 Alessandra Merlini1,2
Alessandra Merlini1,2 Valeria Pavese2Giulia Manessi2
Valeria Pavese2Giulia Manessi2 Martina Rabino2Francesco Tolomeo1Sandra Aliberti1Lorenzo D’Ambrosio2,3*†
Martina Rabino2Francesco Tolomeo1Sandra Aliberti1Lorenzo D’Ambrosio2,3*† Giovanni Grignani1†
Giovanni Grignani1†- 1Candiolo Cancer Institute, IRCCS-FPO, Turin, Italy
- 2Department of Oncology, University of Turin, Turin, Italy
- 3Medical Oncology, Azienda Ospedaliera Universitaria San Luigi Gonzaga, Turin, Italy
Effective treatment of advanced/metastatic bone and soft tissue sarcomas still represents an unmet medical need. Recent advances in targeted therapies have highlighted the potential of cyclin-dependent kinases (CDK) inhibitors in several cancer types, including sarcomas. CDKs are master regulators of the cell cycle; their dysregulation is listed among the “hallmarks of cancer” and sarcomas are no exception to the rule. In this review, we report both the molecular basis, and the potential therapeutic implications for the use of CDK inhibitors in sarcoma treatment. What is more, we describe and discuss the possibility and biological rationale for combination therapies with conventional treatments, target therapy and immunotherapy, highlighting potential avenues for future research to integrate CDK inhibition in sarcoma treatment.
1. Introduction
Sarcomas are a heterogeneous group of rare, mesenchymal malignancies that add up to 1% of all adult cancers and 20% of pediatric cancers (1). The sarcoma family encompasses more than 100 histological subtypes, comprising bone sarcomas and soft tissue sarcomas (BSTS) (2). Standard treatment entails radical surgical resection with (neo)adjuvant radiation therapy and/or chemotherapy in high-risk patients for localized disease, and systemic chemotherapeutic treatment in advanced stages (3). However, prognosis in advanced/metastatic stages remains dismal for the vast majority of sarcoma patients (3, 4). Hence, finding novel, effective treatment strategies for advanced BSTS represents an unmet medical need. Indeed, differently from epithelial cancers, mesenchymal tumors have seldom benefitted from the advent of innovative therapeutic strategies, from targeted therapy to immunotherapy (5, 6). Both the rarity of sarcomas, and the variety of their molecular determinants (7, 8), have represented major challenges for the development of effective, innovative therapeutic strategies in the field in past years. One of the first actionable molecular alterations discovered in sarcomas has been the amplification of the chromosomal region encoding the murine double minute 2 (MDM2) and cyclin dependent kinase 4 (CDK4) genes in a subset of liposarcomas (well-differentiated and dedifferentiated liposarcomas; WDLPS and DDLPS) (9, 10).
However, until recent years, the possibility to safely target master regulators of the cell cycle as MDM2 and CDK4 appeared difficult to translate in the clinical setting, for their potential off-tumor side effects in healthy tissues (11, 12). Targeting CDK4 seemed particularly attractive for WDLPS and DDLPS, in which it has a specific clinical and biological significance, with respect to MDM2 amplified-only liposarcomas. Indeed, CDK4-amplified WDLPS and DDLPS have been associated with worse prognosis with respect to those lacking CDK4 amplification (13). However, the CDK family is involved not only in WDLPS and DDLPS pathobiology, but in many different sarcoma types across BSTS (14). The comprehensive genomic analysis via The Cancer Genome Atlas (TCGA) has shown that approximately one quarter of all sarcomas harbor genetic alterations in the Cyclin-Dependent Kinase Inhibitor 2A (CDKN2A) - Cyclin D (CCND) - CDK4 - retinoblastoma (RB) axis (15, 16), providing strong rationale for targeting this crucial pathway in sarcomas. Hence, better understanding of the role of CDKs in cell biology and cancer, might provide novel avenues of treatment for advanced BSTS (14, 17).
2. CDKs in physiology and cancer
The cell cycle is divided into four distinct phases: a first growth phase (G1), a DNA replication or synthesis phase (S), a second growth phase (G2) and the mitotic phase (M). Cyclin-dependent kinases (CDKs) are members of the serine/threonine protein kinase family; as master regulators of cell cycle control, transcription, and RNA splicing, they are essential for tumor cell proliferation and growth. CDKs do not possess autonomous enzymatic activity and need to be bound to a cyclin subunit to function properly, hence their designation as cyclin-dependent kinases (18). Moreover, a few CDK family members play an important role in RNA transcription and pre-messenger RNA (mRNA) splicing.
The activity of CDKs is respectively up and down regulated by their cyclin partners and cyclin-dependent kinase inhibitors (CKIs). CDKs can phosphorylate the tumor suppressor protein retinoblastoma (Rb). This activity blocks the growth-inhibitory function of Rb: indeed, phospho-Rb (pRb) releases its grip, previously blocking the transactivation domain of the E2F transcription factors, allowing the transcription of genes which are crucial for cell cycle progression to the S-phase (19). In detail, cyclin D-CDK4/6 kinase complexes phosphorylate multiple Rb tumor suppressor protein residues (or its homologs, p107 and p130). As abovementioned, in its hypo-phosphorylated state, Rb actively suppresses G1-S progression by sequestering E2F transcription factors, which transcribe genes needed for DNA replication (20).
The human genome encodes 20 CDKs, divided into two subfamilies: cell cycle-associated CDKs (CDK1−7 and CDK14−18) and transcription-associated CDKs (CDK7−13, 19, and 20). Different CDKs interact with different cyclins to regulate numerous stages of the cell cycle in various cells or to perform other functions. CDK1 is the ancestor of all mitotic kinases; CDK2, CDK4, and CDK6 regulate progression through cell cycle phases. CDK7, instead, is peculiar in that it has been implicated in both transcription processes and cell cycle control (21). CDK8 and CDK9 control the RNA polymerase II (RNA Pol II)-dependent initiation and elongation of transcription (22). Other CDKs (5, 10, 11, 14–18, and 20) do not fit into either canonical roles, exhibiting different functions, often in a tissue-specific fashion. For example, CDK11 has multiple functions in mediating apoptosis, transcription, mitosis, hormone receptor signaling, and autophagy (23, 24). Likewise, CDK5 promotes neuron outgrowth and synaptogenesis in the nervous system, while in pancreatic β cells it reduces insulin secretion (25). As CDKs master fundamental processes required for cell survival and propagation, their hyperactivation (typically through mutation, gene amplification, or altered expression of their regulators) is frequently reported in cancer.
Until a few years ago, CKIs were also classified in two families of cell cycle inhibitors: the CDK family interacting with the CIP/KIP protein and the kinase inhibitor (INK) family. CIP/KIP family members are specific for CDK-cyclin complexes, such as CDK2-cyclin E, A and/or CDK1-cyclin B1, A and/or CDK2,4,6-cyclin D1, D2, D3. Members of the INK family bind CDK4,6 to inhibit formation of CDK4,6-cyclin D1, D2, D3 complexes (26, 27).
3. Partners in life, partners in crime: Key players in cell cycle function and dysregulation beyond CDKs
More recently, additional important regulatory proteins and mechanisms involved in cell cycle control have been discovered, such as members of CDK regulatory subunit (CKS) protein family and new cell cycle regulators. A recent addition to the family is the double homeobox 4 (DUX4) protein, which is of specific interest for sarcoma pathobiology. DUX4 is a transcription factor physiologically expressed during early embryonic development, and it is silenced by epigenetic pathways in most adult somatic cells. Studies revealed that DUX4 binds to CDK1, preventing the formation of CDK1-cyclin B1 complex, thus limiting its kinase activity (28). Aberrant expression of DUX4 in skeletal muscle leads to facioscapulohumeral dystrophy (26, 29). DUX4 rearrangements have been identified in specific types of pediatric B cell acute lymphoblastic leukemia (30, 31), in small round cell bone and soft tissue sarcomas – the so-called CIC-DUX4 rearranged family of sarcomas (32, 33), and rhabdomyosarcoma (34).
The dysregulation of CDK activity through activation of pathways enhancing CDK activity, or through the oncogene-induced inactivation of apoptosis, is a common occurrence in various cancers (35). Identifying and characterizing which cancer types require selected CDK activities for proliferation and survival, might enable to understand which subtypes could benefit more from specific CDK inhibitors (CDKi). However, weighing the importance of each CDK activity to cancer initiation, proliferation and progression is no trivial task, given the individual, multiple roles of each CDK and cyclin beyond cell cycle control (36).
In cancer, CDKs affect multiple targets and phosphorylate relevant transcription factors involved in tumorigenesis. What is more, their pathway can be altered at different stages in various cancer subtypes; even within the same cancer type (and, most importantly, within the same patient), multiple CDK pathway alterations can co-exist and, in some cases, provide escape/resistance to CDK inhibition. Moreover, resistance almost invariably ensues with targeted treatments in cancer, due to both intratumor heterogeneity and tumor evolutionary dynamics, and CDKi treatment is no exception to the rule. The emergence of somatic RB mutations has been identified in the clinic as a relevant resistance mechanism in breast cancer patients treated with CDKi (37); RB mutation/deletion is a frequent event in sarcomas, with deep deletions detected in a significant proportion of STS in the TCGA sarcoma cohort (16).
Another commonly deleted key tumor suppressor gene is Cyclin Dependent Kinase Inhibitor 2A (CDKN2A). CDKN2A encodes two important cell cycle regulatory proteins, p16 (encoded by the INK4A gene) and, in an alternative reading frame, p14 (encoded by the Alternative Reading Frame – ARF - gene). CDKN2A deletions and inactivating mutations seem to have a negative prognostic role across different tumor types, including sarcomas (38–42). p16, a CDK inhibitor, inhibits Rb phosphorylation, while p14 inhibits MDM2, resulting in a positive regulation of p53. p16 expression increases gradually to a sustained, significantly high level in the later stages of cellular senescence.
Indeed, in murine cells, p19/p53 pathway inactivation is generally sufficient to escape senescence, while in human cells disruption of at least both the p53/p21 and the p16/pRb pathways is usually needed. Homozygous deletion of CDKN2A/ARF thus results in inactivation of two major tumor suppressing pathways, mainly acting through Rb and p53 (43).
Hence, a plethora of alterations beyond CDK4/6 genes emerges as highly relevant for sarcoma pathobiology, providing several potential actionable targets at various steps of the CDKN2A-CCND-CDK4-RB axis. Understanding which sarcoma subtypes are most affected by specific alterations in this axis, has provided the rational basis to select those sarcomas which could benefit more from CDK inhibition (14) (Figure 1).
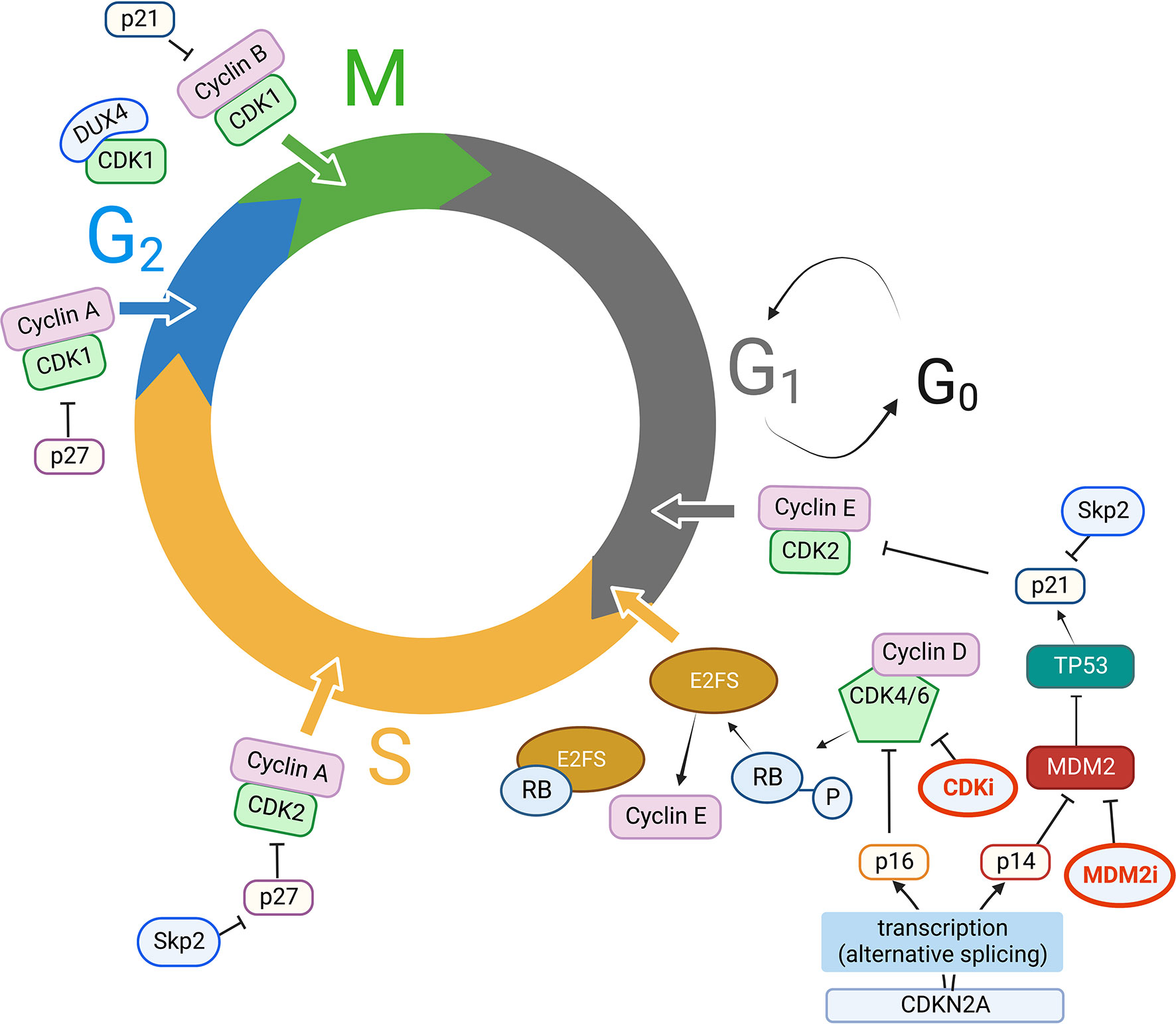
Figure 1 Key players in cell cycle dysregulation in STS. CDK, Cyclins, CKI, and other key molecular players in CDK activity/inhibition. Clockwise, starting from G1 to S phase progression: CDKN2A is transcribed by alternative splicing either into p16 or p14, which respectively inhibit CDK4/6/Cyclin D complexes and MDM2 activity. MDM2, an ubiquitin ligase, ubiquitinates p53 targeting it to the proteasome; p53 has p21 as a direct transcriptional target, and p21 in turn inhibits CDK2/Cyclin E complexes. Cyclin E expression is regulated by E2F transcription factor, which in turn is released from Rb protein grip (usually blocking its transactivation domain) when CDK4/6/Cyclin D complexes phosphorylate Rb, facilitating G1 to S phase progression. The ubiquitin ligase Skp2 targets p21 and p27 for proteasomal degradation, thus promoting CDK2/Cyclin E, CDK2/Cyclin A activity in S phase progression, CDK1/Cyclin A activity for G2 to M transition, which is also fostered by releasing p21 inhibitory activity on CDK1/Cyclin B; DUX4 can also bind CDK1, thus preventing CDK1/Cyclin B interaction. MDM2 activity can also be inhibited with MDM2 inhibitors (MDM2i), while CDK inhibitors (CDKi) currently in use in clinical practice are mainly CDK4/6 inhibitors. MDM2i and CDKi are highlighted in bold (red) in Figure 1. Created with BioRender.com.
4. Actionable targets in CDK signaling across different sarcoma subtypes
Despite the diversity in histotypes, age at presentation, risk of recurrence and prognosis, the most frequently altered genes in sarcomas precisely include genes involved in cell cycle regulation, namely TP53, CDKN2A, RB (44–46). Surprisingly, the only gene whose alterations were associated with worse overall survival across all types of STS was CDKN2A (39). These results confirm the biological importance of the p16INK4a-CDK4/6-pRb pathway and/or ARF signaling pathways in sarcoma (39). Indeed, pinpointing histotype-specific alterations might help to dissect the most appropriate therapeutic challenges and opportunities for each sarcoma subtype.
4.1. Undifferentiated pleomorphic sarcoma
UPS accounts for 15–20% of all STS. Typically, it occurs in the limbs and trunk of adults >40 years of age (47). The development of most UPS is sporadic, but approximately 3% of UPS develop in areas of the body that received radiation therapy to treat an unrelated disease after a median latency of 10 years, and are consequently classified as secondary (or, more appropriately, radiation-induced) UPS (48). The standard of care for patients with localized UPS is surgical resection with (neo)adjuvant chemo/radiotherapy in selected cases; for patients with unresectable or metastatic disease, systemic chemotherapy and/or radiotherapy may be considered with low to moderate response rates in patients with UPS. Remarkably, UPS are also among the most represented sarcoma histotypes with CDKN2A loss (39). About 30% of UPS show MDM2 and CDK4 up-regulation; MDM2 ubiquitinates the tumor suppressor p53 and promotes its proteasomal degradation, while MDM2 overexpression leads to downregulation of the CKI p21. P21 is a transcriptional target of p53, and its downregulation causes hyperactivation of CDKs (49). Up to 78% of UPS tumors carry RB gene deletions, due to losses of different regions within the long arm of chromosome 13 (8). TP53 is also very frequently deleted in UPS, and together with RB and ATRX, is among the few genes recurrently showing pathogenic missense mutations in UPS (16). Intriguingly, S phase kinase-associated protein 2 (Skp2) is required for survival of RB-and TP53-deficient UPS cells, in which it drives cell proliferation by degrading p21 and p27. Hence, the loss of both RB and TP53 renders UPS dependent on Skp2, which could provide the basis for innovative therapeutic strategies in this setting (50). However, there are no experimental studies ongoing or published so far, about the potential of CKIs in UPS patients.
4.2. Liposarcoma
Liposarcomas (LPS) account for a significant proportion (~13–20%) of adult STS (13-20%). LPS are subcategorized into three main groups, including WDLPS/DDLPS, characterized by a typical MDM2 and high-mobility group AT-hook 2 (HMGA2) gene amplification and an inconsistent CDK4 gene amplification (the 12q amplicon can span chromosomal regions from 12q12 up to 12q21); myxoid/round cell liposarcoma (M/RCLPS), carrying a typical t (12, 16)(q13;p111) translocation, and pleomorphic liposarcoma (PLPS), frequently showing TP53 and/or RB gene losses. Roughly 60% of LPS cases are WD/DDLPS, while PLPS is the rarest subtype (~5%). In WDLPS/DDLPS, the CDK4 gene (12q14.1) is within a distinct, inconsistent amplicon that is not present in about 10% of WDLPS/DDLPS (51), and its presence has been associated with a worse prognostic outcome (13). Moreover, patients carrying both gene amplifications (MDM2 and CDK4) have a much higher risk of local recurrence after surgery. The WDLPS/DDLPS genetic signature shows a complex pattern of expression for Cyclin D1, P16INK4a, P14ARF, and RB which is not dependent on CDK4 status. Finally, alterations in CDKN2A/CDKN2B/CDK4/CCND2 axis have been detected in almost all CDK4 amplification-negative WDLPS/DDLPS in a cohort of 104 WDLPS/DDLPS patients (52).
4.3. Malignant peripheral nerve sheath tumors
Malignant peripheral nerve sheath tumors (MPNSTs) add up to 3-10% of all STS diagnoses. They can arise sporadically or in patients affected by neurofibromatosis type I (NF1). MPNSTs are very aggressive and the first cause of oncological death in patients affected by NF1. In those tumors, CDK2 and CDK 4/6 are overexpressed because of the loss of p16 and p27. This causes constant pRb phosphorylation, fostering cell cycle progression (53). Remarkably, up to 80% of MPNST show CDKN2A loss (54). This leads to the upregulation of CDK4/6 and sequentially the initiation of the S phase and promotion of mitosis. Hence, CDK4/6 inhibitors (CKIs) hold promise as a potential innovative treatment for advanced MPNST (55).
4.4. Synovial sarcoma
Synovial sarcoma typically arises in young adults, and is characterized by a typical translocation between chromosome X and chromosome 18 t(X,18;p11,q11), which generates a fusion between SS18 and SSX1/2 or SSX4, disrupting epigenetic regulation within the cancer cell (56, 57). CDKN2A deletion is a highly frequent event in synovial sarcomas (58); moreover, the translocation facilitates repression of CDKN2A activity (59) and increases the expression of CDK4 as well as multiple cyclins (D1, B1, A2, I, and F) (60).
4.5. Other soft tissue sarcomas
Leiomyosarcoma (LMS) accounts for 10-20% of all STS, and can arise at any body site. LMS is characterized by spindle-shaped cells resembling smooth muscle cells and are grouped among the so-called “complex karyotype” STS, as they are not driven by a single translocation or genetic alteration, but are characterized by multiple, various genetic abnormalities. Common genetic alterations include PTEN deletion and/or mutation, TP53 mutations and, importantly, RB loss (16, 61–63). One striking, recent finding is the high frequency of biallelic inactivation of the above mentioned by various mechanisms, in the vast majority of LMS samples analyzed in the study by Chudasama P. and colleagues (61). Rb inactivation casts some doubt on the clinical utility of CDK inhibition, as Rb inactivation affects the CDK pathway downstream of CDKs, presenting a potential mechanism of both primary and secondary resistance to CDKi in LMS, similarly to Rb-mediated resistance mechanisms in CDK-treated breast cancer patients (37).
Intimal sarcomas (INS) are rare STS which can be particularly aggressive also because of their site of origin, most frequently affecting the wall of large vessels or the heart (64–66). INS are characterized by the peculiar presence of large gains/amplifications in the 12q12-15 chromosomal region, encompassing MDM2 and/or CDK4. CDKN2A deletions are also very frequent in INS (65). Taken together, these recent molecular findings might provide the rationale for trials with CDKi in this set of STS patients burdened by very poor prognosis (67).
Rhabdomyosarcoma (RMS) is the most common STS in children and adolescents; alveolar rhabdomyosarcomas (ARMS) are characterized by either PAX3-FOXO1 or PAX7-FOXO1 fusion genes; ARMS with the former fusion most often carry additional 12q13-q14 amplifications, therefore including the CDK4 gene, which has been correlated with poor survival outcomes (68). Disappointingly, in fusion-positive RMS, CDK4 amplification has not been linked to increased sensitivity to CDKi, but, rather counterintuitively, to the opposite condition (resistance to CDKi), at least in in vitro studies (69).
4.6. Bone sarcomas
Osteosarcomas (OS) represent the most common primary malignant tumors of bone. They can arise at any skeletal site, but they more frequently develop in the long bones of the extremities. OS has a bimodal age distribution (adolescents between 14-18 years and older adults, > 40 years old) and, even though it is sensitive to chemotherapy, prognosis in advanced stages remains dismal (2, 70). Intriguingly, TP53 is inactivated in >90% of OS, contributing to cell cycle dysregulation; RB1 is also among genes most frequently mutated in OS (>50%) (71). Indeed, individuals affected by Li-Fraumeni and hereditary retinoblastoma syndromes have an increased risk of developing OS (72). Other genes commonly altered in OS and involved in cell cycle regulation include CDK4, MDM2, PTEN, CDKN2A, CCND3, and CCNE1 (14, 49, 73). The clinical utility of CDKi in OS has not yet been tested in dedicated clinical trials, but advanced osteosarcoma patients with CDK4 overexpression could be included in the phase II PalboSarc trial with the CDKi palbociclib (NCT03242382) (14).
5. Targeting CDKs in cancer
The history and success of CDKi in cancer have now come a long way, with more than 25 years of preclinical and clinical development (74). The first generation of CDKi was constituted by pan-CDKi (e.g. flavopiridol, olomucine, roscovitine) (75, 76), which were designed to halt cell cycle and cell proliferation by inhibiting CDK enzymatic activity. This first generation of pan-CDKi had limited selectivity and was burdened by high toxicity in normal cells, preventing their clinical development. For these reasons, almost all first generation CDKi failed to meet their endpoints in early-stage clinical trials (77, 78). Second-generation CDKi (e.g. dinaciclib, CYC065) have been developed with greater selectivity and fewer side effects (79). Finally, third-generation, selective CDK4/6 inhibitors were the first CDKi which received FDA approval in March 2017, for the treatment of postmenopausal women with hormone receptor (HR)-positive metastatic breast cancer, in combination with an aromatase inhibitor as initial endocrine-based therapy.
Currently, the three FDA- and EMA-approved CDK4/6 inhibitors are palbociclib, ribociclib and abemaciclib. While palbociclib is equally active against CDK4 and CDK6, ribociclib and abemaciclib show higher efficacy in CDK4 inhibition (80). Indeed, all these approved compounds act by inhibiting Rb phosphorylation, thus blocking cell cycle progression from G1 to S phase. However, their action extends beyond simple enzymatic inhibition, with likely direct effects on cell metabolism, senescence, and possibly immune modulation (81, 82).
5.1. Anti-CDK targeted therapy in sarcomas: Ongoing clinical trials and future perspectives
Sarcomas have been included in clinical studies on CDKi since early phase I trials; however, only very few CDKi trials enrolled exclusively BSTS, including an early-phase trial of flavopiridol in association with doxorubicin (14, 75). Among these, two studies are of peculiar interest. In particular, the study “PD0332991 (Palbociclib) in Patients with Advanced or Metastatic Liposarcoma”, NCT01209598, demonstrated a favorable progression free survival (PFS) in a mixed WDLPS/DDLPS patient cohort, which included advanced/metastatic WDLPS/DDLPS patients who had received at least one line of systemic treatment (patients previously untreated for systemic disease were allowed to join the expansion cohort) (83). Another study, NCT02846987, still active although closed to enrollment, has investigated the role of abemaciclib monotherapy in advanced DDLPS, assuming that this novel, more potent CDK inhibitor might achieve better results in the sarcoma population. So far, the study has met its primary endpoint (12-week PFS ≥ 60%) and final results are awaited (84).
One highly attractive combination treatment opportunity in WDLPS/DDLPS is represented by the possibility to combine novel MDM2 inhibitors (85, 86) with CDKi. Preclinical studies demonstrated both evidence of synergism (87), and efficacy of MDM2 inhibitors in overcoming resistance to CDK4/6 inhibitors (88). However, the significant risk of unacceptable combined toxicities of MDM2 and CDK4 inhibitors - especially myelosuppression - casts some doubt over the clinical applicability of their combination.
Concerning other possible targeted treatment combinations, one interesting opportunity could be the association of CDKi with PI3K inhibitors. Indeed, PTEN downregulation and AKT increased phosphorylation were shown to be associated with increased CDK2/cyclin E2 expression in breast cancer cell lines resistant to CDKi, rendering PI3K inhibitors (capable of downregulating cyclin E2) an attractive partner to overcome resistance to CDKi (89).
Finally, studies on the association (combination/sequence) of CDKi with immunotherapy are currently ongoing in many cancer types, including sarcomas (e.g. study NCT04438824, listed in Table 1). Indeed, CDKi seem to have a relevant immune-priming effect (81). Preliminary data are not available yet for BSTS, but similar studies in breast cancer with combination of palbociclib, pembrolizumab and letrozole have yielded promising results (90).

Table 1 Ongoing clinical trials with CDKi in BSTS (source: www.clinicaltrials.gov, accessed on October 20th, 2022).
In Table 1, a list of ongoing, actively recruiting clinical trials with CDKi (alone as monotherapy, or in combination) in BSTS is provided.
6. Conclusions
The presence of molecular alterations affecting the CDKN2A-CDK4-CCND1-RB axis is an important opportunity for innovative targeted treatments for patients with BSTS, typically burdened by dismal prognosis in advanced/metastatic stages. Knowledge of the fine-tuning of these pathways across different sarcoma subtypes is instrumental to develop rationally-based clinical trial proposals in this setting. Indeed, presence of multiple alterations in different steps of cell cycle regulation might provide primary/secondary resistance mechanisms to CDK inhibition; moreover, when present, CDK4 amplification is the main oncogenic driver of only a subset of CDK4-amplified sarcomas. Hence, thorough understanding of the molecular basis of cell cycle dysregulation in each specific histotype, will be crucial for the development of tailored treatment combinations with CDK inhibitors and other innovative targeted therapies or immunotherapeutic strategies.
Author contributions
AM, LA, and GG contributed to conception and design of the review article. AM, VP, LA organized the literature data. AM, VP, LA and GG wrote the first draft of the manuscript. AM, VP, GM, MR, FT, SA, LA, GG wrote sections of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
AIRC IG 2019-ID 23104, to GG.
Conflict of interest
AM: travel expenses from PharmaMar. LA received travel expenses from PharmaMar and Lilly. GG has received fees for consulting and advisory board roles from PharmaMar, Lilly, Novartis, GSK, Bayer, and Eisai.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin (2022) 72:7–33. doi: 10.3322/caac.21708
2. WHO Classification of Tumours Editorial Board. Soft tissue and bone tumours. ed. 5th Vol. vol. 3. . Lyon (France: WHO classification of tumours series (International Agency for Research on Cancer (2020).
3. Gronchi A, Miah AB, Dei Tos AP, Abecassis N, Bajpai J, Bauer S, et al. Soft tissue and visceral sarcomas: ESMO-EURACAN-GENTURIS clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol (2021) 32:1348–65. doi: 10.1016/j.annonc.2021.07.006
4. Lochner J, Menge F, Vassos N, Hohenberger P, Kasper B. Prognosis of patients with metastatic soft tissue sarcoma: Advances in recent years. Oncol Res Treat (2020) 43:613–9. doi: 10.1159/000509519
5. Hall F, Villalobos V, Wilky B. Future directions in soft tissue sarcoma treatment. Curr Probl Cancer (2019) 43:300–7. doi: 10.1016/j.currproblcancer.2019.06.004
6. Rytlewski J, Milhem MM, Monga V. Turning ‘Cold’ tumors ‘Hot’: immunotherapies in sarcoma. Ann Transl Med (2021) 9:1039. doi: 10.21037/atm-20-6041
7. Barretina J, Taylor BS, Banerji S, Ramos AH, Lagos-Quintana M, Decarolis PL, et al. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat Genet (2010) 42:715–21. doi: 10.1038/ng.619
8. Taylor BS, Barretina J, Maki RG, Antonescu CR, Singer S, Ladanyi M. Advances in sarcoma genomics and new therapeutic targets. Nat Rev Cancer (2011) 11:541–57. doi: 10.1038/nrc3087
9. Sirvent N, Forus A, Lescaut W, Burel F, Benzaken S, Chazal M, et al. Characterization of centromere alterations in liposarcomas. Genes Chromosomes Cancer (2000) 29:117–29. doi: 10.1002/1098-2264(2000)9999:9999<::AID-GCC1014>3.0.CO;2-Q
10. Pilotti S, Della Torre G, Lavarino C, Di Palma S, Sozzi G, Minoletti F, et al. Distinct mdm2/p53 expression patterns in liposarcoma subgroups: implications for different pathogenetic mechanisms. J Pathol (1997) 181:14–24. doi: 10.1002/(SICI)1096-9896(199701)181:1<14::AID-PATH730>3.0.CO;2-O
11. Li Q, Lozano G. Molecular pathways: targeting Mdm2 and Mdm4 in cancer therapy. Clin Cancer Res (2013) 19:34–41. doi: 10.1158/1078-0432.CCR-12-0053
12. Jhaveri K, Burris Rd HA, Yap TA, Hamilton E, Rugo HS, Goldman JW, et al. The evolution of cyclin dependent kinase inhibitors in the treatment of cancer. Expert Rev Anticancer Ther (2021) 21:1105–24. doi: 10.1080/14737140.2021.1944109
13. Italiano A, Bianchini L, Gjernes E, Keslair F, Ranchere-Vince D, Dumollard JM, et al. Clinical and biological significance of CDK4 amplification in well-differentiated and dedifferentiated liposarcomas. Clin Cancer Res (2009) 15:5696–703. doi: 10.1158/1078-0432.CCR-08-3185
14. Hsu JY, Seligson ND, Hays JL, Miles WO, Chen JL. Clinical utility of CDK4/6 inhibitors in sarcoma: Successes and future challenges. JCO Precis Oncol (2022) 6:e2100211. doi: 10.1200/PO.21.00211
15. Burns J, Brown JM, Jones KB, Huang PH. The cancer genome atlas: Impact and future directions in sarcoma. Surg Oncol Clin N Am (2022) 31:559–68. doi: 10.1016/j.soc.2022.03.013
16. C. G. A. R. N. E. a. elizabeth.demicco@sinaihealthsystem.ca, C. G. A. R. Network. Comprehensive and integrated genomic characterization of adult soft tissue sarcomas. Cell (2017) 171:950–965.e928. doi: 10.1016/j.cell.2017.10.014
17. Thiel JT, Daigeler A, Kolbenschlag J, Rachunek K, Hoffmann S. The role of CDK pathway dysregulation and its therapeutic potential in soft tissue sarcoma. Cancers (Basel) (2022) 14(14):1–20. doi: 10.3390/cancers14143380
18. Lim S, Kaldis P. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development (2013) 140:3079–93. doi: 10.1242/dev.091744
19. Giacinti C, Giordano A. RB and cell cycle progression. Oncogene (2006) 25:5220–7. doi: 10.1038/sj.onc.1209615
20. Burkhart DL, Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer (2008) 8:671–82. doi: 10.1038/nrc2399
21. Fisher RP. Secrets of a double agent: CDK7 in cell-cycle control and transcription. J Cell Sci (2005) 118:5171–80. doi: 10.1242/jcs.02718
22. Michowski W, Chick JM, Chu C, Kolodziejczyk A, Wang Y, Suski JM, et al. Cdk1 controls global epigenetic landscape in embryonic stem cells. Mol Cell (2020) 78:459–476.e413. doi: 10.1016/j.molcel.2020.03.010
24. Zhou Y, Shen JK, Hornicek FJ, Kan Q, Duan Z. The emerging roles and therapeutic potential of cyclin-dependent kinase 11 (CDK11) in human cancer. Oncotarget (2016) 7:40846–59. doi: 10.18632/oncotarget.8519
25. Wood DJ, Endicott JA. Structural insights into the functional diversity of the CDK-cyclin family. Open Biol (2018) 8(9):1–26. doi: 10.1098/rsob.180112
26. Bury M, Le Calvé B, Ferbeyre G, Blank V, Lessard F. New insights into CDK regulators: Novel opportunities for cancer therapy. Trends Cell Biol (2021) 31:331–44. doi: 10.1016/j.tcb.2021.01.010
27. Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev (1999) 13:1501–12. doi: 10.1101/gad.13.12.1501
28. Bury M, Le Calvé B, Lessard F, Dal Maso T, Saliba J, Michiels C, et al. NFE2L3 controls colon cancer cell growth through regulation of DUX4, a CDK1 inhibitor. Cell Rep (2019) 29:1469–1481.e1469. doi: 10.1016/j.celrep.2019.09.087
29. Dib C, Zakharova V, Popova E, Kiseleva E, Chernyak B, Lipinski M, et al. DUX4 pathological expression: Causes and consequences in cancer. Trends Cancer (2019) 5:268–71. doi: 10.1016/j.trecan.2019.03.001
30. Tanaka Y, Kawazu M, Yasuda T, Tamura M, Hayakawa F, Kojima S, et al. Transcriptional activities of DUX4 fusions in b-cell acute lymphoblastic leukemia. Haematologica (2018) 103:e522–6. doi: 10.3324/haematol.2017.183152
31. Zhang J, McCastlain K, Yoshihara H, Xu B, Chang Y, Churchman ML, et al. Deregulation of DUX4 and ERG in acute lymphoblastic leukemia. Nat Genet (2016) 48:1481–9. doi: 10.1038/ng.3691
32. Gambarotti M, Benini S, Gamberi G, Cocchi S, Palmerini E, Sbaraglia M, et al. CIC-DUX4 fusion-positive round-cell sarcomas of soft tissue and bone: a single-institution morphological and molecular analysis of seven cases. Histopathology (2016) 69:624–34. doi: 10.1111/his.12985
33. Sbaraglia M, Righi A, Gambarotti M, Dei Tos AP. Ewing Sarcoma and Ewing-like tumors. Virchows Arch (2020) 476:109–19. doi: 10.1007/s00428-019-02720-8
34. Sirvent N, Trassard M, Ebran N, Attias R, Pedeutour F. Fusion of EWSR1 with the DUX4 facioscapulohumeral muscular dystrophy region resulting from t(4,22)(q35;q12) in a case of embryonal rhabdomyosarcoma. Cancer Genet Cytogenet (2009) 195:12–8. doi: 10.1016/j.cancergencyto.2009.06.011
35. Sherr CJ, Beach D, Shapiro GI. Targeting CDK4 and CDK6: From discovery to therapy. Cancer Discovery (2016) 6:353–67. doi: 10.1158/2159-8290.CD-15-0894
36. Otto T, Sicinski P. Cell cycle proteins as promising targets in cancer therapy. Nat Rev Cancer (2017) 17:93–115. doi: 10.1038/nrc.2016.138
37. Condorelli R, Spring L, O'Shaughnessy J, Lacroix L, Bailleux C, Scott V, et al. Polyclonal RB1 mutations and acquired resistance to CDK 4/6 inhibitors in patients with metastatic breast cancer. Ann Oncol (2018) 29:640–5. doi: 10.1093/annonc/mdx784
38. Merlini A, Centomo ML, Ferrero G, Chiabotto G, Miglio U, Berrino E, et al. DNA Damage response and repair genes in advanced bone and soft tissue sarcomas: An 8-gene signature as a candidate predictive biomarker of response to trabectedin and olaparib combination. Front Oncol (2022) 12:844250. doi: 10.3389/fonc.2022.844250
39. Bui NQ, Przybyl J, Trabucco SE, Frampton G, Hastie T, van de Rijn M, et al. A clinico-genomic analysis of soft tissue sarcoma patients reveals CDKN2A deletion as a biomarker for poor prognosis. Clin Sarcoma Res (2019) 9:12. doi: 10.1186/s13569-019-0122-5
40. Zhao R, Choi BY, Lee M-H, Bode AM, Dong Z. Implications of genetic and epigenetic alterations of CDKN2A (p16INK4a) in cancer. EBioMedicine (2016) 8:30–9. doi: 10.1016/j.ebiom.2016.04.017
41. Lu VM, O'Connor KP, Shah AH, Eichberg DG, Luther EM, Komotar RJ, et al. The prognostic significance of CDKN2A homozygous deletion in IDH-mutant lower-grade glioma and glioblastoma: a systematic review of the contemporary literature. J Neurooncol (2020) 148:221–9. doi: 10.1007/s11060-020-03528-2
42. Monzon J, Liu L, Brill H, Goldstein AM, Tucker MA, From L, et al. CDKN2A mutations in multiple primary melanomas. New Engl J Med (1998) 338:879–87. doi: 10.1056/NEJM199803263381305
43. Pyeritz R, Korf BGW. Emery and rimoin’s principles and practice of medical genetics and genomics, 7th ed. Academic Press (2020).
44. Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med (2017) 9:34. doi: 10.1186/s13073-017-0424-2
45. Cote GM, He J, Choy E. Next-generation sequencing for patients with sarcoma: A single center experience. Oncologist (2018) 23:234–42. doi: 10.1634/theoncologist.2017-0290
46. Tirrò E, Martorana F, Micale G, Inzerilli N, Carciotto R, Romano C, et al. Next generation sequencing in a cohort of patients with rare sarcoma histotypes: A single institution experience. Pathol Res Pract (2022) 232:153820. doi: 10.1016/j.prp.2022.153820
47. Carvalho SD, Pissaloux D, Crombé A, Coindre JM, Le Loarer F. Pleomorphic sarcomas: The state of the art. Surg Pathol Clin (2019) 12:63–105. doi: 10.1016/j.path.2018.10.004
48. Riad S, Biau D, Holt GE, Werier J, Turcotte RE, Ferguson PC, et al. The clinical and functional outcome for patients with radiation-induced soft tissue sarcoma. Cancer (2012) 118:2682–92. doi: 10.1002/cncr.26543
49. Kohlmeyer JL, Gordon DJ, Tanas MR, Monga V, Dodd RD, Quelle DE. CDKs in sarcoma: Mediators of disease and emerging therapeutic targets. Int J Mol Sci (2020) 21(8):1–30. doi: 10.3390/ijms21083018
50. Li GZ, Okada T, Kim YM, Agaram NP, Sanchez-Vega F, Shen Y, et al. Rb And p53-deficient myxofibrosarcoma and undifferentiated pleomorphic sarcoma require Skp2 for survival. Cancer Res (2020) 80:2461–71. doi: 10.1158/0008-5472.CAN-19-1269
51. Italiano A, Bianchini L, Keslair F, Bonnafous S, Cardot-Leccia N, Coindre JM, et al. HMGA2 is the partner of MDM2 in well-differentiated and dedifferentiated liposarcomas whereas CDK4 belongs to a distinct inconsistent amplicon. Int J Cancer (2008) 122:2233–41. doi: 10.1002/ijc.23380
52. Louis-Brennetot C, Coindre JM, Ferreira C, Pérot G, Terrier P, Aurias A. The CDKN2A/CDKN2B/CDK4/CCND1 pathway is pivotal in well-differentiated and dedifferentiated liposarcoma oncogenesis: an analysis of 104 tumors. Genes Chromosomes Cancer (2011) 50:896–907. doi: 10.1002/gcc.20909
53. Bhalla AD, Landers SM, Singh AK, Landry JP, Yeagley MG, Myerson GSB, et al. Experimental models of undifferentiated pleomorphic sarcoma and malignant peripheral nerve sheath tumor. Lab Invest (2022) 102:658–66. doi: 10.1038/s41374-022-00734-6
54. Chaney KE, Perrino MR, Kershner LJ, Patel AV, Wu J, Choi K, et al. Loss in a model of neurofibroma demonstrates stepwise tumor progression to atypical neurofibroma and MPNST. Cancer Res (2020) 80:4720–30. doi: 10.1158/0008-5472.CAN-19-1429
55. Rhodes SD, He Y, Smith A, Jiang L, Lu Q, Mund J, et al. Cdkn2a (Arf) loss drives NF1-associated atypical neurofibroma and malignant transformation. Hum Mol Genet (2019) 28:2752–62. doi: 10.1093/hmg/ddz095
56. Clark J, Rocques PJ, Crew AJ, Gill S, Shipley J, Chan AM, et al. Identification of novel genes, SYT and SSX, involved in the t(X;18)(p11.2;q11.2) translocation found in human synovial sarcoma. Nat Genet (1994) 7:502–8. doi: 10.1038/ng0894-502
57. Ladanyi M, Antonescu CR, Leung DH, Woodruff JM, Kawai A, Healey JH, et al. Impact of SYT-SSX fusion type on the clinical behavior of synovial sarcoma: a multi-institutional retrospective study of 243 patients. Cancer Res (2002) 62:135–40.
58. Subramaniam MM, Noguera R, Piqueras M, Navarro S, López-Guerrero JA, Llombart-Bosch A. p16INK4A (CDKN2A) gene deletion is a frequent genetic event in synovial sarcomas. Am J Clin Pathol (2006) 126:866–74. doi: 10.1309/E2AAY2XXN431WL81
59. El Beaino M, Araujo DM, Lazar AJ, Lin PP. Synovial sarcoma: Advances in diagnosis and treatment identification of new biologic targets to improve multimodal therapy. Ann Surg Oncol (2017) 24:2145–54. doi: 10.1245/s10434-017-5855-x
60. Haldar M, Hancock JD, Coffin CM, Lessnick SL, Capecchi MR. A conditional mouse model of synovial sarcoma: insights into a myogenic origin. Cancer Cell (2007) 11:375–88. doi: 10.1016/j.ccr.2007.01.016
61. Chudasama P, Mughal SS, Sanders MA, Hübschmann D, Chung I, Deeg KI, et al. Integrative genomic and transcriptomic analysis of leiomyosarcoma. Nat Commun (2018) 9:144. doi: 10.1038/s41467-017-02602-0
62. Guo X, Jo VY, Mills AM, Zhu SX, Lee CH, Espinosa I, et al. Clinically relevant molecular subtypes in leiomyosarcoma. Clin Cancer Res (2015) 21:3501–11. doi: 10.1158/1078-0432.CCR-14-3141
63. Ognjanovic S, Olivier M, Bergemann TL, Hainaut P. Sarcomas in TP53 germline mutation carriers: a review of the IARC TP53 database. Cancer (2012) 118:1387–96. doi: 10.1002/cncr.26390
64. Bode-Lesniewska B, Zhao J, Speel EJ, Biraima AM, Turina M, Komminoth P, et al. Gains of 12q13-14 and overexpression of mdm2 are frequent findings in intimal sarcomas of the pulmonary artery. Virchows Arch (2001) 438:57–65. doi: 10.1007/s004280000313
65. Neuville A, Collin F, Bruneval P, Parrens M, Thivolet F, Gomez-Brouchet A, et al. Intimal sarcoma is the most frequent primary cardiac sarcoma: clinicopathologic and molecular retrospective analysis of 100 primary cardiac sarcomas. Am J Surg Pathol (2014) 38:461–9. doi: 10.1097/PAS.0000000000000184
66. Roszik J, Khan A, Conley AP, Livingston JA, Groisberg R, Ravi V, et al. Unique aberrations in intimal sarcoma identified by next-generation sequencing as potential therapy targets. Cancers (Basel) (2019) 11(9):1–8. doi: 10.3390/cancers11091283
67. Penel N, Taieb S, Ceugnart L, Dansin E, Hoguet D, Vanseymortier L, et al. Report of eight recent cases of locally advanced primary pulmonary artery sarcomas: failure of doxorubicin-based chemotherapy. J Thorac Oncol (2008) 3:907–11. doi: 10.1097/JTO.0b013e318180720d
68. Barr FG, Duan F, Smith LM, Gustafson D, Pitts M, Hammond S, et al. Genomic and clinical analyses of 2p24 and 12q13-q14 amplification in alveolar rhabdomyosarcoma: a report from the children’s oncology group. Genes Chromosomes Cancer (2009) 48:661–72. doi: 10.1002/gcc.20673
69. Olanich ME, Sun W, Hewitt SM, Abdullaev Z, Pack SD, Barr FG. CDK4 amplification reduces sensitivity to CDK4/6 inhibition in fusion-positive rhabdomyosarcoma. Clin Cancer Res (2015) 21:4947–59. doi: 10.1158/1078-0432.CCR-14-2955
70. Mirabello L, Troisi RJ, Savage SA. Osteosarcoma incidence and survival rates from 1973 to 2004: data from the surveillance, epidemiology, and end results program. Cancer (2009) 115:1531–43. doi: 10.1002/cncr.24121
71. Chen X, Bahrami A, Pappo A, Easton J, Dalton J, Hedlund E, et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep (2014) 7:104–12. doi: 10.1016/j.celrep.2014.03.003
72. Fuchs B, Pritchard DJ. Etiology of osteosarcoma. Clin Orthop Relat Res 40-52 (2002). doi: 10.1097/00003086-200204000-00007
73. Zhou Y, Shen JK, Yu Z, Hornicek FJ, Kan Q, Duan Z. Expression and therapeutic implications of cyclin-dependent kinase 4 (CDK4) in osteosarcoma. Biochim Biophys Acta Mol Basis Dis (2018) 1864:1573–82. doi: 10.1016/j.bbadis.2018.02.004
74. Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discovery (2015) 14:130–46. doi: 10.1038/nrd4504
75. Luke JJ, D'Adamo DR, Dickson MA, Keohan ML, Carvajal RD, Maki RG, et al. The cyclin-dependent kinase inhibitor flavopiridol potentiates doxorubicin efficacy in advanced sarcomas: preclinical investigations and results of a phase I dose-escalation clinical trial. Clin Cancer Res (2012) 18:2638–47. doi: 10.1158/1078-0432.CCR-11-3203
76. Cicenas J, Kalyan K, Sorokinas A, Stankunas E, Levy J, Meskinyte I, et al. Roscovitine in cancer and other diseases. Ann Transl Med (2015) 3:135. doi: 10.3978/j.issn.2305-5839.2015.03.61
77. Whittaker SR, Mallinger A, Workman P, Clarke PA. Inhibitors of cyclin-dependent kinases as cancer therapeutics. Pharmacol Ther (2017) 173:83–105. doi: 10.1016/j.pharmthera.2017.02.008
78. Zhang M, Zhang L, Hei R, Li X, Cai H, Wu X, et al. CDK inhibitors in cancer therapy, an overview of recent development. Am J Cancer Res (2021) 11:1913–35.
79. Parry D, Guzi T, Shanahan F, Davis N, Prabhavalkar D, Wiswell D, et al. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol Cancer Ther (2010) 9:2344–53. doi: 10.1158/1535-7163.MCT-10-0324
80. George MA, Qureshi S, Omene C, Toppmeyer DL, Ganesan S. Clinical and pharmacologic differences of CDK4/6 inhibitors in breast cancer. Front Oncol (2021) 11:693104. doi: 10.3389/fonc.2021.693104
81. Goel S, DeCristo MJ, Watt AC, BrinJones H, Sceneay J, Li BB, et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature (2017) 548:471–5. doi: 10.1038/nature23465
82. Laphanuwat P, Jirawatnotai S. Immunomodulatory roles of cell cycle regulators. Front Cell Dev Biol (2019) 7:23. doi: 10.3389/fcell.2019.00023
83. Dickson MA, Schwartz GK, Keohan ML, D'Angelo SP, Gounder MM, Chi P, et al. Progression-free survival among patients with well-differentiated or dedifferentiated liposarcoma treated with CDK4 inhibitor palbociclib: A phase 2 clinical trial. JAMA Oncol (2016) 2:937–40. doi: 10.1001/jamaoncol.2016.0264
84. Dickson MA, Koff A, D'Angelo SP, Gounder MM, Keohan ML, Kelly CM, et al. Phase 2 study of the CDK4 inhibitor abemaciclib in dedifferentiated liposarcoma. J Clin Oncol (2019) 37:11004–4. doi: 10.1200/JCO.2019.37.15_suppl.11004
85. Takahashi S, Fujiwara Y, Nakano K, Shimizu T, Tomomatsu J, Koyama T, et al. Safety and pharmacokinetics of milademetan, a MDM2 inhibitor, in Japanese patients with solid tumors: A phase I study. Cancer Sci (2021) 112:2361–70. doi: 10.1111/cas.14875
86. Cornillie J, Wozniak A, Li H, Gebreyohannes YK, Wellens J, Hompes D, et al. Anti-tumor activity of the MDM2-TP53 inhibitor BI-907828 in dedifferentiated liposarcoma patient-derived xenograft models harboring MDM2 amplification. Clin Transl Oncol (2020) 22:546–54. doi: 10.1007/s12094-019-02158-z
87. Laroche-Clary A, Chaire V, Algeo MP, Derieppe MA, Loarer FL, Italiano A. Combined targeting of MDM2 and CDK4 is synergistic in dedifferentiated liposarcomas. J Hematol Oncol (2017) 10:123. doi: 10.1186/s13045-017-0482-3
88. Vilgelm AE, Saleh N, Shattuck-Brandt R, Riemenschneider K, Slesur L, Chen SC, et al. MDM2 antagonists overcome intrinsic resistance to CDK4/6 inhibition by inducing p21. Sci Transl Med (2019) 11(508):1–30. doi: 10.1126/scitranslmed.aav7171
89. Clark AS, Makhlin I, DeMichele A. Setting the pick: Can PI3K inhibitors circumvent CDK4/6 inhibitor resistance? Clin Cancer Res (2021) 27:371–3. doi: 10.1158/1078-0432.CCR-20-3624
Keywords: sarcoma, cyclin dependent kinases (CDK), cdk inhibitors, target therapy, cell cycle
Citation: Merlini A, Pavese V, Manessi G, Rabino M, Tolomeo F, Aliberti S, D’Ambrosio L and Grignani G (2023) Targeting cyclin-dependent kinases in sarcoma treatment: Current perspectives and future directions. Front. Oncol. 13:1095219. doi: 10.3389/fonc.2023.1095219
Received: 10 November 2022; Accepted: 03 January 2023;
Published: 19 January 2023.
Edited by:
Claudia Fumarola, University of Parma, ItalyReviewed by:
Rossella Rota, Bambino Gesù Children’s Hospital (IRCCS), ItalyMichele Bernasconi, University Children’s Hospital Bern, Switzerland
Copyright © 2023 Merlini, Pavese, Manessi, Rabino, Tolomeo, Aliberti, D’Ambrosio and Grignani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lorenzo D’Ambrosio, lorenzo.dambrosio@unito.it
†These authors share last authorship