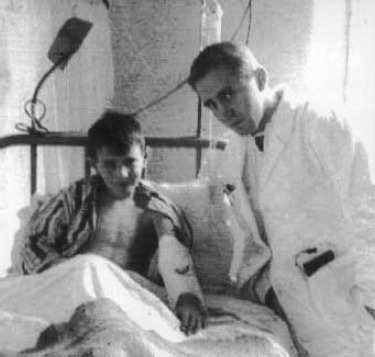
C’est l’histoire d’un homme de 80 ans hospitalisé pour des ecchymoses étendues, d’apparition récente, sur les parties supérieure et inférieure du corps, ainsi qu’au niveau de l’abdomen. Il prend des médicaments depuis des années en raison d’une insuffisance rénale chronique, d’une hypertension, d’une hypercholestérolémie et d’une maladie coronarienne. Il ne présente ni maladie auto-immune, ni cancer. Le seul fait notable est que ce patient prend depuis deux semaines de la doxycycline, un antibiotique, qui lui a été prescrit pour une infection bactérienne cutanée. Quelques jours après la fin du traitement, cet homme a développé des ecchymoses sur les avant-bras, suivies d’un hématome du bras gauche. Il présente également une ecchymose sur la cuisse droite.
Aux urgences, le bilan biologique montre une anémie (hémoglobine à 6,3 g/dl, alors que les valeurs normales sont comprises entre 14 et 18 g/dl), ainsi qu’une anomalie d’un test standard de la coagulation sanguine appelé TCA (temps de céphaline activée). On observe un allongement du TCA. Le taux de plaquettes est normal.
Auto-anticorps inhibiteur du facteur VIII
Par ailleurs, le dosage de l’activité du facteur VIII (facteur anti-hémophilique A), qui agit au niveau de la voie de la coagulation sanguine, indique un taux effondré. Son absence dans le plasma sanguin provoque l’hémophilie A, maladie hémorragique héréditaire dans laquelle le sang ne coagule pas normalement.
Chez ce patient octogénaire, l’activité coagulante du facteur VIII est inférieure à 1 %. Les taux d’autres facteurs de la coagulation (V, IX et XI) sont en revanche normaux. De même, celui du facteur Willebrand (FvW) est normal. L’ensemble de ces résultats fait suspecter la présence dans le plasma sanguin d’une substance qui inhibe spécifiquement l’activité du facteur VIII. Ceci est confirmé en laboratoire*.
1,5 cas par million d’habitants par an
Ce patient présente ce que les hématologistes appellent une hémophilie A acquise. Cette maladie hématologique est due au développement d’un auto-anticorps dirigé contre le facteur VIII.
Il s’agit d’une pathologie rare, dont l’incidence augmente avec l’âge. Elle est estimée à environ 1,5 cas par million d’habitants par an. Elle se manifeste par des hémorragies souvent sévères, dont ne souffre aucun membre de sa famille, et qui apparaissent chez une personne ne présentant pas d’antécédents hémorragiques.
Maladie auto-immune, cancer, grossesse, médicament
L’apparition de l’auto-anticorps anti-facteur VIII peut survenir au cours de maladies auto-immunes, telles que la polyarthrite rhumatoïde (maladie inflammatoire chronique des articulations), le lupus érythémateux systémique (maladie inflammatoire du tissu conjonctif), le syndrome de Gougerot-Sjögren (caractérisé par une sécheresse excessive des yeux, de la bouche et des autres muqueuses), ou encore des maladies dermatologiques comme le psoriasis.
Chez près de 10 % des patients présentant une hémophilie A acquise, la maladie est associée à une tumeur solide (cancer du poumon, de la prostate, du côlon, notamment) ou à un cancer du sang (hémopathies lymphoprolifératives, notamment leucémie lymphoïde chronique, lymphome non-hodgkinien).
La maladie peut également survenir en post-partum (généralement un à quatre mois après la première grossesse), plus rarement au cours de la grossesse. L’hémophilie A acquise du post-partum se manifeste par des hémorragies spontanées (ecchymoses, saignement de nez, sang dans les urines), ou gynécologiques (hémorragie utérine, saignement vaginal). Porter le diagnostic de cette maladie rare est essentiel chez la femme enceinte présentant un allongement isolé du TCA lors du bilan biologique, même lorsque les signes hémorragiques sont minimes ou absents. En effet, cela permet de limiter potentiellement les complications d’ordre hémorragique lors de l’accouchement en mettant préventivement en place des mesures appropriées.
Enfin, l’hémophilie A acquise est parfois associée à la prise de médicaments, tels que la pénicilline et ses dérivés, le triméthoprime/sulfaméthoxazole (antibiotique), la phénytoïne (antiépileptique), la méthyldopa (antihypertenseur), le clopidogrel (antiagrégant plaquettaire), l’interféron-alpha (médicament utilisé en oncologie qui interfère avec le fonctionnement du système immunitaire).
En résumé, le diagnostic de l’hémophilie A acquise est évoqué par la survenue brutale d’un syndrome hémorragique, pouvant potentiellement mettre en jeu le pronostic vital. Dans environ la moitié des cas, l’origine de la maladie est inconnue. On ne trouve pas de pathologie possiblement associée au développement d’auto-anticorps anti-facteur VIII. On parle alors d’hémophilie A acquise idiopathique.
Le premier patient atteint de cette maladie hématologique a été décrit en 1940. Ce n’est qu’en 1961 qu’a été publiée dans la littérature médicale la première synthèse des connaissances sur cette mystérieuse maladie.
La rareté de cette pathologie peut entraîner un retard de diagnostic, d’autant plus que le patient ne présente pas d’antécédent hémorragique personnel et familial. Le patient peut parfois être examiné par plusieurs spécialistes et être soumis à de nombreuses investigations invasives pouvant, elles-mêmes, être à l’origine de graves complications, avant que le diagnostic ne soit correctement posé.
Pour compliquer le tout, les signes cliniques de l’hémophilie A acquise diffèrent de ceux observés dans l’hémophilie « classique » qui est, elle, d’origine génétique et liée au sexe, affectant essentiellement les hommes. L’hémophilie A sévère, caractérisée par un taux de facteur VIII inférieur à 1% de la normale, se manifeste par des hémorragies graves qui débutent habituellement peu après la naissance.
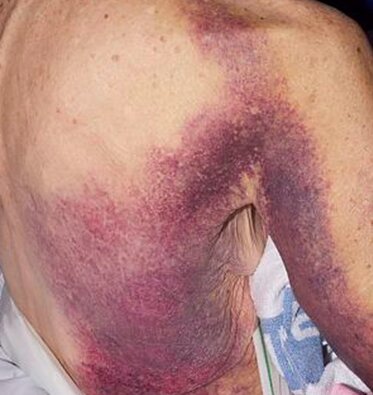
Un tableau clinique qui diffère de celui de l’hémophilie héréditaire
Les symptômes cliniques diffèrent de l’hémophilie classique dans laquelle les saignements prédominent dans les articulations (hémarthroses). En effet, en cas d’hémophilie acquise, les épanchements de sang dans une articulation sont peu fréquents. Les patients développent spontanément des ecchymoses diffuses, des hémorragies musculaires, génito-urinaires, gastro-intestinales ou encore des saignements de nez (épistaxis). La survenue d’une hémorragie intracrânienne est rare mais peut être mortelle. Des signes hémorragiques sévères peuvent apparaître chez des patients ne présentant qu’une baisse modérée d’activité du facteur VIII.
Court-circuiter le facteur VIII
Le traitement a deux objectifs : traiter les complications hémorragiques en même temps que neutraliser l’auto-anticorps anti-facteur VIII. Le traitement antihémorragique repose sur l’utilisation d’agents contournant l’action du facteur VIII.
L’administration de facteur VIII n’est pas efficace en présence d’un taux élevé d’auto-anticorps anti-facteur VIII. Le traitement de l’hémophilie A acquise fait donc appel à des agents qui court-circuitent le facteur VIII dans la cascade de la coagulation, ensemble des nombreuses étapes biochimiques qui aboutissent à l’arrêt du saignement (hémostase). Ces médicaments (on parle d’agents « bypassants » dans le jargon médical) sont des concentrés de facteur VII recombinant activé (rFVIIa) ou des mélanges de facteurs spécifiques de la coagulation (appelés complexes prothrombotiques).
Inhiber la synthèse de l’auto-anticorps
La prise en charge thérapeutique comporte également l’administration précoce d’agents immunosuppresseurs. L’objectif est d’empêcher la production de l’auto-anticorps anti-facteur VIII par le système immunitaire du malade afin de réduire le temps durant lequel le patient est exposé à un risque hémorragique. Les agents immunosuppresseurs utilisés en première intention sont les corticoïdes (prednisone et/ou cyclophosphamide). En l’absence de réponse au traitement, le rituximab (anticorps monoclonal anti-CD20) peut être utilisé. Une étude récente a montré l’intérêt de l’anticorps emicizumab, qui mime certaines fonctions exercées par le facteur VIII.
Les hémorragies sont très rarement la cause directe du décès (environ 3 %). Malgré tout, la mortalité globale associée à cette maladie hématologique est importante. On estime que le taux de mortalité de l’hémophilie A acquise est au moins de 20 % chez les patients de plus de 65 ans et chez ceux souffrant d’un cancer.
Hémophilie induite par un antibiotique
Revenons au patient octogénaire dont le cas est rapporté le 1er octobre 2021 par des internistes et hématologues de la faculté de médecine de l’université du Sud de l’Illinois (Springfield) dans la revue en ligne BMJ Case Reports.
Ce malade a reçu de la prednisone. Trois jours plus tard, l’activité du facteur VIII reste en-deçà de 1 % et le taux d’hémoglobine continue de baisser. Le patient se voit prescrire par voie orale du cyclophosphamide, un immunosuppresseur. Quinze jours plus tard, le taux d’hémoglobine remonte à 9,9 g/dl. Les ecchymoses sur les deux bras et la cuisse droite s’atténuent. Le gonflement dû à l’hématome du bras gauche se résorbe. Le patient quitte l’hôpital le lendemain.
Mais, une semaine plus tard, le patient, toujours traité par prednisone et cyclophosphamide, présente un saignement par le rectum et une hémorragie dans le muscle grand fessier. Ces hémorragies sont probablement dues à la persistance de l’auto-anticorps anti-facteur VIII, associée à une activité coagulante effondrée (inférieure à 1 %).
Alors qu’à son admission à l’hôpital le patient était négatif pour le SARS-CoV-2, virus responsable de la Covid-19, il développe quelque temps après une pneumonie associée à un syndrome de détresse respiratoire aiguë. Son état clinique ne cesse ensuite de se détériorer. Le patient décède de complications respiratoires et rénales.
Selon les auteurs, il s’agit du premier cas d’hémophile acquise associée à la prise récente de doxycycline, aucune autre cause n’ayant été identifiée permettant d’expliquer la survenue de cette maladie hématologique rare chez ce patient. Son hémophilie acquise est due à une réaction auto-immune avec production d’auto-anticorps (immunoglobulines G) reconnaissant certaines régions (épitopes) du facteur VIII et neutralisant son activité coagulante.
Marc Gozlan (Suivez-moi sur Twitter, Facebook, LinkedIn)
* Le test Bethesda confirme la spécificité de l’anticoagulant circulant vis-à-vis du facteur VIII, avec un titre élevé : 115 Bethesda Unit (BU). Le bilan biologique montre un allongement isolé du TCA témoignant de la présence d’un auto-anticorps circulant anti-facteur VIII avec absence de correction du TCA lorsqu’on mélange, à parties égales, le plasma du patient avec un plasma de référence normal. Il existe un déficit isolé en facteur VIII. Le diagnostic de certitude repose sur le titrage du facteur VIII et le dosage de son activité coagulante qui s’avère nettement diminuée, le plus souvent inférieure à 10 % et constamment sous les 30 %, alors que les taux et l’activité des autres facteurs de la coagulation sont en revanche normaux.
Pour en savoir plus :
Shah E, Abro C, Zaidi F, Goel R. Doxycycline-induced acquired haemophilia A. BMJ Case Rep. 2021 Oct 1;14(10):e244748. doi: 10.1136/bcr-2021-244748
Liberman P, Burkholder BM. Adalimumab-associated Acquired Hemophilia in a Patient with Scleritis. Ocul Immunol Inflamm. 2020 Sep 23:1-3. doi: 10.1080/09273948.2020.1808227
Cuilleron J, Mas P, Kiakouama L, et al. Une hémophilie A acquise ayant révélé un cancer du poumon. Rev Mal Respir. 2018 Sep;35(7):727-730. doi: 10.1016/j.rmr.2017.08.005
Franchini M, Vaglio S, Marano G, et al. Acquired hemophilia A: a review of recent data and new therapeutic options. Hematology. 2017 Oct;22(9):514-520. doi: 10.1080/10245332.2017.1319115
Kruse-Jarres R, Kempton CL, et al. Acquired hemophilia A: Updated review of evidence and treatment guidance. Am J Hematol. 2017 Jul;92(7):695-705. doi: 10.1002/ajh.24777
Daumas A, Cauchois R, Massy E, et al. Une hémophilie à 86 ans. Rev Geriatr. 2015 Sep;40(7):433-7.
Tengborn L, Baudo F, Huth-Kühne A, et al; EACH2 registry contributors. Pregnancy-associated acquired haemophilia A: results from the European Acquired Haemophilia (EACH2) registry. BJOG. 2012 Nov;119(12):1529-37. doi: 10.1111/j.1471-0528.2012.03469.x
Collins P, Baudo F, Knoebl P, et al; EACH2 registry collaborators. Immunosuppression for acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2). Blood. 2012 Jul 5;120(1):47-55. doi: 10.1182/blood-2012-02-409185
Chaari M, Sassi M, Galea V, et al. Hémophilie A acquise découverte au cours de la grossesse : à propos d’un cas et revue de la littérature. Rev Med Interne. 2012 Jul;33(7):401-4. doi: 10.1016/j.revmed.2012.04.018
Knoebl P, Marco P, Baudo F, et al; EACH2 Registry Contributors. Demographic and clinical data in acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2). J Thromb Haemost. 2012 Apr;10(4):622-31. doi: 10.1111/j.1538-7836.2012.04654.x
Franchini M, Gandini G, Di Paolantonio T, Mariani G. Acquired hemophilia A: a concise review. Am J Hematol. 2005 Sep;80(1):55-63. doi: 10.1002/ajh.20390
Bossi P, Cabane J, Ninet J, et al. Acquired hemophilia due to factor VIII inhibitors in 34 patients. Am J Med. 1998 Nov;105(5):400-8. doi: 10.1016/s0002-9343(98)00289-7
LIRE aussi : Un patient nommé Christmas