 Linda S. Gutierrez*
Linda S. Gutierrez* Jovita Gutierrez
Jovita Gutierrez- Department of Biology, Wilkes University, Wilkes Barre, PA, United States
The thrombospondin family comprises of five multifunctional glycoproteins, whose best-studied member is thrombospondin 1 (TSP1). This matricellular protein is a potent antiangiogenic agent that inhibits endothelial migration and proliferation, and induces endothelial apoptosis. Studies have demonstrated a regulatory role of TSP1 in cell migration and in activation of the latent transforming growth factor beta 1 (TGFβ1). These functions of TSP1 translate into its broad modulation of immune processes. Further, imbalances in immune regulation have been increasingly linked to pathological conditions such as obesity and diabetes mellitus. While most studies in the past have focused on the role of TSP1 in cancer and inflammation, recently published data have revealed new insights about the role of TSP1 in physiological and metabolic disorders. Here, we highlight recent findings that associate TSP1 and its receptors to obesity, diabetes, and cardiovascular diseases. TSP1 regulates nitric oxide, activates latent TGFβ1, and interacts with receptors CD36 and CD47, to play an important role in cell metabolism. Thus, TSP1 and its major receptors may be considered a potential therapeutic target for metabolic diseases.
Introduction
Thrombospondin 1 (TSP1) was discovered in 1971 (1, 2) as a glycoprotein that is secreted by activated platelets, suggesting that the protein’s main function would be associated with hemostasis. However, pioneering studies have uncovered a variety of functions of TSP1, including regulation of cell migration, apoptosis, and angiogenesis (3–6).
TSP1 has a trimeric 450 kDa complex structure that includes a heparin-binding domain with a procollagen homology domain at the amino terminus (7), and type I, II, and III repeats at the carboxyl-terminal end (2, 8). The type I repeats are also called thrombospondin structural repeats (TSR) (9). Within the TSRs, the sequence (CSVTCG) shows a specific affinity for CD36 (also known as fatty acid translocase, FAT) (10). CD36 is a glycosylated protein, member of the class B scavenger receptor family (11). Through binding with CD36, TSP1 induces apoptosis in endothelial cells (4). Importantly, the sequence RFK is located between the first and second TSRs and mediates the activation of the latent form of transforming growth factor beta1(TGFβ1) (12).
The type III repeats of TSP1 contain domains that interact with neutrophil elastase (13), and inhibit the angiogenic effects of fibroblast growth factor 2 (FGF2) (14). Finally, the carboxyl-terminal domain of TSP1 shows affinity for CD47, also known as integrin associated protein (IAP), an important TSP1 receptor. CD47 regulates the effects of nitric oxide (NO) in metabolic diseases and has major functions in immunity and hemostasis (15). This domain also interacts with integrins modulating cell adhesion and spreading (Figure 1).
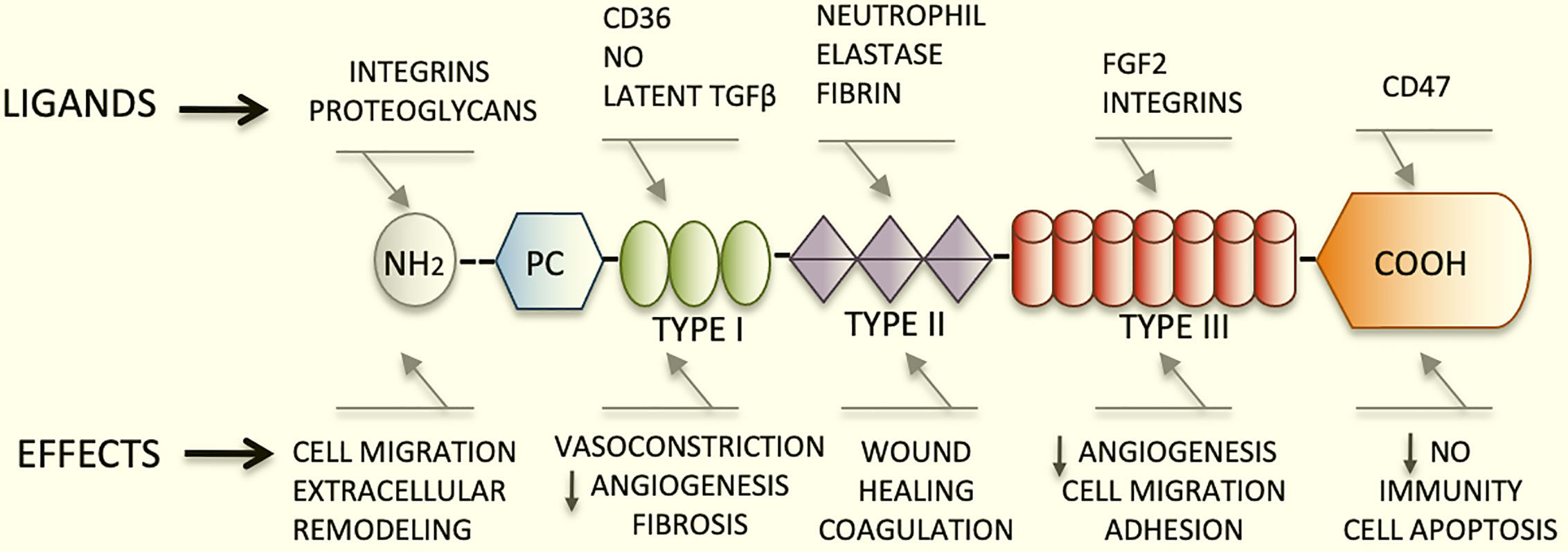
Figure 1 Schematic diagram representing the structure of TSP1, its major ligands and functions. TSP1 displays an amino terminus that interacts with integrins and proteoglycans. The type I, II, and III repeats and the carboxyl-terminal end are also represented herein. The type I repeats also named TSRs contain the binding domain for CD36, responsible for endothelial apoptosis. The sequence RFK that activates the latent form of transforming growth factor beta1(TGFβ1) is also found within these repeats. The type III repeats of TSP1 contain domains that interact with neutrophil elastase and inhibit FGF2. Finally, the carboxyl-terminal domain of TSP1 binds to CD47. This domain interacts with integrins modulating cell adhesion, spreading and migration. TSP1 binds to a diversity of relevant proteins and growth factors not shown in this figure in lieu of clarity.
TSP1 exerts a wide range of functions as its domains can bind to receptors and specific proteins anchored or secreted in the extracellular matrix. TSP1 is an antiangiogenic protein that modulates cell migration and adhesion; it controls the deposition of collagen in the stroma and modulates immunity (2, 5, 16, 17). Recently, there has been an increasing interest in metabolic alterations as major causes of many pathological conditions. This has rekindled the interest in TSP1 and other matricellular proteins as molecules that may be associated with the modulation of metabolic alterations. This review revisits the functions of TSP1, emphasizes its role in the pathophysiology of major metabolic alterations such as obesity, cardiovascular diseases, and diabetes mellitus, and discusses recent advances in metabolomics that are linked to TSP1.
TSP1 in Cardiovascular Diseases
TSP1 is weakly expressed in the normal heart (18). In fact, in mice lacking TSP1 (Thbs1 -/- mice), minimal effects are observed in heart vascularization, ventricular dimensions, and cardiovascular functions as compared to the control mice (18). However, studies in patients with known cardiovascular conditions have indicated that measuring TSP1 level in plasma is useful for the diagnosis and prognosis of these conditions (19, 20). Moreover, polymorphisms in the TSP1 gene are linked to a genetic propensity to myocardial infarction, underlining its importance in this condition (21).
In a myocardial infarction model, Thbs1 -/- mice display more intense and diffuse inflammation surrounding the infarcted area of the heart (22). These effects of TSP1 seem to be sex-dependent. A preclinical study using a myocardial infarction model showed that female mice displayed lesser heart inflammation than the male mice. Leukocytes were isolated and identified as neutrophils, using antibodies specific for nuclear factor (NF)κB-p65 or peroxisome proliferator-activated receptor (PPAR)γ. The lower concentration of TSP1 in the leukocytes of female mice was evident after 7 days as well as lower levels of IL-6 in plasma (23).
Lesser TSP1 and production of ROS would significantly ameliorate inflammation and reduce the binding of TSP1 with the receptor cluster of differentiation, CD36. This multifunctional receptor has relevant functions in metabolic and immune interactions, including the uptake of circulating fatty acids into the cells (24). CD36 is highly expressed in cardiomyocytes and its deletion reduces lipid uptake and storage, and the induction of genes related to fatty acids metabolism (25, 26). CD36 downregulates 5′-adenosine monophosphate-activated protein kinase (AMPK); however, upon binding with fatty acids, CD36 will promote the activation of AMPK enhancing fatty acid oxidation (27).
TSP1 largely contributes to the rapid remodeling of the extracellular matrix. TSP1 directly and indirectly upregulates and binds to MMPs (28); TSP1 promotes the activation of MMPs by inducing profibrotic genes or by activation of latent TGFβ1. This is an important mechanism in cardiovascular pathology (29). TGFβ1 also accelerates the differentiation of inactive fibroblasts into myofibroblasts and induces the transcription of pro-fibrotic genes. In addition, this growth factor exacerbates the inflammatory response (30). An increase in cleaved TSP1 has been detected in adults with dilated cardiomyopathy, wherein fibrosis is more profuse. These results suggest that TSP1 promotes the activation of latent TGFβ1 and MMPs in the adult cardiac heart, and enhances cardiac muscle remodeling and fibrosis (31), thereby indicating a clear role of TSP1 in cardiovascular pathology during the aging process.
Hypoxia could lead to oxidative stress and production of reactive oxygen species (ROS) by mitochondria. In a model of ischemic reperfusion injury in young and aged hearts, ROS production was induced by the damage with concomitant high expression of TSP1 and dynamin-related protein (Drp-1). Drp-1 is a key protein involved in mitochondrial fission and ROS signaling. Inhibitors of TSP1 may potentially reduce cardiomyocyte damage and aging by reducing production of Drp-1 (32).
Oxidative stress and ROS generation lead to accelerated aging. TSP1, along with CD47, contributes to the aging process in cardiac muscle and endothelial cells. CD47 is expressed in many cell types (33). The binding of CD47 to TSP1 is significant in cardiovascular physiology; notably, the hearts of aging mice deficient in TSP1 and CD47 display elevated heart rates and increased cardiac output. These may be compensatory changes but, levels of cAMP were elevated following a nitric oxide (NO) challenge, suggesting an intrinsic ionotropic and chronotropic activity in the cardiac muscle (34).
The role of TSP1 is quite relevant in vascular diseases such as atherosclerosis. This progressive disease is triggered by endothelial injury and inflammation. Damage to endothelial cells occurs because of changes in blood flow, lipid products, and inflammatory mediators that alter the endothelial barrier. Gradual fibrosis and accumulation of leukocytes and lipids within the intima of the vessel wall occur, and smooth muscle cells (SMCs) migrate from the media in response to cytokines released by inflammatory and immune cells. TSP1 is highly expressed in the arterial wall upon endothelial damage (35) that favors the adherence and penetration of monocytes and macrophages to the arterial wall. TSP1 also enhances the migration and adhesion of macrophages, SMCs, and fibroblasts into the vessel wall and promotes the migration and proliferation of SMCs (36).
During inflammatory conditions such as atherosclerosis, hypoxia inducible factor-1 alpha (HIF1α) enhances the release of interleukin-6, tumor necrosis factor alpha, and TSP1, all of which act as profibrotic factors and alter the vascular homeostasis (8, 37, 38). In pathologies such as pulmonary hypertension, hypoxia inducible factor-2 alpha (HIF2α) is also required for the upregulation of TSP1 in pulmonary vasculature under hypoxic conditions (39).
TSP1 also modulates vasodilation and perfusion of tissues via the regulation of NO (34, 40). TSP1 decreases the levels of cAMP and cGMP by inhibiting NO produced by endothelial cells, limiting the smooth muscle cell response, and blocking vasodilation (41–43). Mice lacking TSP1 and CD47 display hypotension, and are more resistant to stress-induced hypertension (15, 44).
TSP1 and CD47 are upregulated in aged arteries (45), and when aortic rings from older individuals were treated with an antibody against CD47, they showed similar sprouting to the aortic rings treated with vascular endothelial growth factor (VEGF). These results revel new and exciting insights into reversing atherosclerosis and the aging process (45).
Lack of TSP1 may contribute to the stabilization of atherosclerotic plaque by inhibiting efferocytosis (46). This function is mediated by the interaction of TSP1 with CD47. CD47 is a surface protein that signals a “Don’t Eat Me” message to phagocytic cells. CD47 inactivates macrophages by binding to the signal regulatory protein alpha (SIRPα) in Mac3+ cells (47) and by inhibiting the activation of integrins (48). Similar to TSP1, CD47 is increasingly upregulated during the entire atherogenic process (49). Mice lacking CD47 are more susceptible to atherosclerosis, and treatment with an inhibitory antibody against CD47 induces the same effect as that of decreasing efferocytosis. CD47 deficiency enhances the activation of natural killer cells (NK) by inducing the release of interferon gamma (INFγ). Additionally, treatment of mice with an anti-NK antibody prevents the formation of plaques (47).
The interaction between TSP1/CD47 is critical for the migration and proliferation of SMCs (50). CD47 can impair the efferocytosis of SMCs thereby, promoting their proliferation (Figure 2). Dedifferentiated SMCs from atheromas overexpress CD47 and complement component 3 (C3). C3 is a key protein of the complement system, whose subunits C3a and C3b act as effectors in opsonization, phagocytosis and inflammation. These CD47+ and C3+ SMCs can evade efferocytosis from polarized M1 murine RAW 264.7 macrophages. Therefore, these SMCs can proliferate and migrate during the process. Additionally, blocking CD47 reestablishes efferocytosis of these SMCs and prevents atherosclerosis (51).
Lack of TSP1 seems to have a protective role in the ApoE-/- mouse model as it is associated with decreased inflammation and improved glucose metabolism (52). However, earlier studies had suggested that TSP1 deficiency promotes atherogenesis and vascular inflammation in the same mouse model (46). It is possible that the functions of TSP1 depend on the vessel type, stage of the lesions, and association with obesity, diabetes, or other metabolic diseases (53). Undoubtedly, the influence of TSP1 on cardiovascular diseases is even more complex and multifactorial due to the variability in activation and functioning of endothelial co-receptors. As an example, TSP1 may activate endothelial receptors CD36 or CD47 differentially or they can induce different responses depending on the type of integrin expressed; for instance, TSP1 inhibits endothelial migration in the microvasculature via binding with CD36 (11, 54), but it could also enhance chemotaxis by binding to integrins α3β1and α6β1in HDMEV (7). In addition, possible interactions between CD47 and CD36 should be considered (33). Undoubtedly, the influence of TSP1 on cardiovascular diseases is complex and multifactorial (Figure 2).
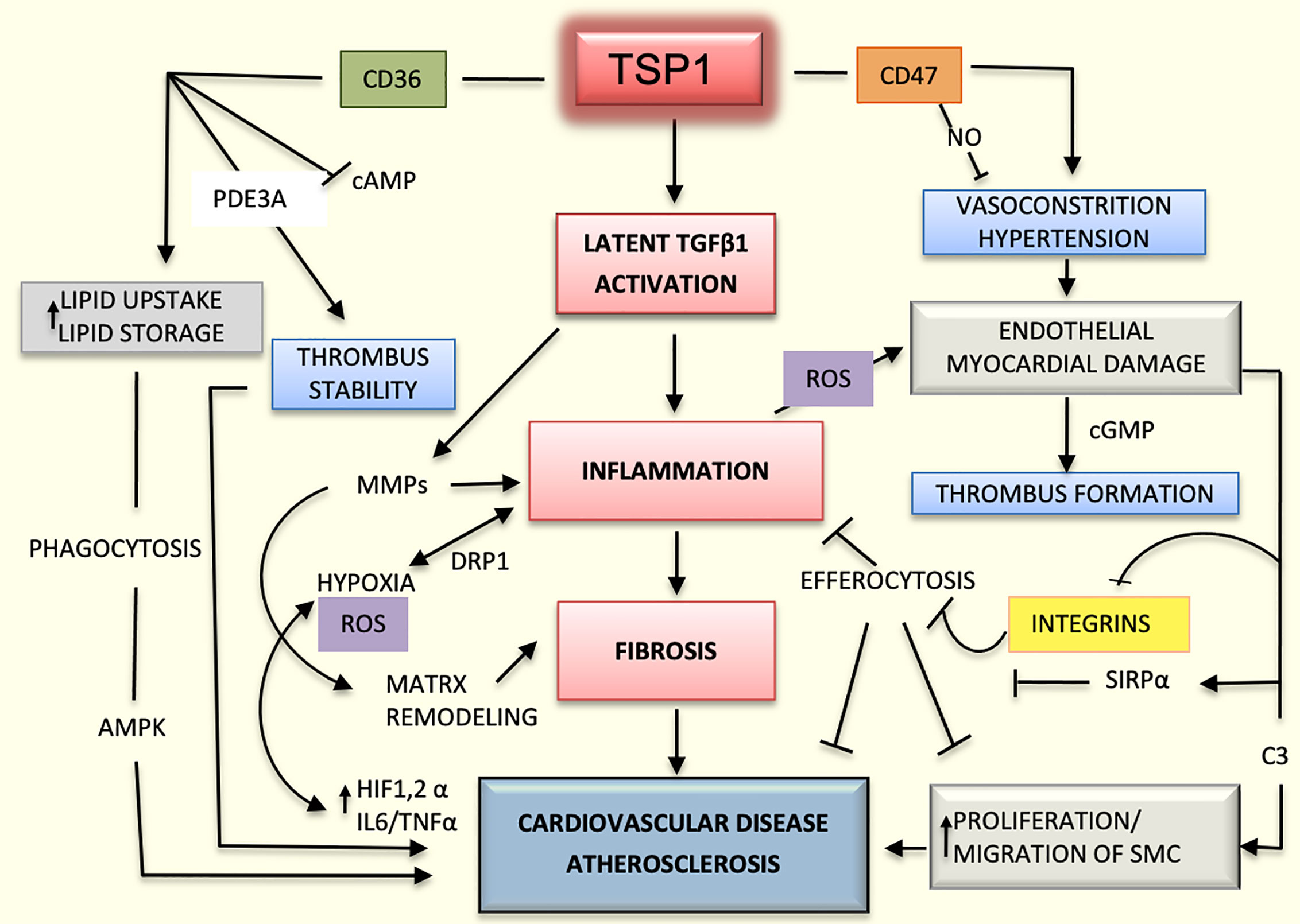
Figure 2 Mechanisms mediated by TSP1 in cardiovascular diseases. The interaction of TSP1 with CD47 inhibits nitric-oxide (NO) levels, thereby decreasing vasodilation and compromising organ perfusion. Hypertension and endothelial injury can in turn, activate the coagulation system, promoting the initiation of thrombus. CD47 suppresses the activation of phagocytic and natural killer (NK) cells during atherogenesis, dampening the inflammatory response and efferocytosis by interacting with signal regulatory protein alpha (SIRPα) and inhibiting integrins. TSP1 activates the latent transforming growth factor beta1 (TGFβ1) enhancing inflammation and fibrosis in the heart and vascular system. Additionally, this growth factor activates matrix metalloproteinases (MMPs), thereby contributing to matrix remodeling in atherosclerosis. Inflammation-induced hypoxia activates hypoxia inducible factor-1 and 2 alpha (HIF1/2α) and increases the levels of interleukin 6 (IL6) and tumor necrosis factor alpha (TNFα) promoting even more the inflammatory process and atherosclerosis. Inflammation also enhances the production of reactive oxygen species (ROS) and cell damage. CD36, as a scavenger receptor B, promotes cellular uptake of lipoproteins and formation of foamy macrophages, which are part of the atherosclerotic plaque. Additionally, by interacting with TSP1, CD36 stabilizes the thrombus in the arterial wall and stimulates the proliferation and migration of smooth muscle cells (SMCs), contributing even further to the atherosclerotic process.
The activation of latent TGFβ1 via TSP1 enhances fibrosis and stiffness in arteries in abnormal conditions without a normal laminar vascular flow (55). These same mechanisms may be implicated in aortic dissection and other arterial pathologies (37, 44, 56). Peptides able to block TSP1 binding to receptors and inhibit its activation of latent TGFβ1may be promising therapeutic agents for cardiovascular diseases and other metabolic diseases (57).
Fibrous plaque leads to ulceration, hemorrhage and scar tissue deposits. Activation of the cascade of coagulation plays an important role in the formation of the atheroma leading to thrombosis and embolism. These complications are first caused by the slowing and turbulence of the blood flow. The next steps involve the adhesion of platelets to the endothelium with the consequent platelet activation and aggregation.
There is evidence of TSP1 as a promoter of platelet aggregation and activation (58). Interestingly, only TSP1 released from alpha granules of activated platelets will suppress cAMP signaling and increase phosphodiesterase 3 (PDE3A) favoring hemostasis (59). CD36 seems to be required, in part, for these processes as platelet-secreted TSP1 can increase the expression of phosphatidylserine by interacting with CD36, enhancing the stabilization of the thrombus (60). Recent studies also show that a CD47-derived peptide, TAX2, inhibits thrombosis by blocking platelet phosphorylation upon contact with collagen. This TAX antibody significantly retarded thrombosis in two models of arterial occlusion (61), and its antithrombotic properties are suspected to be mediated by regulation of NO/cGMP signaling (58). These findings may lead to new therapeutic avenues for metabolic diseases involving thromboembolism.
TSP1 in Glucose Metabolism and Diabetes Mellitus
In a prediabetic condition, glycemic levels are not high enough to establish a diagnosis of diabetes. However, chronic inflammation, endothelial damage, and extracellular remodeling could lead to diabetes and its complications. TSP1 is associated with all these processes and it is also involved in glucose metabolism as observed in animal and clinical models.
The link between TSP1 and glucose metabolism is evidenced by the fact that aged Thbs1 -/- mice show impaired glucose tolerance (62). These mice display hypertrophic pancreatic tissues that produce less proinsulin. Additionally, the pancreas of Thbs1 -/- mice release lesser glucose upon insulin secretion (63). This impaired glucose tolerance could be caused by abnormal oxidation of glucose and mitochondrial dysfunction. Further, the pancreatic tissues of Thbs1 -/- mice show a higher expression of uncoupling protein 2 (UCP2), which is an inner membrane mitochondrial protein involved in oxidation and ROS metabolism (64). Polymorphisms of UCP2 have been linked to human obesity and diabetes. In addition, islets of Thbs1 -/- mice produce more lactate, indicating a switch to glycolysis, which results in reduced production of ATP and proinsulin. This was also confirmed when the islets of Thbs1 -/- mice showed higher production of lactate dehydrogenase A, a key glycolytic enzyme responsible for lactate production. These results indicate that TSP1 is vital to maintain the pancreatic homeostasis during normal glycemic conditions.
Vascular perfusion and regulation of angiogenesis are both crucial for the normal functioning of the pancreas and insulin output. The pancreatic islets have more vascularization and blood flow than the acinar component. Additionally, the islet’s endothelium is highly fenestrated and it is actively involved in the production and secretion of insulin. Endothelial cells of the pancreatic islets produce TSP1 (63); however, they also secrete VEGF (62). Reduction of VEGF in islets impairs the release of insulin into the vascular system and these mice with decreased production of VEGF by their pancreatic islets exhibit impaired glucose tolerance as well (65). Therefore, a balance between both proteins is needed to maintain a healthy secretion of insulin and normal glucose blood levels.
Under normal glycemic conditions, the cGMP-dependent protein kinase (PKG) decreases the gene expression of TSP1 and consequently, the activation of latent TGFβ1 by TSP1. However, during hyperglycemia, TSP1 gene expression is enhanced by the downregulation of PKG (66). This status will lead to the upregulation of the transcription nuclear protein upstream stimulatory factor 2 (USF2). It has been reported that USF2 can bind to a region located in the TSP1 promoter, and enhance TSP1 transcription (67). Glycosylated proteins such as glycated albumin also modulate TSP1 expression by upregulating USF2 (29). This mechanism could promote higher levels of TSP1 in diabetes and lead to complications such as diabetic nephropathy. Interestingly, recent studies show that USF2, is in fact, a tumor suppressor that reduces cell proliferation, migration and oxidative stress (68), protective functions also exerted by TSP1 in some inflammatory diseases and cancers (69, 70).
Undoubtedly, high levels of glucose promote oxidative stress (71, 72). Oxidative stress, insulin resistance, and increased levels of glycosylated products likely lead to endothelial dysfunction in hyperglycemia (73). Glycosylation and the expression of TSP1 in SMCs could be mediated by activation of the hexosamine pathway (Figure 3). Sugars and activators of this pathway may directly enhance the proliferation of SMCs and promote the transcriptional expression of TSP1. The treatment of SMCs with an anti-TSP1 antibody and a TSP1 siRNA reverses their proliferation, thereby suggesting a direct link between the hexosamine catabolic pathway, glycosylation, and TSP1 in diabetes (36, 67).
High expression of TSP1 has been observed in SMCs during hyperglycemia. CD47 regulates the migration of SMCs under hyperglycemic conditions by interacting with SIRPα (50). This binding blocks the migration of SMC mediated by the insulin-like growth factor 1 receptor (IGF1R) signaling (50). When CD47 binds to TSP1, or to a peptide derived from the CD47 domain of TSP1, the binding of CD47 to SIRPα is reduced due activation of the IGF1R signaling (50). However, high levels of glucose protect CD47 from degradation and promote its association with SIRPα (74, 75). Further, protein and mRNA levels of TSP1 and its receptor CD47 were enhanced in endothelial cells isolated from wound lesions of diabetic Wistar rats (76). By using a CD47-blocking siRNA, endothelial cells show increased proliferation, migration, and tube formation. These results suggest that angiogenesis in diabetes may be regulated by the TSP1-CD47 axis (76). As age predisposes the body to insulin resistance and hyperglycemia, CD47 will further inhibit SMCs proliferation and angiogenesis. Indeed, CD47-null aging mice have demonstrated better responses to glucose than control mice (45).
Currently, novel antibodies inhibiting the interaction between SIRPα and CD47 are in preclinical phases, while other anti-CD47 antibodies such as AO-176 are currently under evaluation in clinical trials for the treatments of several types of cancers. Some of these antibodies target tumor macrophages, enhancing efferocytosis of tumor cells (77, 78).
CD36 also plays a significant role in lipid and glucose metabolism, and can promote insulin resistance and hyperinsulinemia (79, 80). However, its functions in these conditions seem contradictory. In a clinical trial that involved patients with a metabolic syndrome, CD36 mRNA was downregulated in the blood mononuclear cells of patients who consumed a healthy low-fat diet (81). In another study, decreased expression of CD36 in muscle was associated with increased risk of type 2 diabetes (82). In fact, deficiency of human CD36 is frequently observed in Asian and African populations, and is linked to insulin resistance (80). In spite of this, mice lacking CD36 are protected from insulin resistance, even under a high-fat diet (25).
CD36 is a multifunctional protein with effects that could be independent of its interaction with TSP1. Instead of having a concerted effect, TSP1 could block the translocase fatty acid activity of CD36 in metabolic syndrome (83). Conversely, the addition of TSP1 or the mimetic peptide ABT526 to hepatocytes cultured under high sucrose levels and insulin significantly decreased hepatic steatosis. In this study, TSP1 inhibited sterol regulatory element-binding protein 1 (SREBP1), an insulin-activated protein with important functions in glucose and lipid metabolism. SREBP1 regulation mediated by TSP1 seems to require CD36 as these results were not reproduced in CD36-deficient hepatocytes (84).
CD36 may have different effects depending on the type of cell and organ in which it is expressed; for example, loss of CD36 reduces the expression of genes related to glucose metabolism in cardiomyocytes but enhances their expression in endothelial cells (85). Additionally, mice lacking CD36, when fed with a low fat diet, show high gluconeogenesis and decreased hepatic glycogen levels; indicating a prediabetic status that seems to be conditional to the type of diet consumed (86). Further research is needed to understand the specific role of CD36 and its interactions with TSP1 in diabetes.
Adding more complexity to the role of TSP1 in hyperglycemia is the fact that its expression could be regulated by microRNAs. Overexpression of miR-467 inhibits TSP1 secretion by endothelial cells during hyperglycemia, and enhances angiogenesis in breast tumors (87). In addition, microRNAs can upregulate TSP1 and genes involved in the TGFβ1 pathway (88). Furthermore, microRNAs can protect pancreatic cells from damage induced by free fatty acids (FFAs). This was demonstrated when a rat insulinoma cell line (INS) was transfected with a TSP1 vector, and the transfected cells became considerably resistant to palmitate (a FFA)-induced apoptosis (89). Conversely, when INS cells were transfected with the 3′-UTR of TSP1 and miR-182-5p, they showed a significant decrease in TSP1 expression, indicating that this microRNA was able to target TSP1 (89).
Diabetes mellitus leads to severe complications involving the cardiovascular system, kidney, retina, and nervous system, resulting in a persistent microvascular injury in tissues and delayed healing (90). The slow wound healing observed in diabetes, could be explained in part by the elevation of TSP1 and inhibition of VEGF observed during capillary regression under hyperglycemic conditions (91). In addition, through its binding to CD36 or even CD47, TSP1 could prevent tube formation and migration of endothelial cells and depress the phagocytic response, hindering the healing process. TSP1 is also involved in the pathophysiology of diabetic cardiomyopathy, obesity, and neuropathy (92–95). Indeed, neuropathy is one of the most devastating sequelae of diabetes, and nervous system damage has been associated with impaired angiogenesis and increased TSP1 expression (96).
One common complication of diabetes is nephropathy. Both TSP1 and TGFβ1 are involved in this condition. TSP1 is highly expressed in glomerular mesangial cells as diabetic nephropathy progresses (97). Activation of latent TGFβ1, mediated by TSP1, is necessary for the normal functioning of pancreatic islets. However during chronic hyperglycemia, TGFβ1 can worsen diabetic nephropathy by inducing fibrosis (98). Thus, blocking this activation could be an alternative mechanism for preventing this complication. For instance, fibrosis in rat peritoneal tissues can be reversed by the treatment with a TSP1-derived peptide. This peptide blocks the activation of latent TGFβ1, and significantly reduces fibrosis both in vivo and in vitro (99).
Clinical studies suggest that TSP1 could be a potential diagnostic marker for diabetes. In patients with glucose intolerance, TSP1 mRNA levels in adipose tissues were significantly lower when patients were treated with pioglitazone, a drug that improves the response to insulin (94). In a randomized study, plasma samples from 398 patients with prediabetes were evaluated, and proteomics studies indicated that TSP1 was correlated with high levels of glucose after 2 h of fasting (100). Reduced levels of TSP1 in serum positively correlated with improved glucose intolerance and diminished liver fat content (84). In another clinical trial evaluating metabolic syndrome, TSP1 serum levels showed a strong association with high HbA1c in males (101). These results confirm TSP1 as a protein linked to hyperglycemic mechanisms but further research is required to better understand its role in glucose metabolism and diabetes (Figure 3).
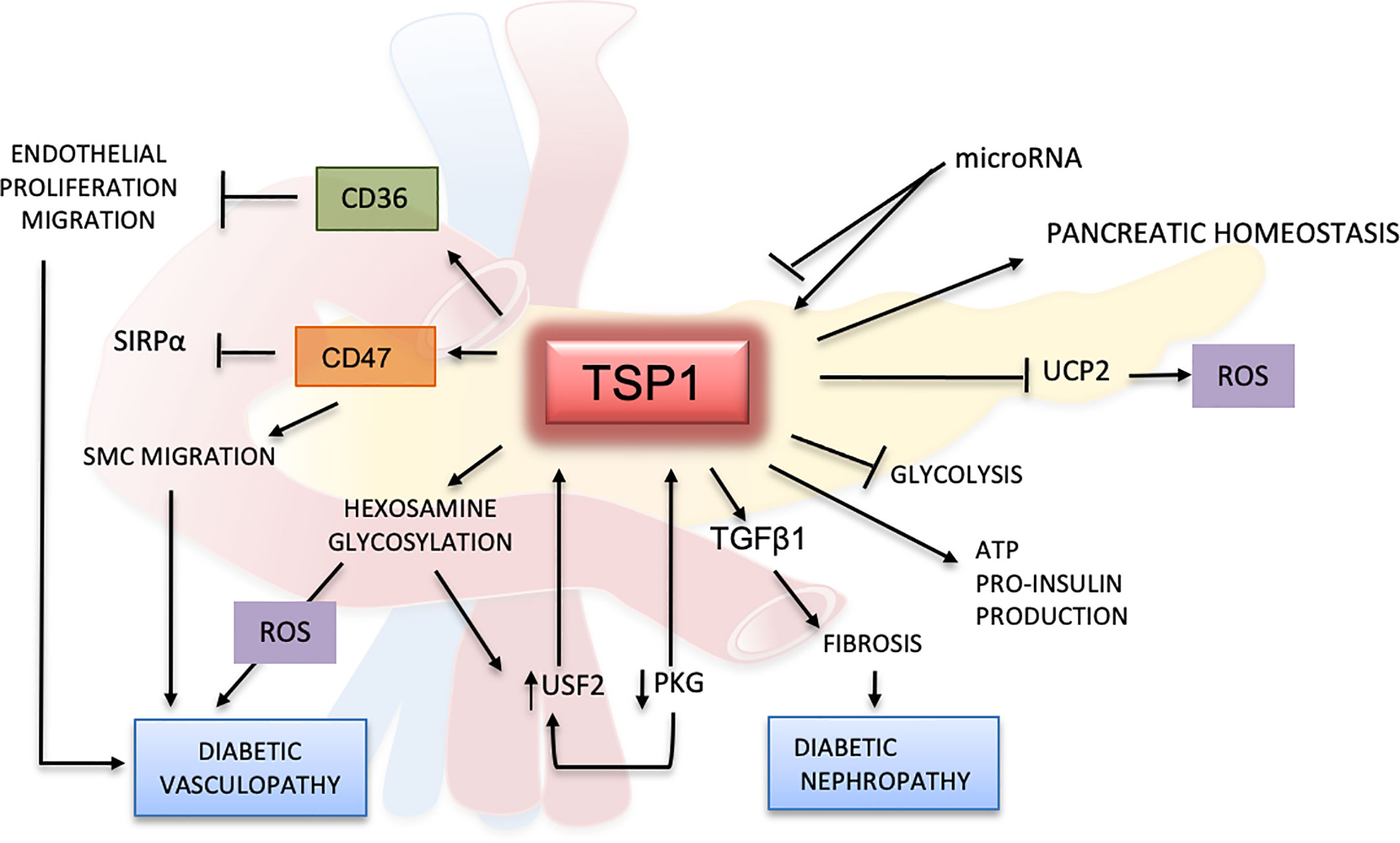
Figure 3 Effects of TSP1 and ligands in glucose metabolism and diabetes mellitus. TSP1 protects the pancreatic endothelium and controls angiogenesis by enhancing insulin production and release. TSP1 also blocks the formation of uncoupling protein 2 (UCP2), a mitochondrial protein involved in oxidation and reactive oxygen species (ROS) metabolism. Under normal glycemic conditions, TSP1 inhibits glycolysis promoting the production of more ATP and pro-insulin. However, under hyperglycemic conditions and by activating TGFβ1, TSP1 accelerates the inflammation and fibrotic changes in multiple organs, especially in the kidney, leading to diabetic nephropathy and other diabetic complications. The TSP1 receptors CD36 and CD47 can inhibit the proliferation and migration of endothelial and smooth muscle cells, thereby contributing to endothelial dysfunction in diabetes. In this context, CD47 regulates SMC migration by binding mainly to TSP1 while limiting its association with SIRP1α. Activation of the hexosamine pathway and glycosylated products can induce further endothelial damage, with generation of ROS and delay of the wound-healing process in diabetes. Hyperglycemia will inhibit PKG and induce upstream stimulatory factor 2 (USF2), promoting the upregulation of TSP1 in diabetes. However, all these effects mediated by TSP1 in the glucose metabolism may be regulated by microRNAs.
TSP1 in Adipocyte Metabolism and Obesity
Adipose tissue is one of the most highly vascularized tissues of the body, and angiogenesis is expected to be involved in its remodeling and metabolism. Besides the well known role of TSP1 in the inhibition of angiogenesis (6, 102), early studies have shown that a lack of TSP1 could decrease abdominal fat without resulting in any change to vascular density (103). Thbs1 -/- mice are resistant to obesity following ingestion of a high-fat diet and display normal levels of leptin, an important adipokine associated with increased body weight (104). However, studies have demonstrated that TSP1 is upregulated in the visceral fat of obese mice; similar findings were reported in clinical studies as well (104–107).
Adipogenesis as well as the growth and distribution of the adipose tissues are associated with angiogenesis. Endothelial cells produce cytokines and growth factors such as VEGF that fuel lipogenesis and adipose tissue expansion (108). These pro-angiogenic factors could also promote the trans differentiation of white adipocytes into brown adipocytes. However, a decrease in vascularity has been observed through the transition of brown fat into white adipose tissue during obesity (109, 110). TSP1 may be involved in these processes by interacting with Argonaute 1(AGO1) a protein required for gene silencing (111). Downregulation of TSP1 was detected in endothelial cells and adipose tissues of mice lacking AGO1 fed with a high fat, high sugar diet. These mice showed lower body weight and normal insulin response (112). Their adipose tissues display higher vascularity and typical features of brown fat compared to control mice. Conversely, TSP1 and AGO1 were upregulated in endothelial cells from obese and diabetic type 2 donors, suggesting that TSP1 may promote obesity and insulin resistance by reducing angiogenesis.
Certainly, TSP1 contributes to adipose tissue metabolism in several ways apart from performing its antiangiogenic functions. The relevance of TSP1 in inflammation and obesity seems to be mediated principally by macrophages. The phenotype of Thbs1-/- macrophages is characterized by lower pro-inflammatory activation and by decreased secretion of cytokines (113, 114). Adipose tissues that lack TSP1 display reduced leukocytic infiltration, and Thbs1 -/- macrophages secrete lower levels of inflammatory cytokines (115). In a diet-induced animal model of obesity, the influx of CD68+ macrophages and angiogenesis in Thbs1 -/- adipose tissues was similar to that of the control tissues (113). Moreover, sensitivity to insulin was also similar to that observed in the Thbs1 +/+ obese mice. However, obese mice with a targeted deletion of TSP1 only in F4/80+ macrophages show impaired leukocytic infiltration in the adipose tissues. Additionally, these adipose tissues display reduced fibrosis, improved insulin resistance, and decreased production of pro-inflammatory cytokines (52). These changes were more evident when obesity was already established, indicating a chronic and progressive mechanism.
These same studies showed a decrease in the activation of latent TGFβ1, both in vitro and in vivo, and indicated that TSP1 deficiency and a decline in latent TGFβ1 activation could be protective against inflammation and obesity. Peptides derived from the 3TSR of TSP1 containing the domain able to activate the latent TGFβ1 have been shown to enhance the influx of CD68+ macrophages in models of inflammation and cancer (116, 117).
As obesity is considered a chronic inflammatory condition, signaling between macrophages and adipocytes occurs during its development and progression. CD36 significantly contributes to enhance the macrophages’ inflammatory functions in adipose tissues (118). The interaction of CD36 with TSP1 may also regulate the damage induced by saturated FFAs during obesity and dyslipidemia. The high expression of TSP1 after the treatment of podocytes with FFA corroborates these results. Additionally, FFA-induced podocyte apoptosis was inhibited in Thbs1 -/- and CD36 -/- podocytes, and they were induced when they were treated with a CD36 peptide (119).
Recent evidence indicates that CD36 signaling is dependent on the activation of STAT3 in tumor cells and adipose tissue (120). High fat diet increases phosphorylation of the signal transducer and activator of transcription 3 (STAT3) via activation of CD36. STAT3 is an important transcription factor in inflammation and immunity. It is also well known that inflammation is a landmark of adipose tissues in obesity. A relationship between inflammation and STAT3 was reported in Thbs1 -/- mice in a model of colitis. These colonic tissues show more inflammation and enhanced phosphorylation of STAT3 as well. A mimetic peptide containing the TSP1 binding domain for CD36 reverted the STAT3 activation (121).
CD47 -/- mice, as well as Thbs1 -/- mice, are resistant to obesity (52, 104). The subcutaneous fat tissue of CD47 -/- mice displays lower expression of TSP1, but higher expression of cGMP (122) which is integral in cardiovascular and adipose metabolism. This signaling regulates the proliferation, differentiation and secretory activity of brown adipocyte depots (123, 124). Moreover, mitochondria from CD47 -/- brown adipocytes display anomalies in shape and high metabolic activity. These results suggest that CD47 may be a potential target for reducing obesity, as increased body distribution and activity of brown fat in adults are regarded as a potential strategy for reducing obesity.
Adipose tissue is an endocrine organ that produces several hormones, including the adipokine leptin, a peptide secreted by adipocytes (125). Leptin receptors are involved in appetite, food intake and insulin secretion. Though, its involvement in obesity is still uncertain (Figure 4), several animal and clinical studies have demonstrated that increased plasma levels of leptin are associated with high body mass and atherosclerosis (126). However, recent studies have shown that ApoE-/-/Thbs1 -/- mice treated with exogenous leptin display significant decreases in body weight and attenuation of atherosclerosis. Levels of VLDL and triglycerides were substantially lower in these double null mice even after being fed with a western high-fat diet. These effects, mediated by TSP1 via leptin, seem to be induced by the upregulation of JAK2 and MAPKs signaling pathways (127). Activation of these pathways has been also implicated in the upregulation of TSP1 by leptin in vascular muscle cells during atherogenesis (128).
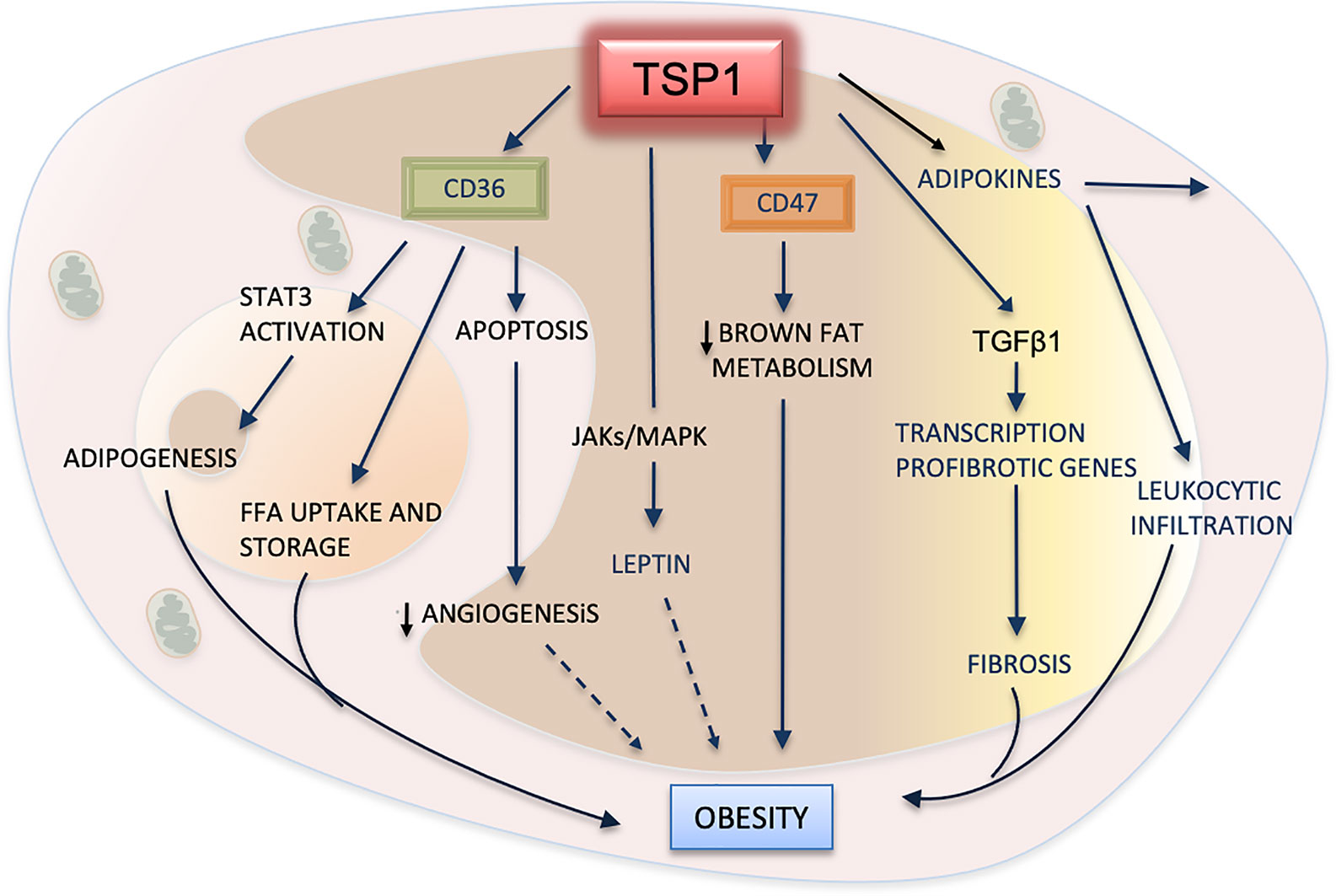
Figure 4 TSP1 in adipose metabolism and obesity. TSP1 activates the latent form of transforming growth factor beta1 (TGFβ1), consequently promoting the recruitment of leukocytes, the influx of macrophages to the adipose tissue, and the release of inflammatory cytokines and adipokines. TGFβ1 will also contribute to fibrosis of adipose tissues by inducing the transcription of pro-fibrotic genes. By interacting with TSP1, CD47 suppresses the mitochondrial metabolism in brown adipose cells, therefore exhibiting a pro-obesity effect. TSP1 interacts with the hormone leptin by activating the JAK/STATs pathway. Increased plasma levels of leptin are correlated with high body fatness. The dashed arrow indicates a probable but yet unexplained association with obesity. CD36 facilitates the uptake of free fatty acids (FFA) into adipose cells, but it can also promote the apoptosis of endothelial cells, decreasing angiogenesis. Perhaps, this could contribute to the lower vascularization observed in white adipose tissues during the progression of this condition (dashed arrow). Finally, CD36 can regulate adipogenesis and expansion of adipose cells by activating the signal transducer and activator of transcription 3 (STAT3) signaling.
TSP1 could be a marker for weight loss and higher basal metabolism. In a clinical study, high TSP1 expression was detected in the visceral fat of patients undergoing a weight loss program consisting of low caloric intake and increased physical activity (101). TSP1 serum levels of these patients were higher, and correlated with abdominal obesity, hypertension, hyperglycemia and high levels of leptin. These associations were observed particularly in premenopausal females. However, these results should be interpreted cautiously as TSP1 levels in serum may result from platelet activation, and may not reflect the high physiological concentration of TSP1 observed in plasma samples. Platelet function can be inhibited in obese patients as well (129). Therefore, low levels of TSP1 could be caused by impairment of platelet activation, which may be enhanced by weight loss. However, the same results were observed in another clinical study when proteomic analyses were performed in peripheral blood mononuclear cells and visceral adipose tissues. TSP1 was again one of the proteins upregulated in obese patients, and TSP1 levels were decreased after the same patients followed an exercise regimen (130). As obesity and its complications are a significant health concern in our society, the premise that TSP1 could be a diagnostic marker for obesity compellingly merits further investigation.
Conclusions and Future Perspectives
TSP1 is a frequently studied and well-characterized matricellular protein. Its antiangiogenic effect in cancer was previously explored in animal studies and clinical trials. As TSP1 contains multiple binding sites to several receptors and growth factors, a more sophisticated understanding of its dynamic functions is necessary. As we have learned more about the intrinsic mechanisms involving TSP1 and its interactions, it has become clear that these mechanisms may be tissue-specific and depend on the affinity and temporal/spatial expression of these cofactors within the tissue environment.
In the past, pioneers in the field developed a foundation for understanding the pathophysiological roles of TSP1 in metabolic diseases. As new scientific tools and more in-depth metabolomics approaches are now possible, new mechanistic insights have emerged about TSP1. Among these exciting breakthroughs is the development of novel antibodies blocking TSP1/CD47 interactions, some of which have progressed to clinical trials for treating several types of cancers. One such antibody, TAX, has shown promising results as an antithrombotic, leading to its potential therapeutic use in cardiovascular diseases and other metabolic conditions.
The potential use of TSP1 as a diagnostic and prognostic indicator of diabetes and obesity is an exciting possibility, as well as the use of microRNAs to mitigate the effects of TSP1 in these diseases. More insights regarding the role of this protein in mitochondrial metabolism and immunity are emerging. These scientific advances position TSP1 and its receptors as important therapeutic targets in heart disease, diabetes, obesity, aging and cancer.
Author Contributions
LG conceived and designed the study, collected and analyzed the data, and wrote the manuscript. JG collected and analyzed the data, contributed to figure creation, and reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Financial support for this work was provided by the Provost Research and Scholarship Funds at Wilkes University.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflict of interest.
Acknowledgments
We would like to thank Editage (www.editage.com) for English language editing.
Abbreviations
TSP1, thrombospondin 1; TGFβ1, transforming growth factor beta 1; Thbs1 -/- mice, mice lacking TSP1 protein; CD47, cluster of differentiation 47; CD36, cluster of differentiation 36; MMP, matrix metallopeptidase; AMPK, 5′-adenosine monophosphate-activated protein kinase; NO, nitric oxide; cAMP, cyclic adenosine monophosphate; HIF1α, hypoxia inducible factor 1 alpha; IL-6, interleukin- 6; TNFα, tumor necrosis factor alpha; ApoE, apolipoprotein E; VEGF, vascular endothelial growth factor; siRNA, small interfering RNA; Mdm2, mouse double minute 2 homolog; let-7d, lethal-7d; ROS, reactive oxygen species; USF2, upstream stimulatory factor 2.
References
1. Baenziger NL, Brodie GN, Majerus PW. A thrombin-sensitive protein of human plateletmembranes. Proc Natl Acad Sci USA (1971)68(1):240–3. doi: 10.1073/pnas.68.1.240
2. Lawler J, Hynes RO. The structure of human thrombospondin, an adhesive glycoprotein withmultiple calcium-binding sites and homologies with several different proteins.J Cell Biol (1986)103(5):1635–48. doi: 10.1083/jcb.103.5.1635
3. Taraboletti G, Roberts DD, Liotta LA. Thrombospondin-induced tumor cell migration: haptotaxis and chemotaxis are mediated by different molecular domains. J Cell Biol (1987) 105(5):2409–15. doi: 10.1083/jcb.105.5.2409
4. Guo N, Krutzsch HC, Inman JK, Roberts DD. Thrombospondin 1 and type I repeat peptides of thrombospondin 1 specifically induce apoptosis of endothelial cells. Cancer Res (1997) 57(9):1735–42.
5. Good DJ, Polverini PJ, Rastinejad F, Le Beau MM, Lemons RS, Frazier WA, et al. A tumor suppressor-dependent inhibitor of angiogenesis is immunologically and functionally indistinguishable from a fragment of thrombospondin. Proc Natl Acad Sci USA (1990) 87(17):6624–8. doi: 10.1073/pnas.87.17.6624
6. Jiménez B, Volpert OV, Crawford SE, Febbraio M, Silverstein RL, Bouck N. Signals leading to apoptosis-dependent inhibition of neovascularization by thrombospondin-1. Nat Med (2000) 6(1):41–8. doi: 10.1038/71517
7. Calzada MJ, Sipes JM, Krutzsch HC, Yurchenco PD, Annis DS, Mosher DF, et al. Recognition of the N-terminal modules of thrombospondin-1 and thrombospondin-2 by alpha6beta1 integrin. J Biol Chem (2003) 278(42):40679–87. doi: 10.1074/jbc.M302014200
8. Lopez-Dee Z, Pidcock K, Gutierrez LS. Thrombospondin-1: multiple paths to inflammation. Mediators Inflamm (2011) 2011:296069. doi: 10.1155/2011/296069
9. Carlson CB, Lawler J, Mosher DF. Structures of thrombospondins. Cell Mol Life Sci (2008) 65(5):672–86. doi: 10.1007/s00018-007-7484-1
10. Asch AS, Silbiger S, Heimer E, Nachman RL. Thrombospondin sequence motif (CSVTCG) is responsible for CD36 binding. Biochem Biophys Res Commun (1992) 182(3):1208–17. doi: 10.1016/0006-291x(92)91860-s
11. Silverstein RL, Febbraio M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci Signal (2009) 2(72):re3. doi: 10.1126/scisignal.272re3
12. Crawford SE, Stellmach V, Murphy-Ullrich JE, Ribeiro SM, Lawler J, Hynes RO, et al. Thrombospondin-1 is a major activator of TGF-beta1 in vivo. Cell (1998) 93(7):1159–70. doi: 10.1016/s0092-8674(00)81460-9
13. Hogg PJ, Owensby DA, Mosher DF, Misenheimer TM, Chesterman CN. Thrombospondin is a tight-binding competitive inhibitor ofneutrophil elastase. J Biol Chem (1993)268(10):7139–46. doi: 10.1016/S0021-9258(18)53157-4
14. Margosio B, Rusnati M, Bonezzi K, Cordes BL, Annis DS, Urbinati C, et al. Fibroblast growth factor-2 binding to the thrombospondin-1 type III repeats, a novel antiangiogenic domain. Int J Biochem Cell Biol (2008) 40(4):700–9. doi: 10.1016/j.biocel.2007.10.002
15. Isenberg JS, Martin-Manso G, Maxhimer JB, Roberts DD. Regulation of nitric oxide signalling by thrombospondin 1: implications for anti-angiogenic therapies. Nat Rev Cancer (2009) 9(3):182–94. doi: 10.1038/nrc2561
16. Mumby SM, Raugi GJ, Bornstein P. Interactions of thrombospondin with extracellular matrix proteins: selective binding to type V collagen. J Cell Biol (1984) 98(2):646–52. doi: 10.1083/jcb.98.2.646
17. Li Z, He L, Wilson K, Roberts D. Thrombospondin-1 inhibits TCR-mediated T lymphocyte early activation. J Immunol (2001) 166(4):2427–36. doi: 10.4049/jimmunol.166.4.2427
18. Malek MH, Olfert IM. Global deletion of thrombospondin-1 increases cardiac and skeletal muscle capillarity and exercise capacity in mice. Exp Physiol (2009) 94(6):749–60. doi: 10.1113/expphysiol.2008.045989
19. Gao JB, Tang WD, Wang HX, Xu Y. Predictive value of thrombospondin-1 for outcomes in patients with acute ischemic stroke. Clin Chim Acta (2015) 450:176–80. doi: 10.1016/j.cca.2015.08.014
20. Choi KY, Kim DB, Kim MJ, Kwon BJ, Chang SY, Jang SW, et al. Higher plasma thrombospondin-1 levels in patients with coronary artery disease and diabetes mellitus. Korean Circ J (2012) 42(2):100–6. doi: 10.4070/kcj.2012.42.2.100
21. Topol EJ, McCarthy J, Gabriel S, Moliterno DJ, Rogers WJ, Newby LK, et al. Single nucleotide polymorphisms in multiple novel thrombospondin genes may be associated with familial premature myocardial infarction. Circulation (2001) 104(22):2641–4. doi: 10.1161/hc4701.100910
22. Frangogiannis NG, Ren G, Dewald O, Zymek P, Haudek S, Koerting A, et al. Critical role of endogenous thrombospondin-1 in preventing expansion of healing myocardial infarcts. Circulation (2005) 111(22):2935–42. doi: 10.1161/CIRCULATIONAHA.104.510354
23. DeLeon-Pennell KY, Mouton AJ, Ero OK, Ma Y, Padmanabhan Iyer R, Flynn ER, et al. LXR/RXR signaling and neutrophil phenotype following myocardial infarction classify sex differences in remodeling. Basic Res Cardiol (2018) 113(5):40. doi: 10.1007/s00395-018-0699-5
24. Ibrahimi A, Bonen A, Blinn WD, Hajri T, Li X, Zhong K, et al. Muscle-specific overexpression of FAT/CD36 enhances fatty acid oxidation by contracting muscle, reduces plasma triglycerides and fatty acids, and increases plasma glucose and insulin. J Biol Chem (1999) 274(38):26761–6. doi: 10.1074/jbc.274.38.26761
25. Febbraio M, Abumrad NA, Hajjar DP, Sharma K, Cheng W, Pearce SF, et al. A null mutation in murine CD36 reveals an important role in fatty acid and lipoprotein metabolism. J Biol Chem (1999) 274(27):19055–62. doi: 10.1074/jbc.274.27.19055
26. Febbraio M, Podrez EA, Smith JD, Hajjar DP, Hazen SL, Hoff HF, et al. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J Clin Invest (2000) 105(8):1049–56. doi: 10.1172/JCI9259
27. Samovski D, Sun J, Pietka T, Gross RW, Eckel RH, Su X, et al. Regulation of AMPK activation by CD36 links fatty acid uptake to β-oxidation. Diabetes (2015) 64(2):353–9. doi: 10.2337/db14-0582
28. Gonzalez-Quesada C, Cavalera M, Biernacka A, Kong P, Lee DW, Saxena A, et al. Thrombospondin-1 induction in the diabetic myocardium stabilizes the cardiac matrix in addition to promoting vascular rarefaction through angiopoietin-2 upregulation. Circ Res (2013) 113(12):1331–44. doi: 10.1161/CIRCRESAHA.113.302593
29. Murphy-Ullrich JE. Thrombospondin 1 and Its Diverse Roles as a Regulator of Extracellular Matrix in Fibrotic Disease. J Histochem Cytochem (2019) 67(9):683–99. doi: 10.1369/0022155419851103
30. Walton KL, Johnson KE, Harrison CA. Targeting TGF-β Mediated SMAD Signaling for the Prevention of Fibrosis. Front Pharmacol (2017) 8:461. doi: 10.3389/fphar.2017.00461
31. Jana S, Zhang H, Lopaschuk GD, Freed DH, Sergi C, Kantor PF, et al. Disparate Remodeling of the Extracellular Matrix and Proteoglycans in Failing Pediatric Versus Adult Hearts. J Am Heart Assoc (2018) 7(19):e010427. doi: 10.1161/JAHA.118.010427
32. Kelm NQ, Beare JE, Weber GJ, LeBlanc AJ. Thrombospondin-1 mediates Drp-1 signaling following ischemia reperfusion in the aging heart. FASEB Bioadv (2020) 2(5):304–14. doi: 10.1096/fba.2019-00090
33. Soto-Pantoja DR, Kaur S, Roberts DD. CD47 signaling pathways controlling cellular differentiation and responses to stress. Crit Rev Biochem Mol Biol (2015) 50(3):212–30. doi: 10.3109/10409238.2015.1014024
34. Isenberg JS, Qin Y, Maxhimer JB, Sipes JM, Despres D, Schnermann J, et al. Thrombospondin-1 and CD47 regulate blood pressure and cardiac responses to vasoactive stress. Matrix Biol (2009) 28(2):110–9. doi: 10.1016/j.matbio.2009.01.002
35. Moura R, Tjwa M, Vandervoort P, Cludts K, Hoylaerts MF. Thrombospondin-1 activates medial smooth muscle cells and triggers neointima formation upon mouse carotid artery ligation. Arterioscler Thromb Vasc Biol (2007) 27(10):2163–9. doi: 10.1161/ATVBAHA.107.151282
36. Raman P, Krukovets I, Marinic TE, Bornstein P, Stenina OI. Glycosylation mediates up-regulation of a potent antiangiogenic and proatherogenic protein, thrombospondin-1, by glucose in vascular smooth muscle cells. J Biol Chem (2007) 282(8):5704–14. doi: 10.1074/jbc.M610965200
37. Kumar R, Mickael C, Kassa B, Gebreab L, Robinson JC, Koyanagi DE, et al. TGF-β activation by bone marrow-derived thrombospondin-1 causes Schistosoma- and hypoxia-induced pulmonary hypertension. Nat Commun (2017) 8:15494. doi: 10.1038/ncomms15494
38. Billaud M, Hill JC, Richards TD, Gleason TG, Phillippi JA. Medial Hypoxia and Adventitial Vasa Vasorum Remodeling in Human Ascending Aortic Aneurysm. Front Cardiovasc Med (2018) 5:124. doi: 10.3389/fcvm.2018.00124
39. Labrousse-Arias D, Castillo-González R, Rogers NM, Torres-Capelli M, Barreira B, Aragonés J, et al. HIF-2α-mediated induction of pulmonary thrombospondin-1 contributes to hypoxia-driven vascular remodelling and vasoconstriction. Cardiovasc Res (2016) 109(1):115–30. doi: 10.1093/cvr/cvv243
40. Roberts DD, Miller TW, Rogers NM, Yao M, Isenberg JS. The matricellular protein thrombospondin-1 globally regulates cardiovascular function and responses to stress via CD47. Matrix Biol (2012) 31(3):162–9. doi: 10.1016/j.matbio.2012.01.005
41. Yao M, Roberts DD, Isenberg JS. Thrombospondin-1 inhibition of vascular smooth muscle cell responses occurs via modulation of both cAMP and cGMP. Pharmacol Res (2011) 63(1):13–22. doi: 10.1016/j.phrs.2010.10.014
42. Stenina-Adognravi O. Invoking the Power of Thrombospondins: Regulation of Thrombospondins Expression. Matrix Biol (2014) 0:69–82. doi: 10.1016/j.matbio.2014.02.001
43. Nevitt C, McKenzie G, Christian K, Austin J, Hencke S, Hoying J, et al. Physiological levels of thrombospondin-1 decrease NO-dependent vasodilation in coronary microvessels from aged rats. Am J Physiol Heart Circ Physiol (2016) 310(11):H1842–1850. doi: 10.1152/ajpheart.00086.2016
44. Rogers NM, Sharifi-Sanjani M, Csányi G, Pagano PJ, Isenberg JS. Thrombospondin-1 and CD47 regulation of cardiac, pulmonary and vascular responses in health and disease. Matrix Biol (2014) 37:92–101. doi: 10.1016/j.matbio.2014.01.002
45. Ghimire K, Li Y, Chiba T, Julovi SM, Li J, Ross MA, et al. CD47 Promotes Age-Associated Deterioration in Angiogenesis, BloodFlow and Glucose Homeostasis. Cells (2020) 9(7):1695. doi: 10.3390/cells9071695
46. Moura R, Tjwa M, Vandervoort P, Van Kerckhoven S, Holvoet P, Hoylaerts MF. Thrombospondin-1 deficiency accelerates atherosclerotic plaque maturation in ApoE-/- mice. Circ Res (2008) 103(10):1181–9. doi: 10.1161/CIRCRESAHA.108.185645
47. Kojima Y, Volkmer JP, McKenna K, Civelek M, Lusis AJ, Miller CL, et al. CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature (2016) 536(7614):86–90. doi: 10.1038/nature18935
48. Morrissey MA, Kern N, Vale RD. CD47 Ligation Repositions the Inhibitory Receptor SIRPA to Suppress Integrin Activation and Phagocytosis. Immunity (2020) 53(2):290–302.e296. doi: 10.1016/j.immuni.2020.07.008
49. Nath PR, Pal-Nath D, Mandal A, Cam MC, Schwartz AL, Roberts DD. Natural Killer Cell Recruitment and Activation Are Regulated by CD47 Expression in the Tumor Microenvironment. Cancer Immunol Res (2019) 7(9):1547–61. doi: 10.1158/2326-6066.CIR-18-0367
50. Maile LA, Clemmons DR. Integrin-associated protein binding domain of thrombospondin-1 enhances insulin-like growth factor-I receptor signaling in vascular smooth muscle cells. Circ Res (2003) 93(10):925–31. doi: 10.1161/01.RES.0000101754.33652.B7
51. Wang Y, Nanda V, Direnzo D, Ye J, Xiao S, Kojima Y, et al. Clonally expanding smooth muscle cells promote atherosclerosis by escaping efferocytosis and activating the complement cascade. Proc Natl Acad Sci USA (2020) 117(27):15818–26. doi: 10.1073/pnas.2006348117
52. Maimaitiyiming H, Clemons K, Zhou Q, Norman H, Wang S. Thrombospondin1 deficiency attenuates obesity-associated microvascular complications in ApoE-/- mice. PLoS One (2015) 10(3):e0121403. doi: 10.1371/journal.pone.0121403
53. Stenina OI, Plow EF. Counterbalancing forces: what is thrombospondin-1 doing in atherosclerotic lesions? Circ Res (2008) 103(10):1053–5. doi: 10.1161/CIRCRESAHA.108.188870
54. Li Z, Calzada MJ, Sipes JM, Cashel JA, Krutzsch HC, Annis DS, et al. Interactions of thrombospondins with alpha4beta1 integrin and CD47 differentially modulate T cell behavior. J Cell Biol (2002) 157(3):509–19. doi: 10.1083/jcb.200109098
55. Kim CW, Pokutta-Paskaleva A, Kumar S, Timmins LH, Morris AD, Kang DW, et al. Disturbed Flow Promotes Arterial Stiffening Through Thrombospondin-1. Circulation (2017) 136(13):1217–32. doi: 10.1161/CIRCULATIONAHA.116.026361
56. Zeng T, Yuan J, Gan J, Liu Y, Shi L, Lu Z, et al. Thrombospondin 1 Is Increased in the Aorta and Plasma of Patients With Acute Aortic Dissection. Can J Cardiol (2019) 35(1):42–50. doi: 10.1016/j.cjca.2018.11.008
57. Emonard H, Duca L, Dedieu S. Editorial: Matricellular Receptors As Potential Targets in Anti-Cancer Therapeutic Strategies. Front Pharmacol (2016) 7:95. doi: 10.3389/fphar.2016.00095
58. Isenberg JS, Romeo MJ, Yu C, Yu CK, Nghiem K, Monsale J, et al. Thrombospondin-1 stimulates platelet aggregation by blocking the antithrombotic activity of nitric oxide/cGMP signaling. Blood (2008) 111(2):613–23. doi: 10.1182/blood-2007-06-098392
59. Aburima A, Berger M, Spurgeon BE, Webb BA, Wraith KS, Febbraio M, et al. Thrombospondin-1 promotes haemostasis through modulation of cAMPsignalling in blood platelets. Blood (2021) 137(5):678–89. doi: 10.1182/blood.2020005382
60. Kuijpers MJ, de Witt S, Nergiz-Unal R, van Kruchten R, Korporaal SJ, Verhamme P, et al. Supporting roles of platelet thrombospondin-1 and CD36 in thrombus formation on collagen. Arterioscler Thromb Vasc Biol (2014) 34(6):1187–92. doi: 10.1161/ATVBAHA.113.302917
61. Jeanne A, Sarazin T, Charlé M, Kawecki C, Kauskot A, Hedtke T, et al. Towards the Therapeutic Use of Thrombospondin 1/CD47 Targeting TAX2 Peptide as an Antithrombotic Agent. Arterioscler Thromb Vasc Biol (2021) 41(1):e1–e17. doi: 10.1161/ATVBAHA.120.314571
62. Drott CJ, Olerud J, Emanuelsson H, Christoffersson G, Carlsson PO. Sustained beta-cell dysfunction but normalized islet mass in aged thrombospondin-1 deficient mice. PLoS One (2012) 7(10):e47451. doi: 10.1371/journal.pone.0047451
63. Olerud J, Mokhtari D, Johansson M, Christoffersson G, Lawler J, Welsh N, et al. Thrombospondin-1: an islet endothelial cell signal of importance for β-cell function. Diabetes (2011) 60(7):1946–54. doi: 10.2337/db10-0277
64. de Souza BM, Michels M, Sortica DA, Bouças AP, Rheinheimer J, Buffon MP, et al. Polymorphisms of the UCP2 Gene Are Associated with Glomerular Filtration Rate in Type 2 Diabetic Patients and with Decreased UCP2 Gene Expression in Human Kidney. PLoS One (2015) 10(7):e0132938. doi: 10.1371/journal.pone.0132938
65. Brissova M, Shostak A, Shiota M, Wiebe PO, Poffenberger G, Kantz J, et al. Pancreatic islet production of vascular endothelial growth factor–a is essential for islet vascularization, revascularization, and function. Diabetes (2006) 55(11):2974–85. doi: 10.2337/db06-0690
66. Wang S, Wu X, Lincoln TM, Murphy-Ullrich JE. Expression of constitutively active cGMP-dependent protein kinase prevents glucose stimulation of thrombospondin 1 expression and TGF-beta activity. Diabetes (2003) 52(8):2144–50. doi: 10.2337/diabetes.52.8.2144
67. Wang S, Skorczewski J, Feng X, Mei L, Murphy-Ullrich JE. Glucose up-regulates thrombospondin 1 gene transcription and transforming growth factor-beta activity through antagonism of cGMP-dependent protein kinase repression via upstream stimulatory factor 2. J Biol Chem (2004) 279(33):34311–22. doi: 10.1074/jbc.M401629200
68. Chi TF, Khoder-Agha F, Mennerich D, Kellokumpu S, Miinalainen I, Kietzmann T, et al. Loss of USF2 promotes proliferation, migration and mitophagy in a redox-dependent manner. Redox Biol (2020) 37:101750. doi: 10.1016/j.redox.2020.101750
69. Lopez-Dee ZP, Chittur SV, Patel H, Chinikaylo A, Lippert B, Patel B, et al. Thrombospondin-1 in a Murine Model of Colorectal Carcinogenesis. PLoS One (2015) 10(10):e0139918. doi: 10.1371/journal.pone.0139918
70. Gutierrez LS, Suckow M, Lawler J, Ploplis VA, Castellino FJ. Thrombospondin 1–a regulator of adenoma growth and carcinoma progression in the APC(Min/+) mouse model. Carcinogenesis (2003) 24(2):199–207. doi: 10.1093/carcin/24.2.199
71. Lan CC, Wu CS, Huang SM, Wu IH, Chen GS. High-glucose environment enhanced oxidative stress and increased interleukin-8 secretion from keratinocytes: new insights into impaired diabetic wound healing. Diabetes (2013) 62(7):2530–8. doi: 10.2337/db12-1714
72. Mohanty P, Hamouda W, Garg R, Aljada A, Ghanim H, Dandona P. Glucose challenge stimulates reactive oxygen species (ROS) generation by leucocytes. J Clin Endocrinol Metab (2000) 85(8):2970–3. doi: 10.1210/jcem.85.8.6854
73. Wasserman DH, Wang TJ, Brown NJ. The Vasculature in Prediabetes. Circ Res (2018) 122(8):1135–50. doi: 10.1161/CIRCRESAHA.118.311912
74. Maile LA, Capps BE, Miller EC, Allen LB, Veluvolu U, Aday AW, et al. Glucose regulation of integrin-associated protein cleavage controls the response of vascular smooth muscle cells to insulin-like growth factor-I. Mol Endocrinol (2008) 22(5):1226–37. doi: 10.1210/me.2007-0552
75. Maile LA, Gollahon K, Wai C, Byfield G, Hartnett ME, Clemmons D. Disruption of the association of integrin-associated protein (IAP) with tyrosine phosphatase non-receptor type substrate-1 (SHPS)-1 inhibits pathophysiological changes in retinal endothelial function in a rat model of diabetes. Diabetologia (2012) 55(3):835–44. doi: 10.1007/s00125-011-2416-x
76. Bitar MS. Diabetes Impairs Angiogenesis and Induces Endothelial CellSenescence by Up-Regulating Thrombospondin-CD47-Dependent Signaling. Int J Mol Sci (2019) 20(3):673. doi: 10.3390/ijms20030673
77. Andrejeva G, Capoccia BJ, Hiebsch RR, Donio MJ, Darwech IM, Puro RJ, et al. Novel SIRPα Antibodies That Induce Single-Agent Phagocytosisof Tumor Cells while Preserving T Cells. J Immunol (2021) 206(4):712–21. doi: 10.4049/jimmunol.2001019
78. Puro RJ, Bouchlaka MN, Hiebsch RR, Capoccia BJ, Donio MJ, Manning PT, et al. Development of AO-176, a Next-Generation Humanized Anti-CD47 Antibody with Novel Anticancer Properties and Negligible Red Blood Cell Binding. Mol Cancer Ther (2020) 19(3):835–46. doi: 10.1158/1535-7163.MCT-19-1079
79. Miquilena-Colina ME, Lima-Cabello E, Sánchez-Campos S, García-Mediavilla MV, Fernández-Bermejo M, Lozano-Rodríguez T, et al. Hepatic fatty acid translocase CD36 upregulation is associated with insulin resistance, hyperinsulinaemia and increased steatosis in non-alcoholic steatohepatitis and chronic hepatitis C. Gut (2011) 60(10):1394–402. doi: 10.1136/gut.2010.222844
80. Miyaoka K, Kuwasako T, Hirano K, Nozaki S, Yamashita S, Matsuzawa Y. CD36 deficiency associated with insulin resistance. Lancet (2001) 357(9257):686–7. doi: 10.1016/s0140-6736(00)04138-6
81. Leder L, Kolehmainen M, Narverud I, Dahlman I, Myhrstad MC, de Mello VD, et al. Effects of a healthy Nordic diet on gene expression changes in peripheral blood mononuclear cells in response to an oral glucose tolerance test in subjects with metabolic syndrome: a SYSDIET sub-study. Genes Nutr (2016) 11:3. doi: 10.1186/s12263-016-0521-4
82. Samovski D, Dhule P, Pietka T, Jacome-Sosa M, Penrose E, Son NH, et al. Regulation of Insulin Receptor Pathway and Glucose Metabolism by CD36 Signaling. Diabetes (2018) 67(7):1272–84. doi: 10.2337/db17-1226
83. Isenberg JS, Jia Y, Fukuyama J, Switzer CH, Wink DA, Roberts DD. Thrombospondin-1 inhibits nitric oxide signaling via CD36 by inhibiting myristic acid uptake. J Biol Chem (2007) 282(21):15404–15. doi: 10.1074/jbc.M701638200
84. Bai J, Xia M, Xue Y, Ma F, Cui A, Sun Y, et al. Thrombospondin 1 improves hepatic steatosis in diet-induced insulin-resistant mice and is associated with hepatic fat content in humans. EBioMedicine (2020) 57:102849. doi: 10.1016/j.ebiom.2020.102849
85. Son NH, Basu D, Samovski D, Pietka TA, Peche VS, Willecke F, et al. Endothelial cell CD36 optimizes tissue fatty acid uptake. J Clin Invest (2018) 128(10):4329–42. doi: 10.1172/JCI99315
86. Yang P, Zeng H, Tan W, Luo X, Zheng E, Zhao L, et al. Loss of CD36 impairs hepatic insulin signaling by enhancing theinteraction of PTP1B with IR. FASEB J (2020) 34(4):4813–5992. doi: 10.1096/fj.201902777RR
87. Bhattacharyya S, Sul K, Krukovets I, Nestor C, Li J, Adognravi OS. Novel tissue-specific mechanism of regulation of angiogenesis and cancer growth in response to hyperglycemia. J Am Heart Assoc (2012) 1(6):e005967. doi: 10.1161/JAHA.112.005967
88. Asama H, Suzuki R, Hikichi T, Takagi T, Masamune A, Ohira H. MicroRNA let-7d targets thrombospondin-1 and inhibits the activationof human pancreatic stellate cells. Pancreatology (2019) 19(1):196–203. doi: 10.1016/j.pan.2018.10.012
89. Liu Y, Dong J, Ren B. MicroRNA-182-5p contributes to the protective effects of thrombospondin 1 against lipotoxicity in INS-1 cells. Exp Ther Med (2018) 16(6):5272–9. doi: 10.3892/etm.2018.6883
90. Xia Y, Dobaczewski M, Gonzalez-Quesada C, Chen W, Biernacka A, Li N, et al. Endogenous thrombospondin 1 protects the pressure-overloaded myocardium by modulating fibroblast phenotype and matrix metabolism. Hypertension (2011) 58(5):902–11. doi: 10.1161/HYPERTENSIONAHA.111.175323
91. Aiken J, Mandel ER, Riddell MC, Birot O. Hyperglycaemia correlates with skeletal muscle capillary regression and is associated with alterations in the murine double minute-2/forkhead box O1/thrombospondin-1 pathway in type 1 diabetic BioBreeding rats. Diabetes Vasc Dis Res (2019) 16(1):28–37. doi: 10.1177/1479164118805928
92. Klaassen I, de Vries EW, Vogels IMC, van Kampen AHC, Bosscha MI, Steel DHW, et al. Identification of proteins associated with clinical and pathological features of proliferative diabetic retinopathy in vitreous and fibrovascular membranes. PLoS One (2017) 12(11):e0187304. doi: 10.1371/journal.pone.0187304
93. Stenina OI, Krukovets I, Wang K, Zhou Z, Forudi F, Penn MS, et al. Increased expression of thrombospondin-1 in vessel wall of diabetic Zucker rat. Circulation (2003) 107(25):3209–15. doi: 10.1161/01.CIR.0000074223.56882.97
94. Varma V, Yao-Borengasser A, Bodles AM, Rasouli N, Phanavanh B, Nolen GT, et al. Thrombospondin-1 is an adipokine associated with obesity, adipose inflammation, and insulin resistance. Diabetes (2008) 57(2):432–9. doi: 10.2337/db07-0840
95. Matsugi K, Hosooka T, Nomura K, Ogawa W. Thrombospondin 1 Suppresses Insulin Signaling in C2C12 Myotubes. Kobe J Med Sci (2016) 62(1):E13–18.
96. Saboory E, Gholizadeh-Ghaleh Aziz S, Samadi M, Biabanghard A, Chodari L. Exercise and IGF-1 supplementation improve angiogenesis and angiogenic cytokines in a rat model of diabetes-induced neuropathy. Exp Physiol (2020) 105(5):783–92. doi: 10.1113/EP088069
97. Wahab NA, Schaefer L, Weston BS, Yiannikouris O, Wright A, Babelova A, et al. Glomerular expression of thrombospondin-1, transforming growth factor beta and connective tissue growth factor at different stages of diabetic nephropathy and their interdependent roles in mesangial response to diabetic stimuli. Diabetologia (2005) 48(12):2650–60. doi: 10.1007/s00125-005-0006-5
98. Lu A, Miao M, Schoeb TR, Agarwal A, Murphy-Ullrich JE. Blockade of TSP1-dependent TGF-β activity reduces renal injury and proteinuria in a murine model of diabetic nephropathy. Am J Pathol (2011) 178(6):2573–86. doi: 10.1016/j.ajpath.2011.02.039
99. Jiang N, Zhang Z, Shao X, Jing R, Wang C, Fang W, et al. Blockade of thrombospondin-1 ameliorates high glucose-induced peritoneal fibrosis through downregulation of TGF-β1/Smad3 signaling pathway. J Cell Physiol (2020) 235(1):364–79. doi: 10.1002/jcp.28976
100. von Toerne C, Huth C, de Las Heras Gala T, Kronenberg F, Herder C, Koenig W, et al. MASP1, THBS1, GPLD1 and ApoA-IV are novel biomarkers associated with prediabetes: the KORA F4 study. Diabetologia (2016) 59(9):1882–92. doi: 10.1007/s00125-016-4024-2
101. Matsuo Y, Tanaka M, Yamakage H, Sasaki Y, Muranaka K, Hata H, et al. Thrombospondin 1 as a novel biological marker of obesity and metabolic syndrome. Metabolism (2015) 64(11):1490–9. doi: 10.1016/j.metabol.2015.07.016
102. Dameron KM, Volpert OV, Tainsky MA, Bouck N. Control of angiogenesis in fibroblasts by p53 regulation of thrombospondin-1. Science (1994) 265(5178):1582–4. doi: 10.1126/science.7521539
103. Kong P, Gonzalez-Quesada C, Li N, Cavalera M, Lee DW, Frangogiannis NG. Thrombospondin-1 regulates adiposity and metabolic dysfunction in diet-induced obesity enhancing adipose inflammation and stimulating adipocyte proliferation. Am J Physiol Endocrinol Metab (2013) 305(3):E439–450. doi: 10.1152/ajpendo.00006.2013
104. Voros G, Lijnen HR. Deficiency of thrombospondin-1 in mice does not affect adipose tissue development. J Thromb Haemost (2006) 4(1):277–8. doi: 10.1111/j.1538-7836.2005.01696.x
105. Hida K, Wada J, Zhang H, Hiragushi K, Tsuchiyama Y, Shikata K, et al. Identification of genes specifically expressed in the accumulatedvisceral adipose tissue of OLETF rats. J Lipid Res (2000) 41(10):1615–22. doi: 10.1016/S0022-2275(20)31994-5
106. Ramis JM, Franssen-van Hal NL, Kramer E, Llado I, Bouillaud F, Palou A, et al. Carboxypeptidase E and thrombospondin-1 are differently expressed in subcutaneous and visceral fat of obese subjects. Cell Mol Life Sci (2002) 59(11):1960–71. doi: 10.1007/pl00012518
107. Efimenko A, Starostina E, Kalinina N, Stolzing A. Angiogenic properties of aged adipose derived mesenchymal stem cells after hypoxic conditioning. J Transl Med (2011) 9:10. doi: 10.1186/1479-5876-9-10
108. Rittig K, Dolderer JH, Balletshofer B, Machann J, Schick F, Meile T, et al. The secretion pattern of perivascular fat cells is different from that of subcutaneous and visceral fat cells. Diabetologia (2012) 55(5):1514–25. doi: 10.1007/s00125-012-2481-9
109. Corvera S, Gealekman O. Adipose tissue angiogenesis: impact on obesity and type-2 diabetes. Biochim Biophys Acta (2014) 1842(3):463–72. doi: 10.1016/j.bbadis.2013.06.003
110. Shimizu I, Aprahamian T, Kikuchi R, Shimizu A, Papanicolaou KN, MacLauchlan S, et al. Vascular rarefaction mediates whitening of brown fat in obesity. J Clin Invest (2014) 124(5):2099–112. doi: 10.1172/JCI71643
111. Wang D, Zhang Z, O’Loughlin E, Lee T, Houel S, O’Carroll D, et al. Quantitative functions of Argonaute proteins in mammalian development. Genes Dev (2012) 26(7):693–704. doi: 10.1101/gad.182758.111
112. Tang X, Miao Y, Luo Y, Sriram K, Qi Z, Lin FM, et al. Suppression of Endothelial AGO1 Promotes Adipose Tissue Browning and Improves Metabolic Dysfunction. Circulation (2020) 142(4):365–79. doi: 10.1161/CIRCULATIONAHA.119.041231
113. Memetimin H, Li D, Tan K, Zhou C, Liang Y, Wu Y, et al. Myeloid-specific deletion of thrombospondin 1 protects against inflammation and insulin resistance in long-term diet-induced obese male mice. Am J Physiol Endocrinol Metab (2018) 315(6):E1194–203. doi: 10.1152/ajpendo.00273.2018
114. Bréchot N, Gomez E, Bignon M, Khallou-Laschet J, Dussiot M, Cazes A, et al. Modulation of macrophage activation state protects tissue from necrosis during critical limb ischemia in thrombospondin-1-deficient mice. PLoS One (2008) 3(12):e3950. doi: 10.1371/journal.pone.0003950
115. Li Y, Tong X, Rumala C, Clemons K, Wang S. Thrombospondin1 deficiency reduces obesity-associated inflammation and improves insulin sensitivity in a diet-induced obese mouse model. PLoS One (2011) 6(10):e26656. doi: 10.1371/journal.pone.0026656
116. Lopez-Dee ZP, Chittur SV, Patel B, Stanton R, Wakeley M, Lippert B, et al. Thrombospondin-1 type 1 repeats in a model of inflammatory bowel disease: transcript profile and therapeutic effects. PLoS One (2012) 7(4):e34590. doi: 10.1371/journal.pone.0034590
117. Matuszewska K, Santry LA, van Vloten JP, AuYeung AWK, Major PP, Lawler J, et al. Combining Vascular Normalization with an Oncolytic Virus Enhances Immunotherapy in a Preclinical Model of Advanced-Stage Ovarian Cancer. Clin Cancer Res (2019) 25(5):1624–38. doi: 10.1158/1078-0432.CCR-18-0220
118. Kennedy DJ, Kuchibhotla S, Westfall KM, Silverstein RL, Morton RE, Febbraio M. A CD36-dependent pathway enhances macrophage and adipose tissue inflammation and impairs insulin signalling. Cardiovasc Res (2011) 89(3):604–13. doi: 10.1093/cvr/cvq360
119. Cui W, Maimaitiyiming H, Zhou Q, Norman H, Zhou C, Wang S. Interaction of thrombospondin1 and CD36 contributes to obesity-associated podocytopathy. Biochim Biophys Acta (2015) 1852(7):1323–33. doi: 10.1016/j.bbadis.2015.03.010
120. Su T, Huang C, Yang C, Jiang T, Su J, Chen M, et al. Apigenin inhibits STAT3/CD36 signaling axis and reduces visceral obesity. Pharmacol Res (2020) 152:104586. doi: 10.1016/j.phrs.2019.104586
121. Gutierrez LS, Ling J, Nye D, Papathomas K, Dickinson C. Thrombospondin peptide ABT-898 inhibits inflammation and angiogenesis in a colitis model. World J Gastroenterol (2015) 21(20):6157–66. doi: 10.3748/wjg.v21.i20.6157
122. Norman-Burgdolf H, Li D, Sullivan P, Wang S. CD47 differentially regulates white and brown fatfunction. Biol Open (2020) 9(12):bio056747. doi: 10.1242/bio.056747
123. Reverte-Salisa L, Sanyal A, Pfeifer A. Role of cAMP and cGMP Signaling in Brown Fat. Handb Exp Pharmacol (2019) 251:161–82. doi: 10.1007/164_2018_117
124. Mitschke MM, Hoffmann LS, Gnad T, Scholz D, Kruithoff K, Mayer P, et al. Increased cGMP promotes healthy expansion and browning of white adipose tissue. FASEB J (2013) 27(4):1621–30. doi: 10.1096/fj.12-221580
125. Kershaw EE, Flier JS. Adipose tissue as an endocrine organ. J Clin Endocrinol Metab (2004) 89(6):2548–56. doi: 10.1210/jc.2004-0395
126. Farr OM, Gavrieli A, Mantzoros CS. Leptin applications in 2015: what have we learned about leptin and obesity? Curr Opin Endocrinol Diabetes Obes (2015) 22(5):353–9. doi: 10.1097/MED.0000000000000184
127. Ganguly R, Khanal S, Mathias A, Gupta S, Lallo J, Sahu S, et al. TSP-1 (Thrombospondin-1) Deficiency Protects ApoE. Arterioscler Thromb Vasc Biol (2021) 41:e112–27. doi: 10.1161/ATVBAHA.120.314962
128. Chavez RJ, Haney RM, Cuadra RH, Ganguly R, Adapala RK, Thodeti CK, et al. Upregulation of thrombospondin-1 expression by leptin in vascular smooth muscle cells via JAK2- and MAPK-dependent pathways. Am J Physiol Cell Physiol (2012) 303(2):C179–191. doi: 10.1152/ajpcell.00008.2012
129. Elaïb Z, Lopez JJ, Coupaye M, Zuber K, Becker Y, Kondratieff A, et al. Platelet Functions are Decreased in Obesity and Restored after Weight Loss: Evidence for a Role of the SERCA3-Dependent ADP Secretion Pathway. Thromb Haemost (2019) 119(3):384–96. doi: 10.1055/s-0038-1677033
Keywords: cardiovascular disease, atherosclerosis, diabetes, angiogenesis, TGFβ1, obesity, nitric oxide
Citation: Gutierrez LS and Gutierrez J (2021) Thrombospondin 1 in Metabolic Diseases. Front. Endocrinol. 12:638536. doi: 10.3389/fendo.2021.638536
Received: 09 December 2020; Accepted: 01 March 2021;
Published: 29 March 2021.
Edited by:
Hiu Yee Kwan, Hong Kong Baptist University, Hong KongReviewed by:
David Soto-Pantoja, Wake Forest School of Medicine, United StatesDavid D. Roberts, National Institutes of Health (NIH), United States
Anthony L. Schwartz, Morphiex Biotherapeutics, United States
Bin Ren, University of Alabama at Birmingham, United States
Gabor Csanyi, Augusta University, United States
Copyright © 2021 Gutierrez and Gutierrez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Linda S. Gutierrez, linda.gutierrez@wilkes.edu