 Angelina T. Regua
Angelina T. Regua Mariana Najjar
Mariana Najjar Hui-Wen Lo
Hui-Wen Lo- 1Department of Cancer Biology, Wake Forest School of Medicine, Winston-Salem, NC, United States
- 2Wake Forest Baptist Comprehensive Cancer Center, Winston-Salem, NC, United States
Rearranged during transfection (RET) receptor tyrosine kinase was first identified over thirty years ago as a novel transforming gene. Since its discovery and subsequent pathway characterization, RET alterations have been identified in numerous cancer types and are most prevalent in thyroid carcinomas and non-small cell lung cancer (NSCLC). In other tumor types such as breast cancer and salivary gland carcinomas, RET alterations can be found at lower frequencies. Aberrant RET activity is associated with poor prognosis of thyroid and lung carcinoma patients, and is strongly correlated with increased risk of distant metastases. RET aberrations encompass a variety of genomic or proteomic alterations, most of which confer constitutive activation of RET. Activating RET alterations, such as point mutations or gene fusions, enhance activity of signaling pathways downstream of RET, namely PI3K/AKT, RAS/RAF, MAPK, and PLCγ pathways, to promote cell proliferation, growth, and survival. Given the important role that mutant RET plays in metastatic cancers, significant efforts have been made in developing inhibitors against RET kinase activity. These efforts have led to FDA approval of Selpercatinib and Pralsetinib for NSCLC, as well as, additional selective RET inhibitors in preclinical and clinical testing. This review covers the current biological understanding of RET signaling, the impact of RET hyperactivity on tumor progression in multiple tumor types, and RET inhibitors with promising preclinical and clinical efficacy.
Introduction
Since the discovery of the first tyrosine kinase in the early 1980s, identification of aberrant tyrosine kinase activity in cancers have made them optimal targets for therapeutic intervention in malignant diseases. Rearranged during transfection (RET) is one such receptor tyrosine kinase first discovered in 1985 as a novel transforming gene in NIH3T3 cells upon transfection with DNA isolated from human lymphoma cells (1), resulting in their oncogenic transformation. Using Southern blot analysis, Takahashi and colleagues determined that the putative transforming gene was a product of two separate DNA sequences that were >25kb apart on the human genome, and therefore was the result of genetic recombination that produced a transcriptionally functional unit. This novel transforming gene was subsequently named for the process through which it was discovered.
Following its discovery, RET has been identified to play critical roles in numerous developmental processes, namely embryonic development of the kidney (2, 3), as well as, the enteric nervous system (2, 4). Accordingly, RET alterations are implicated in numerous disease phenotypes such as Hirschsprung’s disease and cancer (5, 6). Additionally, studies performed over the last three decades have identified numerous alterations in RET that confer constitutive activation of its kinase activity, which is considered a causative factor in many cancer subtypes (7–10). Given that aberrant RET activity is an oncogenic driver in numerous cancers, RET is considered an optimal target for therapeutic intervention, first with multi-kinase inhibitors which were soon followed by the first generation of selective RET inhibitors. This review will summarize the current understanding of canonical RET signaling and its biology, the role of RET signaling in critical developmental processes, and the oncogenic role of aberrant RET activity in multiple cancer types. This review will also discuss the alterations in RET that result in its constitutive and oncogenic activation, the suboptimal clinical benefits of repurposed multi-kinase inhibitors for RET inhibition, and the compounds developed to selectively target RET kinase activity.
Canonical RET signaling
RET is a transmembrane receptor tyrosine kinase with a unique extracellular domain that contains four cadherin-like domains and 16 cysteine residues within a 120 amino acid sequence (11). RET activation occurs upon fulfillment of multiple events: binding of Ca2+ ions to its cadherin-like domains, binding of GDNF family ligands (GFLs; GDNF, ARTN, NRTN, PSPN) to glycosyl phosphatidylinositol-anchored co-receptor GDNF family receptor-α (GFRα1-4), resulting in recruitment of two RET receptors into close proximity and allowing for dimerization and autophosphorylation of several tyrosine residues on the cytoplasmic tails (12). Phosphorylation of these cytoplasmic tyrosine residues of RET allows for recruitment and binding of several adaptor proteins necessary for signal propagation of external stimuli and subsequent activation of downstream signaling cascades, namely PI3K/AKT, RAS/RAF/MEK/ERK, JAK2/STAT3, and PLCγ pathways (Figure 1). RET-Y687 recruits and binds SHP2 phosphatase, allowing for activation of PI3K/AKT which, in turn, promotes cell survival (13). RET-Y752 and Y928 are critical docking sites for Signal Transducer and Activator of Transcription 3 (STAT3), allowing for STAT3 phosphorylation, activation, and nuclear translocation critical for transcription of STAT3 target genes (14). Phosphorylation of RET-Y905 stabilizes the active conformation of RET and is critical for binding of adaptor proteins Grb7/10 (9, 15), whereas RET-Y981 is critical for binding and activation of Src kinase (16). Phospholipase C-γ binds to phospho-RET (Y1015) which, in turn, activates protein kinase C (PKC) pathway (17). RET-Y1062 phosphorylation recruits a number of adaptor proteins imperative for activation of PI3K/AKT, RAS/RAF/MEK/ERK, and MAPK pathways (18). Finally, Grb2 binds to phospho-RET (Y1096) and acts a secondary interaction through which RET can activate RAS/RAF/MEK/ERK pathway and promote cell proliferation and differentiation (19, 20).
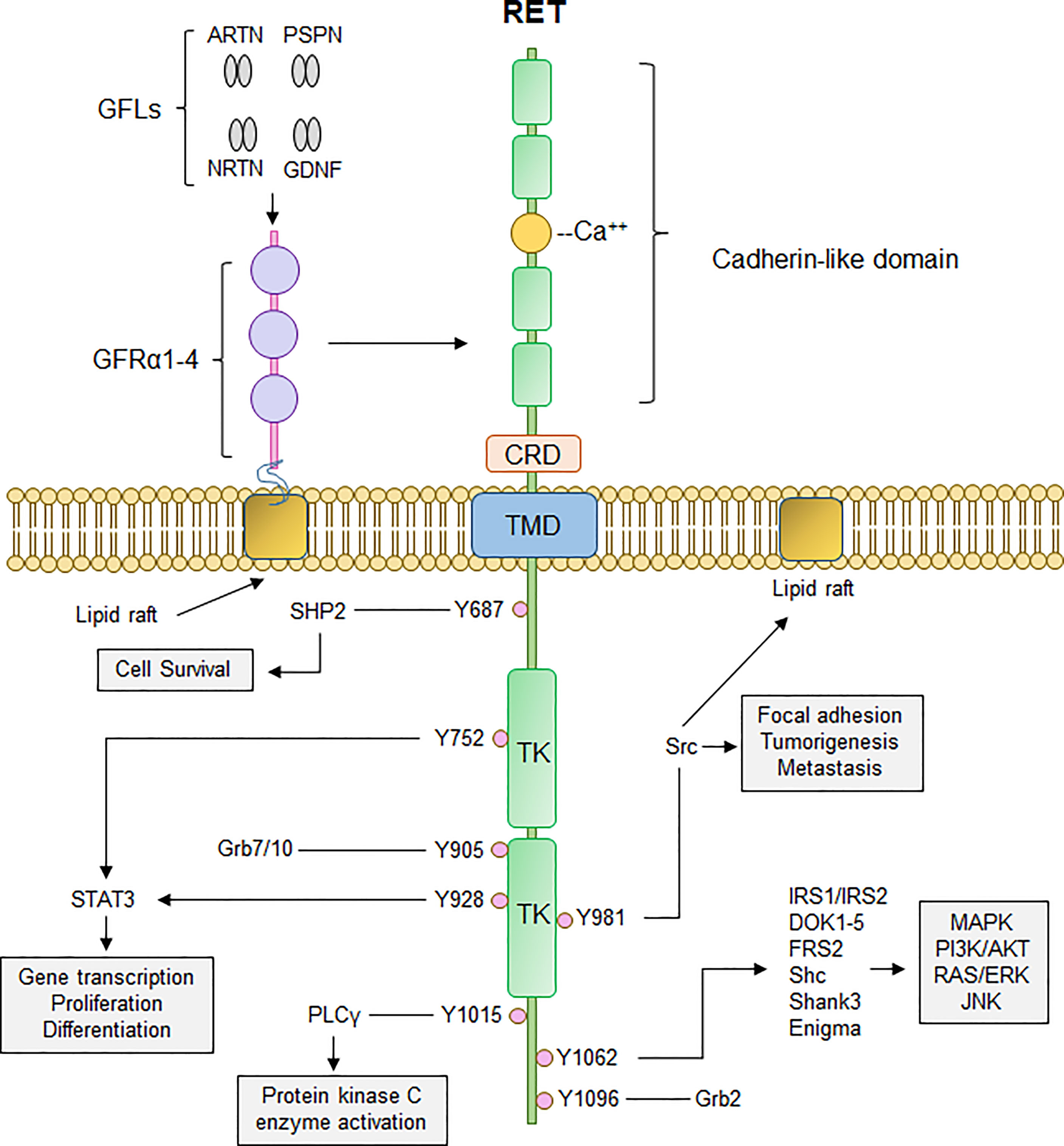
Figure 1 Canonical RET signaling. RET activation occurs upon fulfillment of multiple steps. Binding of GDNF-family ligands (GFLs) such as Artemin, Neurturin, Persephin, (ARTN, NRTN, PSPN, respectively), and GDNF to co-receptor GFRα1-4, concurrently with binding of calcium ions to the calcium binding domain, induces recruitment of RET, forming RET-GFRα complex. Formation of RET-GFRα complex brings two RET monomers in close proximity to induce homodimerization and crossphosphorylation of key RET tyrosine residues that recruit adaptor proteins important for propagation of RET signaling, such as PI3K/AKT, MAPK, PLCγ, and RAS/RAF/ERK. Thus, activation of RET signaling ultimately promotes cell proliferation, growth, and survival through activation of multiple downstream signaling cascades. CRD, cysteine-rich domain; TMD, transmembrane domain; TK, tyrosine kinase domain.
Roles of RET in development of the embryonic kidney, autonomic and enteric nervous systems, and spermatogenesis
In non-tumor tissues, RET signaling is critical for normal embryonic kidney development. Pachnis et al. demonstrated that c-ret is expressed in migrating neural crest cells within the cranial region of E8.5-9.5 mouse embryos, as well as in the cranial ganglia (E10.5) in the autonomic ganglia within the embryonic trunk region of the peripheral nervous system, and the myenteric ganglia of the gut at E13.5 (21). RET expression is also found in the developing Wolffian duct (WD), whereas the GDNF ligand is expressed in the metanephric mesenchyme (MM) adjacent to the WD. RET-expressing cells in the ureteric bud (UB) within the WD respond to GDNF abundance within the MM, promotes invagination of the UB into the MM where it can initiate branching to develop the embryonic kidney (3). Ivanchuk et al. found that RET mRNA is highly expressed in 14-week-old human embryos, however RET expression appears to decrease with gestational age and into adulthood, suggesting that RET is only required for the embryonic development of human kidneys (22). Indeed, Schuchardt et al. determined that, while mice lacking c-ret expression survived until birth, they expire ~24 hours after birth, likely due to severe renal agenesis or dysgenesis, or defective digestive tract function (discussed below) (2). These findings were corroborated by reports by Enomoto et al. (23) and Jain et al. (5) in which loss of the co-receptor GFRα1 or expression of a dominant-negative mutant RET also conferred deficits in embryonic renal development, resulting in renal agenesis and postnatal lethality. Interestingly, c-ret expression is temporally and spatially controlled: the adult salivary gland and adult brain expressed the highest levels of c-ret, but with lower c-ret expression in the heart and spleen, and was largely absent in the adult kidney, liver, lung, ovaries, or testes (21). Thus, RET expression is temporally and spatially regulated within embryonic tissues, suggesting that RET contributes to specific stages in embryonic development.
In addition to its role in embryonic kidney development, RET expression is also imperative for development, maturation, and maintenance of the autonomic and enteric nervous systems (ENS), which underlies proper gastrointestinal (GI) function. Early studies by Enomoto et al. found that RET expression is essential for development and maturation of parasympathetic arm of the murine autonomous nervous system (24), as well as axonal growth and migration of sympathetic neurons (4). Given that mice lacking c-ret expression were found to have reduced numbers, or complete absence, of enteric neurons throughout the digestive tract that can result in neonatal lethality (2), RET expression and activity appears to be critical for embryonic development of the GI tract. While RET expression is abundant in the developing murine intestinal epithelium, RET expression is maintained in a subset of enteroendocrine and enterochromaffin cells in adult tissues (25), which suggests a need for RET signaling in GI tract motility and/or secretion throughout adulthood. The GI phenotypes associated with loss of RET expression or activity are now characteristic of Hirschsprung’s disease (HD or HSCR), a congenital condition in which enteric nerves are missing in portions of the digestive tract (26). Since RET expression is critical for migration of neural crest cells and enteric neurons into the GI tract, it is unsurprising that loss of murine c-ret expression or activity induces GI phenotypes associated with HD, such as intestinal aganglionosis, and is detected in 50% of familial HD and up to 20% of spontaneous HD cases (6).
Several studies also implicated GDNF-RET signaling in spermatogenesis and determination of spermatogonial stem cell (SSC) fate. Spermatogonial maturation is dependent on renewal of SSCs. GDNF ligand is secreted by Sertoli cells (27), is essential for maintenance of SSCs in vitro (28), and SSC cell fate maturation in vivo (29). In addition to defective GI and renal development, Jain and colleagues reported defective spermatogenesis in mice expressing dominant-negative human RET (5). These findings were substantiated by Naughton et al. (30), in which they demonstrated that RET and GFRα1 are co-expressed in germ cells and SSCs during early spermatogenesis, and that the GDNF-RET-GFRα1 signaling axis is required for complete spermatogenesis and SSC self-renewal in a testis-autonomous transplantation mouse model, though this does not affect Sertoli cells (30).
Aberrant RET signaling in tumorigenesis
The proto-oncogenic role of RET has been identified and characterized in numerous cancer types, such as, thyroid, lung, and breast cancers. RET alterations occur in less than 5% of all cancer patients (31, 32), however activating RET alterations, such as genetic amplification and chromosomal rearrangements, are found in increasingly higher proportions of specific cancer types. This section of the review will discuss oncogenic RET alterations and their implications in RET-altered cancers.
Oncogenic RET alterations and RET fusions
RET alterations were first identified in the late 1980s with the detection of an oncogenic RET fusion in papillary thyroid carcinomas (33). Since then, additional RET rearrangements have been identified by multiple groups in a number of solid tumors (34). Oncogenic gene fusions arise from juxtaposition of two otherwise independent genes, resulting from structural rearrangements such as inversions or translocations, transcriptional reading of adjacent genes (35–37), or splicing of pre-mRNA sequences (38, 39). Most RET fusions lack the transmembrane domain, giving rise to chimeric, cytosolic proteins that exert their oncogenic influence through constitutive activation of RET kinase domain (Figure 2A). Since discovery of the first RET fusion in thyroid cancers, at least 13 fusion partners have been identified in papillary thyroid cancers, the most common of which are coiled-coil domain containing 6 (CCDC6-RET) and nuclear receptor co-activator 4 (NCOA4)-RET (40). Similarly, at least 45 RET fusion partners have been identified in lung cancers (34), with the most common being kinesin family member 5B (KIF5B)-RET (70-90%) and CCDC6-RET (10-25%). While RET fusions are detected in other cancers, such as breast cancer and salivary gland carcinomas (discussed later), RET fusions occur in these cancer types at much lower frequencies (0.16% and 3.2%, respectively) (41). Thus, RET fusions present as a clinical and therapeutic challenge in multiple tumor types (Table 1).
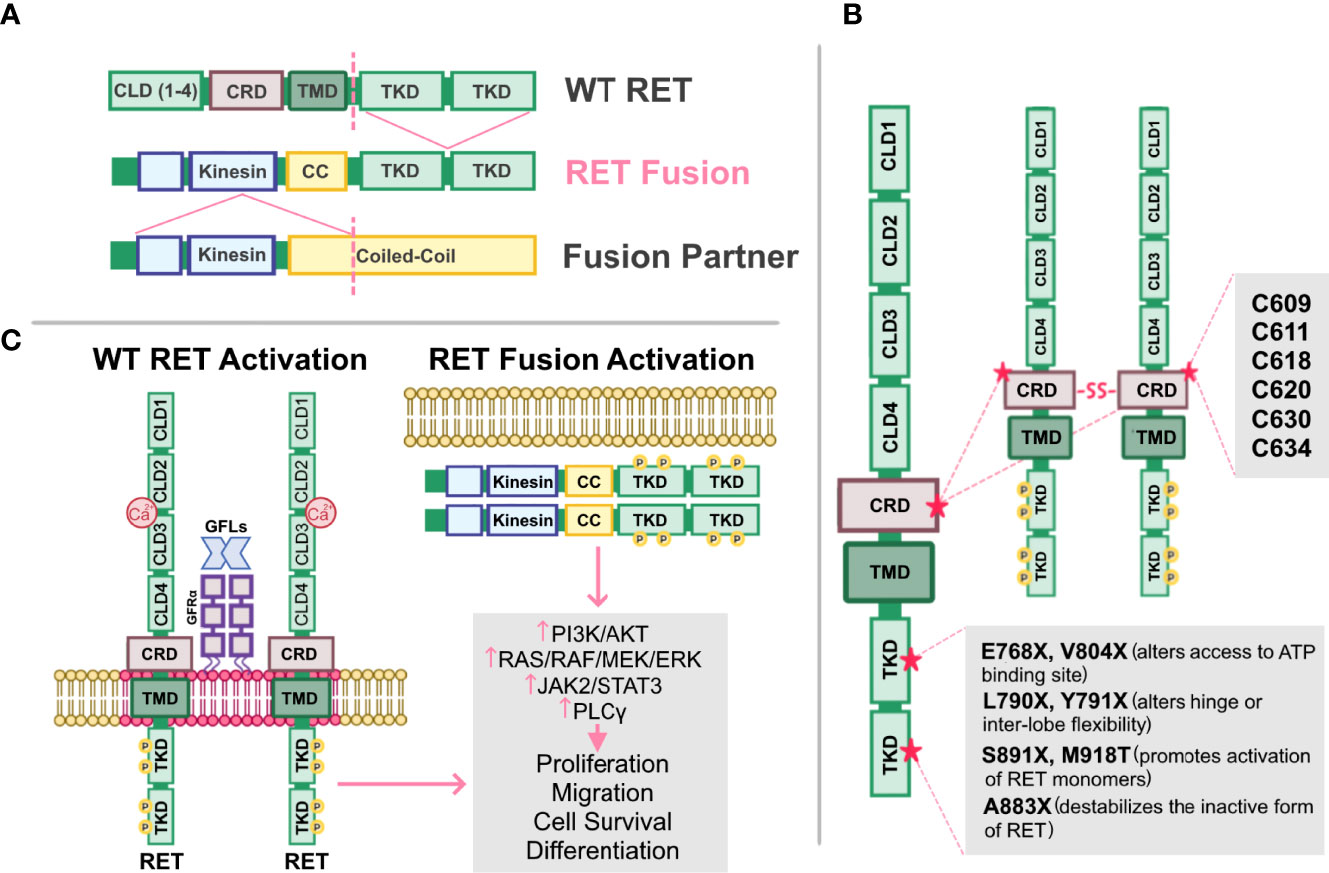
Figure 2 Altered RET and their mechanisms of activation. (A) Schematic representation of the fusion between the tyrosine kinase domain of wild-type (WT) RET receptor, and the kinesin and coiled-coil (CC) domain of a fusion partner. Dashed lines represent fusion sites. (B) Cysteine mutations in RET cysteine-rich domain (CRD) promote formation of intermolecular disulfide bridges, leading to constitutive dimerization and activation of RET that is GDNF-independent. Mutations in RET tyrosine kinase domains (TKD) can elicit steric conformations that regulate access to the ATP binding pocket of RET (E768X, V804X), alter hinge or inter-lobe flexibility (L790X, Y791X), promote activation of RET monomers (S891X, M918T), or destabilize the inactive form of RET (A883X), all of which confer constitutive RET activation albeit with varying activities. (C) Activation of either WT RET or oncogenic RET fusions can promote activation of downstream pathways, PI3K/AKT, RAS/RAF/MEK/ERK, JAK2/STAT3, and PLCγ resulting in enhanced proliferation, migration, cell survival and differentiation, ultimately promoting neoplastic growth and tumorigenesis.
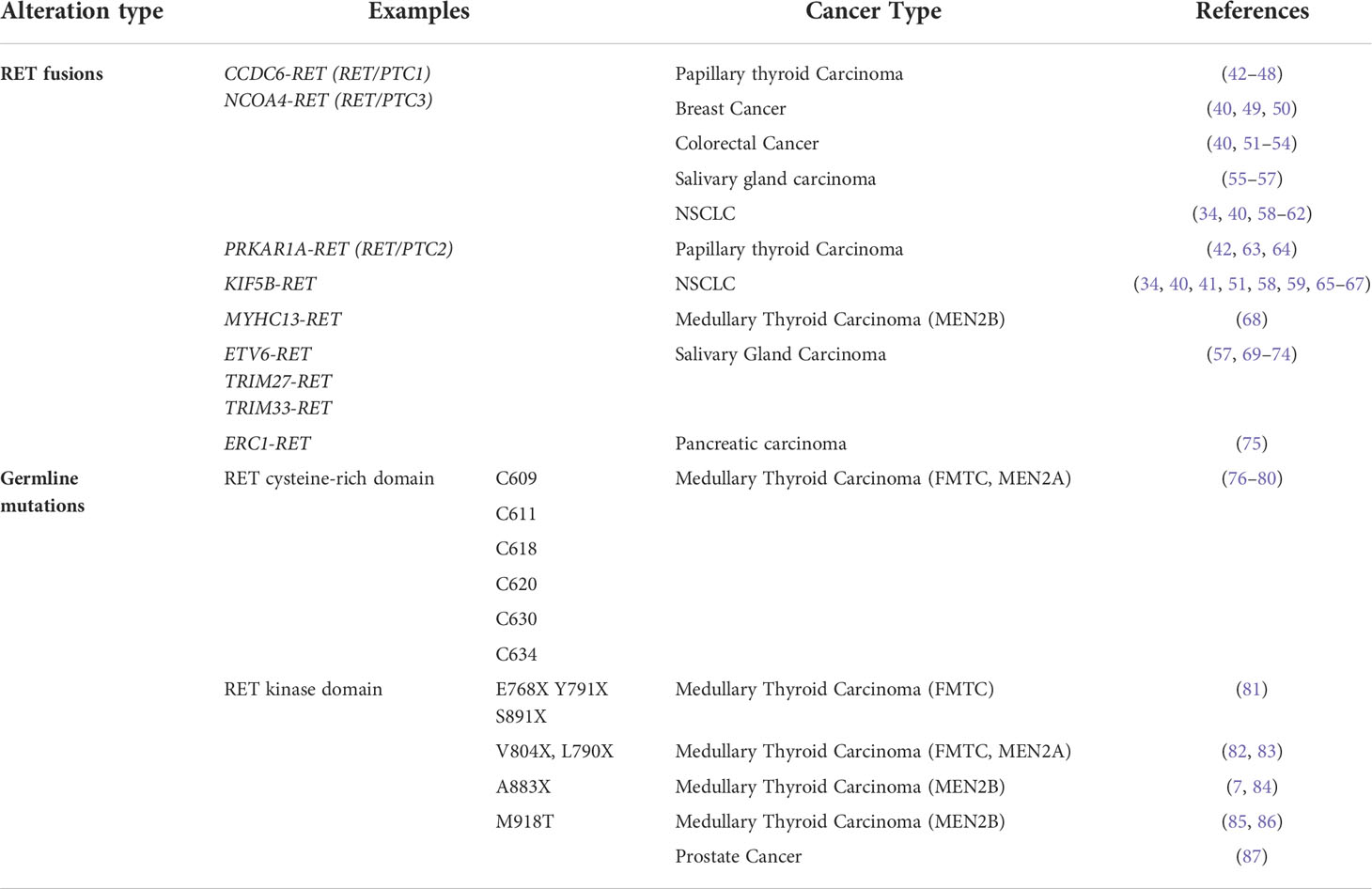
Table 1 Oncogenic RET alterations confer constitutive activation in multiple cancers.
In addition to oncogenic fusions, germline mutations, such as activating point mutations, in RET are also associated with neoplasia and tumorigenesis, namely in multiple endocrine neoplasia type 2 (MEN2; described in 4.2). For example, mutations in the cysteine residues (C609, C611, C618, C620, C630, C634) within the extracellular domain of RET promotes formation of intermolecular disulfide bridges, resulting in constitutive dimerization and activation of RET that is GDNF-independent (Figure 2B) (76). Additionally, mutations in the RET tyrosine kinase domain elicits steric conformations that regulate access to the ATP binding pocket of RET (E768X, V804X) (81–83, 88), alters hinge or inter-lobe flexibility (L790X, Y791X), promotes activation of RET monomers (S891X, M918T) (85), or destabilizes the inactive form of RET (A883X) (7, 84, 89), all of which confer constitutive activation albeit with varying activities (90) (Table 1). Taken together, oncogenic RET alterations can promote constitutive activation of RET signaling, driving tumorigenesis in these affected tissues (Figure 2C).
RET in papillary thyroid carcinomas
Papillary thyroid carcinomas (PTCs) are the most common form of differentiated thyroid cancers, the most common form of radiation exposure-induced thyroid neoplasm, and represents up to 85% of all thyroid cancer cases (91). PTC occurrence was initially associated with activating mutations of RAS and subsequent activation of the RAS/MAPK/ERK pathway (92–94). This paradigm was challenged when Fusco et al. (95) identified oncogenic RET as an unknown transforming oncogene in metastatic PTC and, while the oncogene was analyzed for homology with known kinase genes, including ret, the identity of this novel gene remained unclear and was therefore named ‘PTC’. A follow-up study by the same group determined that PTC is a rearranged form of RET, and rearranged RET DNA sequences were readily detectable in PTC-positive papillary thyroid tumors (33). These findings were corroborated by a study by Ishizaka and colleagues (96), in which they identified aberrant RET transcripts in PTC tumor samples. Further analysis by Lanzi et al. identified two additional RET rearrangements in PTC, named RET/PTC1 and RET/PTC2, which were constitutively activated, tyrosine-phosphorylated, and apparently not bound to the cell membrane (42). Santoro et al. identified a third RET rearrangement, which they named RET/PTC3 (43). In RET/PTC1, RET is fused with a then-unknown gene named H4 (D10S170), which is now known as CCDC6 (33, 97, 98), whereas RET/PTC2 and RET/PTC3 are products of RET kinase domain fusing to the N-terminus of RIα (now known as PRKAR1A) (63, 64) and ELE1 (now known as NCOA4) (43, 99, 100) genes, respectively (Figure 3).
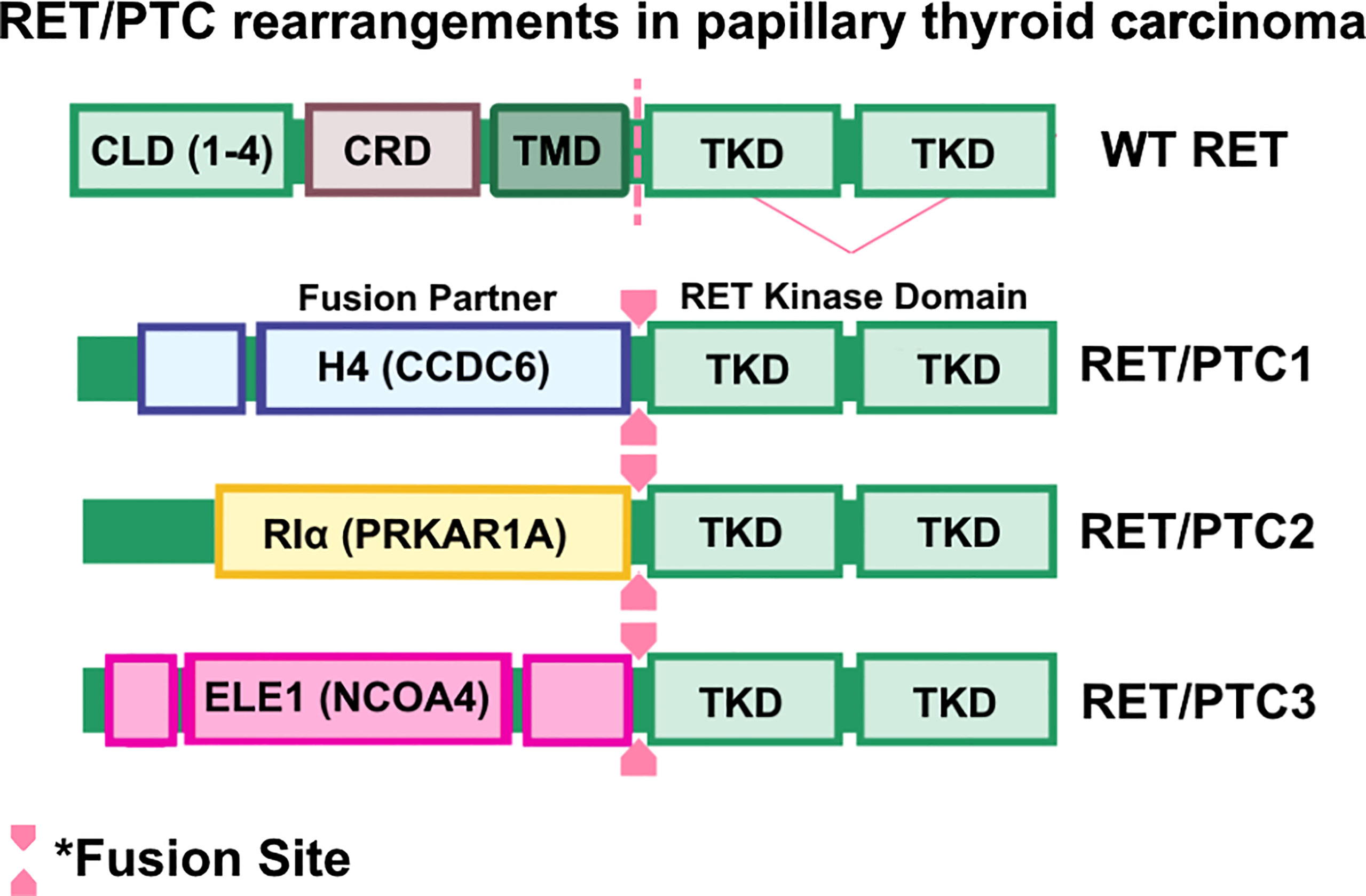
Figure 3 RET/PTC rearrangements in papillary thyroid carcinomas. Wild-type RET and RET rearrangements in papillary thyroid carcinoma (PTC). Coiled-Coil domain containing 6 (CCDC6)-RET, Protein Kinase CAMP-Dependent Type I Regulatory Subunit Alpha (PRKAR1A)-RET, and nuclear receptor coactivator 4 (NCOA4)-RET are frequently found in PTC cases. Oncogenic RET fusions lack the transmembrane domain (TMD) and result in chimeric, cytosolic proteins that are constitutively activated.
While RET/PTC1-3 are considered unique forms of RET/PTC fusions, they share common features such as ubiquitous expression of the 5’ fusion partner (44, 99), altered subcellular localization of RET kinase activity (101), and constitutive activation of RET kinase arising from ligand-independent dimerization of the RET (63, 100, 101). Transgenic overexpression of RET/PTC1 is indeed oncogenic and induces spontaneous and metastatic murine thyroid carcinomas that are histologically similar to human PTC tumors (45, 102), though whether RET/PTC1 overexpression is an initiating factor in human thyroid carcinogenesis remained unclear. Analysis of 26 occult thyroid carcinomas, which are small papillary carcinomas with clinically apparent node metastases but are often microscopic or overlooked by ultrasonograms, determined that RET/PTC activation is found in 42% (11/26) of samples analyzed, suggesting that RET/PTC activity contributes to early papillary thyroid carcinogenesis (103). Importantly, additional studies determined that RET rearrangements are frequently found in individuals exposed to radiation (104), such as the children who experienced the Chernobyl nuclear plant incident (105–107), suggesting an association between radiation and emergence of RET rearrangements. To date, there are at least 12 oncogenic RET fusions identified in PTC (108), suggesting that RET alterations contribute significantly to initiation and progression of PTC. Further characterization of PTC tumors determined that RET proto-oncogene expression is primarily found in PTC subtype of thyroid cancers (109). It is important to note, however, that clonal RET/PTC rearrangements occur in 10-20% of PTC patients and may be induced by varying causative factors (110). For example, RET/PTC rearrangements are found in up to 70% of pediatric PTC cases in areas of high radioisotope contamination (46). However, Rhoden and colleagues showed that RET/PTC rearrangements are found in over 25-30% of spontaneous PTC (47, 111). Additionally, a comparative study by Zhu and colleagues determined that the method by which RET/PTC rearrangements are assessed may alter the reported frequency within the sample cohort (46). Thus, considerations must be made when assessing frequency of RET/PTC rearrangements in PTC patient samples.
RET in medullary thyroid carcinomas
In contrast to PTC, medullary thyroid carcinomas (MTCs) are a rare type of thyroid tumor that arises in parafollicular C cells and represent 2-4% of malignant thyroid neoplasms (91, 112). While the majority of MTC tumors are spontaneous, up to 30% of MTCs are hereditary. MTCs are classified into one of the following major clinical subtypes: classical MEN2A, MEN2A with cutaneous lichen amyloidosis (CLA), MEN2A with Hirschsprung disease, FMTC, and MEN2B. Familial MTC (FMTC) subtype is characterized by occurrence of only MTC, which has relatively delayed latency and is clinically less aggressive compared to MEN2A and MEN2B subtypes (91). MEN2A was first characterized in 1961 by J.H. Sipple after identifying an association between pheochromocytoma and occurrence of a thyroid carcinoma (113). MEN2A subtype represents 70-80% of hereditary MTCs and is characterized by co-occurrence of MTC, pheochromocytoma, and/or primary hyperparathyroidism (may include Hirschsprung’s disease or CLA) (114, 115). MEN2B subtype is characterized by co-occurrence of aggressive MTC and pheochromocytoma, along with intestinal tumors, neuromas, and Marfanoid body habitus (114). The major MTC variants are summarized in Table 2.

Table 2 Major medullary thyroid carcinoma (MTC) variants.
RET rearrangements were first associated with MTC tumors when Santoro et al. detected RET fusion products in pheochromocytomas and MTC samples (123). The following year, a study by Yamamoto and colleagues demonstrated that the mapped chromosomal location of RET is in close proximity with the MEN2A gene locus (116), suggesting a potential genetic correlation between RET expression and MEN2A occurrence. However, Donis-Keller and colleagues proposed a strict association between exon 7/8-specific RET mutations and occurrence of MEN2A and FMTC since these mutations were strictly detected in families with MEN2A and FMTC, not MEN2B (77). These findings were supported by Mulligan et. al, in which they identified germline missense mutations within the RET proto-oncogene in 87% (20/23 cases) of the MEN2A families but not in families without MEN2A (8). Additionally, the Mulligan et al. found that, of the 20 families with germline missense RET mutations, 19 of these families shared a mutation in one of the cysteine residues in the RET extracellular domain, suggesting that mutation of this cysteine residue may predispose patients to MEN2A or FMTC (8). A follow-up study analyzed 118 unrelated families with MEN2A, MEN2B, or FMTC and determined that over 95% of MEN2A families and 86% of FMTC families possessed RET-C634 mutation, which is in the immediate proximity of the transmembrane domain (78). In agreement with their earlier study (8), Mulligan and colleagues (1994) did not detect the RET-C634 mutation in the MEN2B families assessed in this study, however the frequency of RET-C634 mutation was significantly higher in MEN2A families than those with FMTC (84% vs. 50%, respectively) (78). As MEN2A patients develop hyperparathyroidism and/or pheochromocytoma that is not typically detected in FMTC, these findings suggest that the RET-C634 mutation is correlated with increased predisposition to develop MEN2A due to enhanced RET activity.
While MEN2A cases frequently acquire RET-C634 mutations, MEN2B families also share common RET mutations. RET-M918T was first detected in 1994 upon DNA sequence analysis of 9 unrelated MEN2B patients (124), which was corroborated by a separate study that detected the mutation in 34 unrelated MEN2B patients (89). RET-M918T mutation is detected in 95% of MEN2B cases (122) and elicits oncogenic signaling by stabilizing monomeric RET, enhancing binding with ATP, and is activated in absence of GDNF ligand (9, 85, 125). Additionally, RET-A883F (7, 84, 86) is a strongly activating RET mutation that is hypothesized to destabilize the inactive conformation of RET by altering the local conformation of the protein (10).
Until recently, only activating RET mutations were cited as oncogenic events in MTC. However, a 2015 case report identified an MTC patient with Myosin Heavy Chain 13 (MYHC13)-RET fusion (68), suggesting that oncogenic RET rearrangements also occur in MEN2B albeit at lower frequencies. Taken together, MTC subtypes present with unique, activating RET mutations that induce thyroid tumorigenesis.
RET in non-small cell lung cancer
Lung cancers are the second most-commonly diagnosed cancer and the leading cause of cancer-related deaths in American men and women (126). Non-small cell lung cancer (NSCLC) constitutes up to 80-85% of all lung cancers and can further categorized into one of three major subtypes: adenocarcinoma, squamous cell carcinoma, and large cell carcinoma (127). NSCLC subtypes are frequently grouped together due to the similarities in treatment options, namely surgical resection, neoadjuvant or adjuvant chemotherapies, and immunotherapy (for a full review of NSCLC epidemiology and treatments, see Duma et. al) (128).
RET rearrangements occur in 1-2% of NSCLC cases (58, 59, 65). Since identification of the first RET fusion (KIF5B-RET) in lung cancer (51, 65–67), significant efforts have been made to identify additional novel RET fusions in NSCLC. While RET fusions occur in 1-2% of NSCLC cases, there are at least 48 unique RET fusion partners identified in NSCLC, which have been catalogued by Ou and Zhou (34), with the most common being KIF5B-RET and CCDC6-RET. While most NSCLC cases do not present with additional driver mutations, analysis of over 4,871 samples by Kato and colleagues identified co-occurrence of RET fusions with other genetic alterations, such as PI3K-associated genes or MAPK effector genes, in up to 82% (72/88) of NSCLC patients (41). Thus, the clinical and pathological features of RET fusion-positive NSCLC may differ from those observed in NSCLC cases driven by alternative oncogenic aberrations. Indeed, an analysis by Wang and colleagues determined that RET fusion-positive lung carcinomas had more poorly differentiated tumors compared to those with ALK or EGFR alterations, indicating that RET fusions define a unique molecular and clinicopathological subtype of NSCLC (58).
Distant metastases are of a particular concern in NSCLC, with 30-40% of cases metastasizing to the bone, lungs, brain, and adrenal glands (129). At the time of diagnosis, 10% of NSCLC patients present with brain metastases, with 30% of NSCLC patients developing brain metastases over the course of their disease (130). Baseline incidence of brain metastases in RET fusion-positive NSCLC is 27% and is independent of age, smoking status, or 5’ fusion partner status, and this proportion increases to 49% throughout the lifetime of disease (131). Thus, identifying molecular targets in brain-metastatic NSCLC and developing therapeutic compounds with blood-brain-barrier (BBB) permeability is a topic of recent investigation. Multi-kinase inhibitors (MKIs) targeting commonly altered oncogenic drivers, such as EGFR, MET, KIT, and VEGFR2, have shown moderate selectivity against altered RET in NSCLC and were thus repurposed as RET inhibitors (discussed below) (132–136), with many of these inhibitors displaying intracranial activity against brain metastases (137–139). However, a multi-institutional study by Drilon and colleagues determined that 25% of RET fusion-positive stage IV lung cancer patients presented with brain metastases at diagnosis, and less than 20% experienced intracranial response upon treatment with RET-targeting MKIs, suggesting that MKI inhibitors are inadequate for treatment of brain-metastatic lung cancers (140).
RET alterations in other tumor types
While aberrant RET activity is primarily associated with thyroid carcinomas and NSCLC, abnormal RET signaling is also found in other tumor types, albeit at lower frequencies. This section of the review will briefly summarize the role of RET signaling in other solid tumor types and the implications of RET inhibition in these cancers.
Aberrant RET is actionable in breast cancers
RET is altered in ~1.2% of breast cancer cases, of which 66% are RET amplifications and 7% are activating fusions (49). As with other solid tumors types discussed here so far, the most common RET fusions detected in breast cancer are CCDC6-RET and NCOA4-RET, and their constitutive activation induces enhanced cell proliferation and survival that ultimately results in tumorigenesis (58, 141).
Early investigations of RET in breast cancer were primarily focused on estrogen receptor-alpha-positive (ER+) cases due to the prevalent co-overexpression of RET and its co-receptor, GFRα1, in a subset of ER+ breast cancers (142). Additionally, co-overexpression of RET-GFRα1 was inversely correlated with expression of basal markers and positively correlated with enhanced cell proliferation and survival of ER+ cells (143), suggesting that RET pathway activity may be a novel target in this breast cancer subtype. A study by Gattelli and colleagues determined that RET activation promotes growth, proliferation, and migration of ER+ breast cancer cells in vitro and in vivo (144), strongly suggesting that RET is actionable in ER+ breast cancers. Standard-of-care therapies for ER+ breast cancers implement ERα antagonists, such as tamoxifen and fulvestrant (145). ERα antagonists inhibit the estradiol (E2)/ERα interaction to effectively abrogate ERα signaling (146). Unfortunately, up to 50% of breast cancer patients will exhibit endocrine-resistant breast cancers or develop endocrine resistance over the course of prolonged treatment (147). Interestingly, a study from Plaza-Menacho et al. determined that RET regulates sensitivity of ER+ breast cancer cells to endocrine therapies and that activated RET promotes estrogen-independent activation of ERα (148), suggesting that RET and ERα pathway crosstalk in endocrine-resistant breast cancers.
Aromatase inhibition is another therapeutic regimen used for treatment and management of ER+ breast cancers (149). Aromatase, also known as estrogen synthase, is a critical enzyme for estrogen biosynthesis and its increased expression and/or activity can drive estrogen production to sustain growth of ER+ breast cancers (150). As observed with tamoxifen, many ER+ breast cancer cases present with aromatase-resistant breast tumors at the time of diagnosis, or develop aromatase resistance with prolonged treatment (151). In 2013, the Isacke group determined that RET signaling is hyperactivated in aromatase-resistant ER+ breast cancer and that pharmacological RET inhibition impairs GDNF-mediated aromatase inhibitor resistance (152). Additionally, RET-mediated endocrine resistance in ER+ breast cancer cells may be attributed to the finding that RET is an ER target gene and that RET and ERα stimulate unique pathways in luminal breast cancers, thus providing biological rationale for dual targeting of these two receptors to combat hormone resistance in luminal breast cancers (153–157). Thus, RET is actionable in endocrine-resistant ER+ breast cancers and these patients may benefit from combined inhibition of RET and ER pathway. However, a study done by Andreucci and colleagues determined that dual targeting of RET and aromatase yields similar preclinical benefit when compared to RET or aromatase inhibitor monotherapies (158). Moreover, while RET expression is strongly correlated with that of ER in luminal breast cancers, it lacks prognostic significance as an independent biomarker in breast cancer patient tissue (142), suggesting that additional studies are required to determine if targeting RET will yield clinical benefit in luminal breast cancer patients.
Of all breast cancer subtypes, HER2-enriched and triple-negative breast cancers (TNBCs) are the most aggressive subtypes with the highest propensity for metastases (159). RET protein is expressed in HER2-enriched breast cancers and triple-negative breast cancers (153). While trastuzumab is one of the frontline therapies administered to HER2-enriched breast cancers, TNBC patients receive neoadjuvant or adjuvant chemotherapy and surgical resection (160). Despite initial response observed in HER2-positive breast cancer patients receiving trastuzumab, they eventually present with acquired trastuzumab resistance that is difficult to treat (161, 162). A recent report associates trastuzumab resistance with activation of a RET-HER2 signaling axis, whereas trastuzumab-naïve cells rely on a RET-SRC-HER2 signaling (161), suggesting that trastuzumab resistance may be a consequence of Src-independent signaling in HER2-enriched breast cancers that have concurrent RET activation. Similarly, TNBCs lack effective treatments that yield long-term clinical benefit. While RET activity has not been directly identified as a pro-tumorigenic kinase in TNBC, multiple studies demonstrate that MKIs can target RET to inhibit TNBC growth and proliferation in vitro and in vivo. For example, vandetanib can target RET, as well as, vascular endothelial growth factor receptor 2 (VEGFR-2) and epidermal growth factor receptor (EGFR), but exhibits potent anti-tumor effects against TNBC patient-derived xenografts with RET hyperactivity (163), suggesting RET inhibition as a novel treatment modality in TNBC patients. Moreover, recent reports suggest that RET may regulate breast cancer metastases. A recent study identified increased expression of RET in breast cancer brain metastases (BCBM) relative to matched primary breast tumors, and that treatment of these BCBM tissues with MKI cabozantinib reduces RET activation, inhibits BCBM growth, and concurrently induces apoptosis (164). In summary, these findings suggest that RET is actionable in breast cancers, especially those of HER2-enriched and triple-negative subtype.
In addition to oncogenic RET alterations, co-overexpression of wild-type RET and its co-receptor, GFRα1, may also contribute to breast tumorigenesis. RET and co-receptor GFRα1 are overexpressed in 25-75% of breast cancer cases (50). Esseghir and colleagues (2007) demonstrated that co-overexpression of RET and GFRα1 promoted cell proliferation and survival of luminal MCF7 cells in response to stimulation with GDNF, and that GDNF is overexpressed by stromal cells in response to the pro-inflammatory signals within the tumor microenvironment (143). A later study by Boulay et al. (2008) determined that RET and ERα pathway share a functional crosstalk in luminal breast cancer cells and RET expression can be enhanced upon stimulation of the ERα pathway (153), suggesting that RET-positive luminal breast cancers may benefit from RET inhibition. Therefore, in addition to the ~1.2% of breast cancer cases harboring RET alterations (49), breast cancer patients overexpressing RET-GFRα1 may also benefit from RET-targeted therapies.
RET in salivary gland carcinomas
The World Health Organization (WHO) classifies salivary gland tumors into more than 30 malignant and benign subtypes based on cytology rather than the site of the originating tumor (69, 165, 166). Genomic profiling salivary duct carcinomas (SDCs) reveals that RET is frequently altered in this tumor type, with three clinical cases presenting with RET fusion products, CCDC6-RET or NCOA4-RET (55). While this was a retrospective study, the authors also described that patients with NCOA4-RET fusion-positive SDC tumors responded well to cabozantinib, and these patients experienced reduction in chest wall lesions, and improved palpable neck relapse. Intraductal salivary gland carcinomas (ISGCs) are controversially linked to SDCs, with some arguing that these malignancies should be classified as two separate subtypes. ISGCs were first characterized in 1983 as a benign mass that was comprised entirely of intraductal components (167), which was a stark contrast to the infiltrating salivary duct carcinomas first described in the late 1960s (168–170). ISGCs are histologically unique from low grade-intraductal carcinomas (LG-ICs), which were first described by Delgado and colleagues in 1996 as a benign mass that resembled mammary ductal hyperplasia (171). Considered as a rare salivary gland tumor type, LG-ICs typically originate in the parotid gland, are more commonly found in the female population, and are generally asymptomatic (172). Next-generation RNA sequencing of 23 LG-IC samples identified one case with NCOA4-RET fusion, with a total of 47% of ICs presenting with some form of RET rearrangement (56), suggesting that RET is an early driver of salivary gland carcinogenesis. A similar study reported that 8/17 (47%) cases of salivary gland ICs had RET rearrangements (70). Furthermore, several studies have identified oncogenic RET fusions in varying subtypes of salivary gland carcinomas, namely ETV6-RET in mammary analogue secretory carcinomas (71–73, 173), NCOA4-RET in intercalated duct-line SDCs (57, 70, 173), TRIM27-RET fusions in mixed intercalated duct-like and apocrine types of SDC (57, 70), and TRIM33-RET fusions were recently detected in oncocytic intraductal carcinoma (74). Taken together, these findings support RET as a potential therapeutic target in multiple subtypes of salivary gland carcinoma.
RET in prostate cancer tumorigenesis
Prostate cancer is the most commonly diagnosed cancer and second-leading cause of cancer-related deaths in American men (126). While early stage prostate cancers do not usually present with clear symptoms, those that arise are similar to those found in benign prostatic hyperplasias and can include frequent urination, hematuria (blood in urine), dysuria (pain during urination), fatigue, and bone pain (174). Early stage prostate cancers are classified as prostatic intraepithelial neoplasia (PIN) and show enhanced epithelial cell proliferation within benign prostatic acini (175). As PIN satisfies many of the classification requirements of premalignant disease, high-grade PIN is considered the precursor to prostate cancer (176, 177). Malignant prostate cancers progress from PIN to prostatic adenocarcinoma, and eventually to metastatic prostate cancer that may or may not be castration-resistant (178). Prostate cancer screenings include prostate exams as well as blood tests for prostate-specific antigen (PSA) (175), though whether PSA testing improves disease outcome remains to be confirmed.
Overexpression of wild-type RET in prostate cancer was first demonstrated by Robinson and colleagues (179) using a prostate cancer xenograft model. A follow-up study showed that RET protein is overexpressed in high-grade PIN and prostate cancers relative to benign prostatic tissue, and that RET protein expression was positively correlated to the tumor Gleason score (180), suggesting that RET overexpression may contribute to malignant progression of prostate cancers. Concurrent studies by Ban et al. revealed that nearly 19% (61/325) of prostate cancer tissues examined exhibit tumor tissue-specific RET overexpression (181) and that RET expression is critical for prostate cancer growth in vivo (181). However, the increase in RET activity within prostate cancers may also be attributed to increased secretion of GDNF by prostatic stromal cells (182) or GFRα1 by peripheral nerves within the prostate (181, 183, 184), ultimately promoting perineural invasion of prostate cancers.
As discussed in sections 4.2 and 4.3, aberrant RET activation is frequently found in neuroendocrine tumors such as papillary and medullary thyroid cancers. A 2018 case report described a 20-year-old MTC patient that presented with an activating point mutation in RET (RET-M918) four years after total thyroidectomy and presented with prostatic adenocarcinomas twenty years later (87). As the prostate contains epithelial and neuroendocrine cells that express RET protein, this case report suggests an association between RET hyperactivity in neuroendocrine cells and initiation of prostatic neoplasia. In 2020, VanDeusen and colleagues reported that aggressive, androgen-independent prostate cancers can transdifferentiate into neuroendocrine prostate cancers (NEPC), which exhibit tumorigenic RET activity, though this can be suppressed using selective RET inhibitors (185). It is important to note, however, that while RET overexpression is frequently found in prostate cancers (180, 181), RET alterations are found in 1-2% of prostate cancer cases (186). Taken together, these findings suggest that aberrant RET activity may be actionable in prostate cancers, including those associated with neuroendocrine malignancies.
Aberrant RET in colorectal cancers
Colorectal cancer (CRC), also known as colon, bowel, or rectal cancer, represents 7-10% of all cancer cases and is one of the leading causes of cancer-related deaths in American men and women (126, 187). CRC originates from epithelial cells lining the colon and gastrointestinal tract and begin as benign polyps that grow into large polyps, which develop into adenoma, followed by carcinoma (188). Metastatic CRCs frequently metastasize to the liver, lungs, bones, brain, liver, and peritoneum (189).
Ishizaka and colleagues identified a second form of activated RET, ret-II, in human sigmoid colon cancer, which lacked the coding region for the transmembrane domain found in wild-type RET (190). A follow-up study determined that ret-II is likely a gene fusion as the sequences preceding the kinase domain of ret-II were of unknown origin (191). Large-scale genomics analyses demonstrate that RET fusions, including CCDC6-RET and NCOA4-RET, are found in 0.2% (6/3117) of CRC cases (52). Analyses of metastatic CRC (mCRC) cases reveal that RET-rearranged mCRCs have poorer overall survival and may benefit from pharmacological RET inhibition (53). Indeed, patient-derived CRC tumor cells harboring NCOA4-RET fusion were sensitive to the treatment with vandetanib, a multi-kinase inhibitor with non-selective RET activity (54).
Despite the evidence supporting an oncogenic role for RET alterations in CRC, the role of RET in this cancer type remains controversial. A 2013 study by Luo et al. reported enhanced methylation, and therefore downregulation, of RET in CRC samples, suggesting a tumor suppressive function for RET in the CRC samples analyzed (192). The potential tumor suppressive function of RET is further supported by a 2021 study in which immunohistochemical analyses demonstrate a reduction in RET protein expression in CRC tissue when compared to adjacent normal tissue (193), suggesting an inverse correlation between RET expression and CRC development. While the opposing paradigms of RET function may present challenges in the treatment of RET-altered CRC tumors, further analyses are required to determine whether tumor-suppressive or oncogenic RET occurs in specific subpopulations of CRC tumors.
RET in pancreatic and ovarian cancers
RET is altered and potentially actionable in other solid tumor types, albeit at low frequencies. RET alterations occur in ~1.9% of pancreatic cancers (41) whereas wild-type RET, co-receptor GFRα1, and GDNF are overexpressed in 50-70% of pancreatic cancer cases (194, 195). Additionally, Ceyhan and colleagues demonstrated that RET activation by another GFL, Artemin, can promote invasion and migration of pancreatic cancer cells (196). This finding was supported by a later study in which enhanced RET overexpression promotes pancreas tumorigenesis and perineural invasion using a transgenic mouse model of pancreatic cancer (197), suggesting that enhanced activity of wild-type RET is adequate to promote metastatic pancreatic cancer progression. Moreover, enhanced secretion of GDNF by cells within the tumor microenvironment stimulates RET activity in pancreatic tumor cells, thus exerting a paracrine effect on pancreatic cancer cells to drive perineural invasion (197, 198). While RET alterations are rare in pancreatic cancers, exemplified by report of the first RET fusion-positive pancreatic cancer patient in 2021 (75), patients with aberrant RET pathway activity may benefit from RET-targeted therapies. In addition to pancreatic cancers, RET is altered in ~1.2% of ovarian cancers (41). Oncogenic RET variants have been recently identified in epithelial ovarian carcinomas, which were sensitive to treatment with vandetanib, an MKI with non-selective RET activity (199). However, additional studies are required to determine the therapeutic utility of RET inhibition in ovarian cancers.
Multi-kinase inhibitors with non-selective RET activity
Due to the numerous studies implicating aberrant RET kinase activation in varying solid tumors, pharmacological inhibition of RET has become a promising new treatment modality in patients with enhanced RET activity. Targeting RET activity began with repurposing of MKIs that were initially designed to target other kinases, but showed moderate selectivity to RET. This portion of the review will summarize the MKIs with anti-RET activity.
Cabozantinib
Cabozantinib (XL184, sold as Cometriq ® and Cabometyx ®) is an orally available ATP-competitive MKI that can inhibit c-MET, VEGFR2, and a number of other kinases including RET, FLT3, TRK, and KIT (200). Cabozantinib effectively inhibits cell proliferation, growth, migration, and invasion in vitro, and promotes tumor cell and endothelial cell apoptosis in vivo in a number of cancer cell lines (200). Cabozantinib was well-tolerated in a number of clinical trials including MTC (201) and renal cell carcinoma (202). Cabozantinib received FDA approval in 2012 for treatment of MTC, as second line of treatment in kidney cancer in 2016, treatment of renal cell carcinoma in 2017, and again in 2019 for treatment of hepatocellular carcinoma. A phase II clinical trial (NCT01639508) reported that RET-fusion positive lung cancer patients receiving cabozantinib (60mg, p.o. daily) demonstrated partial response (28%; 7/25 patients) and fortifies the notion that RET is actionable in lung cancers (132, 203). Similarly, daily oral administration of cabozantinib (140mg) enhanced overall survival (26.6 vs. 21.1 months in treatment vs. placebo group, respectively) and progression-free survival in phase III clinical trial (NCT00704730) for treatment of metastatic MTC cases harboring RET-M918T with an acceptable safety profile (204, 205). The overall ORR for patients who received cabozantinib was 28%, but increased to 34% when assessing only patients with RET-M918T mutations. While data is limited for use of cabozantinib in treatment of central nervous system (CNS) tumors, such as BCBM, cabozantinib penetrates the BBB and may prove to be effective in targeting brain malignancies arising from RET-altered cancers (137, 206). While cabozantinib received FDA approval for treatment of MTC (207), ongoing clinical trials aim to assess cabozantinib safety and efficacy in RET fusion-positive NSCLC (NCT01639508, NCT04131543).
Lenvatinib
Lenvatinib (E7080; Lenvima ®) is an oral ATP-competitive MKI with activity against FLT-1, FLT-4, KIT, FGFR1, PDGFR, VEGFR1-3, and can inhibit angiogenesis and growth of SCLC and breast cancer cells in vitro and in vivo (208–211). When evaluated in RET fusion-positive preclinical models, lenvatinib effectively inhibits RET fusion kinases and RET pathway activity in vitro, and inhibits growth of RET fusion-positive tumor cells in vivo (212, 213). While a phase II clinical trial (NCT01877083) determined that once-daily oral administration of lenvatinib (24mg) induces relatively low response in RET fusion-positive NSCLC patients (ORR 16%, 4/25 patients), this effect was adequate to improve the median progression-free survival (214). Lenvatinib received FDA approval in 2015 for treatment of differentiated thyroid cancer (215) and 2018 for treatment of unresectable hepatocellular carcinoma (216). Lenvatinib has also received FDA approval as part of combination therapies in renal cell carcinoma (with mTOR inhibitor everolimus) (217) and endometrial carcinoma (with anti-PD-1 antibody pembrolizumab) (218).
Vandetanib
Vandetanib is an orally available inhibitor of KDR/VEGFR-2 tyrosine kinase with moderate activity against FLT-1/VEGFR-1, EGFR, and PDGFRβ (219, 220). Vandetanib potently inhibits VEGF signaling and angiogenesis, resulting in reduced growth of varying tumor cell xenografts, namely human lung, prostate, breast, ovarian, and colon cancer cells, with little to no acute toxicity (221). Within the same year, Carlomagno and colleagues showed that vandetanib potently inhibited kinase activity of RET/PTC, RET/MEN2A, and RET/EGFR in transformed NIH3T3 cells, reverted the morphological phenotype of transformed cells to that of parental cells, and inhibited anchorage-independent growth (222). Vandetanib selectivity for RET was further validated in a D. melanogaster model of MEN2 syndromes and papillary thyroid carcinomas, suggesting that vandetanib can inhibit multiple RET isoforms to treat RET-dependent carcinomas (223). However, a follow-up study by Carlomagno et al. identified RET-V804L or RET-V804M gatekeeper mutations which confer resistance to vandetanib (83), suggesting that vandetanib may not yield clinical efficacy in cases with RET gatekeeper mutations. Clinical trials determined that vandetanib is well-tolerated when administered once daily (300mg, p.o.), elicits anti-tumor response in patients with advanced or metastatic hereditary MTC (224, 225), demonstrated improved therapeutic efficacy, and was predicted to enhance progression-free survival (PFS) in advanced MTC (19.3 months vs. 30.5 months in placebo vs. treatment, respectively) in phase III clinical trial (NCT00410761) (226). As with other MKIs, vandetanib can inhibit many kinases (220–223), thus the anti-tumor effects observed in MTC may not strictly be due to inhibition of RET kinase activity. Nevertheless, vandetanib received FDA approval for treatment of non-resectable advanced or metastatic MTC in 2011 (227).
Sorafenib
Sorafenib (Nexavar ®) is an orally available ATP-competitive inhibitor of Raf kinase (228) with activity against other kinases, such as BRAF, VEGFR-2 and -3, and PDGFRβ, to impede tumor growth and angiogenesis in vivo (229). Subsequent studies illustrated that sorafenib inhibits activity of wild-type RET and oncogenic altered RET with some degree of selectivity to abrogate PTC tumor cell proliferation and growth in vitro and in vivo (230–232). Moreover, RET inhibition by sorafenib is not affected by the RET-V804M gatekeeper mutation, suggesting that sorafenib may yield clinical benefit in MTC patients bearing RET-V804 mutation (231). In addition to being an ATP-competitive inhibitor, Plaza-Menacho and colleagues demonstrated that sorafenib treatment promotes degradation of RET protein independently of proteosomal targeting (231), which suggests a secondary mechanism by which RET activity is depleted upon treatment with this inhibitor. Interestingly, a 2012 study by Mao et al. corroborated the finding that sorafenib affects RET expression and concurrently alters VEGFR2 expression, although these findings are cell-line dependent and require additional mechanistic studies (233). Sorafenib received FDA approval in 2005 for treatment of advanced renal cell carcinoma (234), and was subsequently approved for treatment of unresectable hepatocellular carcinomas (235) and iodine-refractory, metastatic differentiated thyroid cancers (236). Interestingly, an exploratory study by Horiike and colleagues determined that NSCLC patients do not exhibit response to sorafenib (136), suggesting that RET fusion-positive NSCLC patients may not gain clinical benefit from this compound, though this requires additional testing in RET-altered NSCLC patients and other RET-altered cancers.
Regorafenib
Regorafenib (BAY 73-056; Stivarga ®) is an orally bioavailable ATP-competitive inhibitor that targets VEGFR1-3, TIE2, PDGFRβ, KIT, and RET in vitro and in vivo, effectively functioning as an anti-angiogenic, anti-tumorigenic, and pro-apoptotic compound (237). Regorafenib is well-tolerated when administered as a single agent and elicited a partial response or stable disease in patients with advanced or non-resectable solid tumors (238–240), though renal cell carcinoma patients experienced adverse events with prolonged treatment (241). Chen and colleagues illustrated that regorafenib potently inhibits RET activity and RET-mediated PI3K/AKT signaling in neuroblastoma cells to inhibit their growth in vitro and in vivo (242). Additionally, regorafenib targets the RET-Src signaling axis to inhibit JAK1/2-STAT1 and MAPK signaling and concurrently inhibits PD-L1 expression, suggesting inhibition of RET-Src axis may impact immune response to tumor cells in vivo (243). While regorafenib is currently in numerous clinical trials for use in various cancer types (ClinicalTrials.gov), it has not entered trials for use in RET-altered cancers.
Sunitinib
Sunitinib (SU11248; Sutent ®) was developed in 2003 as an orally bioavailable ATP-competitive inhibitor of VEGFR, PDGFR, KIT, and FLT3 receptor tyrosine kinases, and effectively inhibits tumor growth of multiple cancer cell types, namely breast, NSCLC, colon cancer, glioma, and melanoma when administered daily as a single agent (244–247). As a VEGF inhibitor, sunitinib effectively inhibits angiogenesis of tumor cells, reduces endothelial cell migration, and inhibits tubule formation in vivo (248). Sunitinib is potent against PTC tumors harboring RET/PTC3 fusions and effectively inhibits downstream MAPK signaling (249). While sunitinib received FDA approval in 2006 for treatment of GI stromal tumors and advanced renal cell carcinomas, a pilot clinical trial to test safety and efficacy of sunitinib (50mg, p.o. daily) in RET fusion-positive solid tumors (NCT02450123) was placed on hold in 2020 and is of unknown status at the time of this publication.
Alectinib
Alectinib (CH5424802; Alecensa ®) is an orally bioavailable, small molecule competitive ATP inhibitor developed to inhibit ALK alterations and gatekeeper mutations while sparing MET activity (250). Alectinib effectively reduces ALK activation in vitro and in vivo, inhibits activation of STAT3, PI3K/AKT, and MAPK pathways, inhibits growth of tumors harboring ALK alterations (250), and even inhibited ALK-L1196M, a gatekeeper mutation that conferred resistance to earlier generations of ALK inhibitors (251). Alectinib was well-tolerated and highly active in patients with NSCLC patients with ALK alterations (252). Kodama et al. showed that alectinib inhibits wild-type RET and the constitutively active RET-M918T NSCLC cells in vitro and in vivo, and exhibits selectivity against RET with gatekeeper mutations (V804L and V804M) (134). However, while clinical assessment determined that alectinib was well-tolerated in treatment-naïve patients, twice-daily oral administration of alectinib (600mg) exerts minimal activity in RET fusion-positive NSCLC patients (133) and yielded an overall response rate (ORR) of 4% (1/25 patients) and PFS of 3.4 months (135). These findings were corroborated by a recent clinical trial (NCT03131206) in which RET fusion-positive thyroid cancer and NSCLC patients showed minimal to no response to alectinib. Alectinib received accelerated FDA approval in 2015 for treatment of patients with ALK fusion-positive metastatic NSCLC that were unresponsive to crizotinib, an ALK and ROS1 inhibitor (253), and is currently in clinical trials to test efficacy against other solid tumors alone (NCT04116541, NCT02314481, NCT03194893) or in combination with other compounds (NCT03944772).
Selective RET inhibitors
While MKIs show some selectivity in targeting RET in preclinical models, many of these inhibitors have suboptimal responses upon clinical administration to RET fusion-positive patients. A retrospective multi-center analysis by Gautschi et al. found that, of the 165 NSCLC patients with RET alterations who received MKIs, only partial responses were observed in patients who received cabozantinib (37%), sunitinib (22%), or vandetanib (18%) (254). This finding suggests that, despite their activity against RET, MKIs yield suboptimal clinical benefit with prolonged administration to patients. Earlier MKIs that inhibit RET are only moderately potent against RET, and cause toxicity due to stronger selectivity against other kinases, such KDR/VEGFR2, due to structural similarities with RET. Furthermore, MKIs are not selective against gatekeeper mutations that may elicit secondary resistance. Together, these findings have warranted development of selective RET inhibitors with the aim of explicitly inhibiting aberrant RET activity while sparing other kinases. This section summarizes RET-selective inhibitors. (Ongoing clinical trials against RET-altered cancers are summarized in Table 3).
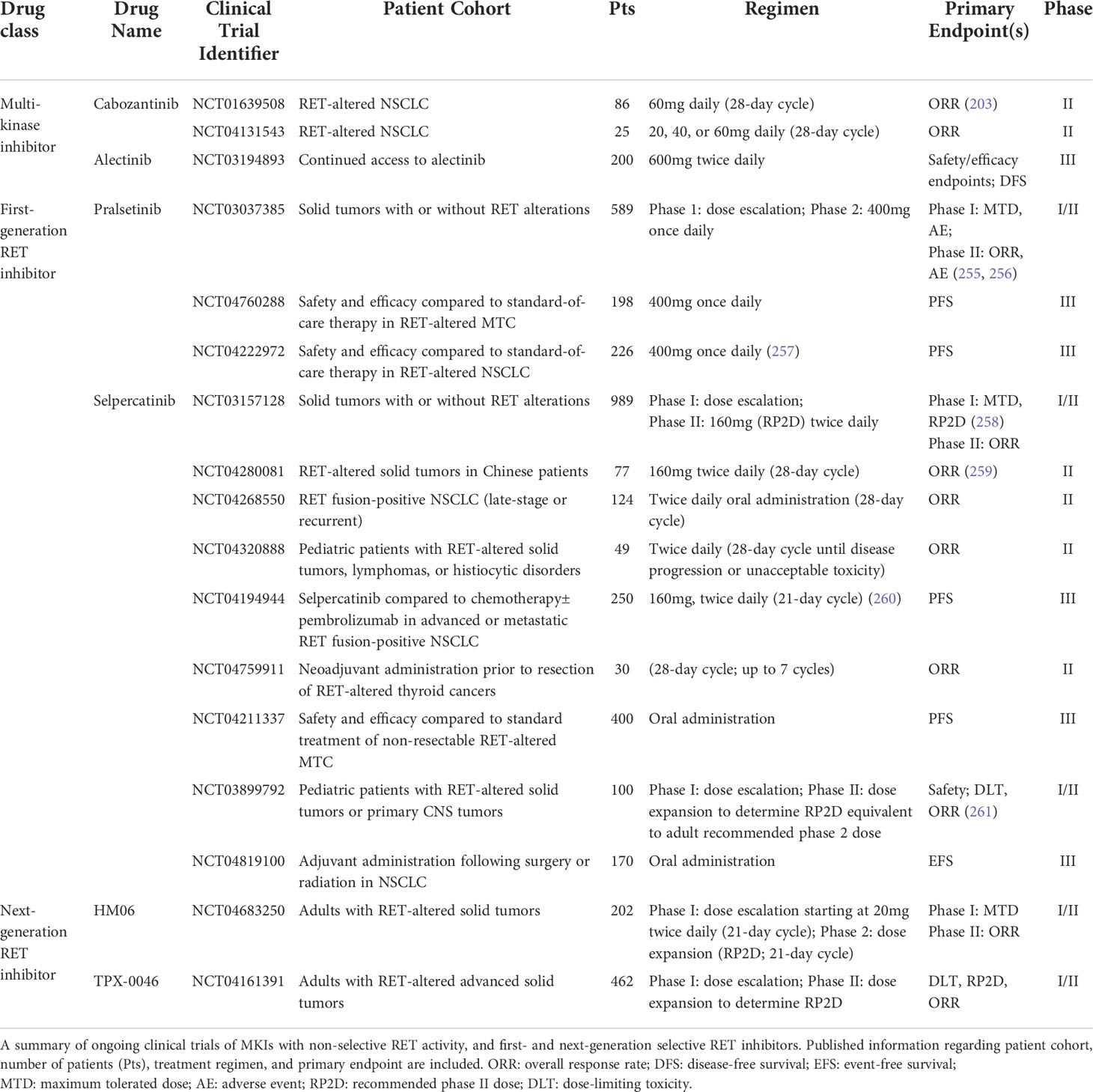
Table 3 Ongoing clinical trials of RET-targeting inhibitors.
Agerafenib
In an effort to overcome the suboptimal clinical benefit of MKIs in RET-altered cancers, agerafenib (CEP-32496; RXDX-105) was developed as an orally available, ATP-competitive, selective inhibitor of RET and BRAF (262). Preclinical assessment in multiple RET-altered cancer types showed that RXDX-105 potently inhibits wild-type and altered RET more potently and selectively than MKIs with non-selective RET activity, and elicited rapid response in the primary tumor and brain metastases of a RET fusion-positive lung cancer patient (262). These findings strongly suggested RXDX-105 as a promising new anti-cancer compound with BBB penetrance. However, a phase I/Ib trial (NCT01877811) revealed that daily oral administration of agerafenib (275mg) does not elicit response in RET fusion-positive NSCLC patients (0/20) and conferred multiple treatment-related adverse events such as diarrhea, fatigue, and hypophosphatemia (263). Ultimately, this clinical trial was discontinued due to lack of clinical response.
Selpercatinib
Selpercatinib (LOXO-292, LY3527723; Retevmo ®) is an orally available RET inhibitor that competitively binds to its ATP binding pocket (264). In vitro studies reveal that selpercatinib selectively inhibits both wild-type and altered RET, spares KDR/VEGFR2 activity, and selectively targets RET-altered cancer cells (264). Selpercatinib also abrogates tumor growth of lung cancer and MTC xenografts in a dose-dependent manner in vivo, highlighting its therapeutic utility in RET-dependent cancers. Further in vitro and in vivo characterization confirms that selpercatinib is selective against RET alterations, including fusions and activating mutations, is well-tolerated upon daily oral administration, and elicited rapid clinical response in two patient cases (MTC and NSCLC) (265). Selpercatinib entered a phase I/II clinical trial (NCT03157128) in patients bearing RET alterations (fusions, mutations, and other alterations). This clinical trial, LIBRETTO-001, remains ongoing but recently reported an objective response of 64% (67/105 patients), with treatment-naïve patients reporting 85% objective response (33/39 patients) with daily oral administration of 20-240mg selpercatinib (266). Since lung cancers frequently metastasize to the brain, effective therapeutic agents should also display BBB penetrance. In the LIBRETTO-001 trial, the authors reported that NSCLC patients with CNS metastases had over 82% objective intracranial response rate (18/22 patients), with two patients undergoing complete response, indicating that selpercatinib can penetrate the BBB to act upon brain metastases of RET-altered lung cancers (265, 267). The same clinical trial also determined an objective response of 69-79% in MTC patients bearing RET alterations (268). Many of these patients (NSCLC or MTC) reported improved or maintained quality of life during treatment with selpercatinib (269, 270). Taken together, these results indicate that selpercatinib elicits durable efficacy, including intracranial activity, with primarily low-grade toxicity effects in patients with RET-altered cancers. Selpercatinib received accelerated approval from the FDA in 2020 for patients with metastatic RET fusion-positive NSCLC, and patients with RET-mutant aggressive or metastatic MTC (271, 272), making it the first FDA approval for RET fusion-positive NSCLC and MTC. In addition to the LIBRETTO-001 trial, ongoing clinical trials are underway to test efficacy of selpercatinib in multiple tumor types, such as pediatric solid tumors (NCT03899792, NCT04320888) and advanced NSCLC (NCT04268550).
Pralsetinib
As with selpercatinib, pralsetinib (BLU-667; Gavreto ®) is an orally available, ATP-competitive inhibitor that selectively inhibits wild-type and altered RET while sparing KDR/VEGFR2 activity (273). Pralsetinib entered Phase I/II clinical trial in 2017 (NCT03037385) to assess safety and efficacy in patients with RET fusion-positive thyroid and lung cancers (274). This clinical trial, entitled ARROW, is a multi-cohort, open-label, first-in-human study that involved 75 sites in 11 countries. An ORR of 61% was observed in NSCLC patients who previously received platinum chemotherapy (53/87 patients), 70% (19/27) in treatment-naïve patients, with a small percentage undergoing complete response (255). Additionally, pralsetinib showed robust intracranial activity in 78% (7/9) of NSCLC patients with CNS metastases and mitigated progression of new CNS lesions, suggesting that pralsetinib crosses the BBB to act on brain metastases (274). Pralsetinib was granted accelerated approval by the FDA in 2020 for adults with metastatic RET fusion-positive NSCLC, and patients with metastatic RET-mutant MTC that may or may not be radioactive iodine-refractory (275). While pralsetinib is well-tolerated in patients included within the ARROW clinical trial, a recent report characterized leptomeningeal progression in a NSCLC patient despite durable extracranial response to pralsetinib, though this patient exhibited clinical CNS response following selpercatinib treatment (276), suggesting that selpercatinib may improve on the intracranial therapeutic efficacy of pralsetinib. In addition to ARROW, another ongoing clinical trial aims to assess efficacy of pralsetinib compared to first-line treatments of advanced NSCLC (NCT04222972), as well as test efficacy compared to standard-of-care therapies in NSCLC (NCT04222972) and MTC (NCT04760288).
Resistance to first-generation RET inhibitors and development of next-generation compounds
While selpercatinib and pralsetinib effectively and selectively inhibit RET in preclinical and clinical settings, recent publications report novel acquired resistance to selpercatinib or pralsetinib with prolonged treatment as well as novel genetic mutations. Analysis of circulating tumor cell DNA, isolated from RET fusion-positive NSCLC or MTC patients who progressed after initial response to selpercatinib, revealed the emergence of three mutations: G810R, G810S, and G810C within the RET solvent front, which are predicted to sterically hinder binding of selpercatinib to RET fusions (277). These findings were corroborated by other studies where, in addition to detection of G810 solvent front mutations, off-target MET (278) or KRAS amplifications were detected in RET fusion-positive NSCLC patients who received either selpercatinib or pralsetinib, albeit at low frequency (60). Additional mutations in the hinge region of RET (Y806) are strongly associated with resistance to selpercatinib or pralsetinib (279). Moreover, Subbiah et al. identified novel KHDRBS1-NTRK3 and KIF5B-RET rearrangements in selpercatinib-resistant tumor samples not previously detected in primary tumor biopsy (280). It is crucial to note that mutation-driven resistance to RET inhibition may vary between inhibitors. For example, Shen and colleagues illustrate that the RET-L730V/I roof mutation confers resistance to pralsetinib, but not selpercatinib (281), which suggests unique mechanisms of binding and inhibition between these two small molecule RET inhibitors. Taken together, while the current generation of selective RET inhibitors elicits potent response in NSCLC and MTC patients, acquired resistance may arise in patients with prolonged administration.
To address the observed acquired resistance to first-generation RET inhibitors, next-generation RET inhibitors are currently being tested in Phase I/II clinical trials. BOS172738 is an investigational compound with potent selectivity against RET, spares VEGFR2 activity, overcomes the RET-G810 resistance mutation, and displays a favorable safety profile when administered as a single-agent to patients with RET fusion-positive NSCLC or MTC tumors (282). Another investigational compound, TPX-0046, showed potent activity and selectivity in RET fusion-positive cancer cells in vitro and in vivo, and can overcome RET-G810 resistance mutations (283). While TPX-0046 cannot circumvent the V804 gatekeeper mutation, a Phase I/II clinical trial (NCT04161391) is ongoing in patients with advanced solid tumors harboring RET alterations. HM06, another investigational compound, selectively inhibits RET across multiple tumor types in vitro and in vivo, circumvents RET-V804X gatekeeper mutation and RET-G810X resistance mutations (284, 285), and is currently in Phase I/II clinical trials in patients with RET-altered tumors (NCT04683250). Lastly, LOX-18228 is a novel compound that potently inhibits RET, RET fusions, and can even inhibit RET-V804X and RET-G810X mutations singly or in tandem (286), suggesting that LOX-18228 may be effective in targeting tumors with resistance to first-generation RET inhibitors. LOX-18228 is expected to enter Phase I clinical trials in 2022 (286).
Concluding remarks
Over the last three decades, cumulative studies have established an important role that RET plays in multiple cancer types. Identification of activating RET alterations and of oncogenic RET fusions in solid tumors has led to the repurposing of MKIs in an effort to inhibit aberrant RET activity (133–136, 139, 203, 212, 230–232, 242, 249). However, these MKIs show suboptimal clinical benefit in patients and warranted the development of selective RET inhibitors that not only inhibit RET activity, but also serve to overcome RET alterations that confer resistance to RET-targeting MKIs. Selpercatinib and pralsetinib are the only selective RET inhibitors FDA-approved for treatment of RET fusion-positive lung and thyroid cancers (271, 275) and show potent intracranial activity (267, 274). While these inhibitors show significantly improved clinical benefit, recent reports describe disease progression in some patients after prolonged treatment with selpercatinib or pralsetinib, some of which are attributed to novel RET mutations that confer this acquired resistance (60, 277–280). These findings strongly support development of second generation selective RET inhibitors that target novel RET mutations as well as the studied oncogenic RET alterations described herein. Moreover, the role of RET in distant metastases remains to be fully examined. While selective RET inhibitors show potent activity in brain metastases of lung and thyroid cancer patients (267, 274), the emergence of therapeutic resistance to RET inhibitors suggests activation of additional or alternative pathways that circumvent pharmacological inhibition of RET. In summary, pharmacological inhibition of RET proto-oncogene is a promising new treatment modality in solid tumors, though additional efforts should be placed in understanding acquired therapeutic resistance to RET inhibition.
Author contributions
Designed the manuscript, AR. Selected the reviewed literature, AR. Compiled the review tables and figures, AR and MN. Wrote the manuscript, AR. Contributed literature search, HWL. Table preparation, AR, MN. Manuscript writing and editing, AR, MN, and H-WL. Supervised, H-WL. Helped design figures, MN and H-WL. Edited, H-WL. All authors have read and agreed to the published version of the manuscript.
Funding
The authors acknowledge funding support by NIH grants 1T32CA247819 (KW, AR), R01CA228137-01A1 (H-WL), and P30CA012197 (BP), as well as, DoD grants, W81XWH-17-1-0044 (H-WL), W81XWH-19-1-0072 (H-WL), W81XWH-19-1-0753 (H-WL), and W81XWH-20-1-0044 (H-WL).
Acknowledgments
The authors would like to thank the Carpenter Library at Wake Forest University School of Medicine for open-access literature support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Takahashi M, Ritz J, Cooper GM. Activation of a novel human transforming gene, ret, by DNA rearrangement. Cell (1985) 42(2):581–8. doi: 10.1016/0092-8674(85)90115-1
2. Schuchardt A, D'Agati V, Larsson-Blomberg L, Costantini F, Pachnis V. Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor ret. Nature (1994) 367(6461):380–3. doi: 10.1038/367380a0
3. Chi X, Michos O, Shakya R, Riccio P, Enomoto H, Licht JD, et al. Ret-dependent cell rearrangements in the Wolffian duct epithelium initiate ureteric bud morphogenesis. Dev Cell (2009) 17(2):199–209. doi: 10.1016/j.devcel.2009.07.013
4. Enomoto H, Crawford PA, Gorodinsky A, Heuckeroth RO, Johnson EM Jr, Milbrandt J. Ret signaling is essential for migration, axonal growth and axon guidance of developing sympathetic neurons. Development (2001) 128(20):3963–74. doi: 10.1242/dev.128.20.3963
5. Jain S, Naughton CK, Yang M, Strickland A, Vij K, Encinas M, et al. Mice expressing a dominant-negative ret mutation phenocopy human hirschsprung disease and delineate a direct role of ret in spermatogenesis. Development (2004) 131(21):5503–13. doi: 10.1242/dev.01421
6. Tomuschat C, Puri P. Ret gene is a major risk factor for hirschsprung's disease: A meta-analysis. Pediatr Surg Int (2015) 31(8):701–10. doi: 10.1007/s00383-015-3731-y
7. Smith DP, Houghton C, Ponder BA. Germline mutation of ret codon 883 in two cases of de novo men 2b. Oncogene (1997) 15(10):1213–7. doi: 10.1038/sj.onc.1201481
8. Mulligan LM, Kwok JB, Healey CS, Elsdon MJ, Eng C, Gardner E, et al. Germ-line mutations of the ret proto-oncogene in multiple endocrine neoplasia type 2a. Nature (1993) 363(6428):458–60. doi: 10.1038/363458a0
9. Iwashita T, Asai N, Murakami H, Matsuyama M, Takahashi M. Identification of tyrosine residues that are essential for transforming activity of the ret proto-oncogene with Men2a or Men2b mutation. Oncogene (1996) 12(3):481–7.
10. Wagner SM, Zhu S, Nicolescu AC, Mulligan LM. Molecular mechanisms of ret receptor-mediated oncogenesis in multiple endocrine neoplasia 2. Clinics (Sao Paulo) (2012) 67(Suppl 1):77–84. doi: 10.6061/clinics/2012(sup01)14
11. Ibanez CF. Structure and physiology of the ret receptor tyrosine kinase. Cold Spring Harb Perspect Biol (2013) 5(2):1–10. doi: 10.1101/cshperspect.a009134
12. Li J, Shang G, Chen YJ, Brautigam CA, Liou J, Zhang X, et al. Cryo-em analyses reveal the common mechanism and diversification in the activation of ret by different ligands. Elife (2019) 8:1–26. doi: 10.7554/eLife.47650
13. Perrinjaquet M, Vilar M, Ibanez CF. Protein-tyrosine phosphatase Shp2 contributes to gdnf neurotrophic activity through direct binding to phospho-Tyr687 in the ret receptor tyrosine kinase. J Biol Chem (2010) 285(41):31867–75. doi: 10.1074/jbc.M110.144923
14. Schuringa JJ, Wojtachnio K, Hagens W, Vellenga E, Buys CH, Hofstra R, et al. Men2a-Ret-Induced cellular transformation by activation of Stat3. Oncogene (2001) 20(38):5350–8. doi: 10.1038/sj.onc.1204715
15. Kawamoto Y, Takeda K, Okuno Y, Yamakawa Y, Ito Y, Taguchi R, et al. Identification of ret autophosphorylation sites by mass spectrometry. J Biol Chem (2004) 279(14):14213–24. doi: 10.1074/jbc.M312600200
16. Encinas M, Crowder RJ, Milbrandt J, Johnson EM Jr. Tyrosine 981, a novel ret autophosphorylation site, binds c-src to mediate neuronal survival. J Biol Chem (2004) 279(18):18262–9. doi: 10.1074/jbc.M400505200
17. Borrello MG, Alberti L, Arighi E, Bongarzone I, Battistini C, Bardelli A, et al. The full oncogenic activity of Ret/Ptc2 depends on tyrosine 539, a docking site for phospholipase cgamma. Mol Cell Biol (1996) 16(5):2151–63. doi: 10.1128/MCB.16.5.2151
18. Asai N, Murakami H, Iwashita T, Takahashi M. A mutation at tyrosine 1062 in Men2a-ret and Men2b-ret impairs their transforming activity and association with shc adaptor proteins. J Biol Chem (1996) 271(30):17644–9. doi: 10.1074/jbc.271.30.17644
19. Liu X, Vega QC, Decker RA, Pandey A, Worby CA, Dixon JE. Oncogenic ret receptors display different autophosphorylation sites and substrate binding specificities. J Biol Chem (1996) 271(10):5309–12. doi: 10.1074/jbc.271.10.5309
20. Alberti L, Borrello MG, Ghizzoni S, Torriti F, Rizzetti MG, Pierotti MA. Grb2 binding to the different isoforms of ret tyrosine kinase. Oncogene (1998) 17(9):1079–87. doi: 10.1038/sj.onc.1202046
21. Pachnis V, Mankoo B, Costantini F. Expression of the c-ret proto-oncogene during mouse embryogenesis. Development (1993) 119(4):1005–17. doi: 10.1242/dev.119.4.1005
22. Ivanchuk SM, Eng C, Cavenee WK, Mulligan LM. The expression of ret and its multiple splice forms in developing human kidney. Oncogene (1997) 14(15):1811–8. doi: 10.1038/sj.onc.1201016
23. Enomoto H, Araki T, Jackman A, Heuckeroth RO, Snider WD, Johnson EM Jr, et al. Gfr Alpha1-deficient mice have deficits in the enteric nervous system and kidneys. Neuron (1998) 21(2):317–24. doi: 10.1016/s0896-6273(00)80541-3
24. Enomoto H, Heuckeroth RO, Golden JP, Johnson EM, Milbrandt J. Development of cranial parasympathetic ganglia requires sequential actions of gdnf and neurturin. Development (2000) 127(22):4877–89. doi: 10.1242/dev.127.22.4877
25. Perea D, Guiu J, Hudry B, Konstantinidou C, Milona A, Hadjieconomou D, et al. Ret receptor tyrosine kinase sustains proliferation and tissue maturation in intestinal epithelia. EMBO J (2017) 36(20):3029–45. doi: 10.15252/embj.201696247
26. Kenny SE, Tam PK, Garcia-Barcelo M. Hirschsprung's disease. Semin Pediatr Surg (2010) 19(3):194–200. doi: 10.1053/j.sempedsurg.2010.03.004
27. Trupp M, Ryden M, Jornvall H, Funakoshi H, Timmusk T, Arenas E, et al. Peripheral expression and biological activities of gdnf, a new neurotrophic factor for avian and mammalian peripheral neurons. J Cell Biol (1995) 130(1):137–48. doi: 10.1083/jcb.130.1.137
28. Kubota H, Avarbock MR, Brinster RL. Growth factors essential for self-renewal and expansion of mouse spermatogonial stem cells. Proc Natl Acad Sci U.S.A. (2004) 101(47):16489–94. doi: 10.1073/pnas.0407063101
29. Meng X, Lindahl M, Hyvonen ME, Parvinen M, de Rooij DG, Hess MW, et al. Regulation of cell fate decision of undifferentiated spermatogonia by gdnf. Science (2000) 287(5457):1489–93. doi: 10.1126/science.287.5457.1489
30. Naughton CK, Jain S, Strickland AM, Gupta A, Milbrandt J. Glial cell-line derived neurotrophic factor-mediated ret signaling regulates spermatogonial stem cell fate. Biol Reprod (2006) 74(2):314–21. doi: 10.1095/biolreprod.105.047365
31. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cbio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discovery (2012) 2(5):401–4. doi: 10.1158/2159-8290.CD-12-0095
32. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cbioportal. Sci Signal (2013) 6(269):pl1. doi: 10.1126/scisignal.2004088
33. Grieco M, Santoro M, Berlingieri MT, Melillo RM, Donghi R, Bongarzone I, et al. Ptc is a novel rearranged form of the ret proto-oncogene and is frequently detected in vivo in human thyroid papillary carcinomas. Cell (1990) 60(4):557–63. doi: 10.1016/0092-8674(90)90659-3
34. Ou SI, Zhu VW. Catalog of 5' fusion partners in ret+ nsclc circa 2020. JTO Clin Res Rep (2020) 1(2):100037. doi: 10.1016/j.jtocrr.2020.100037
35. Nacu S, Yuan W, Kan Z, Bhatt D, Rivers CS, Stinson J, et al. Deep rna sequencing analysis of readthrough gene fusions in human prostate adenocarcinoma and reference samples. BMC Med Genomics (2011) 4:11. doi: 10.1186/1755-8794-4-11
36. Varley KE, Gertz J, Roberts BS, Davis NS, Bowling KM, Kirby MK, et al. Recurrent read-through fusion transcripts in breast cancer. Breast Cancer Res Treat (2014) 146(2):287–97. doi: 10.1007/s10549-014-3019-2
37. Kim HP, Cho GA, Han SW, Shin JY, Jeong EG, Song SH, et al. Novel fusion transcripts in human gastric cancer revealed by transcriptome analysis. Oncogene (2014) 33(47):5434–41. doi: 10.1038/onc.2013.490
38. Li H, Wang J, Ma X, Sklar J. Gene fusions and rna trans-splicing in normal and neoplastic human cells. Cell Cycle (2009) 8(2):218–22. doi: 10.4161/cc.8.2.7358
39. Jividen K, Li H. Chimeric rnas generated by intergenic splicing in normal and cancer cells. Genes Chromosomes Cancer (2014) 53(12):963–71. doi: 10.1002/gcc.22207
40. Li AY, McCusker MG, Russo A, Scilla KA, Gittens A, Arensmeyer K, et al. Ret fusions in solid tumors. Cancer Treat Rev (2019) 81:101911. doi: 10.1016/j.ctrv.2019.101911
41. Kato S, Subbiah V, Marchlik E, Elkin SK, Carter JL, Kurzrock R. Ret aberrations in diverse cancers: Next-generation sequencing of 4,871 patients. Clin Cancer Res (2017) 23(8):1988–97. doi: 10.1158/1078-0432.CCR-16-1679
42. Lanzi C, Borrello MG, Bongarzone I, Migliazza A, Fusco A, Grieco M, et al. Identification of the product of two oncogenic rearranged forms of the ret proto-oncogene in papillary thyroid carcinomas. Oncogene (1992) 7(11):2189–94.
43. Santoro M, Dathan NA, Berlingieri MT, Bongarzone I, Paulin C, Grieco M, et al. Molecular characterization of Ret/Ptc3; a novel rearranged version of the retproto-oncogene in a human thyroid papillary carcinoma. Oncogene (1994) 9(2):509–16.
44. Tong Q, Li Y, Smanik PA, Fithian LJ, Xing S, Mazzaferri EL, et al. Characterization of the promoter region and oligomerization domain of H4 (D10s170), a gene frequently rearranged with the ret proto-oncogene. Oncogene (1995) 10(9):1781–7.
45. Santoro M, Chiappetta G, Cerrato A, Salvatore D, Zhang L, Manzo G, et al. Development of thyroid papillary carcinomas secondary to tissue-specific expression of the Ret/Ptc1 oncogene in transgenic mice. Oncogene (1996) 12(8):1821–6.
46. Zhu Z, Ciampi R, Nikiforova MN, Gandhi M, Nikiforov YE. Prevalence of Ret/Ptc rearrangements in thyroid papillary carcinomas: Effects of the detection methods and genetic heterogeneity. J Clin Endocrinol Metab (2006) 91(9):3603–10. doi: 10.1210/jc.2006-1006
47. Rhoden KJ, Unger K, Salvatore G, Yilmaz Y, Vovk V, Chiappetta G, et al. Ret/Papillary thyroid cancer rearrangement in nonneoplastic thyrocytes: Follicular cells of hashimoto's thyroiditis share low-level recombination events with a subset of papillary carcinoma. J Clin Endocrinol Metab (2006) 91(6):2414–23. doi: 10.1210/jc.2006-0240
48. Santoro M, Moccia M, Federico G, Carlomagno F. Ret gene fusions in malignancies of the thyroid and other tissues. Genes (Basel) (2020) 11(4):424–41. doi: 10.3390/genes11040424
49. Paratala BS, Chung JH, Williams CB, Yilmazel B, Petrosky W, Williams K, et al. Ret rearrangements are actionable alterations in breast cancer. Nat Commun (2018) 9(1):4821. doi: 10.1038/s41467-018-07341-4
50. Morandi A, Plaza-Menacho I, Isacke CM. Ret in breast cancer: Functional and therapeutic implications. Trends Mol Med (2011) 17(3):149–57. doi: 10.1016/j.molmed.2010.12.007
51. Lipson D, Capelletti M, Yelensky R, Otto G, Parker A, Jarosz M, et al. Identification of new alk and ret gene fusions from colorectal and lung cancer biopsies. Nat Med (2012) 18(3):382–4. doi: 10.1038/nm.2673
52. Le Rolle AF, Klempner SJ, Garrett CR, Seery T, Sanford EM, Balasubramanian S, et al. Identification and characterization of ret fusions in advanced colorectal cancer. Oncotarget (2015) 6(30):28929–37. doi: 10.18632/oncotarget.4325
53. Pietrantonio F, Di Nicolantonio F, Schrock AB, Lee J, Morano F, Fuca G, et al. Ret fusions in a small subset of advanced colorectal cancers at risk of being neglected. Ann Oncol (2018) 29(6):1394–401. doi: 10.1093/annonc/mdy090
54. Kim SY, Oh SO, Kim K, Lee J, Kang S, Kim KM, et al. Ncoa4-ret fusion in colorectal cancer: Therapeutic challenge using patient-derived tumor cell lines. J Cancer (2018) 9(17):3032–7. doi: 10.7150/jca.26256
55. Wang K, Russell JS, McDermott JD, Elvin JA, Khaira D, Johnson A, et al. Profiling of 149 salivary duct carcinomas, carcinoma ex pleomorphic adenomas, and adenocarcinomas, not otherwise specified reveals actionable genomic alterations. Clin Cancer Res (2016) 22(24):6061–8. doi: 10.1158/1078-0432.CCR-15-2568
56. Weinreb I, Bishop JA, Chiosea SI, Seethala RR, Perez-Ordonez B, Zhang L, et al. Recurrent ret gene rearrangements in intraductal carcinomas of salivary gland. Am J Surg Pathol (2018) 42(4):442–52. doi: 10.1097/PAS.0000000000000952
57. Skalova A, Ptakova N, Santana T, Agaimy A, Ihrler S, Uro-Coste E, et al. Ncoa4-ret and Trim27-ret are characteristic gene fusions in salivary intraductal carcinoma, including invasive and metastatic tumors: Is "Intraductal" correct? Am J Surg Pathol (2019) 43(10):1303–13. doi: 10.1097/PAS.0000000000001301
58. Wang R, Hu H, Pan Y, Li Y, Ye T, Li C, et al. Ret fusions define a unique molecular and clinicopathologic subtype of non-Small-Cell lung cancer. J Clin Oncol (2012) 30(35):4352–9. doi: 10.1200/JCO.2012.44.1477
59. Tsuta K, Kohno T, Yoshida A, Shimada Y, Asamura H, Furuta K, et al. Ret-rearranged non-Small-Cell lung carcinoma: A clinicopathological and molecular analysis. Br J Cancer (2014) 110(6):1571–8. doi: 10.1038/bjc.2014.36
60. Lin JJ, Liu SV, McCoach CE, Zhu VW, Tan AC, Yoda S, et al. Mechanisms of resistance to selective ret tyrosine kinase inhibitors in ret fusion-positive non-Small-Cell lung cancer. Ann Oncol (2020) 31(12):1725–33. doi: 10.1016/j.annonc.2020.09.015
61. Nelson-Taylor SK, Le AT, Yoo M, Schubert L, Mishall KM, Doak A, et al. Resistance to ret-inhibition in ret-rearranged nsclc is mediated by reactivation of Ras/Mapk signaling. Mol Cancer Ther (2017) 16(8):1623–33. doi: 10.1158/1535-7163.MCT-17-0008
62. Wang H, Wang Z, Zhang G, Zhang M, Zhang X, Li H, et al. Driver genes as predictive indicators of brain metastasis in patients with advanced nsclc: Egfr, alk, and ret gene mutations. Cancer Med (2020) 9(2):487–95. doi: 10.1002/cam4.2706
63. Bongarzone I, Monzini N, Borrello MG, Carcano C, Ferraresi G, Arighi E, et al. Molecular characterization of a thyroid tumor-specific transforming sequence formed by the fusion of ret tyrosine kinase and the regulatory subunit ri alpha of cyclic amp-dependent protein kinase a. Mol Cell Biol (1993) 13(1):358–66. doi: 10.1128/mcb.13.1.358-366.1993
64. Sozzi G, Bongarzone I, Miozzo M, Borrello MG, Blutti MG, Pilotti S, et al. A T(10;17) translocation creates the Ret/Ptc2 chimeric transforming sequence in papillary thyroid carcinoma. Genes Chromosomes Cancer (1994) 9(4):244–50. doi: 10.1002/gcc.2870090404
65. Takeuchi K, Soda M, Togashi Y, Suzuki R, Sakata S, Hatano S, et al. Ret, Ros1 and alk fusions in lung cancer. Nat Med (2012) 18(3):378–81. doi: 10.1038/nm.2658
66. Kohno T, Ichikawa H, Totoki Y, Yasuda K, Hiramoto M, Nammo T, et al. Kif5b-ret fusions in lung adenocarcinoma. Nat Med (2012) 18(3):375–7. doi: 10.1038/nm.2644
67. Ju YS, Lee WC, Shin JY, Lee S, Bleazard T, Won JK, et al. A transforming Kif5b and ret gene fusion in lung adenocarcinoma revealed from whole-genome and transcriptome sequencing. Genome Res (2012) 22(3):436–45. doi: 10.1101/gr.133645.111
68. Grubbs EG, Ng PK, Bui J, Busaidy NL, Chen K, Lee JE, et al. Ret fusion as a novel driver of medullary thyroid carcinoma. J Clin Endocrinol Metab (2015) 100(3):788–93. doi: 10.1210/jc.2014-4153
69. Seethala RR, Stenman G. Update from the 4th edition of the world health organization classification of head and neck tumours: Tumors of the salivary gland. Head Neck Pathol (2017) 11(1):55–67. doi: 10.1007/s12105-017-0795-0
70. Skalova A, Vanecek T, Uro-Coste E, Bishop JA, Weinreb I, Thompson LDR, et al. Molecular profiling of salivary gland intraductal carcinoma revealed a subset of tumors harboring Ncoa4-ret and novel Trim27-ret fusions: A report of 17 cases. Am J Surg Pathol (2018) 42(11):1445–55. doi: 10.1097/PAS.0000000000001133
71. Skalova A, Vanecek T, Martinek P, Weinreb I, Stevens TM, Simpson RHW, et al. Molecular profiling of mammary analog secretory carcinoma revealed a subset of tumors harboring a novel Etv6-ret translocation: Report of 10 cases. Am J Surg Pathol (2018) 42(2):234–46. doi: 10.1097/PAS.0000000000000972
72. Black M, Liu CZ, Onozato M, Iafrate AJ, Darvishian F, Jour G, et al. Concurrent identification of novel egfr-Sept14 fusion and Etv6-ret fusion in secretory carcinoma of the salivary gland. Head Neck Pathol (2020) 14(3):817–21. doi: 10.1007/s12105-019-01074-6
73. Petersson F, Michal M, Ptakova N, Skalova A, Michal M. Salivary gland mucinous adenocarcinoma with minor (Mammary analogue) secretory and low-grade in situ carcinoma components sharing the same Etv6-ret translocation and with no other molecular genetic aberrations detected on ngs analysis. Appl Immunohistochem Mol Morphol (2020) 28(6):e53. doi: 10.1097/PAI.0000000000000806
74. Bishop JA, Nakaguro M, Whaley RD, Ogura K, Imai H, Laklouk I, et al. Oncocytic intraductal carcinoma of salivary glands: A distinct variant with Trim33-ret fusions and braf V600e mutations. Histopathology (2021) 79(3):338–46. doi: 10.1111/his.14296
75. Ma J, Wang B, Meng E, Meng X. Case report: Identification of Erc1-ret fusion in a patient with pancreatic ductal adenocarcinoma. Gland Surg (2021) 10(9):2874–9. doi: 10.21037/gs-21-469
76. Arlt DH, Baur B, Wagner B, Hoppner W. A novel type of mutation in the cysteine rich domain of the ret receptor causes ligand independent activation. Oncogene (2000) 19(30):3445–8. doi: 10.1038/sj.onc.1203688
77. Donis-Keller H, Dou S, Chi D, Carlson KM, Toshima K, Lairmore TC, et al. Mutations in the ret proto-oncogene are associated with men 2a and fmtc. Hum Mol Genet (1993) 2(7):851–6. doi: 10.1093/hmg/2.7.851
78. Mulligan LM, Eng C, Healey CS, Clayton D, Kwok JB, Gardner E, et al. Specific mutations of the ret proto-oncogene are related to disease phenotype in men 2a and fmtc. Nat Genet (1994) 6(1):70–4. doi: 10.1038/ng0194-70
79. Rossel M, Pasini A, Chappuis S, Geneste O, Fournier L, Schuffenecker I, et al. Distinct biological properties of two ret isoforms activated by men 2a and men 2b mutations. Oncogene (1997) 14(3):265–75. doi: 10.1038/sj.onc.1200831
80. Segouffin-Cariou C, Billaud M. Transforming ability of Men2a-ret requires activation of the phosphatidylinositol 3-Kinase/Akt signaling pathway. J Biol Chem (2000) 275(5):3568–76. doi: 10.1074/jbc.275.5.3568
81. Antinolo G, Marcos I, Fernandez RM, Romero M, Borrego S. A novel germline point mutation, C.2304 G–>T, in codon 768 of the ret proto-oncogene in a patient with medullary thyroid carcinoma. Am J Med Genet (2002) 110(1):85–7. doi: 10.1002/ajmg.10399
82. Cranston AN, Carniti C, Oakhill K, Radzio-Andzelm E, Stone EA, McCallion AS, et al. Ret is constitutively activated by novel tandem mutations that alter the active site resulting in multiple endocrine neoplasia type 2b. Cancer Res (2006) 66(20):10179–87. doi: 10.1158/0008-5472.CAN-06-0884
83. Carlomagno F, Guida T, Anaganti S, Vecchio G, Fusco A, Ryan AJ, et al. Disease associated mutations at valine 804 in the ret receptor tyrosine kinase confer resistance to selective kinase inhibitors. Oncogene (2004) 23(36):6056–63. doi: 10.1038/sj.onc.1207810
84. Gimm O, Marsh DJ, Andrew SD, Frilling A, Dahia PL, Mulligan LM, et al. Germline dinucleotide mutation in codon 883 of the ret proto-oncogene in multiple endocrine neoplasia type 2b without codon 918 mutation. J Clin Endocrinol Metab (1997) 82(11):3902–4. doi: 10.1210/jcem.82.11.4508
85. Gujral TS, Singh VK, Jia Z, Mulligan LM. Molecular mechanisms of ret receptor-mediated oncogenesis in multiple endocrine neoplasia 2b. Cancer Res (2006) 66(22):10741–9. doi: 10.1158/0008-5472.CAN-06-3329
86. Iwashita T, Kato M, Murakami H, Asai N, Ishiguro Y, Ito S, et al. Biological and biochemical properties of ret with kinase domain mutations identified in multiple endocrine neoplasia type 2b and familial medullary thyroid carcinoma. Oncogene (1999) 18(26):3919–22. doi: 10.1038/sj.onc.1202742
87. Maqdasy S, Costes-Chalret N, Batisse-Lignier M, Baron S, Tauveron I. Prostate adenocarcinoma in a young patient with multiple endocrine neoplasia 2b. Ann Endocrinol (Paris) (2018) 79(2):86–9. doi: 10.1016/j.ando.2017.09.002
88. Carlomagno F, Guida T, Anaganti S, Provitera L, Kjaer S, McDonald NQ, et al. Identification of tyrosine 806 as a molecular determinant of ret kinase sensitivity to Zd6474. Endocr Relat Cancer (2009) 16(1):233–41. doi: 10.1677/ERC-08-0213
89. Carlson KM, Dou S, Chi D, Scavarda N, Toshima K, Jackson CE, et al. Single missense mutation in the tyrosine kinase catalytic domain of the ret protooncogene is associated with multiple endocrine neoplasia type 2b. Proc Natl Acad Sci U.S.A. (1994) 91(4):1579–83. doi: 10.1073/pnas.91.4.1579
90. Liu X, Shen T, Mooers BHM, Hilberg F, Wu J. Drug resistance profiles of mutations in the ret kinase domain. Br J Pharmacol (2018) 175(17):3504–15. doi: 10.1111/bph.14395
91. Cabanillas ME, McFadden DG, Durante C. Thyroid cancer. Lancet (2016) 388(10061):2783–95. doi: 10.1016/S0140-6736(16)30172-6
92. Lemoine NR, Mayall ES, Wyllie FS, Farr CJ, Hughes D, Padua RA, et al. Activated ras oncogenes in human thyroid cancers. Cancer Res (1988) 48(16):4459–63.
93. Suarez HG, Du Villard JA, Caillou B, Schlumberger M, Tubiana M, Parmentier C, et al. Detection of activated ras oncogenes in human thyroid carcinomas. Oncogene (1988) 2(4):403–6.
94. Lemoine NR, Mayall ES, Wyllie FS, Williams ED, Goyns M, Stringer B, et al. High frequency of ras oncogene activation in all stages of human thyroid tumorigenesis. Oncogene (1989) 4(2):159–64.
95. Fusco A, Grieco M, Santoro M, Berlingieri MT, Pilotti S, Pierotti MA, et al. A new oncogene in human thyroid papillary carcinomas and their lymph-nodal metastases. Nature (1987) 328(6126):170–2. doi: 10.1038/328170a0
96. Ishizaka Y, Itoh F, Tahira T, Ikeda I, Ogura T, Sugimura T, et al. Presence of aberrant transcripts of ret proto-oncogene in a human papillary thyroid carcinoma cell line. Jpn J Cancer Res (1989) 80(12):1149–52. doi: 10.1111/j.1349-7006.1989.tb01645.x
97. Fabien N, Paulin C, Santoro M, Berger N, Grieco M, Galvain D, et al. Detection of ret oncogene activation in human papillary thyroid carcinomas by in situ hybridisation. Br J Cancer (1992) 66(6):1094–8. doi: 10.1038/bjc.1992.416
98. Grieco M, Cerrato A, Santoro M, Fusco A, Melillo RM, Vecchio G. Cloning and characterization of H4 (D10s170), a gene involved in ret rearrangements in vivo. Oncogene (1994) 9(9):2531–5.
99. Jhiang SM, Smanik PA, Mazzaferri EL. Development of a single-step duplex rt-pcr detecting different forms of ret activation, and identification of the third form of in vivo ret activation in human papillary thyroid carcinoma. Cancer Lett (1994) 78(1-3):69–76. doi: 10.1016/0304-3835(94)90033-7
100. Bongarzone I, Butti MG, Coronelli S, Borrello MG, Santoro M, Mondellini P, et al. Frequent activation of ret protooncogene by fusion with a new activating gene in papillary thyroid carcinomas. Cancer Res (1994) 54(11):2979–85.
101. Ishizaka Y, Shima H, Sugimura T, Nagao M. Detection of phosphorylated rettpc oncogene product in cytoplasm. Oncogene (1992) 7(7):1441–4.
102. Jhiang SM, Sagartz JE, Tong Q, Parker-Thornburg J, Capen CC, Cho JY, et al. Targeted expression of the Ret/Ptc1 oncogene induces papillary thyroid carcinomas. Endocrinology (1996) 137(1):375–8. doi: 10.1210/endo.137.1.8536638
103. Viglietto G, Chiappetta G, Martinez-Tello FJ, Fukunaga FH, Tallini G, Rigopoulou D, et al. Ret/Ptc oncogene activation is an early event in thyroid carcinogenesis. Oncogene (1995) 11(6):1207–10.
104. Ito T, Seyama T, Iwamoto KS, Hayashi T, Mizuno T, Tsuyama N, et al. In vitro irradiation is able to cause ret oncogene rearrangement. Cancer Res (1993) 53(13):2940–3.
105. Klugbauer S, Lengfelder E, Demidchik EP, Rabes HM. High prevalence of ret rearrangement in thyroid tumors of children from Belarus after the Chernobyl reactor accident. Oncogene (1995) 11(12):2459–67.
106. Thomas GA, Bunnell H, Cook HA, Williams ED, Nerovnya A, Cherstvoy ED, et al. High prevalence of Ret/Ptc rearrangements in Ukrainian and belarussian post-Chernobyl thyroid papillary carcinomas: A strong correlation between Ret/Ptc3 and the solid-follicular variant. J Clin Endocrinol Metab (1999) 84(11):4232–8. doi: 10.1210/jcem.84.11.6129
107. Bounacer A, Wicker R, Caillou B, Cailleux AF, Sarasin A, Schlumberger M, et al. High prevalence of activating ret proto-oncogene rearrangements, in thyroid tumors from patients who had received external radiation. Oncogene (1997) 15(11):1263–73. doi: 10.1038/sj.onc.1200206
108. Jhiang SM. The ret proto-oncogene in human cancers. Oncogene (2000) 19(49):5590–7. doi: 10.1038/sj.onc.1203857
109. Santoro M, Carlomagno F, Hay ID, Herrmann MA, Grieco M, Melillo R, et al. Ret oncogene activation in human thyroid neoplasms is restricted to the papillary cancer subtype. J Clin Invest (1992) 89(5):1517–22. doi: 10.1172/JCI115743
110. Nikiforov YE, Nikiforova MN. Molecular genetics and diagnosis of thyroid cancer. Nat Rev Endocrinol (2011) 7(10):569–80. doi: 10.1038/nrendo.2011.142
111. Rhoden KJ, Johnson C, Brandao G, Howe JG, Smith BR, Tallini G. Real-time quantitative rt-pcr identifies distinct c-ret, Ret/Ptc1 and Ret/Ptc3 expression patterns in papillary thyroid carcinoma. Lab Invest (2004) 84(12):1557–70. doi: 10.1038/labinvest.3700198
112. Ceolin L, Duval M, Benini AF, Ferreira CV, Maia AL. Medullary thyroid carcinoma beyond surgery: Advances, challenges, and perspectives. Endocr Relat Cancer (2019) 26(9):R499–518. doi: 10.1530/ERC-18-0574
113. Sipple JH. The association of pheochromocytoma with carcinoma of the thyroid gland. Am J Med (1961) 31(1):163–6. doi: 10.1016/0002-9343(61)90234-0
114. van Treijen MJC, de Vries LH, Hertog D, Vriens MR, Verrijn Stuart AA, van Nesselrooij BPM, et al. Multiple endocrine neoplasia type 2. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dhatariya K, et al, editors. Endotext. South Dartmouth MA: MedText.com, Inc. (2000).
115. Verga U, Fugazzola L, Cambiaghi S, Pritelli C, Alessi E, Cortelazzi D, et al. Frequent association between men 2a and cutaneous lichen amyloidosis. Clin Endocrinol (Oxf) (2003) 59(2):156–61. doi: 10.1046/j.1365-2265.2003.01782.x
116. Yamamoto M, Miki T, Tanaka N, Miya A, Shin E, Karakawa K, et al. Tight linkage of the ret proto-oncogene with the multiple endocrine neoplasia type 2a locus. Jpn J Clin Oncol (1991) 21(3):149–52.
117. Santoro M, Carlomagno F, Romano A, Bottaro DP, Dathan NA, Grieco M, et al. Activation of ret as a dominant transforming gene by germline mutations of Men2a and Men2b. Science (1995) 267(5196):381–3. doi: 10.1126/science.7824936
118. Seri M, Celli I, Betsos N, Claudiani F, Camera G, Romeo G. A Cys634gly substitution of the ret proto-oncogene in a family with recurrence of multiple endocrine neoplasia type 2a and cutaneous lichen amyloidosis. Clin Genet (1997) 51(2):86–90. doi: 10.1111/j.1399-0004.1997.tb02425.x
119. Decker RA, Peacock ML. Occurrence of men 2a in familial hirschsprung's disease: A new indication for genetic testing of the ret proto-oncogene. J Pediatr Surg (1998) 33(2):207–14. doi: 10.1016/s0022-3468(98)90433-x
120. Decker RA, Peacock ML, Watson P. Hirschsprung disease in men 2a: Increased spectrum of ret exon 10 genotypes and strong genotype-phenotype correlation. Hum Mol Genet (1998) 7(1):129–34. doi: 10.1093/hmg/7.1.129
121. Moore SW, Zaahl M. Familial associations in medullary thyroid carcinoma with hirschsprung disease: The role of the ret-C620 "Janus" genetic variation. J Pediatr Surg (2010) 45(2):393–6. doi: 10.1016/j.jpedsurg.2009.10.080
122. Mulligan LM, Marsh DJ, Robinson BG, Schuffenecker I, Zedenius J, Lips CJ, et al. Genotype-phenotype correlation in multiple endocrine neoplasia type 2: Report of the international ret mutation consortium. J Intern Med (1995) 238(4):343–6. doi: 10.1111/j.1365-2796.1995.tb01208.x
123. Santoro M, Rosati R, Grieco M, Berlingieri MT, D'Amato GL, de Franciscis V, et al. The ret proto-oncogene is consistently expressed in human pheochromocytomas and thyroid medullary carcinomas. Oncogene (1990) 5(10):1595–8.
124. Hofstra RM, Landsvater RM, Ceccherini I, Stulp RP, Stelwagen T, Luo Y, et al. A mutation in the ret proto-oncogene associated with multiple endocrine neoplasia type 2b and sporadic medullary thyroid carcinoma. Nature (1994) 367(6461):375–6. doi: 10.1038/367375a0
125. Dixit A, Torkamani A, Schork NJ, Verkhivker G. Computational modeling of structurally conserved cancer mutations in the ret and met kinases: The impact on protein structure, dynamics, and stability. Biophys J (2009) 96(3):858–74. doi: 10.1016/j.bpj.2008.10.041
126. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin (2022) 72(1):7–33. doi: 10.3322/caac.21708
127. Molina JR, Yang P, Cassivi SD, Schild SE, Adjei AA. Non-small cell lung cancer: Epidemiology, risk factors, treatment, and survivorship. Mayo Clin Proc (2008) 83(5):584–94. doi: 10.4065/83.5.584
128. Duma N, Santana-Davila R, Molina JR. Non-small cell lung cancer: Epidemiology, screening, diagnosis, and treatment. Mayo Clin Proc (2019) 94(8):1623–40. doi: 10.1016/j.mayocp.2019.01.013
129. Tamura T, Kurishima K, Nakazawa K, Kagohashi K, Ishikawa H, Satoh H, et al. Specific organ metastases and survival in metastatic non-Small-Cell lung cancer. Mol Clin Oncol (2015) 3(1):217–21. doi: 10.3892/mco.2014.410
130. Waqar SN, Samson PP, Robinson CG, Bradley J, Devarakonda S, Du L, et al. Non-Small-Cell lung cancer with brain metastasis at presentation. Clin Lung Cancer (2018) 19(4):e373–e9. doi: 10.1016/j.cllc.2018.01.007
131. Drilon AE, Filleron T, Bergagnini I, Milia J, Hatzoglou V, Velcheti V, et al. Baseline frequency of brain metastases and outcomes with multikinase inhibitor therapy in patients with ret-rearranged lung cancers. J Clin Oncol (2017) 35(15_suppl):9069–. doi: 10.1200/JCO.2017.35.15_suppl.9069
132. Drilon A, Wang L, Hasanovic A, Suehara Y, Lipson D, Stephens P, et al. Response to cabozantinib in patients with ret fusion-positive lung adenocarcinomas. Cancer Discovery (2013) 3(6):630–5. doi: 10.1158/2159-8290.CD-13-0035
133. Lin JJ, Kennedy E, Sequist LV, Brastianos PK, Goodwin KE, Stevens S, et al. Clinical activity of alectinib in advanced ret-rearranged non-small cell lung cancer. J Thorac Oncol (2016) 11(11):2027–32. doi: 10.1016/j.jtho.2016.08.126
134. Kodama T, Tsukaguchi T, Satoh Y, Yoshida M, Watanabe Y, Kondoh O, et al. Alectinib shows potent antitumor activity against ret-rearranged non-small cell lung cancer. Mol Cancer Ther (2014) 13(12):2910–8. doi: 10.1158/1535-7163.MCT-14-0274
135. Takeuchi S, Yanagitani N, Seto T, Hattori Y, Ohashi K, Morise M, et al. Phase 1/2 study of alectinib in ret-rearranged previously-treated non-small cell lung cancer (All-ret). Transl Lung Cancer Res (2021) 10(1):314–25. doi: 10.21037/tlcr-20-549
136. Horiike A, Takeuchi K, Uenami T, Kawano Y, Tanimoto A, Kaburaki K, et al. Sorafenib treatment for patients with ret fusion-positive non-small cell lung cancer. Lung Cancer (2016) 93:43–6. doi: 10.1016/j.lungcan.2015.12.011
137. Klempner SJ, Borghei A, Hakimian B, Ali SM, Ou SI. Intracranial activity of cabozantinib in met exon 14-positive nsclc with brain metastases. J Thorac Oncol (2017) 12(1):152–6. doi: 10.1016/j.jtho.2016.09.127
138. Zou Z, Xing P, Hao X, Wang Y, Song X, Shan L, et al. Intracranial efficacy of alectinib in alk-positive nsclc patients with cns metastases-a multicenter retrospective study. BMC Med (2022) 20(1):12. doi: 10.1186/s12916-021-02207-x
139. Velcheti V, Ahluwalia M. Intracranial and systemic response to alectinib in a patient with ret-Kif5b oncogenic fusion. J Thorac Oncol (2017) 12(7):e98–e9. doi: 10.1016/j.jtho.2017.03.007
140. Drilon A, Lin JJ, Filleron T, Ni A, Milia J, Bergagnini I, et al. Frequency of brain metastases and multikinase inhibitor outcomes in patients with ret-rearranged lung cancers. J Thorac Oncol (2018) 13(10):1595–601. doi: 10.1016/j.jtho.2018.07.004
141. Li F, Feng Y, Fang R, Fang Z, Xia J, Han X, et al. Identification of ret gene fusion by exon array analyses in "Pan-negative" lung cancer from never smokers. Cell Res (2012) 22(5):928–31. doi: 10.1038/cr.2012.27
142. Mechera R, Soysal SD, Piscuoglio S, Ng CKY, Zeindler J, Mujagic E, et al. Expression of ret is associated with oestrogen receptor expression but lacks prognostic significance in breast cancer. BMC Cancer (2019) 19(1):41. doi: 10.1186/s12885-018-5262-0
143. Esseghir S, Todd SK, Hunt T, Poulsom R, Plaza-Menacho I, Reis-Filho JS, et al. A role for glial cell derived neurotrophic factor induced expression by inflammatory cytokines and Ret/Gfr alpha 1 receptor up-regulation in breast cancer. Cancer Res (2007) 67(24):11732–41. doi: 10.1158/0008-5472.CAN-07-2343
144. Gattelli A, Nalvarte I, Boulay A, Roloff TC, Schreiber M, Carragher N, et al. Ret inhibition decreases growth and metastatic potential of estrogen receptor positive breast cancer cells. EMBO Mol Med (2013) 5(9):1335–50. doi: 10.1002/emmm.201302625
145. Ignatiadis M, Sotiriou C. Luminal breast cancer: From biology to treatment. Nat Rev Clin Oncol (2013) 10(9):494–506. doi: 10.1038/nrclinonc.2013.124
146. Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, et al. The structural basis of estrogen Receptor/Coactivator recognition and the antagonism of this interaction by tamoxifen. Cell (1998) 95(7):927–37. doi: 10.1016/s0092-8674(00)81717-1
147. Clarke R, Tyson JJ, Dixon JM. Endocrine resistance in breast cancer–an overview and update. Mol Cell Endocrinol (2015) 418 Pt 3:220–34. doi: 10.1016/j.mce.2015.09.035
148. Plaza-Menacho I, Morandi A, Robertson D, Pancholi S, Drury S, Dowsett M, et al. Targeting the receptor tyrosine kinase ret sensitizes breast cancer cells to tamoxifen treatment and reveals a role for ret in endocrine resistance. Oncogene (2010) 29(33):4648–57. doi: 10.1038/onc.2010.209
149. Sood A, Lang DK, Kaur R, Saini B, Arora S. Relevance of aromatase inhibitors in breast cancer treatment. Curr Top Med Chem (2021) 21(15):1319–36. doi: 10.2174/1568026621666210701143445
150. Zhao H, Zhou L, Shangguan AJ, Bulun SE. Aromatase expression and regulation in breast and endometrial cancer. J Mol Endocrinol (2016) 57(1):R19–33. doi: 10.1530/JME-15-0310
151. Hanamura T, Hayashi SI. Overcoming aromatase inhibitor resistance in breast cancer: Possible mechanisms and clinical applications. Breast Cancer (2018) 25(4):379–91. doi: 10.1007/s12282-017-0772-1
152. Morandi A, Martin LA, Gao Q, Pancholi S, Mackay A, Robertson D, et al. Gdnf-ret signaling in er-positive breast cancers is a key determinant of response and resistance to aromatase inhibitors. Cancer Res (2013) 73(12):3783–95. doi: 10.1158/0008-5472.CAN-12-4265
153. Boulay A, Breuleux M, Stephan C, Fux C, Brisken C, Fiche M, et al. The ret receptor tyrosine kinase pathway functionally interacts with the eralpha pathway in breast cancer. Cancer Res (2008) 68(10):3743–51. doi: 10.1158/0008-5472.CAN-07-5100
154. Frasor J, Stossi F, Danes JM, Komm B, Lyttle CR, Katzenellenbogen BS. Selective estrogen receptor modulators: Discrimination of agonistic versus antagonistic activities by gene expression profiling in breast cancer cells. Cancer Res (2004) 64(4):1522–33. doi: 10.1158/0008-5472.can-03-3326
155. Tozlu S, Girault I, Vacher S, Vendrell J, Andrieu C, Spyratos F, et al. Identification of novel genes that Co-cluster with estrogen receptor alpha in breast tumor biopsy specimens, using a Large-scale real-time reverse transcription-pcr approach. Endocr Relat Cancer (2006) 13(4):1109–20. doi: 10.1677/erc.1.01120
156. Wang C, Mayer JA, Mazumdar A, Brown PH. The rearranged during Transfection/Papillary thyroid carcinoma tyrosine kinase is an estrogen-dependent gene required for the growth of estrogen receptor positive breast cancer cells. Breast Cancer Res Treat (2012) 133(2):487–500. doi: 10.1007/s10549-011-1775-9
157. Stine ZE, McGaughey DM, Bessling SL, Li S, McCallion AS. Steroid hormone modulation of ret through two estrogen responsive enhancers in breast cancer. Hum Mol Genet (2011) 20(19):3746–56. doi: 10.1093/hmg/ddr291
158. Andreucci E, Francica P, Fearns A, Martin LA, Chiarugi P, Isacke CM, et al. Targeting the receptor tyrosine kinase ret in combination with aromatase inhibitors in er positive breast cancer xenografts. Oncotarget (2016) 7(49):80543–53. doi: 10.18632/oncotarget.11826
159. Chen W, Hoffmann AD, Liu H, Liu X. Organotropism: New insights into molecular mechanisms of breast cancer metastasis. NPJ Precis Oncol (2018) 2(1):4. doi: 10.1038/s41698-018-0047-0
160. Waks AG, Winer EP. Breast cancer treatment: A review. JAMA (2019) 321(3):288–300. doi: 10.1001/jama.2018.19323
161. Gardaneh M, Shojaei S, Kaviani A, Behnam B. Gdnf induces ret-Src-Her2-Dependent growth in trastuzumab-sensitive but src-independent growth in resistant breast tumor cells. Breast Cancer Res Treat (2017) 162(2):231–41. doi: 10.1007/s10549-016-4078-3
162. Pohlmann PR, Mayer IA, Mernaugh R. Resistance to trastuzumab in breast cancer. Clin Cancer Res (2009) 15(24):7479–91. doi: 10.1158/1078-0432.CCR-09-0636
163. Hatem R, Labiod D, Chateau-Joubert S, de Plater L, El Botty R, Vacher S, et al. Vandetanib as a potential new treatment for estrogen receptor-negative breast cancers. Int J Cancer (2016) 138(10):2510–21. doi: 10.1002/ijc.29974
164. Vareslija D, Priedigkeit N, Fagan A, Purcell S, Cosgrove N, O'Halloran PJ, et al. Transcriptome characterization of matched primary breast and brain metastatic tumors to detect novel actionable targets. J Natl Cancer Inst (2019) 111(4):388–98. doi: 10.1093/jnci/djy110
166. Thompson L. World health organization classification of tumours: Pathology and genetics of head and neck tumours. Ear Nose Throat J (2006) 85(2):74. doi: 10.1177/014556130608500201
167. Chen KT. Intraductal carcinoma of the minor salivary gland. J Laryngol Otol (1983) 97(2):189–91. doi: 10.1017/s002221510009397x
168. Kleinsasser O, Klein HJ, Hubner G. [Salivary duct carcinoma. a group of salivary gland tumors analogous to mammary duct carcinoma]. Arch Klin Exp Ohren Nasen Kehlkopfheilkd (1968) 192(1):100–5. doi: 10.1007/BF00301495
169. Chen KT, Hafez GR. Infiltrating salivary duct carcinoma. A Clinicopathologic Stud Five Cases Arch Otolaryngol (1981) 107(1):37–9. doi: 10.1001/archotol.1981.00790370039008
170. Fayemi AO, Toker C. Salivary duct carcinoma. Arch Otolaryngol (1974) 99(5):366–8. doi: 10.1001/archotol.1974.00780030378009
171. Delgado R, Klimstra D, Albores-Saavedra J. Low grade salivary duct carcinoma. a distinctive variant with a low grade histology and a predominant intraductal growth pattern. Cancer (1996) 78(5):958–67. doi: 10.1002/(SICI)1097-0142(19960901)78:5<958::AID-CNCR4>3.0.CO;2-8
172. Giovacchini F, Bensi C, Belli S, Laurenti ME, Mandarano M, Paradiso D, et al. Low-grade intraductal carcinoma of salivary glands: A systematic review of this rare entity. J Oral Biol Craniofac Res (2019) 9(1):96–110. doi: 10.1016/j.jobcr.2018.11.003
173. Fisch AS, Laklouk I, Nakaguro M, Nose V, Wirth LJ, Deschler DG, et al. Intraductal carcinoma of the salivary gland with Ncoa4-ret: Expanding the morphologic spectrum and an algorithmic diagnostic approach. Hum Pathol (2021) 114:74–89. doi: 10.1016/j.humpath.2021.05.004
174. Langan RC. Benign prostatic hyperplasia. Prim Care (2019) 46(2):223–32. doi: 10.1016/j.pop.2019.02.003
175. Kim HL, Yang XJ. Prevalence of high-grade prostatic intraepithelial neoplasia and its relationship to serum prostate specific antigen. Int Braz J Urol (2002) 28(5):413–6. discussion 7.
176. Bostwick DG, Liu L, Brawer MK, Qian J. High-grade prostatic intraepithelial neoplasia. Rev Urol (2004) 6(4):171–9. doi: 10.1038/modpathol.3800053
177. Bostwick DG, Brawer MK. Prostatic intra-epithelial neoplasia and early invasion in prostate cancer. Cancer (1987) 59(4):788–94. doi: 10.1002/1097-0142(19870215)59:4<788::aid-cncr2820590421>3.0.co;2-i
178. Wang G, Zhao D, Spring DJ, DePinho RA. Genetics and biology of prostate cancer. Genes Dev (2018) 32(17-18):1105–40. doi: 10.1101/gad.315739.118
179. Robinson D, He F, Pretlow T, Kung HJ. A tyrosine kinase profile of prostate carcinoma. Proc Natl Acad Sci U.S.A. (1996) 93(12):5958–62. doi: 10.1073/pnas.93.12.5958
180. Dawson DM, Lawrence EG, MacLennan GT, Amini SB, Kung HJ, Robinson D, et al. Altered expression of ret proto-oncogene product in prostatic intraepithelial neoplasia and prostate cancer. J Natl Cancer Inst (1998) 90(7):519–23. doi: 10.1093/jnci/90.7.519
181. Ban K, Feng S, Shao L, Ittmann M. Ret signaling in prostate cancer. Clin Cancer Res (2017) 23(16):4885–96. doi: 10.1158/1078-0432.CCR-17-0528
182. Huber RM, Lucas JM, Gomez-Sarosi LA, Coleman I, Zhao S, Coleman R, et al. DNA Damage induces gdnf secretion in the tumor microenvironment with paracrine effects promoting prostate cancer treatment resistance. Oncotarget (2015) 6(4):2134–47. doi: 10.18632/oncotarget.3040
183. He S, Chen CH, Chernichenko N, He S, Bakst RL, Barajas F, et al. Gfralpha1 released by nerves enhances cancer cell perineural invasion through gdnf-ret signaling. Proc Natl Acad Sci U.S.A. (2014) 111(19):E2008–17. doi: 10.1073/pnas.1402944111
184. Paratcha G, Ledda F, Baars L, Coulpier M, Besset V, Anders J, et al. Released Gfralpha1 potentiates downstream signaling, neuronal survival, and differentiation Via a novel mechanism of recruitment of c-ret to lipid rafts. Neuron (2001) 29(1):171–84. doi: 10.1016/s0896-6273(01)00188-x
185. VanDeusen HR, Ramroop JR, Morel KL, Bae SY, Sheahan AV, Sychev Z, et al. Targeting ret kinase in neuroendocrine prostate cancer. Mol Cancer Res (2020) 18(8):1176–88. doi: 10.1158/1541-7786.MCR-19-1245
186. Consortium APG. Aacr project genie: Powering precision medicine through an international consortium. Cancer Discovery (2017) 7(8):818–31. doi: 10.1158/2159-8290.CD-17-0151
187. Cunningham D, Atkin W, Lenz HJ, Lynch HT, Minsky B, Nordlinger B, et al. Colorectal cancer. Lancet (2010) 375(9719):1030–47. doi: 10.1016/S0140-6736(10)60353-4
188. Janne PA, Mayer RJ. Chemoprevention of colorectal cancer. N Engl J Med (2000) 342(26):1960–8. doi: 10.1056/NEJM200006293422606
189. Riihimaki M, Hemminki A, Sundquist J, Hemminki K. Patterns of metastasis in colon and rectal cancer. Sci Rep (2016) 6:29765. doi: 10.1038/srep29765
190. Ishizaka Y, Tahira T, Ochiai M, Ikeda I, Sugimura T, Nagao M. Molecular cloning and characterization of human ret-ii oncogene. Oncogene Res (1988) 3(2):193–7.
191. Ishizaka Y, Ochiai M, Tahira T, Sugimura T, Nagao M. Activation of the ret-ii oncogene without a sequence encoding a transmembrane domain and transforming activity of two ret-ii oncogene products differing in carboxy-termini due to alternative splicing. Oncogene (1989) 4(6):789–94.
192. Luo Y, Tsuchiya KD, Il Park D, Fausel R, Kanngurn S, Welcsh P, et al. Ret is a potential tumor suppressor gene in colorectal cancer. Oncogene (2013) 32(16):2037–47. doi: 10.1038/onc.2012.225
193. Ashkboos M, Nikbakht M, Zarinfard G, Soleimani M. Ret protein expression in colorectal cancer; an immunohistochemical assessment. Asian Pac J Cancer Prev (2021) 22(4):1019–23. doi: 10.31557/APJCP.2021.22.4.1019
194. Zeng Q, Cheng Y, Zhu Q, Yu Z, Wu X, Huang K, et al. The relationship between overexpression of glial cell-derived neurotrophic factor and its ret receptor with progression and prognosis of human pancreatic cancer. J Int Med Res (2008) 36(4):656–64. doi: 10.1177/147323000803600406
195. Amit M, Na'ara S, Fridman E, Vladovski E, Wasserman T, Milman N, et al. Ret, a targetable driver of pancreatic adenocarcinoma. Int J Cancer (2019) 144(12):3014–22. doi: 10.1002/ijc.32040
196. Ceyhan GO, Giese NA, Erkan M, Kerscher AG, Wente MN, Giese T, et al. The neurotrophic factor artemin promotes pancreatic cancer invasion. Ann Surg (2006) 244(2):274–81. doi: 10.1097/01.sla.0000217642.68697.55
197. Amit M, Na'ara S, Leider-Trejo L, Binenbaum Y, Kulish N, Fridman E, et al. Upregulation of ret induces perineurial invasion of pancreatic adenocarcinoma. Oncogene (2017) 36(23):3232–9. doi: 10.1038/onc.2016.483
198. Cavel O, Shomron O, Shabtay A, Vital J, Trejo-Leider L, Weizman N, et al. Endoneurial macrophages induce perineural invasion of pancreatic cancer cells by secretion of gdnf and activation of ret tyrosine kinase receptor. Cancer Res (2012) 72(22):5733–43. doi: 10.1158/0008-5472.CAN-12-0764
199. Guan L, Li Z, Xie F, Pang Y, Zhang C, Tang H, et al. Oncogenic and drug-sensitive ret mutations in human epithelial ovarian cancer. J Exp Clin Cancer Res (2020) 39(1):53. doi: 10.1186/s13046-020-01557-3
200. Yakes FM, Chen J, Tan J, Yamaguchi K, Shi Y, Yu P, et al. Cabozantinib (Xl184), a novel met and Vegfr2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol Cancer Ther (2011) 10(12):2298–308. doi: 10.1158/1535-7163.MCT-11-0264
201. Kurzrock R, Sherman SI, Ball DW, Forastiere AA, Cohen RB, Mehra R, et al. Activity of Xl184 (Cabozantinib), an oral tyrosine kinase inhibitor, in patients with medullary thyroid cancer. J Clin Oncol (2011) 29(19):2660–6. doi: 10.1200/JCO.2010.32.4145
202. Choueiri TK, Pal SK, McDermott DF, Morrissey S, Ferguson KC, Holland J, et al. A phase I study of cabozantinib (Xl184) in patients with renal cell cancer. Ann Oncol (2014) 25(8):1603–8. doi: 10.1093/annonc/mdu184
203. Drilon A, Rekhtman N, Arcila M, Wang L, Ni A, Albano M, et al. Cabozantinib in patients with advanced ret-rearranged non-Small-Cell lung cancer: An open-label, single-centre, phase 2, single-arm trial. Lancet Oncol (2016) 17(12):1653–60. doi: 10.1016/S1470-2045(16)30562-9
204. Schlumberger M, Elisei R, Muller S, Schoffski P, Brose M, Shah M, et al. Overall survival analysis of exam, a phase iii trial of cabozantinib in patients with radiographically progressive medullary thyroid carcinoma. Ann Oncol (2017) 28(11):2813–9. doi: 10.1093/annonc/mdx479
205. Sherman SI, Clary DO, Elisei R, Schlumberger MJ, Cohen EE, Schoffski P, et al. Correlative analyses of ret and ras mutations in a phase 3 trial of cabozantinib in patients with progressive, metastatic medullary thyroid cancer. Cancer (2016) 122(24):3856–64. doi: 10.1002/cncr.30252
206. Ciccarese C, Iacovelli R, Mosillo C, Tortora G. Exceptional response to cabozantinib of rapidly evolving brain metastases of renal cell carcinoma: A case report and review of the literature. Clin Genitourin Cancer (2018) 16(5):e1069–e71. doi: 10.1016/j.clgc.2018.06.005
207. Leavitt J, Copur MS. Fda approved uses of cabozantinib. Oncology (Williston Park) (2019) 33(9):1–2.
208. Matsui J, Yamamoto Y, Funahashi Y, Tsuruoka A, Watanabe T, Wakabayashi T, et al. E7080, a novel inhibitor that targets multiple kinases, has potent antitumor activities against stem cell factor producing human small cell lung cancer H146, based on angiogenesis inhibition. Int J Cancer (2008) 122(3):664–71. doi: 10.1002/ijc.23131
209. Matsui J, Funahashi Y, Uenaka T, Watanabe T, Tsuruoka A, Asada M. Multi-kinase inhibitor E7080 suppresses lymph node and lung metastases of human mammary breast tumor mda-Mb-231 Via inhibition of vascular endothelial growth factor-receptor (Vegf-r) 2 and vegf-R3 kinase. Clin Cancer Res (2008) 14(17):5459–65. doi: 10.1158/1078-0432.CCR-07-5270
210. Glen H, Mason S, Patel H, Macleod K, Brunton VG. E7080, a multi-targeted tyrosine kinase inhibitor suppresses tumor cell migration and invasion. BMC Cancer (2011) 11:309. doi: 10.1186/1471-2407-11-309
211. Yamamoto Y, Matsui J, Matsushima T, Obaishi H, Miyazaki K, Nakamura K, et al. Lenvatinib, an angiogenesis inhibitor targeting Vegfr/Fgfr, shows broad antitumor activity in human tumor xenograft models associated with microvessel density and pericyte coverage. Vasc Cell (2014) 6:18. doi: 10.1186/2045-824X-6-18
212. Okamoto K, Kodama K, Takase K, Sugi NH, Yamamoto Y, Iwata M, et al. Antitumor activities of the targeted multi-tyrosine kinase inhibitor lenvatinib (E7080) against ret gene fusion-driven tumor models. Cancer Lett (2013) 340(1):97–103. doi: 10.1016/j.canlet.2013.07.007
213. Tohyama O, Matsui J, Kodama K, Hata-Sugi N, Kimura T, Okamoto K, et al. Antitumor activity of lenvatinib (E7080): An angiogenesis inhibitor that targets multiple receptor tyrosine kinases in preclinical human thyroid cancer models. J Thyroid Res (2014) 2014:638747. doi: 10.1155/2014/638747
214. Hida T, Velcheti V, Reckamp KL, Nokihara H, Sachdev P, Kubota T, et al. A phase 2 study of lenvatinib in patients with ret fusion-positive lung adenocarcinoma. Lung Cancer (2019) 138:124–30. doi: 10.1016/j.lungcan.2019.09.011
215. Lenvatinib approved for certain thyroid cancers. Cancer Discovery (2015) 5(4):338. doi: 10.1158/2159-8290.CD-NB2015-029
216. Nair A, Reece K, Donoghue MB, Yuan WV, Rodriguez L, Keegan P, et al. Fda supplemental approval summary: Lenvatinib for the treatment of unresectable hepatocellular carcinoma. Oncologist (2021) 26(3):e484–e91. doi: 10.1002/onco.13566
217. Fda approves drug combo for kidney cancer. Cancer Discovery (2016) 6(7):687–8. doi: 10.1158/2159-8290.CD-NB2016-067
218. Arora S, Balasubramaniam S, Zhang W, Zhang L, Sridhara R, Spillman D, et al. Fda approval summary: Pembrolizumab plus lenvatinib for endometrial carcinoma, a collaborative international review under project orbis. Clin Cancer Res (2020) 26(19):5062–7. doi: 10.1158/1078-0432.CCR-19-3979
219. Hennequin LF, Stokes ES, Thomas AP, Johnstone C, Ple PA, Ogilvie DJ, et al. Novel 4-anilinoquinazolines with c-7 basic side chains: Design and structure activity relationship of a series of potent, orally active, vegf receptor tyrosine kinase inhibitors. J Med Chem (2002) 45(6):1300–12. doi: 10.1021/jm011022e
220. Ciardiello F, Caputo R, Damiano V, Caputo R, Troiani T, Vitagliano D, et al. Antitumor effects of Zd6474, a small molecule vascular endothelial growth factor receptor tyrosine kinase inhibitor, with additional activity against epidermal growth factor receptor tyrosine kinase. Clin Cancer Res (2003) 9(4):1546–56.
221. Wedge SR, Ogilvie DJ, Dukes M, Kendrew J, Chester R, Jackson JA, et al. Zd6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration. Cancer Res (2002) 62(16):4645–55.
222. Carlomagno F, Vitagliano D, Guida T, Ciardiello F, Tortora G, Vecchio G, et al. Zd6474, an orally available inhibitor of kdr tyrosine kinase activity, efficiently blocks oncogenic ret kinases. Cancer Res (2002) 62(24):7284–90.
223. Vidal M, Wells S, Ryan A, Cagan R. Zd6474 suppresses oncogenic ret isoforms in a drosophila model for type 2 multiple endocrine neoplasia syndromes and papillary thyroid carcinoma. Cancer Res (2005) 65(9):3538–41. doi: 10.1158/0008-5472.CAN-04-4561
224. Robinson BG, Paz-Ares L, Krebs A, Vasselli J, Haddad R. Vandetanib (100 mg) in patients with locally advanced or metastatic hereditary medullary thyroid cancer. J Clin Endocrinol Metab (2010) 95(6):2664–71. doi: 10.1210/jc.2009-2461
225. Wells SA Jr, Gosnell JE, Gagel RF, Moley J, Pfister D, Sosa JA, et al. Vandetanib for the treatment of patients with locally advanced or metastatic hereditary medullary thyroid cancer. J Clin Oncol (2010) 28(5):767–72. doi: 10.1200/JCO.2009.23.6604
226. Wells SA Jr, Robinson BG, Gagel RF, Dralle H, Fagin JA, Santoro M, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: A randomized, double-blind phase iii trial. J Clin Oncol (2012) 30(2):134–41. doi: 10.1200/JCO.2011.35.5040
227. Thornton K, Kim G, Maher VE, Chattopadhyay S, Tang S, Moon YJ, et al. Vandetanib for the treatment of symptomatic or progressive medullary thyroid cancer in patients with unresectable locally advanced or metastatic disease: U.S. food and drug administration drug approval summary. Clin Cancer Res (2012) 18(14):3722–30. doi: 10.1158/1078-0432.CCR-12-0411
228. Lyons JF, Wilhelm S, Hibner B, Bollag G. Discovery of a novel raf kinase inhibitor. Endocr Relat Cancer (2001) 8(3):219–25. doi: 10.1677/erc.0.0080219
229. Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, et al. Bay 43-9006 exhibits broad spectrum oral antitumor activity and targets the Raf/Mek/Erk pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res (2004) 64(19):7099–109. doi: 10.1158/0008-5472.CAN-04-1443
230. Carlomagno F, Anaganti S, Guida T, Salvatore G, Troncone G, Wilhelm SM, et al. Bay 43-9006 inhibition of oncogenic ret mutants. J Natl Cancer Inst (2006) 98(5):326–34. doi: 10.1093/jnci/djj069
231. Plaza-Menacho I, Mologni L, Sala E, Gambacorti-Passerini C, Magee AI, Links TP, et al. Sorafenib functions to potently suppress ret tyrosine kinase activity by direct enzymatic inhibition and promoting ret lysosomal degradation independent of proteasomal targeting. J Biol Chem (2007) 282(40):29230–40. doi: 10.1074/jbc.M703461200
232. Henderson YC, Ahn SH, Kang Y, Clayman GL. Sorafenib potently inhibits papillary thyroid carcinomas harboring Ret/Ptc1 rearrangement. Clin Cancer Res (2008) 14(15):4908–14. doi: 10.1158/1078-0432.CCR-07-1772
233. Mao WF, Shao MH, Gao PT, Ma J, Li HJ, Li GL, et al. The important roles of ret, Vegfr2 and the Raf/Mek/Erk pathway in cancer treatment with sorafenib. Acta Pharmacol Sin (2012) 33(10):1311–8. doi: 10.1038/aps.2012.76
234. Kane RC, Farrell AT, Saber H, Tang S, Williams G, Jee JM, et al. Sorafenib for the treatment of advanced renal cell carcinoma. Clin Cancer Res (2006) 12(24):7271–8. doi: 10.1158/1078-0432.CCR-06-1249
235. Lang L. Fda approves sorafenib for patients with inoperable liver cancer. Gastroenterology (2008) 134(2):379. doi: 10.1053/j.gastro.2007.12.037
236. McFarland DC, Misiukiewicz KJ. Sorafenib in radioactive iodine-refractory well-differentiated metastatic thyroid cancer. Onco Targets Ther (2014) 7:1291–9. doi: 10.2147/OTT.S49430
237. Wilhelm SM, Dumas J, Adnane L, Lynch M, Carter CA, Schutz G, et al. Regorafenib (Bay 73-4506): A new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int J Cancer (2011) 129(1):245–55. doi: 10.1002/ijc.25864
238. Mross K, Frost A, Steinbild S, Hedbom S, Buchert M, Fasol U, et al. A phase I dose-escalation study of regorafenib (Bay 73-4506), an inhibitor of oncogenic, angiogenic, and stromal kinases, in patients with advanced solid tumors. Clin Cancer Res (2012) 18(9):2658–67. doi: 10.1158/1078-0432.CCR-11-1900
239. Strumberg D, Scheulen ME, Schultheis B, Richly H, Frost A, Buchert M, et al. Regorafenib (Bay 73-4506) in advanced colorectal cancer: A phase I study. Br J Cancer (2012) 106(11):1722–7. doi: 10.1038/bjc.2012.153
240. George S, Wang Q, Heinrich MC, Corless CL, Zhu M, Butrynski JE, et al. Efficacy and safety of regorafenib in patients with metastatic and/or unresectable gi stromal tumor after failure of imatinib and sunitinib: A multicenter phase ii trial. J Clin Oncol (2012) 30(19):2401–7. doi: 10.1200/JCO.2011.39.9394
241. Eisen T, Joensuu H, Nathan PD, Harper PG, Wojtukiewicz MZ, Nicholson S, et al. Regorafenib for patients with previously untreated metastatic or unresectable renal-cell carcinoma: A single-group phase 2 trial. Lancet Oncol (2012) 13(10):1055–62. doi: 10.1016/S1470-2045(12)70364-9
242. Chen Z, Zhao Y, Yu Y, Pang JC, Woodfield SE, Tao L, et al. Small molecule inhibitor regorafenib inhibits ret signaling in neuroblastoma cells and effectively suppresses tumor growth in vivo. Oncotarget (2017) 8(61):104090–103. doi: 10.18632/oncotarget.22011
243. Wu RY, Kong PF, Xia LP, Huang Y, Li ZL, Tang YY, et al. Regorafenib promotes antitumor immunity Via inhibiting pd-L1 and Ido1 expression in melanoma. Clin Cancer Res (2019) 25(14):4530–41. doi: 10.1158/1078-0432.CCR-18-2840
244. Mendel DB, Laird AD, Xin X, Louie SG, Christensen JG, Li G, et al. In vivo antitumor activity of Su11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: Determination of a Pharmacokinetic/Pharmacodynamic relationship. Clin Cancer Res (2003) 9(1):327–37.
245. O'Farrell AM, Abrams TJ, Yuen HA, Ngai TJ, Louie SG, Yee KW, et al. Su11248 is a novel Flt3 tyrosine kinase inhibitor with potent activity in vitro and in vivo. Blood (2003) 101(9):3597–605. doi: 10.1182/blood-2002-07-2307
246. Abrams TJ, Lee LB, Murray LJ, Pryer NK, Cherrington JM. Su11248 inhibits kit and platelet-derived growth factor receptor beta in preclinical models of human small cell lung cancer. Mol Cancer Ther (2003) 2(5):471–8.
247. Abrams TJ, Murray LJ, Pesenti E, Holway VW, Colombo T, Lee LB, et al. Preclinical evaluation of the tyrosine kinase inhibitor Su11248 as a single agent and in combination with ”Standard of care” therapeutic agents for the treatment of breast cancer. Mol Cancer Ther (2003) 2(10):1011–21.
248. Osusky KL, Hallahan DE, Fu A, Ye F, Shyr Y, Geng L. The receptor tyrosine kinase inhibitor Su11248 impedes endothelial cell migration, tubule formation, and blood vessel formation in vivo, but has little effect on existing tumor vessels. Angiogenesis (2004) 7(3):225–33. doi: 10.1007/s10456-004-3149-y
249. Jeong WJ, Mo JH, Park MW, Choi IJ, An SY, Jeon EH, et al. Sunitinib inhibits papillary thyroid carcinoma with Ret/Ptc rearrangement but not braf mutation. Cancer Biol Ther (2011) 12(5):458–65. doi: 10.4161/cbt.12.5.16303
250. Sakamoto H, Tsukaguchi T, Hiroshima S, Kodama T, Kobayashi T, Fukami TA, et al. Ch5424802, a selective alk inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell (2011) 19(5):679–90. doi: 10.1016/j.ccr.2011.04.004
251. Lu L, Ghose AK, Quail MR, Albom MS, Durkin JT, Holskin BP, et al. Alk mutants in the kinase domain exhibit altered kinase activity and differential sensitivity to small molecule alk inhibitors. Biochemistry (2009) 48(16):3600–9. doi: 10.1021/bi8020923
252. Seto T, Kiura K, Nishio M, Nakagawa K, Maemondo M, Inoue A, et al. Ch5424802 (Ro5424802) for patients with alk-rearranged advanced non-Small-Cell lung cancer (Af-001jp study): A single-arm, open-label, phase 1-2 study. Lancet Oncol (2013) 14(7):590–8. doi: 10.1016/S1470-2045(13)70142-6
253. Larkins E, Blumenthal GM, Chen H, He K, Agarwal R, Gieser G, et al. Fda approval: Alectinib for the treatment of metastatic, alk-positive non-small cell lung cancer following crizotinib. Clin Cancer Res (2016) 22(21):5171–6. doi: 10.1158/1078-0432.CCR-16-1293
254. Gautschi O, Milia J, Filleron T, Wolf J, Carbone DP, Owen D, et al. Targeting ret in patients with ret-rearranged lung cancers: Results from the global, multicenter ret registry. J Clin Oncol (2017) 35(13):1403–10. doi: 10.1200/JCO.2016.70.9352
255. Gainor JF, Curigliano G, Kim DW, Lee DH, Besse B, Baik CS, et al. Pralsetinib for ret fusion-positive non-Small-Cell lung cancer (Arrow): A multi-cohort, open-label, phase 1/2 study. Lancet Oncol (2021) 22(7):959–69. doi: 10.1016/S1470-2045(21)00247-3
256. Subbiah V, Hu MI, Wirth LJ, Schuler M, Mansfield AS, Curigliano G, et al. Pralsetinib for patients with advanced or metastatic ret-altered thyroid cancer (Arrow): A multi-cohort, open-label, registrational, phase 1/2 study. Lancet Diabetes Endocrinol (2021) 9(8):491–501. doi: 10.1016/S2213-8587(21)00120-0
257. Besse B, Felip E, Kim ES, Clifford C, Louie-Gao M, Yagui-Beltran A, et al. Pul01.02 acceleret lung: A phase 3 study of first-line pralsetinib in patients with ret-fusion+ Advanced/Metastatic nsclc. J Thorac Oncol (2021) 16:S44–S5. doi: 10.1016/j.jtho.2020.10.087
258. Goto K, Oxnard GR, Tan DS-W, Loong HHF, Bauer TM, Gainor JF, et al. Selpercatinib (Loxo-292) in patients with ret-fusion+ non-small cell lung cancer. J Clin Oncol (2020) 38(15_suppl):3584. doi: 10.1200/JCO.2020.38.15_suppl.3584
259. Zheng X, Ji Q, Sun Y, Ge M, Zhang B, Cheng Y, et al. 29p libretto-321, a phase ii study of the efficacy and safety of selpercatinib in Chinese patients with advanced ret-altered thyroid cancer (Tc). Ann Oncol (2022) 33:S14. doi: 10.1016/j.annonc.2022.01.038
260. Solomon BJ, Zhou CC, Drilon A, Park K, Wolf J, Elamin Y, et al. Phase iii study of selpercatinib versus chemotherapy +/- pembrolizumab in untreated ret positive non-Small-Cell lung cancer. Future Oncol (2021) 17(7):763–73. doi: 10.2217/fon-2020-0935
261. Morgenstern DA, Mascarenhas L, Campbell M, Ziegler DS, Nysom K, Casanova M, et al. Oral selpercatinib in pediatric patients (Pts) with advanced ret-altered solid or primary cns tumors: Preliminary results from the phase 1/2 libretto-121 trial. J Clin Oncol (2021) 39(15_suppl):10009. doi: 10.1200/JCO.2021.39.15_suppl.10009
262. Li GG, Somwar R, Joseph J, Smith RS, Hayashi T, Martin L, et al. Antitumor activity of rxdx-105 in multiple cancer types with ret rearrangements or mutations. Clin Cancer Res (2017) 23(12):2981–90. doi: 10.1158/1078-0432.CCR-16-1887
263. Drilon A, Fu S, Patel MR, Fakih M, Wang D, Olszanski AJ, et al. A phase I/Ib trial of the vegfr-sparing multikinase ret inhibitor rxdx-105. Cancer Discovery (2019) 9(3):384–95. doi: 10.1158/2159-8290.CD-18-0839
264. Brandhuber B, Haas J, Tuch B, Ebata K, Bouhana K, McFaddin E, et al. eds. Ena-0490 the development of loxo-292, a potent, Kdr/Vegfr2-sparing ret kinase inhibitor for treating patients with ret-dependent cancers. In: AACR-NCI-EORTC international conference on molecular targets and cancer therapeutics. AACR: Munich, Germany.
265. Subbiah V, Velcheti V, Tuch BB, Ebata K, Busaidy NL, Cabanillas ME, et al. Selective ret kinase inhibition for patients with ret-altered cancers. Ann Oncol (2018) 29(8):1869–76. doi: 10.1093/annonc/mdy137
266. Drilon A, Oxnard GR, Tan DSW, Loong HHF, Johnson M, Gainor J, et al. Efficacy of selpercatinib in ret fusion-positive non-Small-Cell lung cancer. N Engl J Med (2020) 383(9):813–24. doi: 10.1056/NEJMoa2005653
267. Subbiah V, Gainor JF, Oxnard GR, Tan DSW, Owen DH, Cho BC, et al. Intracranial efficacy of selpercatinib in ret fusion-positive non-small cell lung cancers on the libretto-001 trial. Clin Cancer Res (2021) 27(15):4160–7. doi: 10.1158/1078-0432.CCR-21-0800
268. Wirth LJ, Sherman E, Robinson B, Solomon B, Kang H, Lorch J, et al. Efficacy of selpercatinib in ret-altered thyroid cancers. N Engl J Med (2020) 383(9):825–35. doi: 10.1056/NEJMoa2005651
269. Wirth LJ, Robinson B, Boni V, Tan DSW, McCoach C, Massarelli E, et al. Patient-reported outcomes with selpercatinib treatment among patients with ret-mutant medullary thyroid cancer in the phase I/Ii libretto-001 trial. Oncologist (2022) 27(1):13–21. doi: 10.1002/onco.13977
270. Minchom A, Tan AC, Massarelli E, Subbiah V, Boni V, Robinson B, et al. Patient-reported outcomes with selpercatinib among patients with ret fusion-positive non-small cell lung cancer in the phase I/Ii libretto-001 trial. Oncologist (2022) 27(1):22–9. doi: 10.1002/onco.13976
271. Bradford D, Larkins E, Mushti SL, Rodriguez L, Skinner AM, Helms WS, et al. Fda approval summary: Selpercatinib for the treatment of lung and thyroid cancers with ret gene mutations or fusions. Clin Cancer Res (2021) 27(8):2130–5. doi: 10.1158/1078-0432.CCR-20-3558
272. Markham A. Selpercatinib: First approval. Drugs (2020) 80(11):1119–24. doi: 10.1007/s40265-020-01343-7
273. Rahal R, Maynard M, Hu W, Brubaker JD, Cao Q, Kim JL, et al. eds. (2017). Blu-667 is a potent and highly selective ret inhibitor being developed for ret-driven cancers, in: AACR-NCI-EORTC International Conference: Molecular Targets and Cancer Therapeutics,. AACR, Philadelphia, PA.
274. Gainor JF, Lee DH, Curigliano G, Doebele RC, Kim D-W, Baik CS, et al. Clinical activity and tolerability of blu-667, a highly potent and selective ret inhibitor, in patients with advanced ret-fusion+ non-small cell lung cancer. J Clin Oncol (2019) 37:9008. doi: 10.1200/JCO.2019.37.15_suppl.9008
275. Kim J, Bradford D, Larkins E, Pai-Scherf LH, Chatterjee S, Mishra-Kalyani PS, et al. Fda approval summary: Pralsetinib for the treatment of lung and thyroid cancers with ret gene mutations or fusions. Clin Cancer Res (2021) 27(20):5452–6. doi: 10.1158/1078-0432.CCR-21-0967
276. Tsui DCC, Kavanagh BD, Honce JM, Rossi C, Patil T, Camidge DR. Central nervous system response to selpercartinib in patient with ret-rearranged non-small cell lung cancer after developing leptomeningeal disease on pralsetinib. Clin Lung Cancer (2022) 23(1):e5–8. doi: 10.1016/j.cllc.2021.06.005
277. Solomon BJ, Tan L, Lin JJ, Wong SQ, Hollizeck S, Ebata K, et al. Ret solvent front mutations mediate acquired resistance to selective ret inhibition in ret-driven malignancies. J Thorac Oncol (2020) 15(4):541–9. doi: 10.1016/j.jtho.2020.01.006
278. Zhu VW, Madison R, Schrock AB, Ou SI. Emergence of high level of met amplification as off-target resistance to selpercatinib treatment in Kif5b-ret nsclc. J Thorac Oncol (2020) 15(7):e124–e7. doi: 10.1016/j.jtho.2020.03.020
279. Subbiah V, Shen T, Terzyan SS, Liu X, Hu X, Patel KP, et al. Structural basis of acquired resistance to selpercatinib and pralsetinib mediated by non-gatekeeper ret mutations. Ann Oncol (2021) 32(2):261–8. doi: 10.1016/j.annonc.2020.10.599
280. Subbiah V, Shen T, Tetzlaff M, Weissferdt A, Byers LA, Cascone T, et al. Patient-driven discovery and post-clinical validation of Ntrk3 fusion as an acquired resistance mechanism to selpercatinib in ret fusion-positive lung cancer. Ann Oncol (2021) 32(6):817–9. doi: 10.1016/j.annonc.2021.02.010
281. Shen T, Hu X, Liu X, Subbiah V, Mooers BHM, Wu J. The L730v/I ret roof mutations display different activities toward pralsetinib and selpercatinib. NPJ Precis Oncol (2021) 5(1):48. doi: 10.1038/s41698-021-00188-x
282. Schoffski P, Cho BC, Italiano A, Loong HHF, Massard C, Rodriguez LM, et al. Bos172738, a highly potent and selective ret inhibitor, for the treatment of ret-altered tumors including ret-fusion+ nsclc and ret-mutant mtc: Phase 1 study results. J Clin Oncol (2021) 39(15_suppl):3008. doi: 10.1200/JCO.2021.39.15_suppl.3008
283. Drilon AE, Zhai D, Rogers E, Deng W, Zhang X, Ung J, et al. The next-generation ret inhibitor tpx-0046 is active in drug-resistant and naïve ret-driven cancer models. J Clin Oncol (2020) 38(15_suppl):3616. doi: 10.1200/JCO.2020.38.15_suppl.3616
284. Miyazaki I, Ishida K, Kato M, Suzuki T, Fujita H, Ohkubo S, et al. Abstract P06-02: Discovery of Tas0953/Hm06, a novel next generation ret-specific inhibitor capable of inhibiting ret solvent front mutations. Mol Cancer Ther (2021) 20(12_Supplement):P06–2-P-2. doi: 10.1158/1535-7163.TARG-21-P06-02%J Molecular Cancer Therapeutics
285. Odintsov I, Kurth RI, Ishizawa K, Delasos L, Lui AJW, Khodos I, et al. Abstract P233: Tas0953/Hm06 is effective in preclinical models of diverse tumor types driven by ret alterations. Mol Cancer Ther (2021) 20(12_Supplement):P233–P. doi: 10.1158/1535-7163.TARG-21-P233 %J Molecular Cancer Therapeutics
Keywords: RET, cancer, therapeutics, lung cancer, thyroid cancer
Citation: Regua AT, Najjar M and Lo H-W (2022) RET signaling pathway and RET inhibitors in human cancer. Front. Oncol. 12:932353. doi: 10.3389/fonc.2022.932353
Received: 29 April 2022; Accepted: 27 June 2022;
Published: 25 July 2022.
Edited by:
Dharmendra Kumar Yadav, Gachon University, South KoreaReviewed by:
Shiva Prasad Kollur, Amrita Vishwa Vidyapeetham University, IndiaVan-An Duong, Gachon University, South Korea
Lucieli Ceolin, Federal University of São Paulo, Bazil
Copyright © 2022 Regua, Najjar and Lo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui-Wen Lo, hlo@wakehealth.edu