthesis in non-alcoholic fatty liver disease through SCD-1. World J Gastroenterol. (2017) 23:8140–51. doi: 10.3748/wjg.v23.i46.8140
105. Yuan X, Wang J, Tang X, Li Y, Xia P, Gao X. Berberine ameliorates nonalcoholic fatty liver disease by a global modulation of hepatic mRNA and lncRNA expression profiles. J Transl Med. (2015) 13:24. doi: 10.1186/s12967-015-0383-6
106. Nie H, Song C, Wang D, Cui S, Ren T, Cao Z, et al. MicroRNA-194 inhibition improves dietary-induced non-alcoholic fatty liver disease in mice through targeting on FXR. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:3087–94. doi: 10.1016/j.bbadis.2017.09.020
107. Wang X. Down-regulation of lncRNA-NEAT1 alleviated the non-alcoholic fatty liver disease via mTOR/S6K1 signaling pathway. J Cell Biochem. (2018) 119:1567–74. doi: 10.1002/jcb.26317
108. Chen Q, Xiong C, Jia K, Jin J, Li Z, Huang Y, et al. Hepatic transcriptome analysis from HFD-fed mice defines a long noncoding RNA regulating cellular cholesterol levels. J Lipid Res. (2019) 60:341–52. doi: 10.1194/jlr.M086215
109. Li Y, Luan Y, Li J, Song H, Li Y, Qi H, et al. Exosomal miR-199a-5p promotes hepatic lipid accumulation by modulating MST1 expression and fatty acid metabolism. Hepatol Int. (2020) 14:1057–74. doi: 10.1007/s12072-020-10096-0
110. Ma TT, Huang C, Ni Y, Yang Y, Li J. ATP citrate lyase and LncRNA NONMMUT010685 play crucial role in nonalcoholic fatty liver disease based on analysis of microarray data. Cell Physiol Biochem. (2018) 51:871–85. doi: 10.1159/000495384
111. Feng YY, Xu XQ, Ji CB, Shi CM, Guo XR, Fu JF. Aberrant hepatic microRNA expression in nonalcoholic fatty liver disease. Cell Physiol Biochem. (2014) 34:1983–97. doi: 10.1159/000366394
112. Hu Y, Ye H, Shi LX. MicroRNA-205 ameliorates lipid accumulation in non-alcoholic fatty liver disease through targeting NEU1. Eur Rev Med Pharmacol Sci. (2019) 23:10072–82. doi: 10.26355/eurrev_201911_19575
113. Gong Z, Tang J, Xiang T, Lin J, Deng C, Peng Y, et al. Genomewide identification of long noncoding RNAs in CCl4induced liver fibrosis via RNA sequencing. Mol Med Rep. (2018) 18:299–307. doi: 10.3892/mmr.2018.8986
114. Jiang X, Jiang L, Shan A, Su Y, Cheng Y, Song D, et al. Targeting hepatic miR-221/222 for therapeutic intervention of nonalcoholic steatohepatitis in mice. EBioMedicine. (2018) 37:307–21. doi: 10.1016/j.ebiom.2018.09.051
115. Zheng J, Yu F, Dong P, Wu L, Zhang Y, Hu Y, et al. Long non-coding RNA PVT1 activates hepatic stellate cells through competitively binding microRNA-152. Oncotarget. (2016) 7:62886–97. doi: 10.18632/oncotarget.11709
116. He Y, Hwang S, Cai Y, Kim SJ, Xu M, Yang D, et al. MicroRNA-223 ameliorates nonalcoholic steatohepatitis and cancer by targeting multiple inflammatory and oncogenic genes in hepatocytes. Hepatology. (2019) 70:1150–67. doi: 10.1002/hep.30645
117. Di Mauro S, Scamporrino A, Petta S, Urbano F, Filippello A, Ragusa M, et al. Serum coding and non-coding RNAs as biomarkers of NAFLD and fibrosis severity. Liver Int. (2019) 39:1742–54. doi: 10.1111/liv.14167
118. Chen C, Wu CQ, Zhang ZQ, Yao DK, Zhu L. Loss of expression of miR-335 is implicated in hepatic stellate cell migration and activation. Exp Cell Res. (2011) 317:1714–25. doi: 10.1016/j.yexcr.2011.05.001
119. Kaur S, Rawal P, Siddiqui H, Rohilla S, Sharma S, Tripathi DM, et al. Increased expression of RUNX1 in liver correlates with NASH activity score in patients with non-alcoholic steatohepatitis (NASH). Cells. (2019) 8:1277. doi: 10.3390/cells8101277
120. Lei L, Zhou C, Yang X, Li L. Down-regulation of microRNA-375 regulates adipokines and inhibits inflammatory cytokines by targeting AdipoR2 in non-alcoholic fatty liver disease. Clin Exp Pharmacol Physiol. (2018) 45:819–31. doi: 10.1111/1440-1681.12940
121. Chen G, Yu D, Nian X, Liu J, Koenig RJ, Xu B, et al. LncRNA SRA promotes hepatic steatosis through repressing the expression of adipose triglyceride lipase (ATGL). Sci Rep. (2016) 6:35531. doi: 10.1038/srep35531
122. Zhang T, Hu J, Wang X, Zhao X, Li Z, Niu J, et al. MicroRNA-378 promotes hepatic inflammation and fibrosis via modulation of the NF-kappaB-TNFalpha pathway. J Hepatol. (2019) 70:87–96. doi: 10.1016/j.jhep.2018.08.026
123. Zeng N, Huang R, Li N, Jiang H, Li R, Wang F, et al. MiR-451a attenuates free fatty acids-mediated hepatocyte steatosis by targeting the thyroid hormone responsive spot 14 gene. Mol Cell Endocrinol. (2018) 474:260–71. doi: 10.1016/j.mce.2018.03.016
124. Moran-Salvador E, Mann J. Epigenetics and liver fibrosis. Cell Mol Gastroenterol Hepatol. (2017) 4:125–34. doi: 10.1016/j.jcmgh.2017.04.007
125. Ishimoto T, Lanaspa MA, Rivard CJ, Roncal-Jimenez CA, Orlicky DJ, Cicerchi C, et al. High-fat and high-sucrose (western) diet induces steatohepatitis that is dependent on fructokinase. Hepatology. (2013) 58:1632–43. doi: 10.1002/hep.26594
126. Lytle KA, Jump DB. Is western diet-induced nonalcoholic steatohepatitis in Ldlr-/- mice reversible? PLoS ONE. (2016) 11:e0146942. doi: 10.1371/journal.pone.0146942
127. Zhang H, Leveille M, Courty E, Gunes A, B NN, Estall JL. Differences in metabolic and liver pathobiology induced by two dietary mouse models of nonalcoholic fatty liver disease. Am J Physiol Endocrinol Metab. (2020) 319:E863–76. doi: 10.1152/ajpendo.00321.2020
128. Schuster S, Cabrera D, Arrese M, Feldstein AE. Triggering and resolution of inflammation in NASH. Nat Rev Gastroenterol Hepatol. (2018) 15:349–64. doi: 10.1038/s41575-018-0009-6
129. Neuschwander-Tetri BA. Carbohydrate intake and nonalcoholic fatty liver disease. Curr Opin Clin Nutr Metab Care. (2013) 16:446–52. doi: 10.1097/MCO.0b013e328361c4d1
130. Jensen T, Abdelmalek MF, Sullivan S, Nadeau KJ, Green M, Roncal C, et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J Hepatol. (2018) 68:1063–75. doi: 10.1016/j.jhep.2018.01.019
131. DiStefano JK, Shaibi GQ. The relationship between excessive dietary fructose consumption and paediatric fatty liver disease. Pediatr Obes. (2021) 16:e12759. doi: 10.1111/ijpo.12759
132. Asgharpour A, Cazanave SC, Pacana T, Seneshaw M, Vincent R, Banini BA, et al. A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J Hepatol. (2016) 65:579–88. doi: 10.1016/j.jhep.2016.05.005
133. Clapper JR, Hendricks MD, Gu G, Wittmer C, Dolman CS, Herich J, et al. Diet-induced mouse model of fatty liver disease and nonalcoholic steatohepatitis reflecting clinical disease progression and methods of assessment. Am J Physiol Gastrointest Liver Physiol. (2013) 305:G483–95. doi: 10.1152/ajpgi.00079.2013
134. Kim Y, Chang Y, Cho YK, Ahn J, Shin H, Ryu S. Obesity and weight gain are associated with progression of fibrosis in patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. (2019) 17:543–50.e2. doi: 10.1016/j.cgh.2018.07.006
135. Polyzos SA, Kountouras J, Mantzoros CS. Obesity and nonalcoholic fatty liver disease: From pathophysiology to therapeutics. Metabolism. (2019) 92:82–97. doi: 10.1016/j.metabol.2018.11.014
136. Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, et al. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American association for the study of liver diseases. Hepatology. (2018) 67:328–57. doi: 10.1002/hep.29367
137. Targher G, Corey KE, Byrne CD, Roden M. The complex link between NAFLD and type 2 diabetes mellitus - mechanisms and treatments. Nat Rev Gastroenterol Hepatol. (2021) 18:599–612. doi: 10.1038/s41575-021-00448-y
138. Katsiki N, Mikhailidis DP, Mantzoros CS. Non-alcoholic fatty liver disease and dyslipidemia: An update. Metabolism. (2016) 65:1109–23. doi: 10.1016/j.metabol.2016.05.003
139. Arslan N. Obesity, fatty liver disease and intestinal microbiota. World J Gastroenterol. (2014) 20:16452–63. doi: 10.3748/wjg.v20.i44.16452
140. Leung C, Rivera L, Furness JB, Angus PW. The role of the gut microbiota in NAFLD. Nat Rev Gastroenterol Hepatol. (2016) 13:412–25. doi: 10.1038/nrgastro.2016.85
141. Bashiardes S, Shapiro H, Rozin S, Shibolet O, Elinav E. Non-alcoholic fatty liver and the gut microbiota. Mol Metab. (2016) 5:782–94. doi: 10.1016/j.molmet.2016.06.003
142. Gerard P. Gut microbiota and obesity. Cell Mol Life Sci. (2016) 73:147–62. doi: 10.1007/s00018-015-2061-5
143. Boursier J, Diehl AM. Implication of gut microbiota in nonalcoholic fatty liver disease. PLoS Pathog. (2015) 11:e1004559. doi: 10.1371/journal.ppat.1004559
144. Aron-Wisnewsky J, Warmbrunn MV, Nieuwdorp M, Clement K. Nonalcoholic fatty liver disease: modulating gut microbiota to improve severity? Gastroenterology. (2020) 158:1881–98. doi: 10.1053/j.gastro.2020.01.049
145. Brandl K, Schnabl B. Intestinal microbiota and nonalcoholic steatohepatitis. Curr Opin Gastroenterol. (2017) 33:128–33. doi: 10.1097/MOG.0000000000000349
146. Mouries J, Brescia P, Silvestri A, Spadoni I, Sorribas M, Wiest R, et al. Microbiota-driven gut vascular barrier disruption is a prerequisite for non-alcoholic steatohepatitis development. J Hepatol. (2019) 71:1216–28. doi: 10.1016/j.jhep.2019.08.005
147. Brown K, DeCoffe D, Molcan E, Gibson DL. Diet-induced dysbiosis of the intestinal microbiota and the effects on immunity and disease. Nutrients. (2012) 4:1095–119. doi: 10.3390/nu4081095
148. Boursier J, Mueller O, Barret M, Machado M, Fizanne L, Araujo-Perez F, et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology. (2016) 63:764–75. doi: 10.1002/hep.28356
149. Shen F, Zheng RD, Sun XQ, Ding WJ, Wang XY, Fan JG. Gut microbiota dysbiosis in patients with non-alcoholic fatty liver disease. Hepatobiliary Pancreat Dis Int. (2017) 16:375–81. doi: 10.1016/S1499-3872(17)60019-5
150. Zhu L, Baker SS, Gill C, Liu W, Alkhouri R, Baker RD, et al. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology. (2013) 57:601–9. doi: 10.1002/hep.26093
151. Nalbantoglu IL, Brunt EM. Role of liver biopsy in nonalcoholic fatty liver disease. World J Gastroenterol. (2014) 20:9026–37. doi: 10.3748/wjg.v20.i27.9026
152. Sebastiani G, Alberti A. Non invasive fibrosis biomarkers reduce but not substitute the need for liver biopsy. World J Gastroenterol. (2006) 12:3682–94. doi: 10.3748/wjg.v12.i23.3682
153. Sumida Y, Nakajima A, Itoh Y. Limitations of liver biopsy and non-invasive diagnostic tests for the diagnosis of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J Gastroenterol. (2014) 20:475–85. doi: 10.3748/wjg.v20.i2.475
154. Stern C, Castera L. Non-invasive diagnosis of hepatic steatosis. Hepatol Int. (2017) 11:70–8. doi: 10.1007/s12072-016-9772-z
155. Sviklane L, Olmane E, Dzerve Z, Kupcs K, Pirags V, Sokolovska J. Fatty liver index and hepatic steatosis index for prediction of non-alcoholic fatty liver disease in type 1 diabetes. J Gastroenterol Hepatol. (2018) 33:270–6. doi: 10.1111/jgh.13814
156. Wong VW, Adams LA, de Ledinghen V, Wong GL, Sookoian S. Noninvasive biomarkers in NAFLD and NASH - current progress and future promise. Nat Rev Gastroenterol Hepatol. (2018) 15:461–78. doi: 10.1038/s41575-018-0014-9
157. Loomba R. Role of imaging-based biomarkers in NAFLD: Recent advances in clinical application and future research directions. J Hepatol. (2018) 68:296–304. doi: 10.1016/j.jhep.2017.11.028
158. Hernaez R, Lazo M, Bonekamp S, Kamel I, Brancati FL, Guallar E, et al. Diagnostic accuracy and reliability of ultrasonography for the detection of fatty liver: a meta-analysis. Hepatology. (2011) 54:1082–90. doi: 10.1002/hep.24452
159. Tobari M, Hashimoto E, Yatsuji S, Torii N, Shiratori K. Imaging of nonalcoholic steatohepatitis: advantages and pitfalls of ultrasonography and computed tomography. Intern Med. (2009) 48:739–46. doi: 10.2169/internalmedicine.48.1869
160. Cam I, Koc U, Genez S, Gunes A. Computed tomography measurements of hepatic steatosis in cholelitihiasis and cholecystectomy cases using unenhanced images. J Med Imaging Radiat Sci. (2020) 51:137–44. doi: 10.1016/j.jmir.2019.12.003
161. Desai NK, Harney S, Raza R, Al-Ibraheemi A, Shillingford N, Mitchell PD, et al. Comparison of controlled attenuation parameter and liver biopsy to assess hepatic steatosis in pediatric patients. J Pediatr. (2016) 173:160–4.e1. doi: 10.1016/j.jpeds.2016.03.021
162. Middleton MS, Heba ER, Hooker CA, Bashir MR, Fowler KJ, Sandrasegaran K, et al. Agreement between magnetic resonance imaging proton density fat fraction measurements and pathologist-assigned steatosis grades of liver biopsies from adults with nonalcoholic steatohepatitis. Gastroenterology. (2017) 153:753–61. doi: 10.1053/j.gastro.2017.06.005
163. Shi KQ, Tang JZ, Zhu XL, Ying L, Li DW, Gao J, et al. Controlled attenuation parameter for the detection of steatosis severity in chronic liver disease: a meta-analysis of diagnostic accuracy. J Gastroenterol Hepatol. (2014) 29:1149–58. doi: 10.1111/jgh.12519
164. Caussy C, Alquiraish MH, Nguyen P, Hernandez C, Cepin S, Fortney LE, et al. Optimal threshold of controlled attenuation parameter with MRI-PDFF as the gold standard for the detection of hepatic steatosis. Hepatology. (2018) 67:1348–59. doi: 10.1002/hep.29639
165. Bannas P, Kramer H, Hernando D, Agni R, Cunningham AM, Mandal R, et al. Quantitative magnetic resonance imaging of hepatic steatosis: validation in ex vivo human livers. Hepatology. (2015) 62:1444–55. doi: 10.1002/hep.28012
166. Resino S, Bellon JM, Asensio C, Micheloud D, Miralles P, Vargas A, et al. Can serum hyaluronic acid replace simple non-invasive indexes to predict liver fibrosis in HIV/Hepatitis C coinfected patients? BMC Infect Dis. (2010) 10:244. doi: 10.1186/1471-2334-10-244
167. Kumagai E, Mano Y, Yoshio S, Shoji H, Sugiyama M, Korenaga M, et al. Serum YKL-40 as a marker of liver fibrosis in patients with non-alcoholic fatty liver disease. Sci Rep. (2016) 6:35282. doi: 10.1038/srep35282
168. Yoshiji H, Kuriyama S, Miyamoto Y, Thorgeirsson UP, Gomez DE, Kawata M, et al. Tissue inhibitor of metalloproteinases-1 promotes liver fibrosis development in a transgenic mouse model. Hepatology. (2000) 32:1248–54. doi: 10.1053/jhep.2000.20521
169. Benyon RC, Iredale JP, Goddard S, Winwood PJ, Arthur MJ. Expression of tissue inhibitor of metalloproteinases 1 and 2 is increased in fibrotic human liver. Gastroenterology. (1996) 110:821–31. doi: 10.1053/gast.1996.v110.pm8608892
170. Thiele ND, Wirth JW, Steins D, Koop AC, Ittrich H, Lohse AW, et al. TIMP-1 is upregulated, but not essential in hepatic fibrogenesis and carcinogenesis in mice. Sci Rep. (2017) 7:714. doi: 10.1038/s41598-017-00671-1
171. Gudowska M, Gruszewska E, Panasiuk A, Cylwik B, Swiderska M, Flisiak R, et al. High serum N-terminal propeptide of procollagen type III concentration is associated with liver diseases. Prz Gastroenterol. (2017) 12:203–7. doi: 10.5114/pg.2017.70474
172. Tanwar S, Trembling PM, Guha IN, Parkes J, Kaye P, Burt AD, et al. Validation of terminal peptide of procollagen III for the detection and assessment of nonalcoholic steatohepatitis in patients with nonalcoholic fatty liver disease. Hepatology. (2013) 57:103–11. doi: 10.1002/hep.26030
173. Hallsworth K, Adams LA. Lifestyle modification in NAFLD/NASH: Facts and figures. JHEP Rep. (2019) 1:468–79. doi: 10.1016/j.jhepr.2019.10.008
174. Oseini AM, Sanyal AJ. Therapies in non-alcoholic steatohepatitis (NASH). Liver Int. (2017) 37:97–103. doi: 10.1111/liv.13302
175. Vilar-Gomez E, Martinez-Perez Y, Calzadilla-Bertot L, Torres-Gonzalez A, Gra-Oramas B, Gonzalez-Fabian L, et al. Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology. (2015) 149:367–78.e5. doi: 10.1053/j.gastro.2015.04.005
176. Pacana T, Sanyal AJ. Vitamin E and nonalcoholic fatty liver disease. Curr Opin Clin Nutr Metab Care. (2012) 15:641–8. doi: 10.1097/MCO.0b013e328357f747
177. Chen Z, Tian R, She Z, Cai J, Li H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic Biol Med. (2020) 152:116–41. doi: 10.1016/j.freeradbiomed.2020.02.025
178. Phung N, Pera N, Farrell G, Leclercq I, Hou JY, George J. Pro-oxidant-mediated hepatic fibrosis and effects of antioxidant intervention in murine dietary steatohepatitis. Int J Mol Med. (2009) 24:171–80. doi: 10.3892/ijmm_00000220
179. Vilar-Gomez E, Vuppalanchi R, Gawrieh S, Ghabril M, Saxena R, Cummings OW, et al. Vitamin E improves transplant-free survival and hepatic decompensation among patients with nonalcoholic steatohepatitis and advanced fibrosis. Hepatology. (2020) 71:495–509. doi: 10.1002/hep.30368
180. Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. (2010) 362:1675–85. doi: 10.1056/NEJMoa0907929
181. Bril F, Biernacki DM, Kalavalapalli S, Lomonaco R, Subbarayan SK, Lai J, et al. Role of vitamin e for nonalcoholic steatohepatitis in patients with type 2 diabetes: a randomized controlled trial. Diabetes Care. (2019) 42:1481–8. doi: 10.2337/dc19-0167
182. Lian J, Fu J. Pioglitazone for NAFLD patients with prediabetes or type 2 diabetes mellitus: a meta-analysis. Front Endocrinol (Lausanne). (2021) 12:615409. doi: 10.3389/fendo.2021.615409
183. Lee JI, Lee HW, Lee KS, Lee HS, Park JY. Effects of statin use on the development and progression of nonalcoholic fatty liver disease: a nationwide nested case-control study. Am J Gastroenterol. (2021) 116:116–24. doi: 10.14309/ajg.0000000000000845
184. Cusi K. Pioglitazone for the treatment of NASH in patients with prediabetes or type 2 diabetes mellitus. Gut. (2018) 67:1371. doi: 10.1136/gutjnl-2017-313958
185. Majima T, Komatsu Y, Doi K, Shigemoto M, Takagi C, Fukao A, et al. Safety and efficacy of low-dose pioglitazone (7.5 mg/day) vs. standard-dose pioglitazone (15 mg/day) in Japanese women with type 2 diabetes mellitus. Endocr J. (2006) 53:325–30. doi: 10.1507/endocrj.K05-067
186. Athyros VG, Boutari C, Stavropoulos K, Anagnostis P, Imprialos KP, Doumas M, et al. Statins: An under-appreciated asset for the prevention and the treatment of NAFLD or NASH and the related cardiovascular risk. Curr Vasc Pharmacol. (2018) 16:246–53. doi: 10.2174/1570161115666170621082910
187. Doumas M, Imprialos K, Dimakopoulou A, Stavropoulos K, Binas A, Athyros VG. The role of statins in the management of nonalcoholic fatty liver disease. Curr Pharm Des. (2018) 24:4587–92. doi: 10.2174/1381612825666190117114305
188. Nadkarni P, Chepurny OG, Holz GG. Regulation of glucose homeostasis by GLP-1. Prog Mol Biol Transl Sci. (2014) 121:23–65. doi: 10.1016/B978-0-12-800101-1.00002-8
189. Meier JJ. GLP-1 receptor agonists for individualized treatment of type 2 diabetes mellitus. Nat Rev Endocrinol. (2012) 8:728–42. doi: 10.1038/nrendo.2012.140
190. Bernsmeier C, Meyer-Gerspach AC, Blaser LS, Jeker L, Steinert RE, Heim MH, et al. Glucose-induced glucagon-like Peptide 1 secretion is deficient in patients with non-alcoholic fatty liver disease. PLoS ONE. (2014) 9:e87488. doi: 10.1371/journal.pone.0087488
191. Lv X, Dong Y, Hu L, Lu F, Zhou C, Qin S. Glucagon-like peptide-1 receptor agonists (GLP-1 RAs) for the management of nonalcoholic fatty liver disease (NAFLD): A systematic review. Endocrinol Diabetes Metab. (2020) 3:e00163. doi: 10.1002/edm2.163
192. Ghazanfar H, Kandhi SD, Nawaz I, Javed N, Abraham MC, Farag M, et al. Role of glucagon-like peptide-1 receptor agonists in the management of non-alcoholic steatohepatitis: a clinical review article. Cureus. (2021) 13:e15141. doi: 10.7759/cureus.15141
193. Seghieri M, Christensen AS, Andersen A, Solini A, Knop FK, Vilsboll T. Future perspectives on GLP-1 receptor agonists and GLP-1/glucagon receptor co-agonists in the treatment of NAFLD. Front Endocrinol. (2018) 9:649. doi: 10.3389/fendo.2018.00649
194. Chinetti G, Fruchart JC, Staels B. Peroxisome proliferator-activated receptors (PPARs): nuclear receptors at the crossroads between lipid metabolism and inflammation. Inflamm Res. (2000) 49:497–505. doi: 10.1007/s000110050622
195. Ratziu V, Harrison SA, Francque S, Bedossa P, Lehert P, Serfaty L, et al. Elafibranor, an agonist of the peroxisome proliferator-activated receptor-alpha and -delta, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology. (2016) 150:1147–59.e5. doi: 10.1053/j.gastro.2016.01.038
196. Jiao Y, Lu Y, Li XY. Farnesoid X receptor: a master regulator of hepatic triglyceride and glucose homeostasis. Acta Pharmacol Sin. (2015) 36:44–50. doi: 10.1038/aps.2014.116
197. Abenavoli L, Falalyeyeva T, Boccuto L, Tsyryuk O, Kobyliak N. Obeticholic acid: a new era in the treatment of nonalcoholic fatty liver disease. Pharmaceuticals (Basel). (2018) 11:40104. doi: 10.3390/ph11040104
198. Harrison SA, Bashir MR, Lee KJ, Shim-Lopez J, Lee J, Wagner B, et al. A structurally optimized FXR agonist, MET409, reduced liver fat content over 12 weeks in patients with non-alcoholic steatohepatitis. J Hepatol. (2021) 75:25–33. doi: 10.1016/j.jhep.2021.01.047
199. An P, Wei G, Huang P, Li W, Qi X, Lin Y, et al. A novel non-bile acid FXR agonist EDP-305 potently suppresses liver injury and fibrosis without worsening of ductular reaction. Liver Int. (2020) 40:1655–69. doi: 10.1111/liv.14490
200. Krenkel O, Puengel T, Govaere O, Abdallah AT, Mossanen JC, Kohlhepp M, et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology. (2018) 67:1270–83. doi: 10.1002/hep.29544
201. Smith-Cortinez N, Fagundes RR, Gomez V, Kong D, de Waart DR, Heegsma J, et al. Collagen release by human hepatic stellate cells requires vitamin C and is efficiently blocked by hydroxylase inhibition. FASEB J. (2021) 35:e21219. doi: 10.1096/fj.202001564RR
202. Marra F, Tacke F. Roles for chemokines in liver disease. Gastroenterology. (2014) 147:577–94.e1. doi: 10.1053/j.gastro.2014.06.043
203. Friedman SL, Ratziu V, Harrison SA, Abdelmalek MF, Aithal GP, Caballeria J, et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology. (2018) 67:1754–67. doi: 10.1002/hep.29477
204. Henderson NC, Mackinnon AC, Farnworth SL, Poirier F, Russo FP, Iredale JP, et al. Galectin-3 regulates myofibroblast activation and hepatic fibrosis. Proc Natl Acad Sci USA. (2006) 103:5060–5. doi: 10.1073/pnas.0511167103
205. Traber PG, Zomer E. Therapy of experimental NASH and fibrosis with galectin inhibitors. PLoS ONE. (2013) 8:e83481. doi: 10.1371/journal.pone.0083481
206. Chalasani N, Abdelmalek MF, Garcia-Tsao G, Vuppalanchi R, Alkhouri N, Rinella M, et al. Belapectin study, effects of belapectin, an inhibitor of galectin-3, in patients with nonalcoholic steatohepatitis with cirrhosis and portal hypertension. Gastroenterology. (2020) 158:1334–45.e5. doi: 10.1053/j.gastro.2019.11.296
207. Schuster-Gaul S, Geisler LJ, McGeough MD, Johnson CD, Zagorska A, Li L, et al. ASK1 inhibition reduces cell death and hepatic fibrosis in an Nlrp3 mutant liver injury model. JCI Insight. (2020) 5:123294. doi: 10.1172/jci.insight.123294
208. Loomba R, Lawitz E, Mantry PS, Jayakumar S, Caldwell SH, Arnold H, et al. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: A randomized, phase 2 trial. Hepatology. (2018) 67:549–59. doi: 10.1002/hep.29514
209. Harrison SA, Wong VW, Okanoue T, Bzowej N, Vuppalanchi R, Younes Z, et al. Selonsertib for patients with bridging fibrosis or compensated cirrhosis due to NASH: Results from randomized phase III STELLAR trials. J Hepatol. (2020) 73:26–39. doi: 10.1016/j.jhep.2020.02.027
210. Saltzman ET, Palacios T, Thomsen M, Vitetta L. Intestinal microbiome shifts, dysbiosis, inflammation, and non-alcoholic fatty liver disease. Front Microbiol. (2018) 9:61. doi: 10.3389/fmicb.2018.00061
211. Tripathi A, Debelius J, Brenner DA, Karin M, Loomba R, Schnabl B, et al. The gut-liver axis and the intersection with the microbiome. Nat Rev Gastroenterol Hepatol. (2018) 15:397–411. doi: 10.1038/s41575-018-0011-z
212. Heck AM, Yanovski JA, Calis KA. Orlistat, a new lipase inhibitor for the management of obesity. Pharmacotherapy. (2000) 20:270–9. doi: 10.1592/phco.20.4.270.34882
213. Ke J, An Y, Cao B, Lang J, Wu N, Zhao D. Orlistat-induced gut microbiota modification in obese mice. Evid Based Complement Alternat Med. (2020) 2020:9818349. doi: 10.1155/2020/9818349
214. Rotman Y, Sanyal AJ. Current and upcoming pharmacotherapy for non-alcoholic fatty liver disease. Gut. (2017) 66:180–90. doi: 10.1136/gutjnl-2016-312431
215. Sumida Y, Yoneda M. Current and future pharmacological therapies for NAFLD/NASH. J Gastroenterol. (2018) 53:362–76. doi: 10.1007/s00535-017-1415-1
216. Polyzos SA, Kang ES, Tsochatzis EA, Kechagias S, Ekstedt M, Xanthakos S, et al. Commentary: Nonalcoholic or metabolic dysfunction-associated fatty liver disease? The epidemic of the 21st century in search of the most appropriate name. Metabolism. (2020) 113:154413. doi: 10.1016/j.metabol.2020.154413
217. Rinella ME, Green RM. The methionine-choline deficient dietary model of steatohepatitis does not exhibit insulin resistance. J Hepatol. (2004) 40:47–51. doi: 10.1016/j.jhep.2003.09.020
Keywords: non-alcohol fatty liver disease, non-alcoholic steatohepatitis, pathogenesis, diagnosis, treatment
Citation: Zhu B, Chan S-L, Li J, Li K, Wu H, Cui K and Chen H (2021) Non-alcoholic Steatohepatitis Pathogenesis, Diagnosis, and Treatment. Front. Cardiovasc. Med. 8:742382. doi: 10.3389/fcvm.2021.742382
Received: 16 July 2021; Accepted: 13 August 2021;
Published: 07 September 2021.
Edited by:
Changcheng Zhou, University of California, Riverside, United StatesReviewed by:
Jun Yu, Temple University, United StatesYanqiao Zhang, Northeast Ohio Medical University, United States
Copyright © 2021 Zhu, Chan, Li, Li, Wu, Cui and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hong Chen, hong.chen@harvard.childrens.edu