 Wenjie Zhou1,2†Yingying Li2,3,4†Zhuobo Li2,3,5Bo Ma2,3,5
Wenjie Zhou1,2†Yingying Li2,3,4†Zhuobo Li2,3,5Bo Ma2,3,5 Xiao Jiang2,3
Xiao Jiang2,3 Chaoqun Hu2,3
Chaoqun Hu2,3 Yongxing Ai1*
Yongxing Ai1* Peng Luo*2,3
Peng Luo*2,3- 1College of Animal Science, Jilin University, Changchun, China
- 2CAS Key Laboratory of Tropical Marine Bio-Resources and Ecology (LMB), Guangdong Provincial Key Laboratory of Applied Marine Biology (LAMB), South China Sea Institute of Oceanology, Chinese Academy of Sciences, Guangzhou, China
- 3Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou), Guangzhou, China
- 4Geological Survey Institute of Guangzhou, Guangzhou, China
- 5University of Chinese Academy of Sciences, Beijing, China
Bacteriophages (phages) and their bacterial hosts were the most abundant and genetically highly diverse organisms on the earth. In this study, a series of phage-resistant mutant (PRM) strains derived from Vibrio alginolyticus were isolated and Infrequent-restriction-site PCR (IRS-PCR) was used to investigate the genetic diversity of the PRM strains. Phenotypic variations of eight PRM strains were analyzed using profiles of utilizing carbon sources and chemical sensitivity. Genetic variations of eight PRM strains and coevolved V. alginolyticus populations with phages were analyzed by whole-genome sequencing and resequencing, respectively. The results indicated that eight genetically discrepant PRM stains exhibited abundant and abundant phenotypic variations. Eight PRM strains and coevolved V. alginolyticus populations (VE1, VE2, and VE3) contained numerous single nucleotide variations (SNVs) and insertions/indels (InDels) and exhibited obvious genetic divergence. Most of the SNVs and InDels in coding genes were related to the synthesis of flagellar, extracellular polysaccharide (EPS), which often served as the receptors of phage invasion. The PRM strains and the coevolved cell populations also contained frequent mutations in tRNA and rRNA genes. Two out of three coevolved populations (VE1 and VE2) contained a large mutation segment severely deconstructing gene nrdA, which was predictably responsible for the booming of mutation rate in the genome. In summary, numerous mutations and genetic divergence were detected in the genomes of V. alginolyticus PRM strains and in coevolved cell populations of V. alginolyticus under phage infection stress. The phage infection stress may provide an important force driving genomic evolution of V. alginolyticus.
Introduction
Bacteriophages (phages) and their bacterial hosts are the most abundant and genetically highly diverse organisms on the earth (Suttle, 2005; Weitz et al., 2013; Batinovic et al., 2019). Phages propagate through hijacking replication and metabolic mechanisms of bacterial hosts and induce the lysis of bacterial hosts, and thus bring a very strong selection stress for bacterial survival and evolution (Thurber, 2009; Gómez and Buckling, 2013; Warwick-Dugdale et al., 2019), which results in the occurrence of phage-resistant mutant (PRM) population. For a long time, bacteria–phage interaction is believed as an important driver of ecological and evolutionary processes for both communities (Fuhrman, 1999; Koskella and Brockhurst, 2014; Shabbir et al., 2016; Hampton et al., 2020), however, studies on genetic variations of PRM strains and genomic evolution of coevolved bacterial populations with phages are very rare. Some studies on comparative genomics between PRM and ancestor strains revealed some genetic mutations, mainly focusing on the effect of mutations on the expression of genes related to cell surface structure, such as LPS and flagella (Kashiwagi and Yomo, 2011; Castillo et al., 2015; Lis and Connerton, 2016; Gonzalez et al., 2018). Little is known about detailed genetic variations of PRM strains on whole-genomic scale under phage infection stress. Scanlan et al. (2015) first revealed that coevolution with phage Phi2 drives genomic-wide host evolution and constrains the acquisition of abiotic-beneficial mutations in Pseudomonas fluorescens through whole-genome sequencing, and the isolates with discrepant phenotypes in coevolved populations exhibited much more numerous mutations and much higher genetic divergence compared to these in evolved populations (Scanlan et al., 2015). Resequencing provides a new approach to probe into the genetic variations and mutation frequencies within cell populations. However, so far as now, the technique was only used to investigate occurrence frequencies of mutation sites from the perspective of phage φ2 when it was coevolved with its host P. fluorescens (Paterson et al., 2010).
Phages are the most abundant viruses in ocean, and it is estimated that phages kill and lyse between 15 and 40% of ocean’s bacteria every day, which poses a huge pressure on the survival of marine bacteria (Danovaro et al., 2011). Ocean contains the most abundant bacteria and phages on the Earth, however, the research on the genomic evolution of marine bacteria under phage infection stress has so far been completely blank. Vibrio alginolyticus is one of the most common bacterial species in estuary and marine environments (Luo et al., 2012; Fu et al., 2016), and it is also a conditional pathogen for human beings (Daniels and Shafaie, 2000; Slifka et al., 2017) and many kinds of marine fish, shellfish, and crustaceans (Pruzzo et al., 2006; Abdelaziza et al., 2017). In this study, we first isolated numerous PRM strains derived from an ancestor strain V. alginolyticus E06333 under phage infection stress. The whole genomic sequences of several genetically divergent mutants were analyzed focusing on single nucleotide variations (SNVs) and insertions/deletions (InDels) and their potential functions. Besides, resequencing of the evolved populations of V. alginolyticus E06333 and the coevolved populations with enriched phages at different periods were carried out to characterize mutation sites and their frequencies of occurrence.
Results
Screening of Phage-Resistant Mutant Strains by Double-Layer Plate Method
A large number of PRM colonies derived from V. alginolyticus E06333 appeared on double-layer plates, and the sizes and surface morphologies of PRM colonies were very different from these grown normally on LB plates without phages (Figure 1), which implied that genetic variations harbored among these PRM colonies. Totally 80 PRM strains of V. alginolyticus were acquired.
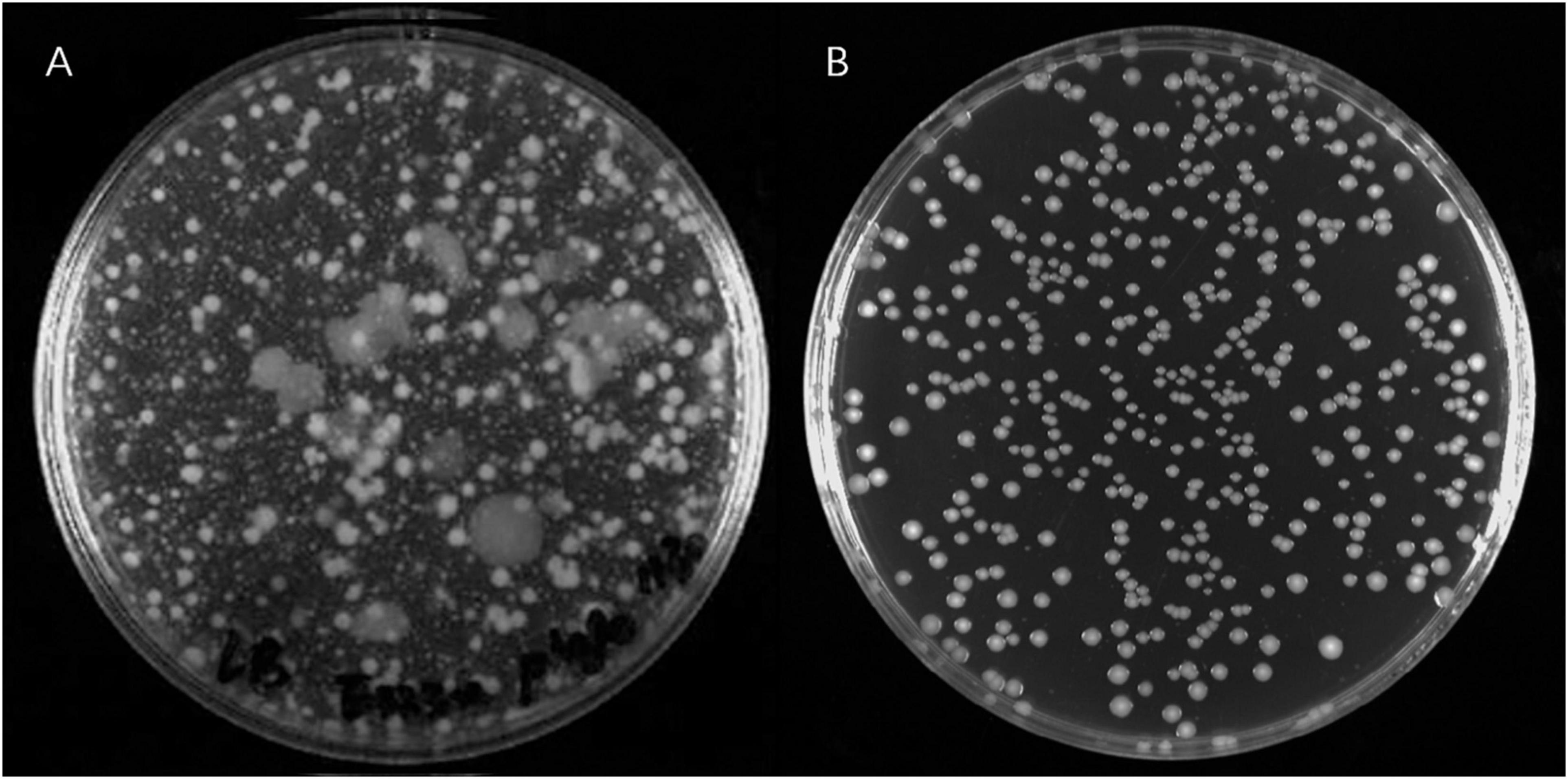
Figure 1. The phage resistant colonies derived from V. alginolyticus E06333 and normal colonies V. alginolyticus E06333. (A) The phage resistant colonies occurred on a double-layer plate with enriched lytic phages. (B) The normal colonies of V. alginolyticus E06333 on a LB plate.
Infrequent-Restriction-Site-PCR Exhibited Extensive Genetic Variations in the Phage-Resistant Mutant Strains
Genotypes of 80 PRM strains were determined by IRS-PCR, and the results indicated most PRM strains did not show obvious genetic variations in this method except that 15 PRM strains generated obvious discrepant and bright bands within the range of 1500–3000 bp (Figure 2). Among them, 8 PRM strains were singled out for subsequent experiments, and they were renamed as VAM01–VAM08 (Figure 2).
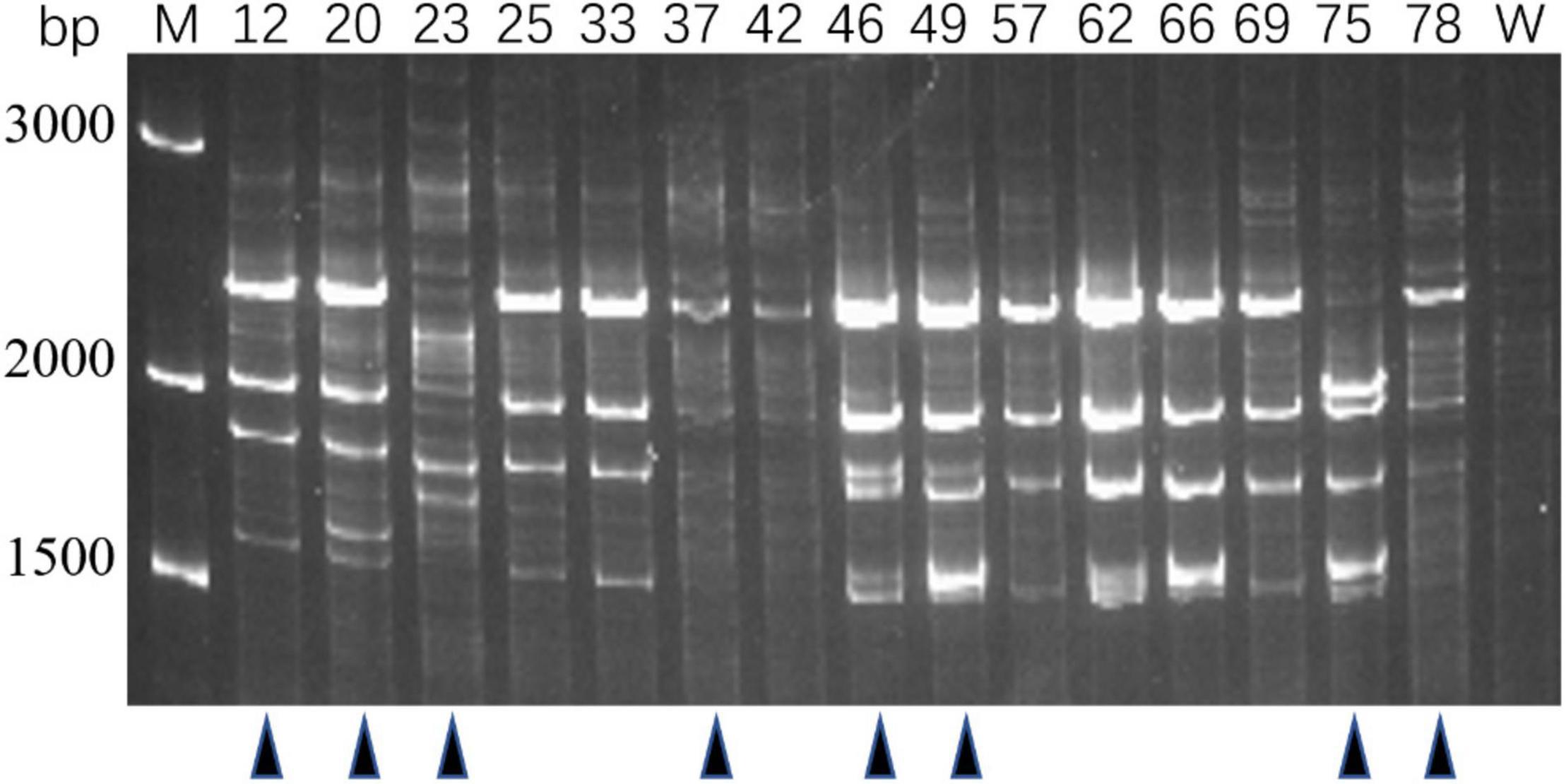
Figure 2. Genotyping of typical PRM strains derived from V. alginolyticus E06333 by IRS-PCR. These PRM strains exhibit obviously different fingerprints from the primary V. alginolyticus E06333. The numbers on the top represent the strain numbers. The black triangles on the bottom present eight PRM strains that are further selected for subsequent analysis. Eight PRM strains were renamed (according to the order from left to right) as VAM01, VAM02, VAM03, VAM04, VAM05, VAM06, VAM07, and VAM08, respectively.
Phenotypic Variations of the Phage-Resistant Mutant Strains Revealed by Biolog Assays
Biolog assays with GEN III microplates indicated that eight PRM strains generated 32 differentiated reactions compared with the ancestor strain E06333, among which 27 reactions were related to carbon utilization and 5 reactions were related to chemical sensitivity (Figure 3). Each PRM strain had a unique reaction profile, but all PRM strains tended to lose some phenotypes rather than to acquire some phenotypes (Figure 3). Among them, PRM strain VAM01 lost the most 19 positive phenotypes of utilizing 19 carbon sources when compared with wild strain V. alginolyticus E06333. All the PRM strains lost the ability to utilize L-Arginine and became sensitive to sodium bromate. Eight PRM strains only shifted to acquire 1–3 positive phenotypes of utilizing carbon sources, and 6 out of 8 PRM strains obtained the ability utilizing α-hydroxy-butyric Acid (Figure 3). Biolog assays clearly indicated that eight PRM strains generated rich and obvious phenotypic changes and these changes were not significantly associated with anti-phage infection, which suggested the genetic variations harbored in these PRM strains.
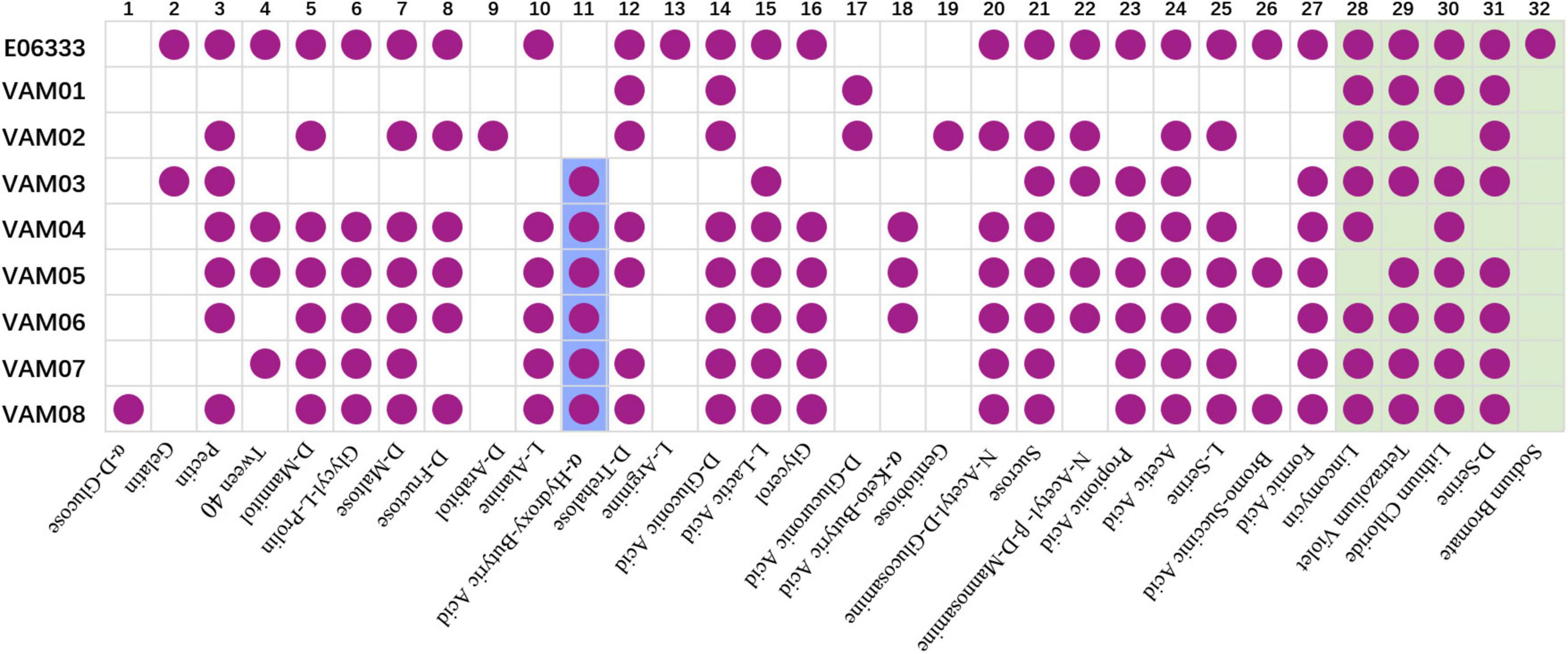
Figure 3. Biochemical profiles of eight PRM strains and the primary strain E06333 from V. alginolyticus revealed by Biolog assays. The purple circles in boxes represent a positive reaction (growth). The purple circles with blue background represent six PRM strains obtained the ability of utilizing α-Hydroxy-Butyric Acid. The boxes with light green background represent five tests of chemical sensitivity.
Discrepant Mutation Sites and Their Related Genes in the Phage-Resistant Mutant Strains of V. alginolyticus
Comparative genomics of V. alginolyticus E06333 and eight PRM strains revealed numerous SNVs and InDels in eight PRM strains. Eight PRM strains had discrepant numbers of SNVs and InDels with different sizes (Figure 4). The PRM strain VAM04 harbored the most SNVs and InDels including 242 SNVs and 42 InDels while the PRM strain VAM05 harbored the least SNVs and InDels including 20 SNVs and 15 InDels (Figure 4). The PRM strain VAM03 harbored an only >10-bp insertion (11 bp), and each PRM strain contained 1 or 2 big deletions that exceeded 100 bp (actually >400 bp) except for the strain VAM03 (Figure 4).
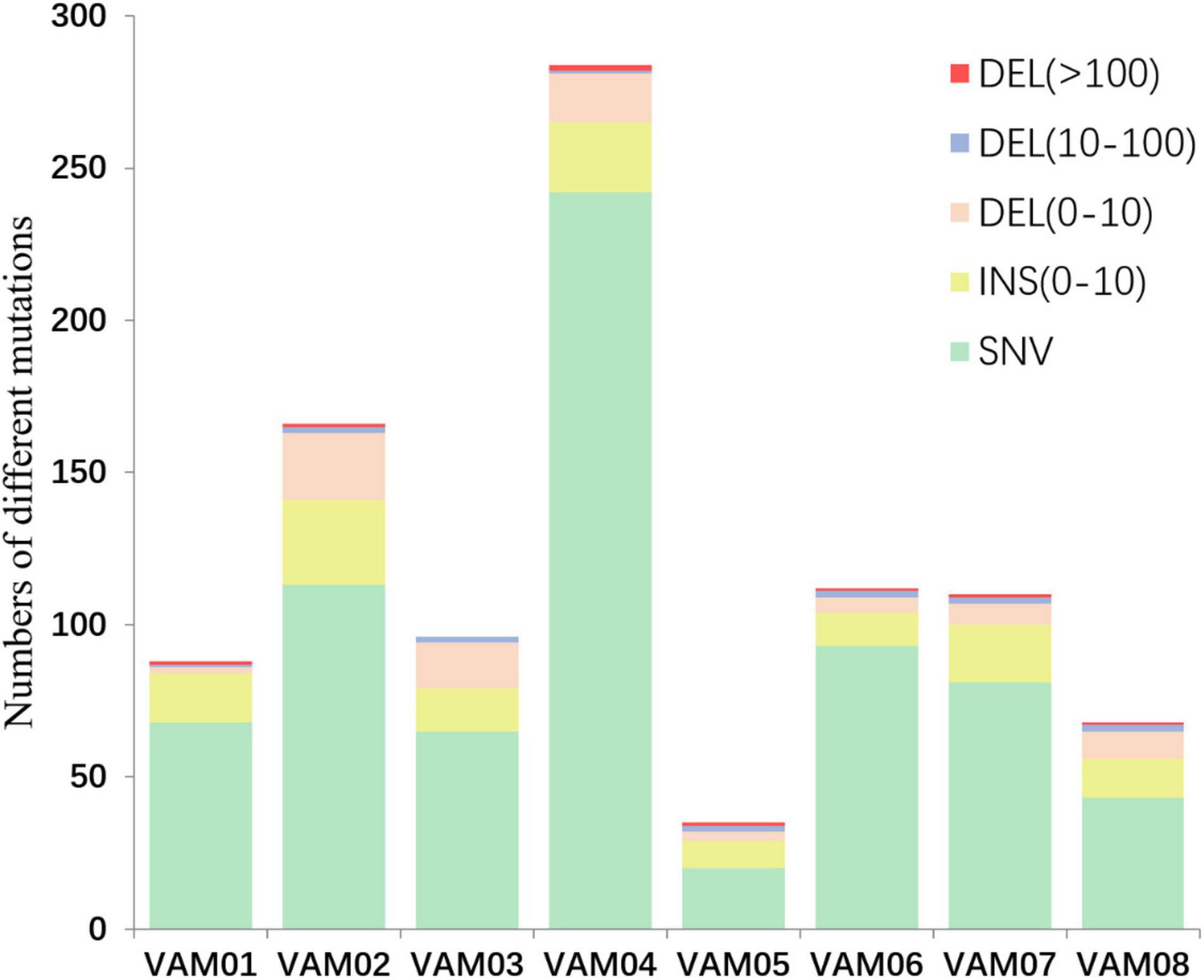
Figure 4. The numbers of different mutations in eight PRM strains from V. alginolyticus. The mutations were classed into insertion (INS), deletions (DEL), and single nucleotide variations (SNVs) according to the mutation types and sizes. An 11-bp insertion in the PRM strain VAM03 was not shown in this figure.
Further identification of these numerous mutation sites revealed that 15 mutation sites occurred within coding genes of eight PRM strains, and these intragenic mutation sites included 11 scattered non-synonymous SNVs, 3 InDels and one large region containing concentrated SNVs, LCM1 (Table 1), wherein 8 PRMs had discrepant SNV distribution. The PRM strains, VAM01–VAM08, contained 3, 5, 7, 7, 4, 8, 4, and 4 SNVs and InDels within coding genes, respectively, and each PRM strain had a unique mutation profile (Table 1). 15 mutation sites covered 12 genes involving in different functions, including secretion of lectins and toxins, bacterial outer membrane translocation and assembly, polysaccharide synthesis, flagella synthesis and regulation, and aerotolerance operon (Table 1). Among 12 genes, the mutations of fliF, flhA, fleQ, epsD, ugdH, glyT, and ytfM were predictably related to the phage resistance of the PRM strains in varying degrees (Table 1), and they participated in the synthesis of flagellar, extracellular polysaccharide (EPS) including capsular polysaccharides and lipopolysaccharides (LPS). The mutations in flhA and fliF were non-sense mutations, leading to the premature termination of peptide synthesis. A 11-bp insertion gave rise to the change of 24 amino acids residues at the end of the espD gene. The SNVs at nt2573434 and nt2573439 resulted the changes of two amino acids residues (P/L and I/Y) in the translated protein, UGDH, coded by ugdH, two changed amino acids located on conserved sites of NADB Rossmann Superfamily. A 18-bp deletion in batB of aerotolerance operon was the only mutation region shared by eight PRM strains. The end of batB contained 55 core repeats of “GCAACA,” and the deletion contained three core repeats that resulted in truncated and altered amino acids in predicted gene translocation. Six PRM strains (VAM02, VAM03, VAM04, VAM06, VAM07, and VAM08) contained SNVs in omp1 coding for a putative calcium-binding outer membrane-like protein in T1SS system. Four strains (VAM02, VAM04, VAM06, and VAM07) and 6 PRM strains (VAM01, VAM03, VAM04, VAM05, VAM06, and VAM08) harbored large fragment mutations in rtx1, and rtx2, respectively. The gene rtx1 encoded for a tandem-95 repeat protein containing a cadherin-like domain, and the gene rtx2 encoded a putative RTX toxin in T1SS system. Three genes, omp1, rtx1, and rtx2, were featured by many tandem repeats and large size (>10000 bp). Besides, it is little known about the function of a gene, hyp1, in V. alginolyticus and SNVs in batB were predicted not to have direct relations to phage resistance.
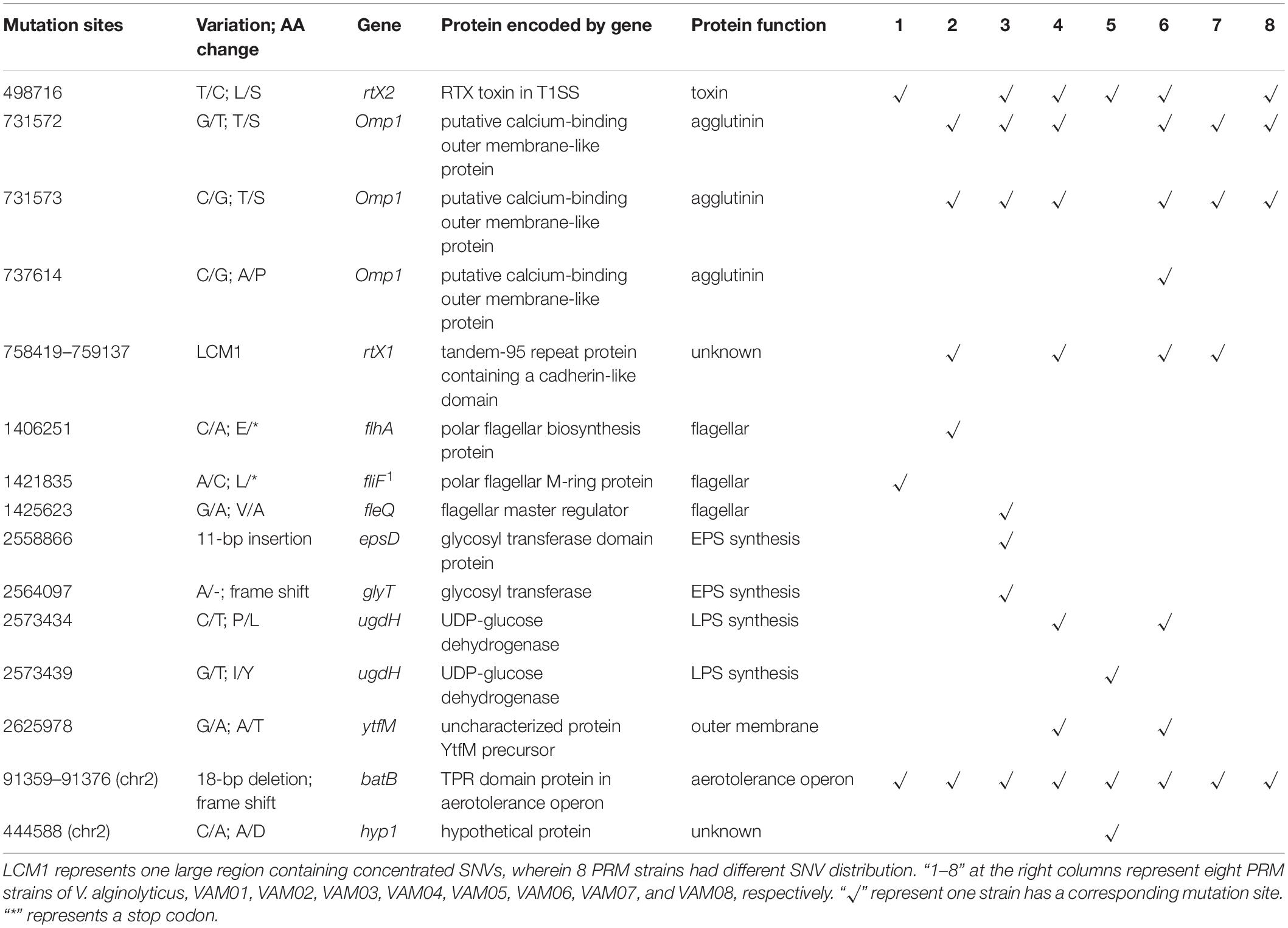
Table 1. Mutation sites in coding genes of eight PRM strains derived from V. alginolyticus E06333.
Mutations in DNA Regions Related to Non-coding RNA Genes of the Phage-Resistant Mutant Strains
It is notable that eight PRM strains also harbored 12 SNVs and InDels in non-coding RNA genes (Table 2), including 5 big deletion regions (422–677 bp) and 3 concentrated SNV regions (SNVs > 5 within the range of 400 bp), and 4 scattered SNV regions, respectively (Table 2). Five big deletion regions all occurred in the tandem repeat regions of tRNA genes. In addition, 2 concentrated SNV regions and 4 SNVs occurred in the 16S or 23S rRNA genes. Each PRM strain had a different mutation profile in non-coding RNA genes, which further exhibited genetic divergence among eight PRM strains.

Table 2. Mutation sites in non-coding RNA genes of eight PRM strains derived from V. alginolyticus E06333.
Divergent Evolution of Coevolved V. alginolyticus E06333 Cell Populations Revealed by Genomic Resequencing
Resequencing facilitates to reveal of the frequencies of mutations in each bacterial cell population. The SNVs and InDels detected by resequencing data were shown in Supplementary Table 1. Totally, 14 SNVs and InDels were detected in the evolved cell populations (VC1, VC2, and VC3) and the coevolved cell populations (VE1, VE2, and VE3), including 4 SNVs, 10 InDels, and 1 large region of 1260 bp containing concentrated point mutations (LCM2) (Figure 5). Coevolved cell populations of V. alginolyticus contained 8 unique SNVs and InDels (53% of total numbers) while evolved cell populations only had 2 unique SNVs (13% of total numbers of mutation sites), which manifested that most of mutation sites occurred in coevolved cell populations with phages. It clearly indicated that coexisting phages markedly raised detectable SNVs and InDels in V. alginolyticus cell populations. Each coevolved cell populations at different time points (VE1 at 2 h, VE2 at 13 h, VE3 at 22 h) had a unique mutation profile, and they were also very different from the mutation profiles of evolved cell populations (Figure 5). The result manifested that phage infection stress drove and accelerated the divergent evolution of V. alginolyticus cell populations.
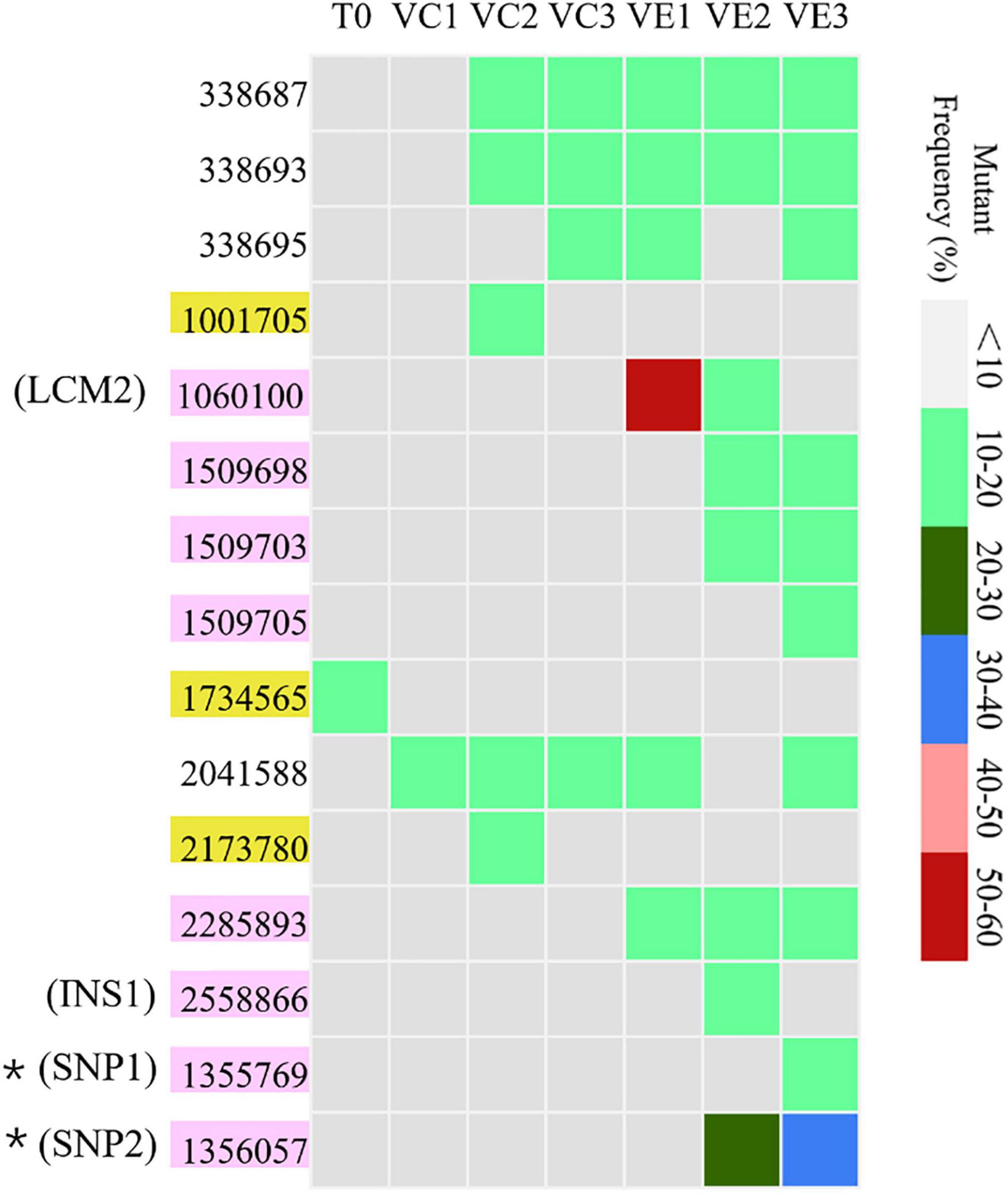
Figure 5. The distribution and the mutation frequencies of SNVs and InDels in evolved and coevolved V. alginolyticus cell populations revealed by resequencing. T0: the collected sample before grouping at the early exponential stage (OD600 nm = 0.6–1); VC1: collected samples from evolved cell populations at 2 h; VC2: collected samples from evolved cell populations at 13 h; VC3: collected samples from evolved cell populations at 22 h; VE1: collected samples from coevolved cell populations at 2 h; VE2: collected samples from coevolved cell populations at 13 h; VE3: collected samples from coevolved cell populations at 22 h. Mutation sites with yellow background represent 2 unique SNVs in the evolved cell populations. Positions of mutation sites with purple background represent 8 unique mutations in the coevolved cell populations. Mutation frequencies of these sites are shown with different colors.
Unique Mutation Sites in Coevolved Cell Populations of V. alginolyticus and Their Functions of Related Genes
Though the evolved cell populations harbored 2 unique SNVs and 4 shared SNVs (Figure 5), here we only focused on the unique mutation sites in coevolved cell populations of V. alginolyticus and their predicted functions of related genes (Table 3). Among 8 unique mutation sites in the coevolved cell populations of V. alginolyticus, 3 SNVs occurred in tsaA coding for a tRNA (Thr-GGU) A37N-methylase, and one InDel occurred in the upstream of glyS coding for a Glycyl-tRNA synthetase β chain, and therefore these four mutation sites are closely related to tRNA synthesis. An insertion of 11 bp (INS1) at nt2558866 resulted in a frameshift mutation of epsD, and this 11-bp insertion also occurred in the PRM strain VAM03 (Table 1). The insertion gave rise to the change of 24 amino acids residues at the end. The gene epsD is predicted related to the synthesis of extracellular polysaccharides and catalyze the transfer of UDP-glucose or UDP-galactose to lipid carriers, which is the first step in the synthesis of oligosaccharides. The coevolved cell populations also contained 2 SNVs (SNP1 and SNP2) in galE, and SNP2 caused the premature termination of transcription of galE. The large mutation region of 1260 bp (LCM2) in VE1 and VE2 populations severely deconstructed gene nrdA, which blocked the synthesis of ribonucleotide reductase (RNR). The mutation frequency of this gene at the stage of VE1 reached at as high as 55% and then dramatically declined to 13% at the stage of VE2 and below detection limitation (<10%) at the stage of VE3.
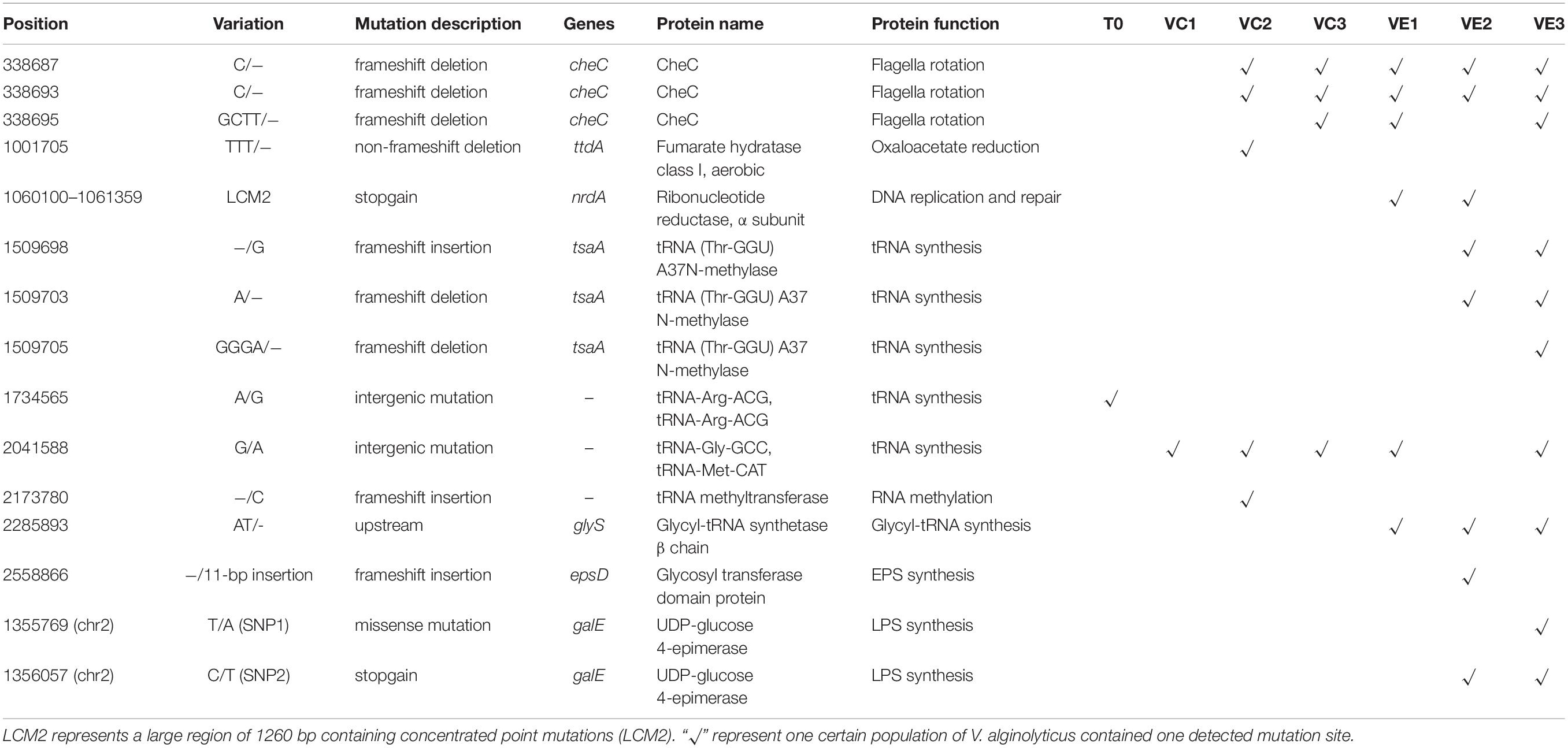
Table 3. Mutant sites and related genes in evolved and coevolved V. alginolyticus cell populations.
Discussion
Exploring the adaptive evolution of environmental microbes represents an increasing interest under the background of the extensively used high-through technologies, which facilitates us to understand the history of bacterial genomic evolution and bacterial genetic diversity. The arms race between bacteria and their phage has been long believed to be an important force for mutual evolution (Hampton et al., 2020; Safari et al., 2020), but few details in terms of bacterial whole-genome scale have been revealed. In three PRM strains, three variant genes related to flagella synthesis and regulation, flhA, fliF, and fleQ, were detected, among which the SNVs in flhA and fliF resulted in the premature termination of flagella synthesis. Flagella are often used as a receptor for phages to invade bacterial hosts (Kropinski et al., 2012; Choi et al., 2013; Silva et al., 2016), and therefore the inhibition of flagellin synthesis in the PRM strains may affect the phage adsorption process. Besides, the PRM strains displayed various mutations in ugdH and epsD, and coevolved V. alginolyticus populations harbored unique mutations in galE and epsD. The product of galE, GalE, was an important enzyme for the synthesis of active carbohydrates. It was frequently shared in a variety of EPS biosynthetic pathways (Bahat-Samet et al., 2004; Schaper et al., 2019) and affected the biosynthesis of a variety of bacterial lipopolysaccharides (Bengoechea et al., 2002). Four genes involved in the synthesis of extracellular polysaccharides including capsular polysaccharides and lipopolysaccharides were thought to be responsible for phage adsorption (Baldvinsson et al., 2014; Dang and Lovell, 2015; Ha et al., 2019). Mutations in the genes encoding for flagellin synthesis and extracellular polysaccharides were often reported in phage-resistant mutants since these cell structures served as the receptors of phages (Nesper et al., 2001; Gonzalez et al., 2018; Cai et al., 2019; Schaper et al., 2019). From this regard, generating mutations in the genes encoding for flagellin synthesis and extracellular polysaccharides is likely a common strategy to cope with phage infection in V. alginolyticus. Reduced virulence was often observed in phage-resistance mutants as mutant genes changed surface antigens that are crucial for both phage adsorption and cellular invasiveness (Laanto et al., 2012; Castillo et al., 2015; Sumrall et al., 2019; Markwitz et al., 2021). Except these virulent genes involving in phage adsorption generated mutations, a virulence-associated gene rtx2 coding for RTX toxin in T1SS also generated mutations. Therefore, mutations under phage infection stress are not limited within genes responsible for phage adsorption. Besides, the functions of some genes bearing mutations in the PRM strains and in the coevolved cell populations remain unknown, and it is worthy to explore their potential roles in phage infections in future.
It is generally recognized that rRNA genes unlikely generate rapid and frequent changes as they perform the most fundamental and important functions in life. Therefore, the rRNA genes were once considered the ideal chronometer to record the evolutionary history of life, potentially all the way back to the last common ancestor (Woese, 1987, 1998), and rRNA sequence comparisons led to the construction of a “universal tree of life,” dividing all life on the Earth into three equidistant domains: eukarya, bacteria, and archaea (Woese et al., 1990). Until now, 16S and 23S rRNA gene mutations were often observed in some antibiotic-resistant bacterial strains (Bachir et al., 2018; Chen et al., 2018; Lauener et al., 2019), wherein antibiotics generally impact the normal functions of 16S and 23S rRNA or their coupling ribosomes. It is obvious that the mutations of 16S and 23S rRNA genes have not contributed to bacterial resistance against phage. This study first demonstrated that phage infection stress can change the sequences of highly conserved rRNA genes, which complicate bacterial phylogenetics based on rRNA genes, and thus phage infection stress has a possibility to drive bacterial rRNA molecular evolution. In addition, few mutations in tRNA genes or related DNA regions were noticed in bacteria. However, it’s surprising that frequent mutations of tRNA genes were also observed in eight PRM strains. The rRNA- and tRNA-related mutations in PRM strains of V. alginolyticus under experimental conditions urge us to speculate that similar incidents may occur with a certain frequency in natural bacterial communities due to the prevalence of phages and strong selective stress by them. In this study, we observed the reduced copy numbers of tRNA genes (Table 2). For instance, the chromosomal site (Chr1: nt12887–13817) of the primary strain E06333 contains six tRNA-Tyr genes, but in the PRM VAM01 and VAM02 only remained two tRNA genes. By Blast, we found that conspecific strains not only vary copy numbers of tRNA genes [e.g., the strains VIO5 (AP022861.1) and YM19 (AP022863.1)] at this chromosomal site but also contained variable sequences. Yona et al. (2013) first carried out a systematic search in hundreds of genomes that revealed tRNA gene mutations occur throughout the tree of life. They further demonstrate that mutation in tRNA genes is a common adaptive mechanism when meeting new translational demands (Yona et al., 2013). Until now, there are no evidences indicating direct correlation between phage-resistant mechanisms and rRNA and tRNA gene mutations. Hence, we speculated the variations in tRNA pool as an adaptive mechanism to meet the new translational demands in the PRM strains. Together, these results indicated that phage infection stress can drive bacterial rRNA and tRNA gene evolution.
It is notable that variations in coding genes and rRNA- and tRNA- genes among these PRM strains lacked significant correlation with the phenotypical shifts revealed by Biolog assays. Therefore, it is reasonable to speculate some genetic changes affecting phenotypic characteristics were not discovered. On the other hand, though many mutation sites were detected in the draft genomes of eight PRM strains, the gaps between scaffolds potentially contained some mutation sites not detected. As a result, the actual numbers of SNVs and InDels may exceed our calculation. Numerous genetic variations in the PRM strains and the coevolved populations also raised a question whether phage infection stress induced the generation of most observed mutations in V. alginolyticus and boost the mutation rate. Through whole-genome sequencing, it is estimated that spontaneous mutations in Escherichia coli, as a typical representation of prokaryotes is 1.0 × 10–3 mutations per genome per generation (Lee et al., 2016). According to this spontaneous mutation rate, it is impossible to accumulate numerous genetic variations in the genomes of the PRM strains and coevolved populations of V. alginolyticus even at the longest generation time of 22 h (about 66 generations) for VE3 populations. Therefore, increased genetic variations (mutations) in the PRM strains and coevolved populations were not generated by spontaneous mutations but mainly induced by phage infection stress, which provides one important force driving genomic evolution of V. alginolyticus and accelerate genetic divergence via boosting mutation rate. Swings et al. (2017) found that mutation rates of E. coli populations rise when cells experience higher stress and decline again once cells are adapted, and they identified cellular mortality as the major force driving the quick evolution of mutation rates (Swings et al., 2017). Increased mutation rates under environmental stress were also observed in some cases (Bjedov, 2003; Galhardo et al., 2007; MacLean et al., 2013). All these findings strongly supported the idea that mutation rate variability plays a key role in adaptive evolution under selective pressure (Engelhardt and Shakhnovich, 2019). Together, these studies suggested that the role of environmental stress is not limited to naturally select preexisting mutants and it is not the whole story that all mutations are random and evolution is slow. Therefore, it shed some light on the acknowledges of microbial genomic evolution in a natural environment full of competition and adverseness.
Interestingly, the coevolved populations VE1 were observed to have a high-frequency (53%) large mismatch mutation in nrdA coding for a ribonucleotide reductase (RNR) alpha subunit, RNR1. RNR catalyzes the conversion of nucleoside diphosphates (NDPs) into deoxynucleotides and is a key enzyme for DNA replication and repair in all organisms (Cotruvo and Stubbe, 2008). The levels of the cellular dNTPs are tightly controlled, in large part through allosteric control of RNR (Ahluwalia et al., 2012; Zhu et al., 2017). Ahluwalia et al. (2012) found that a set of RNR1 mutants of E. coli changed dNTP pools and the mutation rate of bacteria also increases with the increase of the dNTP change range. They further found that even relatively modest dNTP pool deviations caused by one set of RNR1 mutants gave rise to exceptionally strong mutator phenotypes (>1,000-fold increases) (Ahluwalia et al., 2012). From this case, we speculated that high-frequency and large mutation in nrdA likely changed the dNTPs pool in V. alginolyticus and mainly contribute to the booming of the mutations. The mutation frequency of nrdA dramatically declined to 13% and below the detection limit (<10%). This sharp decline in coevolved populations was likely due to the adaptive evolution of V. alginolyticus under phage stress at different stages. Once numerous phage-resistant mutator phenotypes merged, it is necessary to regain the normal intracellular environment to ensure accurate DNA replication and stable propagation. Therefore, we speculated that the occurrence of PRM strains containing numerous mutation sites is likely the consequence of the dynamic change of nrdA in a certain stage though the PRM strains did not harbor nrdA mutation. The functions of RNR in V. alginolyticus need to be further explored in future.
Bacteria and phages are locked in a constant arm race and both are perpetually changing their tactics to overcome each other (Hampton et al., 2020; Safari et al., 2020). Bacteria use various strategies to overcome the invading phages, including adsorption inhibition, restriction-modification (R/E) systems, CRISPR–Cas (clustered regularly interspaced short palindromic repeats–CRISPR-associated proteins) systems, abortive infection (Abi), etc (Safari et al., 2020). To counteract, phages employ intelligent tactics for the nullification of bacterial defense systems, such as accessing host receptors, evading R/E systems, and anti-CRISPR proteins (Safari et al., 2020). However, if these defense systems in bacteria do not exist or work, change themselves through accelerated molecular evolution under phage infection stress should be another strategy. Conversely, phages can also generate mutation to increase their invasion ability. For instance, the rate of molecular evolution in phage φ2 was far higher when both P. fluorescens and the phage coevolved with each other than when phage evolved against a constant host genotype (Paterson et al., 2010). The most rapidly evolving phage genes under coevolution were those involved in host infection (Paterson et al., 2010). Continual natural selection for adaptation and counter-adaptation drive molecular evolution of interacting bacteria and their phages (Gómez and Buckling, 2011; Koskella and Brockhurst, 2014). In this study, the distribution profiles of SNVs in coevolved cell populations were discrepant at different development stages, and it implied that coevolved cell populations adopted different adaptation strategies to adapt to variable infection stress which may be caused by genetically changed phages.
At last, we should point out that though totally 80 PRM strains of V. alginolyticus were acquired mainly based on the morphologies of resistant colonies, similar morphologies of resistant colonies may harbor divergent mutants as some genetic changes cannot be exhibited by the differences of common phenotype characteristics. Therefore, genotyping by IRS-PCR was adopted to first screen the genetically variable PRM strains as the method is simple and time-saving. However, genotyping by IRS-PCR cannot show all genetic changes of tested strains due to the limitation of this method such as short amplification fragments and the selection of restriction endonucleases, and thus it is likely that some PRM strains that were not selected for subsequent analysis may also harbor many genetic variations.
In summary, V. alginolyticus PRM strains exhibited abundant genetic changes and genetic divergence under phage infection stress. Resequencing revealed that coevolved V. alginolyticus populations with phages had discrepant profiles of SNVs and InDels at three coevolved stages, while most detected SNVs and InDels occurred in the coevolved populations. Mutations were observed within rRNA and tRNA genes in the PRM strains and coevolved cell populations. Our results combining with other findings indicated that phage infection stress can boost mutation rates in V. alginolyticus genome and accelerate the genetic divergence, and thus it is likely one driving force for the bacterial genome evolution.
Materials and Methods
The Primary V. alginolyticus Strain E06333
Vibrio alginolyticus strain E06333 was isolated from marine fish, Epinephelus coioides, in 2006, in Guangdong province, China, and stored in our laboratory (Luo et al., 2018). The strain E06333 was cultured in LB plates and single colony was picked out for subsequent experiments.
Enrichment of Mixed Phages Infecting V. alginolyticus
Wastewater samples were collected from an outfall of shrimp aquafarm in Maoming, Guangdong, China, and the water samples were used to enrich mixed phages that can infect and lyse V. alginolyticus E06333 cells. The samples were centrifuged at 10,000 g for 10 min, and then the supernatants were filtered using a sterile 0.45-μm filter. The filtrates were mixed with the same volume of 2 × LB broth and then were added with 1% overnight culture of V. alginolyticus E06333 cells followed by incubation at 30°C for 12 h. The lytic culture samples (which remained transparent) were centrifuged, the supernatant was pipetted out, and was then filtered through a sterile 0.22-μm filter. The filtrate was used as enriched phages and was stored at 4°C.
Isolation of V. alginolyticus Phage-Resistant Mutant Strains From Phage-Resistant Colonies
The double-layer plates were used to isolate PRM colonies (Carlson, 2005). Briefly, each of 0.5-ml cultures of V. alginolyticus at OD600 nm = 0.6 was mixed with 0.5 ml of abovementioned enriched phage fluid at an approximative multiplicity of infection (MOI) of 30, and added into 4-ml LB broth (supplemented with 0.7% agar) preheated at 45°C, and then immediately poured onto the lower LB agar plate. The plates were incubated overnight at 30°C and then PRM colonies with different morphologies were picked and restreaked for the isolation of individual colonies more than four times.
Genotyping of V. alginolyticus Phage-Resistant Mutant Strains With Infrequent-Restriction-Site-PCR
The HhaI adaptors (AH1 and AH2) and the XbaI adaptors (XbaI-ad1 and AX2) were prepared as described previously (Ren et al., 2008). The Primer PN-X was constructed to complement XbaI-ad1. Sequences of the other four primers (PN-A, PN-T, PN-C, or PN-G) were identical to that of PN-X except that an additional base (A, T, C, or G, respectively) was placed at their 3’ ends. The digestion and ligation of template DNA were carried out as described previously (Mazurek et al., 1996). Less than 1 μg genomic DNA was digested with 10 U of HhaI (TaKaRa) and 10 U of XbaI (TaKaRa) in 1 × T + BSA buffer (final volume, 20 μl) for 2 h at 37°C. T4 DNA ligase (525 U), 10 × T4 buffer (3 μl), the XbaI adaptor (10 pmol), the HhaI adaptor (10 pmol), and water were added for a total volume of 20 μl. The mixture was incubated at 16°C for 1 h to ligate the adaptors to the digested DNA and then at 65°C for 20 min to inactivate T4 DNA ligase. The sample was digested with 5 U of XbaI and 5 U of HhaI at 37°C for 15 min to cleave any restriction sites reformed by ligation. Amplification was performed in the optimized conditions in which a 50 μl PCR mixture included 25 ul of PrimeSTAR Max Premix (2×), 5 μl of restricted–ligated DNA, 1 μl (10 μM) AH1 and 1 ul (10 μM) either PN-X, PN-A, PN-T, PN-C, or PN-G, then sterilized distilled water was added to 50 ul. Amplification condition consisted of an initial denaturation of 98°C for 3 min and 38 cycles of 10 s at 98°C, 5 s at 60°C, 30 s at 72°C and a final extension of 10 min at 72°C. All the enzymes were bought from TaKaRa Company (Japan). The sequences of primers and adaptors are shown in Supplementary Table 2. The PCR products were loaded into wells of a 8% polyacrylamide gel prepared from a 30% acrylamide–bisacrylamide (29:1) solution in 1 × TBE buffer (0.045 M Tris-borate, 0.001 M EDTA). After electrophoreses for 5 h at 200 V, the gel was stained with ethidium bromide (0.5 mg/ml) for 30–45 min, destained in water for 25 min, and photographed with UV illumination.
The Analysis on Biochemical Profiles of Selected V. alginolyticus Phage-Resistant Mutant Strains
The GEN III microplates (BIOLOG, United States) assaying the utilization of 71 carbon sources and the sensitivity to 23 chemical substrates were adopted to exhibit the potential shift of biochemical profile in PRM strains singled out by IRS-PCR. The cells of tested strains were suspended in inoculating fluids, and the aliquots of 100-μl suspended cells were added into the microplates. After 24–36 of hours incubation, the microplates were analyzed by a Biolog Microstation System (Biolog, United States).
Whole-Genome Sequencing of the Selected Phage-Resistant Mutant Strains
Genomic DNAs of PRM strains were sheared, then approximately 500-bp DNA fragments were selected. DNA fragments were end repaired and ligated with Illumina universal adapters. After adapter ligation, DNA fragments were further size-selected on an agarose gel and PCR-amplified for 6–8 cycles using the Illumina P1 and Index primer pair and Phusion® High-Fidelity PCR Master Mix (NEB, China). The final library was purified using Agencourt AMPure XP beads and quality-assessed by Agilent Bioanalyzer 2100 (DNA 7500 kit) to determine library quantity and fragment size distribution before sequencing. The genomes of the selected PRM strains were de novo sequenced using the Illumina HiSeq2500 platform (GENEWIZ, China), and the sequencing coverage depth ranged from 385× to 420×. Reads by quality control were assembled using velvet (v1.2.10) (Zerbino and Birney, 2008; Zerbino et al., 2009) and gap-filled with SSPACE (v3.0) (Boetzer et al., 2011) and GapFiller (v1–10) (Boetzer and Pirovano, 2012). Genome comparison between the PRM strains and ancestor V. alginolyticus E06333 (GenBank: CP071058-CP071059) was carried out by MAUVE (Darling et al., 2004) software. The coding genes were annotated by Glimmer (v3.02) (Delcher et al., 2007). GO (Gene Ontology) database (Harris et al., 2004) and KEGG (Kyoto Encyclopedia of Genes and Genomes) database (Kanehisa and Goto, 2000) were used to annotate the functions of genes and pathways, respectively. Non-coding RNA genes were predicted by Rfam database (Nawrock et al., 2015).
Evolved and Coevolved Cell Populations and Resequencing
Vibrio alginolyticus E06333 was cultured at the early exponential stage (OD600 nm = 0.6–1), 5 ml of culture fluid was collected and labeled as sample T0. At the same time, 40 replicates of 5 ml LB broth were inoculated with 60 μl of V. alginolyticus E06333 culture fluid. Among them, 20 replicates were inoculated with 20 μl of phage fluid at a multiplicity of infection (MOI) about 10 to serve as coevolved populations, wherein bacterial cells coevolved with enriched phages, and the other 20 replicates were inoculated with 20 μl of LB broth without the addition of phages to serve as evolved populations. Coevolved and evolved populations were incubated at 30°C in a shaker.
When OD600 nm of cultures in coevolved populations reached about 0.4, 1.0, and 1.5, five replicates in coevolved populations were mixed and labeled as samples VE1, VE2, and VE3, respectively, and at the same timepoints five replicates in evolved populations were mixed and labeled as samples VC1, VC2, and VC3, respectively. The cells of all the samples were centrifuged and collected for DNA extraction.
Genome resequencing of V. alginolyticus cell populations from all the samples was carried out by Illumina HiSeq × 10 and the sequencing coverage depth for each base site exceeded 230× (most exceed 400×) Pass filter data were removed adaptors and bases of low quality by Cutadapt (v1.9.1) to obtain clean data for continuous data analysis. Alignment software BWA (v0.7.12) (Li and Durbin, 2009) was used to map clean data to the reference genome of V. alginolyticus E06333. The detection of SNV (single base variation) and InDel (insert or deletion mutation) was performed using Samtools (v1.1) (Li et al., 2009) and the Unified Genotyper module of GATK (v3.4.6) software (DePristo et al., 2011). Annotation for SNV/InDel was performed by Annovar (Wang et al., 2010). Besides, InDels were detected and their frequencies were calculated no matter how many bases they consisted. Considering the accuracy of resequencing, only the mutation frequencies >10% were counted.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, CP071058; https://www.ncbi.nlm.nih.gov/genbank/, CP071059.
Author Contributions
All authors contributed to the writing of the manuscript. PL and YA conceptualized and designed the experiments. WZ and YL performed the experiments including IRS-PCR, Biolog assays, and DNA extraction and carried out bioinformatic analysis. ZL isolated PRM strains. XJ isolated the phages infecting V. alginolyticus. CH reviewed the manuscript and offered partial funding. PL and WZ wrote the manuscript.
Funding
This work was supported by Natural Science Foundation of Guangdong Province, China (2019A1515011492), National Key Research and Development Program: Blue Granary Scientific and Technological Innovation (2020YFD0901104), and Key Special Project for Introduced Talents Team of Southern Marine Science and Engineering Guangdong Laboratory (GML2019ZD0402).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.710262/full#supplementary-material
References
Abdelaziza, M., Ibrahemab, M. D., Ibrahimc, M. A., Abu-Elala, N. M., and Abdel-Moneama, D. A. (2017). Monitoring of different Vibrio species affecting marine fishes in Lake Qarun and Gulf of Suez: phenotypic and molecular characterization. Egypt. J. Aquat. Res. 43, 141–146. doi: 10.1016/j.ejar.2017.06.002
Ahluwalia, D., Bienstock, R. J., and Schaaper, R. M. (2012). Novel mutator mutants of E. coli nrdAB ribonucleotide reductase: insight into allosteric regulation and control of mutation rates. DNA Repair 11, 480–487. doi: 10.1016/j.dnarep.2012.02.001
Bachir, M., Allem, R., Benejat, L., Tifrit, A., Medjekane, M., Drici, A. E., et al. (2018). Molecular detection of mutations involved in Helicobacter pylori antibiotic resistance in Algeria. J. Antimicrob. Chemother. 73, 2034–2038. doi: 10.1093/jac/dky167
Bahat-Samet, E., Castro-Sowinski, S., and Okon, Y. (2004). Arabinose content of extracellular polysaccharide plays a role in cell aggregation of Azospirillum brasilense. FEMS Microbiol. Lett. 237, 195–203. doi: 10.1016/j.femsle.2004.06.036
Baldvinsson, S. B., Sorensen, M. C., Vegge, C. S., Clokie, M. R., and Brondsted, L. (2014). Campylobacter jejuni motility is required for infection of the flagellotropic bacteriophage F341. Appl. Environ. Microbiol. 80, 7096–7106. doi: 10.1128/AEM.02057-14
Batinovic, S., Wassef, F., Knowler, S. A., Rice, D. T. F., Stanton, C. R., Rose, J., et al. (2019). Bacteriophages in natural and artificial environments. Pathogens 8:100. doi: 10.3390/pathogens8030100
Bengoechea, J. A., Pinta, E., Salminen, T., Oertelt, C., Holst, O., Radziejewska-Lebrecht, J., et al. (2002). Functional characterization of Gne (UDP-N-acetylglucosamine-4-epimerase), Wzz (chain length determinant), and Wzy (O-antigen polymerase) of Yersinia enterocolitica serotype O:8. J. Bacteriol. 184, 4277–4287. doi: 10.1128/jb.184.15.4277-4287.2002
Bjedov, I. (2003). Stress-induced mutagenesis in bacteria. Science 300, 1404–1409. doi: 10.1126/science.1082240
Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D., and Pirovano, W. (2011). Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579. doi: 10.1093/bioinformatics/btq683
Boetzer, M., and Pirovano, W. (2012). Toward almost closed genomes with GapFiller. Genome Biol. 13:R56. doi: 10.1186/gb-2012-13-6-r56
Cai, R., Wang, G., Le, S., Wu, M., Cheng, M., Guo, Z., et al. (2019). Three capsular polysaccharide synthesis-related glucosyltransferases, GT-1, GT-2 and WcaJ, are associated with virulence and phage sensitivity of Klebsiella pneumoniae. Front. Microbiol. 10:1189. doi: 10.3389/fmicb.2019.01189
Carlson, K. (2005). “Working with bacteriophages: common techniques and methodological approaches” in Bacteriophage: biology and Applications. eds E. Kutter and A. Sulakvelidze (USA: CRC Press). 437–494.
Castillo, D., Christiansen, R. H., Dalsgaard, I., Madsen, L., and Middelboe, M. (2015). Bacteriophage resistance mechanisms in the fish pathogen Flavobacterium psychrophilum: linking genomic mutations to changes in bacterial virulence factors. Appl. Environ. Microbiol. 81, 1157–1167. doi: 10.1128/AEM.03699-14
Chen, J., Ye, L., Jin, L., Hu, X., Xu, P., Wang, X., et al. (2018). Application of next-generation sequencing to characterize novel mutations in clarithromycin-susceptible Helicobacter pylori strains with A2143G of 23S rRNA gene. Ann. Clin. Microbiol. Antimicrob. 17:10. doi: 10.1186/s12941-018-0259-8
Choi, Y., Shin, H., Lee, J. H., and Ryu, S. (2013). Identification and characterization of a novel flagellum-dependent Salmonella-infecting bacteriophage, iEPS5. Appl. Environ. Microbiol. 79, 4829–4837. doi: 10.1128/AEM.00706-13
Cotruvo, J. A., and Stubbe, J. (2008). NrdI, a flavodoxin involved in maintenance of the diferric-tyrosyl radical cofactor in Escherichia coli class Ib ribonucleotide reductase. Proc. Natl. Acad. Sci. U. S. A. 105, 14383–14388. doi: 10.1073/pnas.0807348105
Dang, H., and Lovell, C. R. (2015). Microbial surface colonization and biofilm development in marine environments. Microbiol. Mol. Biol. Rev. 80, 91–138. doi: 10.1128/MMBR.00037-15
Daniels, N. A., and Shafaie, A. (2000). A review of pathogenic Vibrio infections for clinicians. Infect. Med. 17, 665–685. doi: 10.1086/501713
Danovaro, R., Corinaldesi, C., Dell’anno, A., Fuhrman, J. A., Middelburg, J. J., Noble, R. T., et al. (2011). Marine viruses and global climate change. FEMS Microbiol. Rev. 35, 993–1034. doi: 10.1111/j.1574-6976.2010.00258.x
Darling, A. C., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Delcher, A. L., Bratke, K. A., Powers, E. C., and Salzberg, S. L. (2007). Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23, 673–679. doi: 10.1093/bioinformatics/btm009
DePristo, M. A., Banks, E., Poplin, R., Garimella, K. V., Maguire, J. R., Hartl, C., et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498. doi: 10.1038/ng.806
Engelhardt, D., and Shakhnovich, E. I. (2019). Mutation rate variability as a driving force in adaptive evolution. Phys. Rev. E. 99:022424. doi: 10.1101/354712
Fu, K., Li, J., Wang, Y., Liu, J., Yan, H., Shi, L., et al. (2016). An innovative method for rapid identification and detection of Vibrio alginolyticus in different infection models. Front. Microbiol. 7:651. doi: 10.3389/fmicb.2016.00651
Fuhrman, J. A. (1999). Marine viruses and their biogeochemical and ecological effects. Nature 399, 541–548. doi: 10.1038/21119
Galhardo, R. S., Hastings, P. J., and Rosenberg, S. M. (2007). Mutation as a stress response and the regulation of evolvability. Crit. Rev. Biochem. Mol. 42, 399–435. doi: 10.1080/10409230701648502
Gómez, P., and Buckling, A. (2011). Bacteria-phage antagonistic coevolution in soil. Science 332, 106–109. doi: 10.1126/science.1198767
Gómez, P., and Buckling, A. (2013). Coevolution with phages does not influence the evolution of bacterial mutation rates in soil. ISME J. 7, 2242–2244. doi: 10.1038/ismej.2013.105
Gonzalez, F., Helm, R. F., Broadway, K. M., and Scharf, B. E. (2018). More than rotating flagella: lipopolysaccharide as a secondary receptor for flagellotropic phage 7-7-1. J. Bacteriol. 200, e00363–18. doi: 10.1128/JB.00363-18
Ha, E., Chun, J., Kim, M., and Ryu, S. (2019). Capsular polysaccharide is a receptor of a Clostridium perfringens bacteriophage CPS1. Viruses 11:1002. doi: 10.3390/v11111002
Hampton, H. G., Watson, B. N. J., and Fineran, P. C. (2020). The arms race between bacteria and their phage foes. Nature 577, 327–336. doi: 10.1038/s41586-019-1894-8
Harris, M. A., Clark, J., Ireland, A., Lomax, J., Ashburner, M., Foulger, R., et al. (2004). The gene ontology (GO) database and informatics resource. Nucleic Acids Res. 32, D258–D261. doi: 10.1093/nar/gkh036
Kanehisa, M., and Goto, S. (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. doi: 10.1093/nar/28.1.27
Kashiwagi, A., and Yomo, T. (2011). Ongoing phenotypic and genomic changes in experimental coevolution of RNA bacteriophage Qbeta and Escherichia coli. PLoS Genet. 7:e1002188. doi: 10.1371/journal.pgen.1002188
Koskella, B., and Brockhurst, M. A. (2014). Bacteria-phage coevolution as a driver of ecological and evolutionary processes in microbial communities. FEMS Microbiol. Rev. 38, 916–931. doi: 10.1111/1574-6976.12072
Kropinski, A. M., Van Den Bossche, A., Lavigne, R., Noben, J. P., Babinger, P., and Schmitt, R. (2012). Genome and proteome analysis of 7-7-1, a flagellotropic phage infecting Agrobacterium sp H13-3. Virol. J. 9:102. doi: 10.1186/1743-422X-9-102
Laanto, E., Bamford, J. K. H., Laakso, J., and Sundberg, L. R. (2012). Phage-driven loss of virulence in a fish pathogenic bacterium. PLoS One 7:e53157. doi: 10.1371/journal.pone.0053157
Lauener, F. N., Imkamp, F., Lehours, P., Buissonniere, A., Benejat, L., and Zbinden, R. (2019). Genetic determinants and prediction of antibiotic resistance phenotypes in Helicobacter pylori. J. Clin. Med. 8:53. doi: 10.3390/jcm8010053
Lee, H., Doak, T. G., Popodi, E., Foster, P. L., and Tang, H. (2016). Insertion sequence-caused large-scale rearrangements in the genome of Escherichia coli. Nucleic Acids Res. 44, 7109–7119. doi: 10.1093/nar/gkw647
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map (SAM) format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Lis, L., and Connerton, I. F. (2016). The Minor Flagellin of Campylobacter jejuni (FlaB) Confers Defensive Properties against Bacteriophage Infection. Front. Microbiol. 7:1908. doi: 10.3389/fmicb.2016.01908
Luo, P., Jiang, H., Wang, Y., Su, T., Hu, C., Ren, C., et al. (2012). Prevalence of mobile genetic elements and transposase genes in Vibrio alginolyticus from the southern coastal region of China and their role in horizontal gene transfer. Int. Microbiol. 15, 201–210. doi: 10.2436/20.1501.01.172
Luo, P., Liu, Q. T., Deng, Y. Q., Tian, Y. S., Yun, L., and Hu, C. Q. (2018). Strand-specific RNA-Seq analysis provides first insight into transcriptome response of Vibrio alginolyticus to phage infection. Mar. Genom. 38, 5–8. doi: 10.1016/j.margen.2017.05.011
MacLean, R. C., Torres-Barceló, C., and Moxon, R. (2013). Evaluating evolutionary models of stress-induced mutagenesis in bacteria. Nat. Rev. Genet. 14, 221–227.
Markwitz, P., Olszak, T., Gula, G., Kowalska, M., Arabski, M., and Drulis-Kawa, Z. (2021). Emerging phage resistance in Pseudomonas aeruginosa PAO1 is accompanied by an enhanced heterogeneity and reduced virulence. Viruses 13:1332. doi: 10.3390/v13071332
Mazurek, G. H., Reddy, V., Marston, B. J., Haas, W. H., and Crawford, J. T. (1996). DNA fingerprinting by infrequent-restriction-site amplification. J. Clin. Microbiol. 34, 2386–2390. doi: 10.1128/JCM.34.10.2386-2390.1996
Nawrock, E. P., Burge, S. W., Bateman, A., Daub, J., Eberhardt, R. Y., Eddy, S. R., et al. (2015). Rfam 12.0: updates to the RNA families database. Nucleic Acids Res. 43, D130–D137. doi: 10.1093/nar/gku1063
Nesper, J., Lauriano, C. M., Klose, K. E., Kapfhammer, D., Kraiss, A., and Reidl, J. (2001). Characterization of Vibrio cholerae O1 El tor galU and galE mutants: influence on lipopolysaccharide structure, colonization, and biofilm formation. Infect. Immun. 69, 435–445. doi: 10.1128/IAI.69.1.435-445.2001
Paterson, S., Vogwill, T., Buckling, A., Benmayor, R., Spiers, A. J., Thomson, N. R., et al. (2010). Antagonistic coevolution accelerates molecular evolution. Nature 464, 275–278. doi: 10.1038/nature08798
Pruzzo, C., Huq, A., Colwell, R. R., and Donelli, G. (2006). “Pathogenic Vibrio Species in the marine and estuarine environment” in Oceans and Health: pathogens in the Marine Environment. eds S. Belkin and R. R. Colwell (Boston: Springer). 217–252.
Ren, C. H., Hu, C. Q., Luo, P., Chen, C., Jiang, X., and Wang, Q. B. (2008). Genotyping of Vibrio alginolyticus isolates from Daya Bay by infrequent-restriction-site PCR and pulsed-field gel electrophoresis. Mol. Cell Probes 22, 267–271. doi: 10.1016/j.mcp.2008.05.003
Safari, F., Sharifi, M., Farajnia, S., Akbari, B., Karimi Baba Ahmadi, M., Negahdaripour, M., et al. (2020). The interaction of phages and bacteria: the co-evolutionary arms race. Crit. Rev. Biotechnol. 40, 119–137. doi: 10.1080/07388551.2019.1674774
Scanlan, P. D., Hall, A. R., Blackshields, G., Friman, V. P., Davis, M. R. Jr., Goldberg, J. B., et al. (2015). Coevolution with bacteriophages drives genome-wide host evolution and constrains the acquisition of abiotic-beneficial mutations. Mol. Biol. Evol. 32, 1425–1435. doi: 10.1093/molbev/msv032
Schaper, S., Wendt, H., Bamberger, J., Sieber, V., Schmid, J., and Becker, A. (2019). A Bifunctional UDP-Sugar 4-Epimerase Supports Biosynthesis of Multiple Cell Surface Polysaccharides in Sinorhizobium meliloti. J. Bacteriol. 201, e00801–18.
Shabbir, M. A., Hao, H., Shabbir, M. Z., Wu, Q., Sattar, A., and Yuan, Z. (2016). Bacteria vs. bacteriophages: parallel evolution of immune arsenals. Front. Microbiol. 7:1292. doi: 10.3389/fmicb.2016.01292
Silva, J. B., Storms, Z., and Sauvageau, D. (2016). Host receptors for bacteriophage adsorption. FEMS Microbiol. Lett. 363:fnw002. doi: 10.1093/femsle/fnw002
Slifka, K. M. J., Newton, A. E., and Mahon, B. E. (2017). Vibrio alginolyticus infections in the USA, 1988-2012. Epidemiol. Infect. 145, 1491–1499. doi: 10.1017/S0950268817000140
Sumrall, E. T., Shen, Y., Keller, A. P., Rismondo, J., Pavlou, M., Eugster, M. R., et al. (2019). Phage resistance at the cost of virulence: listeria monocytogenes serovar 4b requires galactosylated teichoic acids for InlB-mediated invasion. PLoS Pathog. 15:e1008032. doi: 10.1371/journal.ppat.1008032
Swings, T., den Bergh, B. V., Wuyts, S., Oeye, E., Voordeckers, K., Verstrepen, K. J., et al. (2017). Adaptive tuning of mutation rates allows fast response to lethal stress in Escherichia coli. Elife 6:e22939. doi: 10.7554/eLife.22939
Thurber, R. V. (2009). Current insights into phage biodiversity and biogeography. Curr. Opin. Microbiol. 12, 582–587. doi: 10.1016/j.mib.2009.08.008
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38:e164. doi: 10.1093/nar/gkq603
Warwick-Dugdale, J., Buchholz, H. H., Allen, M. J., and Temperton, B. (2019). Host-hijacking and planktonic piracy: how phages command the microbial high seas. Virol. J. 16:15. doi: 10.1186/s12985-019-1120-1
Weitz, J. S., Poisot, T., Meyer, J. R., Flores, C. O., Valverde, S., Sullivan, M. B., et al. (2013). Phage-bacteria infection networks. Trends. Microbiol. 21, 82–91. doi: 10.1016/j.tim.2012.11.003
Woese, C. R. (1987). Bacterial evolution. Microbiol. Rev. 51, 221–271. doi: 10.1128/MMBR.51.2.221-271.1987
Woese, C. R. (1998). The universal ancestor. Proc. Natl. Acad. Sci. U. S. A. 95, 6854–6859. doi: 10.1073/pnas.95.12.6854
Woese, C. R., Kandler, O., and Wheelis, M. L. (1990). Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc. Natl. Acad. Sci. U. S. A. 87, 4576–4579. doi: 10.1073/pnas.87.12.4576
Yona, A. H., Bloom-Ackermann, Z., Frumkin, I., Hanson-Smith, V., Charpak-Amikam, Y., Feng, Q., et al. (2013). tRNA genes rapidly change in evolution to meet novel translational demands. Elife 2:e01339. doi: 10.7554/eLife.01339
Zerbino, D. R., and Birney, E. (2008). Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829. doi: 10.1101/gr.074492.107
Zerbino, D. R., Mcewen, G. K., Margulies, E. H., and Birney, E. (2009). Pebble and rock band: heuristic resolution of repeats and scaffolding in the velvet short-read de novo assembler. PLoS One 4:e8407. doi: 10.1371/journal.pone.0008407
Keywords: Vibrio alginolyticus, phages, genetic divergence, phage-resistant mutations, genomic changes
Citation: Zhou W, Li Y, Li Z, Ma B, Jiang X, Hu C, Ai Y and Luo P (2021) Genomic Changes and Genetic Divergence of Vibrio alginolyticus Under Phage Infection Stress Revealed by Whole-Genome Sequencing and Resequencing. Front. Microbiol. 12:710262. doi: 10.3389/fmicb.2021.710262
Received: 15 May 2021; Accepted: 14 September 2021;
Published: 04 October 2021.
Edited by:
Francesca Leoni, Istituto Sperimentale di Zooprofilassi dell’Umbria e delle Marche (IZSUM), ItalyReviewed by:
Michio Homma, Nagoya University, JapanEmmanouil Flemetakis, Agricultural University of Athens, Greece
Jesus L. Romalde, University of Santiago de Compostela, Spain
Copyright © 2021 Zhou, Li, Li, Ma, Jiang, Hu, Ai and Luo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongxing Ai, aiyx@jlu.edu.cn; Peng Luo, luopeng@scsio.ac.cn
†These authors have contributed equally to this work