
Therapeutic Approaches Targeting PAX3-FOXO1 and Its Regulatory and Transcriptional Pathways in Rhabdomyosarcoma

Laboratory of Pathology, National Cancer Institute, 10 Center Drive, Bethesda, MD 20892, USA
*
Author to whom correspondence should be addressed.
Molecules 2018, 23(11), 2798; https://doi.org/10.3390/molecules23112798
Submission received: 13 September 2018
/
Revised: 24 October 2018
/
Accepted: 26 October 2018
/
Published: 28 October 2018
(This article belongs to the Special Issue Transcription Factors as Therapeutic Targets)
Abstract
:Rhabdomyosarcoma (RMS) is a family of soft tissue cancers that are related to the skeletal muscle lineage and predominantly occur in children and young adults. A specific chromosomal translocation t(2;13)(q35;q14) that gives rise to the chimeric oncogenic transcription factor PAX3-FOXO1 has been identified as a hallmark of the aggressive alveolar subtype of RMS. PAX3-FOXO1 cooperates with additional molecular changes to promote oncogenic transformation and tumorigenesis in various human and murine models. Its expression is generally restricted to RMS tumor cells, thus providing a very specific target for therapeutic approaches for these RMS tumors. In this article, we review the recent understanding of PAX3-FOXO1 as a transcription factor in the pathogenesis of this cancer and discuss recent developments to target this oncoprotein for treatment of RMS.
1. Introduction
Rhabdomyosarcoma (RMS) is a heterogeneous group of malignant soft tissue tumors that share biological features with skeletal myogenesis. Although considered a rare malignancy, RMS is one of the most common cancers in children, accounting for approximately 50% of all soft tissue sarcomas or ~3–8% of all pediatric cancers [1,2,3,4,5].
RMS tumors are traditionally divided into two major subtypes, embryonal RMS (ERMS) and alveolar RMS (ARMS), based on their histologic features [6,7,8,9]. ERMS is the most common subtype, accounting for approximately 70–80% of RMS cases, while ARMS represents about 20–30% of RMS cases. Each histologic subtype is associated with distinct genetic alterations (Table 1). Point mutations, often involving genes encoding proteins in the RAS signaling pathway, are frequently found in ERMS tumors whereas PAX3-FOXO1 (~60%) and PAX7-FOXO1 (~20%) gene fusions are hallmarks of ARMS tumors [10,11,12,13,14,15] and reviewed in [9,16]. Other fusions of PAX3 with nuclear proteins such as FOXO4 and NCOA1 have been found in rare cases of ARMS tumors [17,18,19,20,21,22,23]. The remaining 20% of ARMS cases do not have any detectable gene fusions, and show mutations characteristic of ERMS tumors [18,24]. In contrast, these recurrent point mutations are rare in fusion-positive ARMS cases. In recent literature, RMS tumors are often divided into PAX gene fusion-positive (FP) and fusion-negative (FN) categories [9,17]. This fusion-based classification seems to reflect better the tumor genetics, which may eventually guide targeted therapeutic decisions, and clinical course of RMS tumors. Several clinical studies provide evidence that this genetic stratification more accurately predicts the clinical progression, treatment response and prognosis of RMS patients [12,18,24,25,26,27,28]. ERMS tumors and FN tumors with an ARMS appearance are generally associated with a good outcome whereas FP tumors are more aggressive, often metastatic, less responsive to chemotherapies, frequently recurrent and have a worse prognosis [12,24,26,29,30,31]. Furthermore, in the setting of FP tumors, patients with PAX3-FOXO1-positive tumors have a worse outcome than those harboring the PAX7-FOXO1 fusion [27,28].
Current management of RMS is based on multimodal treatment that includes surgery and traditional chemotherapy with or without radiotherapy [32,33,34,35]. During the last few decades, effective regimens were identified and have contributed to significantly improved survival. The current chemotherapy regimen for RMS patients relies on a three-drug backbone of vincristine, actinomycin D and cyclophosphamyide [36]. Over the last 30–40 years, the 5-year survival rate of RMS patients has increased to over 60% [2,37]. However, the improved outcome mostly benefits ERMS patients, who have a 5-year survival over 70%, in contrast to ARMS patients, who have a 5-year survival of less than 50% [2]. In addition, though these cytotoxic drugs effectively control most localized RMS cases, the treatment is not very effective against metastatic cases. Approximately 12% of all RMS and 24% of ARMS cases present with distant metastases at the time of diagnosis [38]. In the group of metastatic ARMS tumors, the 5-year overall survival falls below 20% [38]. Many ARMS patients may be initially responsive to multimodal therapy, but later recur and develop metastases. In addition to the ineffectiveness of current therapies in several RMS subsets, many RMS survivors suffer from immediate and long-term treatment-associated toxicities [39,40], underlining the need for more specific drugs.
Molecularly targeted chemotherapy has proven to be successful for treatment of many types of cancer. The finding of RMS subsets harboring distinct genetic alterations provides the starting point for the development of targeted therapy that can be individualized for these RMS subsets. In this article, we focus our review on PAX3-FOXO1 and its signaling as molecular points for targeting FP RMS tumors.
2. PAX-FOXO1 Fusions are Oncogenic Drivers and Therapeutic Targets in FP RMS Tumors
The PAX3-FOXO1 and PAX7-FOXO1 fusion genes are products of the characteristic translocations t(2;13)(q35;q14) or t(1;13)(p36;q14), respectively. The fusion genes are transcribed into fusion transcripts that are in turn translated in fusion proteins. These events result in an in-frame fusion of the DNA binding domains of paired box family protein PAX3 or PAX7 to the transcriptional activating domain of the forkhead family protein FOXO1 (Figure 1) [16,41,47,48].
PAX3 and PAX7 are transcription factors that play essential roles during myogenesis [49]. The PAX3-FOXO1 and PAX7-FOXO1 fusions, which contain intact DNA binding domains from these PAX proteins, are much more potent than wild-type PAX3 or PAX7 in activating transcriptional target expression [50] (Figure 1). Transcriptomic analysis revealed that PAX3 and PAX3-FOXO1 regulate overlapping sets of target genes, although some genes may be preferentially regulated by the fusion [51]. Several lines of evidence indicate that the PAX gene fusions, in cooperation with other genetic changes, are oncogenic drivers in FP RMS tumors. First, the fusion transcription factors induce expression of a number of transcriptional targets that promote oncogenic transformation, including MET, ALK1, MYCN, IGFR1 and FGFR4. Second, FP RMS tumors have a significantly lower number of somatic mutations overall, and few if any recurrent mutations, as compared to FN RMS samples [17]. Third, PAX3-FOXO1 expression is capable of driving oncogenic transformation in cell culture and animal models. Targeted conditional expression of PAX3-FOXO1 in the myogenic lineage of mice induces the formation of tumors resembling human ARMS [52,53]. Furthermore, inactivation of CDNK2A and/or TP53 substantially augments the frequency and progression of these PAX3-FOXO1-driven tumors [52], indicating a possible cooperation of these genetic elements during tumorigenesis. In human cells, PAX3-FOXO1 stimulates cell growth and proliferation in a number of cell culture and xenograft tumor models [54,55,56,57,58,59]. Although expression of PAX3-FOXO1 alone is not able to induce transformation of normal human cells [60,61,62], it can cooperate with additional events, including inactivation of CDKN2A and overexpression of TERT and MYCN to promote tumorigenesis [52,55,62,63]. Similar changes are observed in human RMS tumors [64,65,66,67,68]. In addition to its tumorigenic activity, PAX3-FOXO1 also enhances cell invasion and migration [53,54,57,69]. Fewer experiments have been conducted to examine the role of the closely related PAX7-FOXO1 protein. However, given the similarity in protein structure and activity of the PAX fusions [70], it is expected that the two fusions exert similar effects in promoting tumorigenesis. In addition to the above-described experiments with enforced aberrant PAX3-FOXO1 expression, complementary experiments (described below) in which PAX3-FOXO1 is depleted have shown that PAX3-FOXO1 loss leads to growth arrest and stimulates terminal myogenic differentiation and apoptosis of FP RMS cell lines [54,55,58,59,71]. These results confirm the pro-cancer activity of PAX3-FOXO1 and provide a proof of principle that it is a good cancer target.
The formation of an invariant PAX3-FOXO1 fusion in the majority of ARMS tumors further enhances its value as an attractive target for cancer treatment. It is interesting to note that the PAX3-FOXO1 fusion has been found to be temporarily expressed at the mRNA and protein level during some early stages of normal myogenesis, perhaps as a result of trans-splicing of the PAX3 and FOXO1 transcripts [72,73]. However, it appears that PAX3-FOXO1 is only capable of interfering with normal myogenesis and promoting oncogenic transformation when constantly expressed as a result of a genetic rearrangement [72]. Furthermore, this aberrant fusion has not been detected in normal myogenic cells in children and adults, who would be the patients treated with such targeted therapy. Therefore, the specificity of targeting only tumor cells in these patients is another important aspect of this approach since normal cells will not be significantly affected by many of the proposed interventions.
3. Pharmaceutical Targeting of PAX3-FOXO1 Regulatory and Transcriptional Pathways
3.1. Direct Inhibition of PAX3-FOXO1 by Small Molecules
Design and development of small molecule inhibitors have resulted in remarkable progress for treatment of certain cancers, particularly with drugs targeting protein kinases. However, little progress has been made in directly targeting many transcription factors, some of which appear to be potentially attractive cancer targets, such as PAX3-FOXO1. Direct inhibitors are expected to work by (i) specific binding and promotion of degradation and/or (ii) specific binding and blocking sites required for target protein activation or interaction with other critical effector proteins. The inability to date to design direct inhibitors for wild-type and fusion transcription factors can be attributed in part to the large protein-protein interaction interfaces and absence of deep protein pockets that are common targetable sites for drug design in the case of protein kinases [74,75]. Furthermore, the greater post-translational protein stability of PAX3-FOXO1 compared to the wild type PAX3 [76] can also be an additional obstacle for strategies promoting degradation of the PAX3-FOXO1 protein.
3.2. Inhibition of PAX3-FOXO1 Regulatory Networks
Given the challenges associated with modeling and designing direct inhibitors for PAX3-FOXO1, other strategies have been explored. There has been significant progress in several approaches, such as targeting signal transduction pathways that enhance stability and activity of PAX3-FOXO1, and inhibiting its transcriptional co-activators. Disruption of these pathways or coactivators leads to selective suppression of cell growth and proliferation of PAX3-FOXO1-expressing ARMS cell lines, and thus represents attractive therapeutic strategies.
3.2.1. Targeting Phosphorylation of PAX3-FOXO1
Phosphorylation of PAX3-FOXO1 modulates its transcriptional activity and protein stability (Figure 2) [77,78,79,80]. This modification is carried out by protein kinases, which is a class of proteins that represents a large proportion of actionable cancer targets and for which inhibitors have been intensively screened and developed. The widespread generality of these phosphorylation pathways is particularly important to provide potential drugs to interrogate FP RMS and other rare cancers with critical targets regulated by phosphorylation. Thus, understanding the biology of PAX3-FOXO1 may provide the opportunity to utilize the existing pool of specific inhibitors or drugs to suppress PAX3-FOXO1 activity. Towards this goal, RNA interference and chemical screens have been conducted to identify kinases that promote PAX3-FOXO1 activity and their inhibitors (Table 2).
Several serine/threonine protein kinases phosphorylate PAX3-FOXO1 at serine residues and influence its transcriptional activity (Figure 2). Serine residue 201 (S201), which is located within the PAX3 DNA binding domain, is phosphorylated by GSK3β [81], while neighboring residues S205 and S209 are modified by casein kinase II (CK2) [82]. Serine 430, which is located within the FOXO1 region, is phosphorylated by CDK4 [79]. These modifications lead to enhanced transcriptional activity of the fusion protein, promoting the expression of its downstream targets and oncogenic effects. In addition, inhibition of GSK3β, CK2 and CDK4 suppresses transcriptional activity of PAX3-FOXO1, and in the case of serine 430, inhibition of phosphorylation has been reported to lead to cytoplasmic localization of the protein [78,79,81,82,83]. Similarly, a kinome-targeted siRNA screen found Polo-like kinase-1 and 4 (PLK1 and PLK4) as PAX3-FOXO1 upstream regulators. These Polo-like kinases phosphorylate PAX3-FOXO1 at serine 503 (S503), resulting in enhanced protein stability and oncogenic activity [80]. Concurrently, a parallel small molecule screen identified that PLK1 inhibitors, BI-2536 and BI-6727, strongly inhibited transcriptional activity of PAX3-FOXO1.
These findings raise the possibility that inhibition of PAX3-FOXO1-modifying kinases may be a useful strategy in the treatment of FP RMS. It should be noted that inhibitors of these kinases are already being actively studied in other cancers [89,90,91]. Several GSK3 inhibitors, including TWS119, SB216763, TDZD-8 and roscovitine, suppress PAX3-FOXO1 transcriptional activity, inhibit proliferation and induce apoptosis of PAX3-FOXO1-expressing cells [78]. Similarly, fascaplysin, a selective inhibitor of CDK4/Cyclin D1, also inhibits PAX3-FOXO1 transcriptional activity, and exerts a superior growth inhibitory effect in FP compared to FN RMS cells [79]. Another small molecule, PKC412, which targets a group of protein kinases, inhibits phosphorylation of PAX3-FOXO1 at multiple serine residues (S187, S193, S197, S201, S205 and S209), and suppresses growth in FP RMS lines more effectively than in FN RMS lines in both in vitro and in vivo assays [77]. The PLK1 inhibitors, BI-2536 and BI-6727, promote degradation of the PAX3-FOXO1 protein and induce regression of FP RMS xenogratft tumors [80]. In general, though these kinase studies suggest potential utility as a strategy for FP RMS treatment, the specificity and selectivity for PAX3-FOXO1-expressing cells need to be more fully investigated and independently reproduced in additional preclinical studies.
The sarco/endoplasmic reticulum Ca2+ ATPase inhibitor, thapsigargin, is another potentially useful drug that suppresses expression of PAX3-FOXO1 targets and induces apoptosis in FP RMS cell lines both in vitro and in vivo [84]. The inhibitory effect of thapsigargin on PAX3-FOXO1 transcriptional activity is accompanied by AKT activation, increased phosphorylation of PAX3-FOXO1 and decreased PAX3-FOXO1 protein expression. Although PAX3-FOXO1 contains two of the three serine residues phosphorylated by AKT1 in wild-type FOXO1 (S256 and 319), a previous study showed that AKT1 decreased the transcriptional activity of wild-type FOXO1 but did not affect the transcriptional activity of PAX3-FOXO1 [92]. Thus, it remains unclear how thapsigargin suppresses PAX3-FOXO1 activity. Moreover, the thapsigargin-induced activation of the usually pro-oncogenic AKT signaling pathway might be a concern for cancer therapy.
3.2.2. Targeting Transcriptional Co-Activators of PAX3-FOXO1
Recent studies suggest that targeting co-transcriptional activators of PAX3-FOXO1 is another potential approach for inhibiting PAX3-FOXO1 activity. The function of most transcription factors depends on direct interactions with cofactors as a means of influencing the transcription machinery to promote expression of target genes [93]. Several studies provide evidence that PAX3-FOXO1 needs binding partners to activate transcription of its downstream effectors. One such important co-regulator of PAX3-FOXO1 transcriptional activity is the chromodomain helicase DNA binding protein CHD4 [94]. PAX3-FOXO1 recruits CHD4 to activate expression of a subset of PAX3-FOXO1 target genes that promote cell proliferation. Inhibition of CHD4 reduces viability of FP but not FN RMS cells in vitro and in xenograft tumors [94]. In addition, PAX3-FOXO1 partners with several other proteins such as BRD4, MED1 and p300. Through interactions with these proteins, PAX3-FOXO1 co-occupies and establishes myogenic super enhancers on target genes, such as FGFR4, MYCN, ALK and MET, whose expression has been shown to be responsible for oncogenic activities of PAX3-FOXO1. BRD4 interaction is required for stability and functionality of PAX3-FOXO1 at these key super enhancers, and BRD4 inhibition suppresses PAX3-FOXO1-driven cell growth in FP RMS cell lines in vitro and in xenograft tumors [85].
It should further be noted that both CHD4 and BRD4 are epigenetic chromatin modifiers. CHD4 is an integral component of the nucleosome remodeling deacetylase (NuRD) complex that possesses both chromatin remodeling activity with histone deacetylase (HDAC) and demethylase functions involved in transcriptional repression [95]. Based on the differing effect of CHD4 inhibition on PAX3-FOXO1 target gene expression compared to other members of the NuRD complex, it is hypothesized that CHD4 may also act in a pathway independent of the NuRD complex. BRD4 belongs to the bromodomain and extra-terminal domain (BET) protein family in which each member contains two conserved N-terminal bromodomains. These domains are chromatin interaction modules that recognize acetylated lysines on nuclear proteins such as histones and transcription factors. BRD4 acts to recruit transcriptional regulatory complexes to the acetylated chromatin region [96]. As activation of mammalian gene expression requires a concerted action of transcription factors and chromatin modifiers [97], PAX3-FOXO1 is likely to gain access to the binding sites on its transcription targets within the chromatin landscape created by CHD4, BRD4 and their functioning complexes. Once bound to specific DNA binding motifs within target genes, PAX3-FOXO1 may also further modify the chromatin landscape to promote expression of its oncogenic targets.
The experimental results of CHD4 and BRD4 inhibition in FP RMS cells resemble the effect of PAX3-FOXO1 depletion, providing a rationale for targeting transcriptional co-activators of PAX3-FOXO1, such as CHD4 and BRD4, as potential therapeutic targets in ARMS. However, it should be noted that the effects of BRD4 inhibitors and other epigenetic-based drugs on PAX3-FOXO1-positive RMS cell lines may also be mediated through other mechanisms. The finding of a prominent decrease in PAX3-FOXO1 mRNA expression caused by the HDAC inhibitor entinostat suggests that this drug is acting at the level of PAX3-FOXO1 gene transcription instead of PAX3-FOXO1 protein function [87]. The HDAC inhibitor JNJ-2648185 induces mitochondria-mediated apoptosis in both FP and FN cell lines [98]. JNJ-2648185 was also shown to synergistically enhance apoptotic effects of common anticancer drugs for RMS such as vincristine, actinomycin D, cyclophosphamide, etoposide and doxurubicin in both FP and FN cell lines [99,100]. Combinations of BRD4 and HDAC inhibitors also effectively and synergistically induce apoptosis in both FP and FN cell lines [101]. Thus, these inhibitors may exert antitumor effects by generally disrupting the interplay between transcription factors and the epigenome. Still, there appears to be some level of anti-growth selectivity for FP RMS cells treated with these BET and HDAC inhibitors suggesting a dominant role of transcriptional pathways involving the PAX3-FOXO1 protein and a resulting dependency of these lines on the fusion protein for cell growth and proliferation. Therefore, these inhibitors are considered as important potential drugs for treating FP RMS.
3.2.3. Targeting the Acetylation of PAX3-FOXO1
Another potential therapeutic target in FP RMS is the lysine acetyltransferase (KAT) domain-containing enzyme P/CAF (p300/CBP). PAX3-FOXO1 interacts with P/CAF in FP RMS cells, and the interaction induces acetylation of PAX3-FOXO1 at lysine residues K426 and K429, which are located within the transactivation domain, resulting in increased PAX3-FOXO1 stability and transcriptional activity [86] (Figure 2). Knockdown of P/CAF or inhibition of its acetyltransferase activity by the small molecule embelin reduces PAX3-FOXO1 protein expression and suppresses growth and proliferation of FP RMS lines and xenograft tumors [86]. Embelin specifically interacts with the CoA binding pocket within the KAT domain of P/CAF, and by juxtaposing the 11-carbon alkyl chain of embelin into close contact with the C574 catalytic residue, this small molecule inhibits the KAT activity of P/CAF [102]. Of note, acetylation of the equivalent lysine on FOXO1 (K245) and a nearby residue (K262) was previously shown to enhance AKT1-mediated phosphorylation of FOXO1 and attenuate its binding affinity with cognate DNA targets [103]. In this context, it is possible that AKT is responsible for the contrasting effects of acetylation on the transcriptional activity of FOXO1 and PAX3-FOXO1; AKT1 was previously reported to phosphorylate FOXO1, leading to its cytoplasmic retention and inactivation [104], but it had no effect on the PAX3-FOXO1 fusion protein [92]. Finally, it should also be noted that P/CAF inhibition might cause additional effects in FP RMS cells. For example, P/CAF is a required cofactor for transcriptional activity of the myogenic differentiation factor MyoD, whose corresponding gene is a transcriptional target of PAX3-FOXO1. P/CAF acetylates histones 3 and 4 at the promoter sites of MyoD target genes, creating a chromatin state that activates its transcriptional activity in muscle cells [105,106].
3.3 Targeting Downstream Effectors of PAX3-FOXO1
PAX3-FOXO1 exerts its oncogenic effect through transcriptional activation of downstream targets whose expression is proposed to promote tumorigenesis. High-throughput technologies such as DNA microarray, RNA sequencing and chromatin immunoprecipitation sequencing (ChIP-Seq) have enabled generation of comprehensive signatures of downstream targets expressed in PAX3-FOXO1-expressing tumors. Several of these downstream effectors have activities that promote cancer development, such as stimulating cell proliferation and inhibiting apoptosis [16,42,71,107,108], and thus a subset of these targets may confer the tumorigenic effects of PAX3-FOXO1. These findings provide a rationale for potentially targeting one or more of these downstream targets as a potential therapeutic intervention in FP RMS. Specific inhibitors have been identified and characterized for some oncogenic targets of PAX3-FOXO1 (Table 3). These inhibitors selectively suppress cell growth and proliferation in vitro and suppress tumor growth in vivo in preclinical models of FP RMS [99,109,110,111,112,113,114,115,116]. Previously, we reviewed potential targets such as FGFR4, MYCN and MET that have been explored for FP RMS treatment [16]. Recent studies have provided more evidence regarding the effects resulting from inhibition of these targets and have identified new targets for treatment of FP RMS. We update the list of potential candidates that have been or could be explored for treatment of FP RMS patients (Table 3, [69,117,118,119,120,121,122,123]).
In these studies of downstream targets of PAX3-FOXO1, the oncogenic activity of PAX3-FOXO1 is postulated to result from the collective effects of multiple downstream targets. Indeed, individual expression of each of these downstream targets does not recapitulate the full oncogenic effect of PAX3-FOXO1. These findings suggest that each of the selected downstream targets may be necessary but not sufficient for the oncogenic effect of PAX3-FOXO1. In contrast, as described above and in Table 3, multiple studies have reported that individual inhibition of one of several downstream targets can induce tumor regression in preclinical models. Thus, there may be limitations in the interpretation or overall validity of the experimental models utilized in these studies.
4. Immunotherapy Applications to Target PAX3-FOXO1 and Downstream Pathways
4.1. Targeting PAX3-FOXO1 by Immunotherapy
Immunotherapies such as neutralizing antibodies, cancer vaccines and T cells modified to express a chimeric antigen receptor (CAR) have been successfully applied to treat multiple types of human cancer. Efforts have been made to develop cancer vaccines that specifically target PAX3-FOXO1, a nuclear oncoprotein with potential neoantigens [146,147,148,149]. Though one study did not find a peptide antigen in the fusion breakpoint region that could effectively stimulate a cytotoxic lymphocyte response, a second study identified a peptide from the PAX3-FOXO1 breakpoint area that is able to elicit such an immune response [147,148]. In particular, this latter study pulsed autologous dendritic cells from a normal donor with the breakpoint peptide to generate a cytotoxic lymphocyte line that can lyse RMS tumor cells expressing PAX3-FOXO1 [148]. A subsequent clinical pilot study found that 39% of patients with advanced RMS or Ewing’s sarcoma receiving dendritic cells pulsed with peptides derived from the fusion breakpoint region showed measurable immune response to the corresponding fusion peptides [149]. However, the finding that all these patients were capable of developing an immune response to the influenza vaccine, which was concurrently administered, indicated that the fusion peptides were not consistently immunogenic. In addition, the immune response to the fusion peptides was found to be short-lived and was not associated with a difference in outcome. Additional technological improvements are thus needed to induce a strong, sustained and clinically effective immune response to the PAX3-FOXO1 fusion peptides in RMS patients.
4.2. Targeting Cell Surface Targets of PAX3-FOXO1 by Monoclonal Antibodies
In addition to targeting oncogenic effectors, some of the PAX3-FOXO1 targets, such as FGFR4, CXCR4 and IGF1R, are cell surface antigens, and thus constitute attractive molecular targets for immunotherapy. The high expression levels of these surface antigens in FP tumor cells allow a possible dose titration of immunologic agents to specifically target the cancer cells while causing minimum effects on normal body cells.
Monoclonal antibodies (MAB) are perhaps the most common and effective immunologic agents used to target cell surface antigens. These agents often act by binding to the extracellular domains of antigens, and then inhibiting ligand binding and/or promoting intracellular internalization. Several MABs against surface antigens in FP RMS have been developed and tested for their effect on RMS tumors. IGF1R MABs are very effective in inhibiting growth of RMS cell lines that express high levels of the IGF1R protein and inducing regression of xenograft tumors generated from these cell lines [150]. Several clinical trials using IGF1R MAB also recruited RMS patients [132,151] and ongoing NCT03041701; however, the low number of FP RMS patients recruited to date does not permit any conclusion about their utility for treating FP RMS. Similarly, CXCR4 MABs are efficient in suppressing cell growth, invasion and metastasis of FP RMS cells [141,152]. Neutralizing MABs targeting PDGFRα inhibit cell growth of PAX3-FOXO1-expressing cells both in vitro and in vivo [119]. Currently, several clinical trials are being conducted to evaluate the effect of MABs against these surface antigens in human cancer, and these trials include FP RMS patients (Table 3).
4.3. Targeting Cell Surface Targets of PAX3-FOXO1 by CAR T-Cells
Recently, T-cell-mediated immunotherapy has been developed as a novel approach for cancer treatment [153,154]. In this therapy, T cells are genetically modified to express chimeric antigen receptors (CAR) that target cell surface markers on cancer cells. Therapy with CAR T cells has been shown to be highly effective in certain types of leukemia and lymphoma; for example CAR T cells targeting CD19 have strong activity in the treatment of refractory B-cell malignancies [155,156]. In addition, other types of CAR T cell therapy are being investigated in clinical trials of hematological malignancies as well as solid tumors [156,157,158,159] and reviewed in [160,161,162]. Preclinical data suggest that T cell therapy could be employed to eradicate cells expressing surface antigens encoded by transcriptional targets of PAX3-FOXO1. In particular, T cells genetically modified to express a CAR that targets FGFR4 have been developed for treatment of RMS tumors [151]. Testing of these FGFR4-specific CAR T cells in vitro and in mouse models revealed promising effects of these T cells in killing tumor cells and suggested that these agents could be beneficial for treatment of high-risk, refractory and relapsed RMS [126,127,163]. In addition, IGFR1-specific CAR T cells were recently developed, and initial results indicated that they suppress growth of several sarcoma categories expressing this receptor in both cell line and xenograft systems [164].
5. Future Approaches in the Treatment of RMS
5.1. Inhibition of PAX3-FOXO1 Expression by Oligonucleotide-Based Technologies
Synthetic oligonucleotide-mediated targeting technologies, including antisense oligo-nucleotides (ASO), RNA interference (RNAi) and more recently CRISPR/Cas9-based genome editing, have proven to be effective strategies to inhibit in vitro expression of many cellular targets. For a difficult-to-target protein like PAX3-FOXO1, in which pharmaceutically targeted approaches may not be readily available, the oligonucleotide-based approaches appear to be desirable alternatives due to their high specificity. Although considered highly specific, these oligonucleotide-based technologies can have off-target effects, often due to inadequate sequence specificity. These effects can be detected in most cases by comparing the effects of independent reagents and performing rescue experiments.
Specific ASOs against the PAX3 gene were used successfully to inhibit PAX3-FOXO1 expression and induce apoptosis in a FP RMS cell line [165], though the number of studies using this technology for PAX3-FOXO1 suppression are very limited. The advancement in silencing approaches represented by RNAi and CRISPR technologies has contributed to the phasing out of ASO-mediated approaches. Specific RNAi approaches designed against PAX3-FOXO1 (based on isolated oligonucleotides or shRNA-containing expression constructs) have been used successfully in a number of studies in cell culture experiments and xenograft tumors. These approaches significantly decrease PAX3-FOXO1 expression, and induce myogenic differentiation, growth arrest and apoptosis in FP RMS lines [54,58].
During the last few years, CRISPR/Cas9 has become a method of choice over RNAi and ASOs in many in vitro and in vivo studies due to its relatively simple design and the ability to completely inhibit target expression compared to the uncertain degree of silencing often obtained with the other methods. The CRISPR/Cas9 construct can be delivered by viral transduction to tumor cells to abolish PAX3-FOXO1 expression in both cell culture and xenograft models of FP RMS. In particular, CRISPR-mediated knockout of PAX3-FOXO1 expression resulted in a significant regression of FP RMS xenograft tumors [166]. Similar inhibitory effects were observed using CRISPR-mediated knockout of exogenous PAX3-FOXO1 expression in a myoblast model of FP RMS [55].
Currently, specific tools targeting PAX3-FOXO1 such as RNAi or CRISPR, which can be very effective in preclinical models, are still not applicable in the clinical setting, mainly due to the lack of an efficient delivery system for human use. One proposed means for delivery involves use of nanoparticles to encase the therapeutic agent. Despite advances in manufacturing nanoparticles for use in humans, multiple structural features need to be engineered and optimized to deliver safe therapeutic doses of these nanoparticle-packed agents and to minimize clearance by the body. An encouraging finding in several recent studies suggests that endogenous nanoparticles (exosomes), nano-sized extracellular vesicles with a membrane lipid bilayer that are released by all types of body cells, can be highly effective as a delivery vehicle for clinical use. Importantly, the lipid bilayer exosomes have been found to contain immune modulating factors that protect them from being targeted by the immune system and can efficiently enter target cells [167]. A recent study provided an example in which siRNA-packaged exosomes efficiently target oncogenic KRAS in a mouse model of pancreatic cancer [168]. These studies hold promise for a new class of vehicles that can be safely and efficiently utilized to deliver specific agents for clinical use.
5.2. Identification of New Therapeutic Targets for RMS
A potential avenue for identifying new approaches to seemingly “undruggable” oncogenic targets is the elucidation of novel gene dependencies by synthetic lethality screening [75,169]. In the context of oncogene addiction, the gain-of-function mutations that contribute to tumorigenesis often leads to cellular dependence on the expression of another gene and thus confers tumor-specific vulnerabilities if that second gene is inhibited. For example, investigation of synthetic lethality interactions involving the RAS or MYC oncogenes has pointed to new directions for potential therapy of tumor cells whose growth is addicted to one of these intractable oncogenes [170,171,172]. Such approaches could also be considered for PAX3-FOXO1 and similar fusion oncoproteins. Recent advances in genome-scale technologies such as whole genome CRISPR-based genetic screens, which can allow comprehensive interrogations of functional genomics, are expected to escalate the identification of synthetic lethality interactions or dependencies for many oncoproteins.
6. Ongoing Challenges in the Development of Therapies for RMS
A challenge complicating the development of therapies targeting the fusion oncoproteins in FP RMS and other sarcomas is the relatively small number of patients afflicted with such cancers. For a tumor-specific oncoprotein in a rare disease, there is not much financial motivation for pharmaceutical companies to invest the considerable resources required for drug development to identify and optimize specific inhibitors. In contrast, oncogenic changes that are present in common tumors or in multiple tumor categories provide a larger potential market and are thus much more attractive targets for commercial drug development.
In the studies of PAX3-FOXO1 described above, more experimental data are needed to establish whether the results obtained from the selected preclinical models are sufficient to infer a similar effect in humans. It is important to acknowledge that there are clear issues involved in using long-term tumor-derived cell lines that have been selected for optimal growth in cell culture conditions. There is a body of evidence indicating genetic and epigenetic alterations in such cell lines when compared to primary human tumors. At the present time, patient-derived xenografts (PDX), which are directly established from primary human tumors and not passaged in cell culture, more faithfully reflect the behavior of the original tumors and thus appear to be a superior system for preclinical studies (reviewed in [173,174,175]). Therefore, it is recommended that the promising pre-clinical results for targeted therapeutics of FP RMS be validated in PDX models before proceeding to investigate the effect of these therapies in clinical trials. However, it must also be appreciated that these PDX studies are conducted in immunocompromised mice so that the interactions of the immune system are not investigated, and thus there is also a role for other animal models with intact immune systems to fully understand the effects of these targeted agents.
If successfully developed and implemented, therapeutic targeting of PAX3-FOXO1 may provide new opportunities for treating FP RMS patients. However, as has been found for other targeted therapies, treatment with a PAX3-FOXO1-targeting drug as a single agent will most likely fail to provide a long-term cure due to the development of tumor cells resistant to this therapy. There is certainly heterogeneity within any FP RMS tumor and there are likely to be rare cells within the population that possess an ability to evade any single therapeutic mechanism. In addition, though the FP RMS tumor cells appear to be addicted to the PAX3-FOXO1 fusion protein, we recently demonstrated in a myoblast model of FP RMS that PAX3-FOXO1-independent tumors can recur after fusion protein expression is inhibited [52]. Multidrug chemotherapy together with surgery and radiotherapy generally results in a greater long-term therapeutic impact than a single modality or agent. The combined synergistic or additive effect of multiple drugs and therapeutic modalities substantially reduces the probability of resistance and ensures that both local and distal disease are managed. Furthermore, the combination of drugs may allow reduced doses of each drug, thus avoiding toxicity associated with higher dosages of a single agent. Therefore, as with conventional cancer therapies, the combination of several drugs and treatment modalities continues to be a cornerstone in cancer therapy.
7. Conclusions
Molecularly targeted therapies have become a reality for treatment of many human cancers. The success is attributed to accumulated understanding of tumor genetics, including identification of cancer driver genes, specific expression signatures and essential signaling networks specific for particular groups or subgroups of cancer. Gene fusions emerge as an attractive category of cancer genes, because the chimeric molecules encoded by these gene fusions are often dominant-acting drivers of tumorigenesis and usually expressed only in tumor cells. Successful therapies have been developed to directly target several fusion proteins involved in signal transduction, such as BCR-ABL in leukemia and EML4-ALK1 in lung carcinoma. However, the development of therapeutic tools to suppress the oncogenic activity of other fusion proteins, such as fusion transcription factors, has encountered numerous challenges, including protein structural constraints and access to the nuclear localization.
Though efforts will continue to identify drugs that specifically target various tumor-specific fusion oncoproteins, investigations of the larger biological context of these fusion transcription factors has led to a larger set of therapeutic opportunities that can take advantage of pre-existing drugs. For FP RMS, an understanding of the signaling pathways controlling the biological functions of PAX3-FOXO1 has pointed to inhibitors of these pathways as promising therapeutic tools. The recent identification of kinases regulating PAX3-FOXO1 and discovery of its essential cofactors such as CHD4 and BRD4 has contributed to new conceptual strategies for targeting PAX3-FOXO1. Targeting selected downstream effectors of PAX3-FOXO1 may be another promising approach for treatment of FP RMS tumors. In addition to small molecule inhibitors of these downstream targets, a subgroup of these targets are cell surface molecules that are potential targets for immunotherapies using neutralizing antibodies or CAR T cells.
Funding
This research was supported by the Intramural Research Program of the National Cancer Institute and the Joanna McAfee Childhood Cancer Foundation.
Conflicts of Interest
Authors declare no conflict of interest.
References
- Linet, M.S.; Ries, L.A.; Smith, M.A.; Tarone, R.E.; Devesa, S.S. Cancer surveillance series: Recent trends in childhood cancer incidence and mortality in the united states. J. Natl. Cancer Inst. 1999, 91, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Ognjanovic, S.; Linabery, A.M.; Charbonneau, B.; Ross, J.A. Trends in childhood rhabdomyosarcoma incidence and survival in the united states, 1975–2005. Cancer 2009, 115, 4218–4226. [Google Scholar] [CrossRef] [PubMed]
- Weihkopf, T.; Blettner, M.; Dantonello, T.; Jung, I.; Klingebiel, T.; Koscielniak, E.; Lückel, M.; Spix, C.; Kaatsch, P. Incidence and time trends of soft tissue sarcomas in german children 1985–2004–A report from the population-based german childhood cancer registry. Eur. J. Cancer 2008, 44, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Ward, E.; DeSantis, C.; Robbins, A.; Kohler, B.; Jemal, A. Childhood and adolescent cancer statistics, 2014. CA: Cancer J. Clin. 2014, 64, 83–103. [Google Scholar] [CrossRef] [PubMed]
- Gatta, G.; Trama, A.; Capocaccia, R.; Hackl, M.; Eycken, E.V.; Henau, K.; Dimitrova, N.; Sekerija, M.; Dušek, L.; Mägi, M.; et al. Epidemiology of rare cancers and inequalities in oncologic outcomes. Eur. J. Surg. Oncol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Pediatric Bone and Soft Tissue Sarcomas; Pappo, A.S. (Ed.) Springer Science & Business Media: Berlin/Heidelberg, Germany, 2006; ISBN 978-3-540-40843-7. [Google Scholar]
- Yang, L.; Takimoto, T.; Fujimoto, J. Prognostic model for predicting overall survival in children and adolescents with rhabdomyosarcoma. BMC Cancer 2014, 14, 395–397. [Google Scholar] [CrossRef] [PubMed]
- O'Neill, J.P.; Bilsky, M.H.; Kraus, D. Head and neck sarcomas. Neurosurg. Clin. N. Am. 2013, 24, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Parham, D.M.; Barr, F.G. Classification of rhabdomyosarcoma and its molecular basis. Adv. Anatomic Pathol. 2013, 20, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Barr, F.G.; Nauta, L.E.; Davis, R.J.; Schafer, B.W.; Nycum, L.M.; Biegel, J.A. In Vivo amplification of the pax3-fkhr and pax7-fkhr fusion genes in alveolar rhabdomyosarcoma. Hum. Mol. Genet. 1996, 5, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.J.; Barr, F.G. Fusion genes resulting from alternative chromosomal translocations are overexpressed by gene-specific mechanisms in alveolar rhabdomyosarcoma. Proc. Natl. Acad. Sci. USA 1997, 94, 8047–8051. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, P.H.B.; Lynch, J.C.; Qualman, S.J.; Tirabosco, R.; Lim, J.F.; Maurer, H.M.; Bridge, J.A.; Crist, W.M.; Triche, T.J.; Barr, F.G. PAX3-FKHRand PAX7-FKHR gene fusions are prognostic indicators in alveolar rhabdomyosarcoma: a report from the children’s oncology group. J. Clin. Oncol. 2002, 20, 2672–2679. [Google Scholar] [CrossRef] [PubMed]
- de Alava, E.; Ladanyi, M.; Rosai, J.; Gerald, W.L. Detection of chimeric transcripts in desmoplastic small round cell tumor and related developmental tumors by reverse transcriptase polymerase chain reaction. A specific diagnostic assay. Am. J. Pathol. 1995, 147, 1584–1591. [Google Scholar] [PubMed]
- Frascella, E.; Toffolatti, L.; Rosolen, A. Normal and rearranged PAX3 expression in human rhabdomyosarcoma. Cancer Genet. Cytogenet. 1998, 102, 104–109. [Google Scholar] [CrossRef]
- Barr, F.G.; Smith, L.M.; Lynch, J.C.; Strzelecki, D.; Parham, D.M.; Qualman, S.J.; Breitfeld, P.P. Examination of gene fusion status in archival samples of alveolar rhabdomyosarcoma entered on the intergroup rhabdomyosarcoma study-III trial. J. Mol. Diagn. 2006, 8, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Olanich, M.E.; Barr, F.G. A Call to ARMS: Targeting the PAX3-FOXO1gene in Alveolar Rhabdomyosarcoma. Expert Opin. Ther. Targets 2013, 17, 607–623. [Google Scholar] [CrossRef] [PubMed]
- Shern, J.F.; Chen, L.; Chmielecki, J.; Wei, J.S.; Patidar, R.; Rosenberg, M.; Ambrogio, L.; Auclair, D.; Wang, J.; Song, Y.K.; et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. 2014, 4, 216–231. [Google Scholar] [CrossRef] [PubMed]
- Barr, F.G.; Qualman, S.J.; Macris, M.H.; Melnyk, N.; Lawlor, E.R.; Strzelecki, D.M.; Triche, T.J.; Bridge, J.A.; Sorensen, P.H.B. Genetic heterogeneity in the alveolar rhabdomyosarcoma subset without typical gene fusions. Cancer Res. 2002, 62, 4704–4710. [Google Scholar] [PubMed]
- Wachtel, M.; Dettling, M.; Koscielniak, E.; Stegmaier, S.; Treuner, J.; Simon-Klingenstein, K.; Bühlmann, P.; Niggli, F.K.; Schäfer, B.W. Gene expression signatures identify rhabdomyosarcoma subtypes and detect a novel T(2;2)(Q35;P23) translocation fusing PAX3 to NCOA1. Cancer Res. 2004, 64, 5539–5545. [Google Scholar] [CrossRef] [PubMed]
- Chmielecki, J.; Bailey, M.; He, J.; Elvin, J.; Vergilio, J.-A.; Ramkissoon, S.; Suh, J.; Frampton, G.M.; Sun, J.X.; Morley, S.; et al. Genomic profiling of a large set of diverse pediatric cancers identifies known and novel mutations across tumor spectra. Cancer Res. 2017, 77, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois, J.M.; Knezevich, S.R.; Mathers, J.A.; Sorensen, P.H. Molecular detection of the ETV6-NTRK3 gene fusion differentiates congenital fibrosarcoma from other childhood spindle cell tumors. Am. J. Surg. Pathol. 2000, 24, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, G.; Guanciali Franchi, P.; Stuppia, L.; Rossi, C.; Bianchi, C.; Antonucci, A.; Palka, G. Translocation (8;11)(Q12–13;Q21) in embryonal rhabdomyosarcoma. Cancer Genet. Cytogenet. 1992, 58, 210–211. [Google Scholar] [CrossRef]
- Sirvent, N.; Trassard, M.; Ebran, N.; Attias, R.; Pedeutour, F. Fusion of EWSR1 with the DUX4 facioscapulohumeral muscular dystrophy region resulting from T(4;22)(Q35;Q12) in a case of embryonal rhabdomyosarcoma. Cancer Genet. Cytogenet. 2009, 195, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Williamson, D.; Missiaglia, E.; de Reyniès, A.; Pierron, G.; Thuille, B.; Palenzuela, G.; Thway, K.; Orbach, D.; LAÉ, M.; Fréneaux, P.; et al. Fusion gene–negative alveolar rhabdomyosarcoma is clinically and molecularly indistinguishable from embryonal rhabdomyosarcoma. J. Clin. Oncol. 2010, 28, 2151–2158. [Google Scholar] [CrossRef] [PubMed]
- Davicioni, E.; Anderson, M.J.; Finckenstein, F.G.; Lynch, J.C.; Qualman, S.J.; Shimada, H.; Schofield, D.E.; Buckley, J.D.; Meyer, W.H.; Sorensen, P.H.B.; et al. Molecular classification of rhabdomyosarcoma-genotypic and phenotypic determinants of diagnosis: A report from the children's oncology group. Am. J. Pathol. 2009, 174, 550–564. [Google Scholar] [CrossRef] [PubMed]
- Skapek, S.X.; Anderson, J.; Barr, F.G.; Bridge, J.A.; Gastier-Foster, J.M.; Parham, D.M.; Rudzinski, E.R.; Triche, T.; Hawkins, D.S. PAX-FOXO1 fusion status drives unfavorable outcome for children with rhabdomyosarcoma: A children’s oncology group report. Pediatr. Blood Cancer 2013, 60, 1411–1417. [Google Scholar] [CrossRef] [PubMed]
- Missiaglia, E.; Williamson, D.; Chisholm, J.; Wirapati, P.; Pierron, G.; Petel, F.; Concordet, J.-P.; Thway, K.; Oberlin, O.; Pritchard-Jones, K.; et al. PAX3/FOXO1 fusion gene status is the key prognostic molecular marker in rhabdomyosarcoma and significantly improves current risk stratification. J. Clin. Oncol. 2012, 30, 1670–1677. [Google Scholar] [CrossRef] [PubMed]
- Kubo, T.; Shimose, S.; Fujimori, J.; Furuta, T.; Ochi, M. Prognostic value of PAX3/7–FOXO1 fusion status in alveolar rhabdomyosarcoma: systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 2015, 96, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Duan, F.; Smith, L.M.; Gustafson, D.M.; Zhang, C.; Dunlevy, M.J.; Gastier-Foster, J.M.; Barr, F.G. Genomic and clinical analysis of fusion gene amplification in rhabdomyosarcoma: a report from the children's oncology group. Genes Chromosom. Cancer 2012, 51, 662–674. [Google Scholar] [CrossRef] [PubMed]
- Selfe, J.; Olmos, D.; Al-Saadi, R.; Thway, K.; Chisholm, J.; Kelsey, A.; Shipley, J. Impact of fusion gene status versus histology on risk-stratification for rhabdomyosarcoma: retrospective analyses of patients on uk trials. Pediatr. Blood Cancer 2016, 64, e26386. [Google Scholar] [CrossRef] [PubMed]
- Eguía-Aguilar, P.; López-Martínez, B.; Retana-Contreras, C.; Perezpeña-Diazconti, M. Alveolar Rhabdomyosarcoma: Origin and prognostic implications of molecular findings. Boletín Médico del Hospital Infantil de México 2017, 73, 405–410. [Google Scholar]
- Huh, W.; Egas Bejar, D. Rhabdomyosarcoma in adolescent and young adult patients: Current perspectives. Adolesc. Health Med. Ther. 2014, 5, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Hosoi, H. Current status of treatment for pediatric rhabdomyosarcoma in the usa and japan. Pediatr. Int. 2016, 58, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Van der Graaf, W.T.A.; Orbach, D.; Judson, I.R.; Ferrari, A. Soft tissue sarcomas in adolescents and young adults: A comparison with their paediatric and adult counterparts. Lancet Oncol. 2017, 18, e166–e175. [Google Scholar] [CrossRef]
- Ray, A.; Huh, W.W. Current state-of-the-art systemic therapy for pediatric soft tissue sarcomas. Curr. Oncol. Rep. 2012, 14, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Crist, W.M.; Garnsey, L.; Beltangady, M.S.; Gehan, E.; Ruymann, F.; Webber, B.; Hays, D.M.; Wharam, M.; Maurer, H.M. Prognosis in children with rhabdomyosarcoma: a report of the intergroup rhabdomyosarcoma studies I and II. Intergroup rhabdomyosarcoma committee. J. Clin. Oncol. 1990, 8, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Hibbitts, E.; Hawkins, D.S.; Arndt, C.; Chi, Y.Y. Risk stratification including FOXO1 fusion status (FOXO1) in patients with rhabdomyosarcoma (RMS) treated on six recent frontline trials: A report from the children's oncology group. J. Clin. Oncol. 2017, 35 (Suppl. S15), 10528. [Google Scholar] [CrossRef]
- Breneman, J.C.; Lyden, E.; Pappo, A.S.; Link, M.P.; Anderson, J.R.; Parham, D.M.; Qualman, S.J.; Wharam, M.D.; Donaldson, S.S.; Maurer, H.M.; et al. Prognostic factors and clinical outcomes in children and adolescents with metastatic rhabdomyosarcoma—A report from the intergroup rhabdomyosarcoma study IV. J. Clin. Oncol. 2003, 21, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Arndt, C.A.S.; Stoner, J.A.; Hawkins, D.S.; Rodeberg, D.A.; Hayes-Jordan, A.A.; Paidas, C.N.; Parham, D.M.; Teot, L.A.; Wharam, M.D.; Breneman, J.C.; et al. Vincristine, actinomycin, and cyclophosphamide compared with vincristine, actinomycin, and cyclophosphamide alternating with vincristine, topotecan, and cyclophosphamide for intermediate-risk rhabdomyosarcoma: Children’s oncology group study D9803. J. Clin. Oncol. 2009, 27, 5182–5188. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.J.; Schwartz, C. Survivorship. J. Surg. Oncol. 2015, 111, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Barr, F.G. Gene fusions involving PAX and FOX family members in alveolar rhabdomyosarcoma. Oncogene 2001, 20, 5736–5746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linardic, C.M. PAX3–FOXO1 fusion gene in rhabdomyosarcoma. Cancer Lett. 2008, 270, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Barr, F.G.; Duan, F.; Smith, L.M.; Gustafson, D.; Pitts, M.; Hammond, S.; Gastier-Foster, J.M. Genomic and clinical analyses of 2p24 and 12q13-Q14 amplification in alveolar rhabdomyosarcoma: A report from the children’s oncology group. Genes Chromosom. Cancer 2009, 48, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Olanich, M.E.; Sun, W.; Hewitt, S.M.; Abdullaev, Z.; Pack, S.D.; Barr, F.G. CDK4 amplification reduces sensitivity to cdk4/6 inhibition in fusion-positive rhabdomyosarcoma. Clin. Cancer Res. 2015, 21, 4947–4959. [Google Scholar] [CrossRef] [PubMed]
- Bridge, J.A.; Liu, J.; Qualman, S.J.; Suijkerbuijk, R.; Wenger, G.; Zhang, J.; Wan, X.; Baker, K.S.; Sorensen, P.; Barr, F.G. Genomic gains and losses are similar in genetic and histologic subsets of rhabdomyosarcoma, whereas amplification predominates in embryonal with anaplasia and alveolar subtypes. Genes Chromosom. Cancer 2002, 33, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Seki, M.; Nishimura, R.; Yoshida, K.; Shimamura, T.; Shiraishi, Y.; Sato, Y.; Kato, M.; Chiba, K.; Tanaka, H.; Hoshino, N.; et al. Integrated genetic and epigenetic analysis defines novel molecular subgroups in rhabdomyosarcoma. Nat. Commun. 2015, 6, 3395–3398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barr, F.G.; Galili, N.; Holick, J.; Biegel, J.A.; Rovera, G.; Emanuel, B.S. Rearrangement of the PAX3 paired box gene in the paediatric solid tumour alveolar rhabdomyosarcoma. Nat. Genet. 1993, 3, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Galili, N.; Davis, R.J.; Fredericks, W.J.; Mukhopadhyay, S.; Rauscher, F.J.; Emanuel, B.S.; Rovera, G.; Barr, F.G. Fusion of a fork head domain gene to pax3 in the solid tumour alveolar rhabdomyosarcoma. Nat. Genet. 1993, 5, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, M.; Relaix, F. PAX3 and PAX7 as upstream regulators of myogenesis. Semin. Cell Dev. Biol. 2015, 44, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Fredericks, W.J.; Galili, N.; Mukhopadhyay, S.; Rovera, G.; Bennicelli, J.; Barr, F.G.; Rauscher, F.J., III. The PAX3-FKHR fusion protein created by the T(2;13) translocation in alveolar rhabdomyosarcomas is a more potent transcriptional activator than PAX3. Mol. Cell. Biol. 1995, 15, 1522–1535. [Google Scholar] [CrossRef] [PubMed]
- Begum, S.; Emani, N.; Cheung, A.; Wilkins, O.; Der, S.; Hamel, P.A. Cell-Type-Specific regulation of distinct sets of gene targets by Pax3 and Pax3/FKHR. Oncogene 2005, 24, 1860–1872. [Google Scholar] [CrossRef] [PubMed]
- Keller, C.; Arenkiel, B.R.; Coffin, C.M.; El-Bardeesy, N.; DePinho, R.A.; Capecchi, M.R. Alveolar rhabdomyosarcomas in conditional pax3:fkhr mice: Cooperativity of Ink4a/ARF and Trp53 loss of function. Genes Dev. 2004, 18, 2614–2626. [Google Scholar] [CrossRef] [PubMed]
- Nishijo, K.; Chen, Q.-R.; Zhang, L.; McCleish, A.T.; Rodriguez, A.; Cho, M.J.; Prajapati, S.I.; Gelfond, J.A.L.; Chisholm, G.B.; Michalek, J.E.; et al. Credentialing a preclinical mouse model of alveolar rhabdomyosarcoma. Cancer Res. 2009, 69, 2902–2911. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, K.; Tsuchiya, K.; Otabe, O.; Gotoh, T.; Tamura, S.; Katsumi, Y.; Yagyu, S.; Tsubai-Shimizu, S.; Miyachi, M.; Iehara, T.; et al. Effects of PAX3-FKHR on malignant phenotypes in alveolar rhabdomyosarcoma. Biochem. Biophys. Res. Commun. 2008, 365, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.R.; Chatterjee, B.; Olanich, M.E.; Khan, J.; Miettinen, M.M.; Hewitt, S.M.; Barr, F.G. PAX3-FOXO1 is essential for tumour initiation and maintenance but not recurrence in a human myoblast model of rhabdomyosarcoma. J. Pathol. 2017, 241, 626–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, M.H.; Zhao, H.; Womer, R.B.; Barr, F.G. Proliferative and apoptotic differences between alveolar rhabdomyosarcoma subtypes: A comparative study of tumors containing PAX3-FKHR or PAX7-FKHR gene fusions. Med. Pediatr. Oncol. 2001, 37, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.; Ramsay, A.; Gould, S.; Pritchard-Jones, K. PAX3-FKHR induces morphological change and enhances cellular proliferation and invasion in rhabdomyosarcoma. Am. J. Pathol. 2001, 159, 1089–1096. [Google Scholar] [CrossRef]
- Bernasconi, M.; Remppis, A.; Fredericks, W.J.; Rauscher, F.J.; Schafer, B.W. Induction of Apoptosis in Rhabdomyosarcoma Cells Through Down-Regulation of PAX Proteins. Proc. Natl. Acad. Sci. USA 1996, 93, 13164–13169. [Google Scholar] [CrossRef] [PubMed]
- Ayyanathan, K.; Fredericks, W.J.; Berking, C.; Herlyn, M.; Balakrishnan, C.; Gunther, E.; Rauscher, F.J. Hormone-dependent tumor regression in vivo by an inducible transcriptional repressor directed at the PAX3-FKHR oncogene. Cancer Res. 2000, 60, 5803–5814. [Google Scholar] [PubMed]
- Keller, C.; Capecchi, M.R. New genetic tactics to model alveolar rhabdomyosarcoma in the mouse. Cancer Res. 2005, 65, 7530–7532. [Google Scholar] [CrossRef] [PubMed]
- Scheidler, S.; Fredericks, W.J.; Rauscher, F.J.; Barr, F.G.; Vogt, P.K. The hybrid PAX3-FKHR fusion protein of alveolar rhabdomyosarcoma transforms fibroblasts in culture. Proc. Natl. Acad. Sci. USA 1996, 93, 9805–9809. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.-X.; Finckenstein, F.G.; Abdueva, D.A.; Shahbazian, V.; Chung, B.; Weinberg, K.I.; Triche, T.J.; Shimada, H.; Anderson, M.J. Mouse mesenchymal stem cells expressing PAX-FKHR form alveolar rhabdomyosarcomas by cooperating with secondary mutations. Cancer Res. 2008, 68, 6587–6597. [Google Scholar] [CrossRef] [PubMed]
- Naini, S.; Etheridge, K.T.; Adam, S.J.; Qualman, S.J.; Bentley, R.C.; Counter, C.M.; Linardic, C.M. Defining the cooperative genetic changes that temporally drive alveolar rhabdomyosarcoma. Cancer Res. 2008, 68, 9583–9588. [Google Scholar] [CrossRef] [PubMed]
- Diller, L.; Sexsmith, E.; Gottlieb, A.; Li, F.P.; Malkin, D. Germline P53 mutations are frequently detected in young children with rhabdomyosarcoma. J. Clin. Investig. 1995, 95, 1606–1611. [Google Scholar] [CrossRef] [PubMed]
- Obana, K.; Yang, H.-W.; Piao, H.-Y.; Taki, T.; Hashizume, K.; Hanada, R.; Yamamoto, K.; Tanaka, Y.; Toyoda, Y.; Takita, J.; et al. Aberrations of p16INK4A, p14ARF and p15INK4B genes in pediatric solid tumors. Int. J. Oncol. 2003, 23, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Iolascon, A.; Faienza, M.F.; Coppola, B.; Rosolen, A.; Basso, G.; Della Ragione, F.; Schettini, F. Analysis of cyclin-dependent kinase inhibitor genes (CDKN2A, CDKN2B, and CDKN2C) in childhood rhabdomyosarcoma. Genes Chromosom. Cancer 1996, 15, 217–222. [Google Scholar] [CrossRef]
- Hodgson, D.R.; Clayton, S.J.; Girdler, F.; Brotherick, I.; Shenton, B.; Browell, D.; Stuart, M.; Fox, J.C.; Ceuppens, P.; Foy, C.A.; et al. ARMS allele-specific amplification-based detection of mutant P53 DNA and mRNA in tumors of the breast. Clin. Chem. 2001, 47, 774–778. [Google Scholar] [PubMed]
- Linardic, C.M.; Naini, S.; Herndon, J.E.; Kesserwan, C.; Qualman, S.J.; Counter, C.M. The PAX3-FKHR fusion gene of rhabdomyosarcoma cooperates with loss of p16INK4A to promote bypass of cellular senescence. Cancer Res. 2007, 67, 6691–6699. [Google Scholar] [CrossRef] [PubMed]
- Marshall, A.D.; Lagutina, I.; Grosveld, G.C. PAX3-FOXO1 induces cannabinoid receptor 1 to enhance cell invasion and metastasis. Cancer Res. 2011, 71, 7471–7480. [Google Scholar] [CrossRef] [PubMed]
- Marshall, A.D.; Grosveld, G.C. Alveolar rhabdomyosarcoma–the molecular drivers of PAX3/7-FOXO1-induced tumorigenesis. Skelet Muscle 2012, 2, 25. [Google Scholar] [CrossRef] [PubMed]
- Ebauer, M.; Wachtel, M.; Niggli, F.K.; Schafer, B.W. Comparative expression profiling identifies an in vivo target gene signature with TFAP2B as a mediator of the survival function of PAX3/FKHR. Oncogene 2007, 26, 7267–7281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, H.; Qin, F.; Movassagh, M.; Park, H.; Golden, W.; Xie, Z.; Zhang, P.; Sklar, J.; Li, H. A Chimeric RNA characteristic of rhabdomyosarcoma in normal myogenesis process. Cancer Discov. 2013, 3, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Babiceanu, M.; Kumar, S.; Jia, Y.; Qin, F.; Barr, F.G.; Li, H. Fusion transcriptome profiling provides insights into alveolar rhabdomyosarcoma. Proc. Natl. Acad. Sci. USA 2016, 113, 13126–13131. [Google Scholar] [CrossRef] [PubMed]
- Redell, M.S.; Tweardy, D.J. Targeting transcription factors in cancer: challenges and evolving strategies. Drug Discov Today Technol. 2006, 3, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; Reddy, E.P.; Shokat, K.M.; Soucek, L. Drugging the“Undruggable”Cancer Targets. Nat. Rev. Cancer 2017, 17, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Miller, P.J.; Hollenbach, A.D. The oncogenic fusion protein Pax3–FKHR has a greater post-translational stability relative to pax3 during early myogenesis. Biochim. Biophys. Acta 2007, 1770, 1450–1458. [Google Scholar] [CrossRef] [PubMed]
- Amstutz, R.; Wachtel, M.; Troxler, H.; Kleinert, P.; Ebauer, M.; Haneke, T.; Oehler-Janne, C.; Fabbro, D.; Niggli, F.K.; Schafer, B.W. Phosphorylation regulates transcriptional activity of PAX3/FKHR and reveals novel therapeutic possibilities. Cancer Res. 2008, 68, 3767–3776. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.-Y.; Dong, H.; Cui, J.; Liu, L.; Chen, T. Glycogen synthase kinase 3 regulates PAX3–FKHR-mediated cell proliferation in human alveolar rhabdomyosarcoma cells. Biochem. Biophys. Res. Commun. 2010, 391, 1049–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Wu, J.; Ong, S.S.; Chen, T. Cyclin-dependent kinase 4 phosphorylates and positively regulates PAX3-FOXO1 in human alveolar rhabdomyosarcoma cells. PLoS ONE 2013, 8, e58193. [Google Scholar] [CrossRef] [PubMed]
- Thalhammer, V.; Lopez-Garcia, L.A.; Herrero-Martin, D.; Hecker, R.; Laubscher, D.; Gierisch, M.E.; Wachtel, M.; Bode, P.; Nanni, P.; Blank, B.; et al. PLK1 phosphorylates PAX3-FOXO1, the inhibition of which triggers regression of alveolar rhabdomyosarcoma. Cancer Res. 2015, 75, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Loupe, J.M.; Miller, P.J.; Ruffin, D.R.; Stark, M.W.; Hollenbach, A.D. Inhibiting phosphorylation of the oncogenic PAX3-FOXO1 reduces alveolar rhabdomyosarcoma phenotypes identifying novel therapy options. Oncogenesis 2015, 4, e145. [Google Scholar] [CrossRef] [PubMed]
- Dietz, K.N.; Miller, P.J.; Iyengar, A.S.; Loupe, J.M.; Hollenbach, A.D. Identification of serines 201 and 209 as sites of Pax3 phosphorylation and the altered phosphorylation status of Pax3-FOXO1 during early myogenic differentiation. Int. J. Biochem. Cell Biol. 2011, 43, 936–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyengar, A.S.; Loupe, J.M.; Miller, P.J.; Hollenbach, A.D. Identification of CK2 as the kinase that phosphorylates Pax3 at Ser209 in early myogenic differentiation. Biochem. Biophys. Res. Commun. 2012, 428, 24–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jothi, M.; Mal, M.; Keller, C.; Mal, A.K. Small molecule inhibition of PAX3-FOXO1 through AKT activation suppresses malignant phenotypes of alveolar rhabdomyosarcoma. Mol. Cancer Ther. 2013, 12, 2663–2674. [Google Scholar] [CrossRef] [PubMed]
- Gryder, B.E.; Yohe, M.E.; Chou, H.-C.; Zhang, X.; Marques, J.; Wachtel, M.; Schaefer, B.; Sen, N.; Song, Y.; Gualtieri, A.; et al. PAX3–FOXO1 establishes myogenic super enhancers and confers BET bromodomain vulnerability. Cancer Discov. 2017, 7, 884–899. [Google Scholar] [CrossRef] [PubMed]
- Bharathy, N.; Suriyamurthy, S.; Rao, V.K.; Ow, J.R.; Lim, H.J.; Chakraborty, P.; Vasudevan, M.; Dhamne, C.A.; Chang, K.T.E.; Min, V.L.K.; et al. P/CAF mediates PAX3-FOXO1-dependent oncogenesis in alveolar rhabdomyosarcoma. J. Pathol. 2016, 240, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Abraham, J.; Nuñez-Álvarez, Y.; Hettmer, S.; Carrió, E.; Chen, H.-I.H.; Nishijo, K.; Huang, E.T.; Prajapati, S.I.; Walker, R.L.; Davis, S.; et al. Lineage of origin in rhabdomyosarcoma informs pharmacological response. Genes Dev. 2014, 28, 1578–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrero Martín, D.; Boro, A.; Schäfer, B.W. Cell-based small-molecule compound screen identifies fenretinide as potential therapeutic for translocation-positive rhabdomyosarcoma. PLoS ONE 2013, 8, e55072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Lee, B.H.; Williams, I.R.; Kutok, J.L.; Mitsiades, C.S.; Duclos, N.; Cohen, S.; Adelsperger, J.; Okabe, R.; Coburn, A.; et al. FGFR3 as a therapeutic target of the small molecule inhibitor PKC412 in hematopoietic malignancies. Oncogene 2005, 24, 8259–8267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gleixner, K.V.; Mayerhofer, M.; Aichberger, K.J.; Derdak, S.; Sonneck, K.; Böhm, A.; Gruze, A.; Samorapoompichit, P.; Manley, P.W.; Fabbro, D.; et al. PKC412 inhibits in vitro growth of neoplastic human mast cells expressing the D816V-mutated variant of KIT: Comparison with AMN107, imatinib, and cladribine (2CdA) and evaluation of cooperative drug effects. Blood 2006, 107, 752–759. [Google Scholar] [CrossRef] [PubMed]
- Bahlis, N.J.; Miao, Y.; Koc, O.N.; Lee, K.; Boise, L.H.; Gerson, S.L. N-Benzoylstaurosporine (PKC412) inhibits akt kinase inducing apoptosis in multiple myeloma cells. Leuk. Lymphoma 2009, 46, 899–908. [Google Scholar] [CrossRef] [PubMed]
- del Peso, L.; González, V.M.; Hernández, R.; Barr, F.G.; Núñez, G. Regulation of the forkhead transcription factor FKHR, but not the PAX3-FKHR fusion protein, by the serine/threonine kinase Akt. Oncogene 1999, 18, 7328–7333. [Google Scholar] [CrossRef] [PubMed]
- Zabidi, M.A.; Stark, A. Regulatory enhancer–core-promoter communication via transcription factors and cofactors. Trends Genet. 2016, 32, 801–814. [Google Scholar] [CrossRef] [PubMed]
- Böhm, M.; Wachtel, M.; Marques, J.G.; Streiff, N.; Laubscher, D.; Nanni, P.; Mamchaoui, K.; Santoro, R.; Schäfer, B.W. Helicase CHD4 is an epigenetic coregulator of PAX3-FOXO1 in alveolar rhabdomyosarcoma. J. Clin. Investig. 2016, 126, 4237–4249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, Y.; Wong, J.; Moreno, G.T.; Young, M.K.; Côté, J.; Wang, W. NURD, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities. Mol. Cell 1998, 2, 851–861. [Google Scholar] [CrossRef]
- Zhou, M.-M.; Dhalluin, C.; Carlson, J.E.; Zeng, L.; He, C.; Aggarwal, A.K.; Zhou, M.-M. Structure and ligand of a histone acetyltransferase bromodomain. Nature 1999, 39, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Spiegelman, B.M.; Heinrich, R. Biological control through regulated transcriptional coactivators. Cell 2004, 119, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Heinicke, U.; Kupka, J.; Fichter, I.; Fulda, S. Critical role of mitochondria-mediated apoptosis for JNJ-26481585-induced antitumor activity in rhabdomyosarcoma. Oncogene 2015, 35, 3729–3741. [Google Scholar] [CrossRef] [PubMed]
- Heinicke, U.; Kupka, J.; Fulda, S. JNJ-26481585 primes rhabdomyosarcoma cells for chemotherapeutics by engaging the mitochondrial pathway of apoptosis. Oncotarget 2015, 6, 37836–37851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haydn, T.; Metzger, E.; Schuele, R.; Fulda, S. Concomitant epigenetic targeting of LSD1 and HDAC synergistically induces mitochondrial apoptosis in rhabdomyosarcoma cells. Cell Death Dis. 2017, 8, e2879. [Google Scholar] [CrossRef] [PubMed]
- Enßle, J.C.; Boedicker, C.; Wanior, M.; Vogler, M.; Knapp, S.; Fulda, S. Co-targeting of BET proteins and hdacs as a novel approach to trigger apoptosis in rhabdomyosarcoma cells. Cancer Lett. 2018, 428, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Modak, R.; Basha, J.; Bharathy, N.; Maity, K.; Mizar, P.; Bhat, A.V.; Vasudevan, M.; Rao, V.K.; Kok, W.K.; Natesh, N.; et al. Probing P300/CBP associated factor (PCAF)-dependent pathways with a small molecule inhibitor. ACS Chem. Biol. 2013, 8, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, H.; Daitoku, H.; Hatta, M.; Aoyama, H.; Yoshimochi, K.; Fukamizu, A. Acetylation of Foxo1 alters its DNA-binding ability and sensitivity to phosphorylation. Proc. Natl. Acad. Sci. USA 2005, 102, 11278–11283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]
- Dilworth, F.J.; Seaver, K.J.; Fishburn, A.L.; Htet, S.L.; Tapscott, S.J. In Vitro Transcription system delineates the distinct roles of the coactivators pCAF and P300 during MyoD/E47-dependent transactivation. Proc. Natl. Acad. Sci. USA 2004, 101, 11593–11598. [Google Scholar] [CrossRef] [PubMed]
- Sartorelli, V.; Puri, P.L.; Hamamori, Y.; Ogryzko, V.; Chung, G.; Nakatani, Y.; Wang, J.Y.; Kedes, L. Acetylation of myod directed by PCAF is necessary for the execution of the muscle program. Mol. Cell 1999, 4, 725–734. [Google Scholar] [CrossRef]
- Davicioni, E.; Finckenstein, F.G.; Shahbazian, V.; Buckley, J.D.; Triche, T.J.; Anderson, M.J. Identification of a PAX-FKHR gene expression signature that defines molecular classes and determines the prognosis of alveolar rhabdomyosarcomas. Cancer Res. 2006, 66, 6936–6946. [Google Scholar] [CrossRef] [PubMed]
- Laé, M.; Ahn, E.H.; Mercado, G.E.; Chuai, S.; Edgar, M.; Pawel, B.R.; Olshen, A.; Barr, F.G.; Ladanyi, M. Global gene expression profiling of PAX-FKHR fusion-positive alveolar and PAX-FKHR fusion-negative embryonal rhabdomyosarcomas. J. Pathol. 2007, 212, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Crose, L.E.S.; Etheridge, K.T.; Chen, C.; Belyea, B.; Talbot, L.J.; Bentley, R.C.; Linardic, C.M. FGFR4 blockade exerts distinct antitumorigenic effects in human embryonal versus alveolar rhabdomyosarcoma. Clin. Cancer Res. 2012, 18, 3780–3790. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Dong, J.; Sun, L.; Geng, L.; Wang, J.; Zheng, J.; Li, Y.; Bridge, J.; Hinrichs, S.H.; Ding, S.-J. Inhibition of phosphorylated C-Met in rhabdomyosarcoma cell lines by a small molecule inhibitor SU11274. J. Transl. Med. 2011, 9, 64. [Google Scholar] [CrossRef] [PubMed]
- Abraham, J.; Prajapati, S.I.; Nishijo, K.; Schaffer, B.S.; Taniguchi, E.; Kilcoyne, A.; McCleish, A.T.; Nelon, L.D.; Giles, F.G.; Efstratiadis, A.; et al. Evasion mechanisms to Igf1r inhibition in rhabdomyosarcoma. Mol. Cancer Ther. 2011, 10, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Scotlandi, K.; Manara, M.C.; Nicoletti, G.; Lollini, P.-L.; Lukas, S.; Benini, S.; Croci, S.; Perdichizzi, S.; Zambelli, D.; Serra, M.; et al. Antitumor activity of the insulin-like growth factor-I receptor kinase inhibitor NVP-AEW541 in musculoskeletal tumors. Cancer Res. 2005, 65, 3868–3876. [Google Scholar] [CrossRef] [PubMed]
- García-Echeverría, C.; Pearson, M.A.; Marti, A.; Meyer, T.; Mestan, J.; Zimmermann, J.; Gao, J.; Brueggen, J.; Capraro, H.-G.; Cozens, R.; et al. In vivo antitumor activity of NVP-AEW541—A novel, potent, and selective inhibitor of the IGF-IR kinase. Cancer Cell 2004, 5, 231–239. [Google Scholar] [CrossRef] [Green Version]
- Faqar-Uz-Zaman, S.F.; Heinicke, U.; Meister, M.T.; Vogler, M.; Fulda, S. BCL-xl–Selective BH3 mimetic sensitizes rhabdomyosarcoma cells to chemotherapeutics by activation of the mitochondrial pathway of apoptosis. Cancer Lett. 2018, 412, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Peron, M.; Lovisa, F.; Poli, E.; Basso, G.; Bonvini, P. Understanding the interplay between expression, mutation and activity of ALK receptor in rhabdomyosarcoma cells for clinical application of small-molecule inhibitors. PLoS ONE 2015, 10, e0132330. [Google Scholar] [CrossRef] [PubMed]
- Megiorni, F.; McDowell, H.P.; Camero, S.; Mannarino, O.; Ceccarelli, S.; Paiano, M.; Losty, P.D.; Pizer, B.; Shukla, R.; Pizzuti, A.; et al. Crizotinib-induced antitumour activity in human alveolar rhabdomyosarcoma cells is not solely dependent on ALK and MET inhibition. J. Exp. Clin. Cancer Res. 2015, 34, 112. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Yu, Y.; Bilke, S.; Walker, R.L.; Mayeenuddin, L.H.; Azorsa, D.O.; Yang, F.; Pineda, M.; Helman, L.J.; Meltzer, P.S. Genome-wide identification of PAX3-FKHR binding sites in rhabdomyosarcoma reveals candidate target genes important for development and cancer. Cancer Res. 2010, 70, 6497–6508. [Google Scholar] [CrossRef] [PubMed]
- Epstein, J.A.; Song, B.; Lakkis, M.; Wang, C. Tumor-specific PAX3-FKHR transcription factor, but not PAX3, activates the platelet-derived growth factor alpha receptor. Mol. Cell. Biol. 1998, 18, 4118–4130. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, E.; Nishijo, K.; McCleish, A.T.; Michalek, J.E.; Grayson, M.H.; Infante, A.J.; Abboud, H.E.; LeGallo, R.D.; Qualman, S.J.; Rubin, B.P.; et al. PDGFR-a is a therapeutic target in alveolar rhabdomyosarcoma. Oncogene 2008, 27, 6550–6560. [Google Scholar] [CrossRef] [PubMed]
- Mercado, G.E.; Xia, S.J.; Zhang, C.; Ahn, E.H.; Gustafson, D.M.; Laé, M.; Ladanyi, M.; Barr, F.G. Identification of PAX3-FKHR-regulated genes differentially expressed between alveolar and embryonal rhabdomyosarcoma: focus on MYCN as a biologically relevant target. Genes Chromosom. Cancer 2008, 47, 510–520. [Google Scholar] [CrossRef] [PubMed]
- Walters, Z.S.; Villarejo-Balcells, B.; Olmos, D.; Buist, T.W.S.; Missiaglia, E.; Allen, R.; Al-Lazikani, B.; Garrett, M.D.; Blagg, J.; Shipley, J. JARID2 is a direct target of the PAX3-FOXO1 fusion protein and inhibits myogenic differentiation of rhabdomyosarcoma cells. Oncogene 2014, 33, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- Oesch, S.; Walter, D.; Wachtel, M.; Pretre, K.; Salazar, M.; Guzmán, M.; Velasco, G.; Schäfer, B.W. Cannabinoid receptor 1 is a potential drug target for treatment of translocation-positive rhabdomyosarcoma. Mol. Cancer Ther. 2009, 8, 1838–1845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Wang, Y.-D.; Wu, J.; Cui, J.; Chen, T. Carnitine palmitoyltransferase 1A (CPT1A): A transcriptional target of PAX3-FKHR and mediates PAX3-FKHR-dependent motility in alveolar rhabdomyosarcoma cells. BMC Cancer 2012, 12, 154. [Google Scholar] [CrossRef] [PubMed]
- Li, S.Q.; Cheuk, A.T.; Shern, J.F.; Song, Y.K.; Hurd, L.; Liao, H.; Wei, J.S.; Khan, J. Targeting wild-type and mutationally activated FGFR4 in rhabdomyosarcoma with the inhibitor Ponatinib (AP24534). PLoS ONE 2013, 8, e76551–e76559. [Google Scholar] [CrossRef] [PubMed]
- VI, J.G.T.; Cheuk, A.T.; Tsang, P.S.; Chung, J.-Y.; Song, Y.K.; Desai, K.; Yu, Y.; Chen, Q.-R.; Shah, K.; Youngblood, V.; et al. Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. J. Clin. Investig. 2009, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Baskar, S. Targeting FGFR4 with monoclonal antibodies as therapeutic agents for the treatment of rhabdomyosarcoma. Cancer Res. 2016, 76 (Suppl. S14), 4996. [Google Scholar] [CrossRef]
- Shivaprasad, N.; Xiong, Y.; Yohe, M.; Schneider, D.; Shern, J.; Baskar, S.; Dimitrov, D.; Sorenson, P.; Orentas, R.; Khan, J. Developing FGFR4 chimeric antigen receptor CAR T cell therapy against rhabdomyosarcoma. Mol. Ther. 2016, 24, S257–S258. [Google Scholar] [CrossRef]
- Dolgikh, N.; Fulda, S. Rhabdomyosarcoma cells are susceptible to cell death by LDK378 alone or in combination with sorafenib independently of anaplastic lymphoma kinase status. Anti-Cancer Drugs 2017, 28, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- van Erp, A.E.M.; Hillebrandt-Roeffen, M.H.S.; van Houdt, L.; Fleuren, E.D.G.; van der Graaf, W.T.A.; Versleijen-Jonkers, Y.M.H. Targeting Anaplastic Lymphoma Kinase (ALK) in Rhabdomyosarcoma (RMS) with the Second-Generation ALK Inhibitor Ceritinib. Target. Oncol. 2017, 12, 815–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beltran, P.J.; Chung, Y.A.; Moody, G.; Mitchell, P.; Cajulis, E.; Vonderfecht, S.; Kendall, R.; Radinsky, R.; Calzone, F.J. Efficacy of Ganitumab (AMG 479), alone and in combination with rapamycin, in Ewing's and osteogenic sarcoma models. J. Pharmacol. Exp. Ther. 2011, 337, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Atzori, F.; Tabernero, J.; Cervantes, A.; Prudkin, L.; Andreu, J.; Rodríguez-Braun, E.; Domingo, A.; Guijarro, J.; Gamez, C.; Rodon, J.; et al. A phase I pharmacokinetic and pharmacodynamic study of Dalotuzumab (MK-0646), an anti-insulin-like growth factor-1 receptor monoclonal antibody, in patients with advanced solid tumors. Clin. Cancer Res. 2011, 17, 6304–6312. [Google Scholar] [CrossRef] [PubMed]
- Pappo, A.S.; Vassal, G.; Crowley, J.J.; Bolejack, V.; Hogendoorn, P.C.W.; Chugh, R.; Ladanyi, M.; Grippo, J.F.; Dall, G.; Staddon, A.P.; et al. A phase 2 trial of R1507, a monoclonal antibody to the insulin-like growth factor-1 receptor (IGF-1R), in patients with recurrent or refractory rhabdomyosarcoma, osteosarcoma, synovial sarcoma, and other soft tissue sarcomas: results of a sarcoma alliance. Cancer 2014, 120, 2448–2456. [Google Scholar] [CrossRef] [PubMed]
- Tarnowski, M.; Tkacz, M.; Zgutka, K.; Bujak, J.; Kopytko, P.; Pawlik, A. Picropodophyllin (PPP) is a potent rhabdomyosarcoma growth inhibitor both in vitro and in vivo. BMC Cancer 2017, 1–11. [Google Scholar] [CrossRef] [PubMed]
- McDermott, U.; Ames, R.Y.; Iafrate, A.J.; Maheswaran, S.; Stubbs, H.; Greninger, P.; McCutcheon, K.; Milano, R.; Tam, A.; Lee, D.Y.; et al. Ligand-dependent platelet-derived growth factor receptor (PDGFR)- activation sensitizes rare lung cancer and sarcoma cells to PDGFR kinase inhibitors. Cancer Res. 2009, 69, 3937–3946. [Google Scholar] [CrossRef] [PubMed]
- Abraham, J.; Chua, Y.X.; Glover, J.M.; Tyner, J.W.; Loriaux, M.M.; Kilcoyne, A.; Giles, F.J.; Nelon, L.D.; Carew, J.S.; Ouyang, Y.; et al. An adaptive Src–PDGFRA–Raf axis in rhabdomyosarcoma. Biochem. Biophys. Res. Commun. 2012, 426, 363–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roskoski, R., Jr. The role of small molecule platelet-derived growth factor receptor (PDGFR) inhibitors in the treatment of neoplastic disorders. Pharmacol. Res. 2018, 129, 65–83. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.P.; Todd, J.R.; Finetti, M.A.; McCarthy, F.; Broncel, M.; Vyse, S.; Luczynski, M.T.; Crosier, S.; Ryall, K.A.; Holmes, K.; et al. Dual targeting of PDGFRα and FGFR1 displays synergistic efficacy in malignant rhabdoid tumors. Cell Rep. 2016, 17, 1265–1275. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, C.; Leruste, A.; Tauziede-Espariat, A.; Andrianteranagna, M.; Surdez, D.; Lescure, A.; Han, Z.-Y.; Anthony, E.; Richer, W.; Baulande, S.; et al. High-throughput drug screening identifies Pazopanib and Clofilium Tosylate as promising treatments for malignant rhabdoid tumors. Cell Rep. 2017, 21, 1737–1745. [Google Scholar] [CrossRef] [PubMed]
- Lowery, C.D.; Blosser, W.; Dowless, M.; Knoche, S.; Stephens, J.; Li, H.; Surguladze, D.; Loizos, N.; Luffer-Atlas, D.; Oakley, G.J.; et al. Olaratumab exerts antitumor activity in preclinical models of pediatric bone and soft tissue tumors through inhibition of platelet-derived growth factor receptor Α. Clin. Cancer Res. 2018, 24, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Miekus, K.; Lukasiewicz, E.; Jarocha, D.; Sekula, M.; Drabik, G.; Majka, M. The decreased metastatic potential of rhabdomyosarcoma cells obtained through MET receptor downregulation and the induction of differentiation. Cell Death Dis. 2013, 4, e459. [Google Scholar] [CrossRef] [PubMed]
- Kashima, K.; Watanabe, M.; Sato, Y.; Hata, J.; Ishii, N.; Aoki, Y. Inhibition of metastasis of rhabdomyosarcoma by a novel neutralizing antibody to CXC chemokine receptor-4. Cancer Sci. 2014, 105, 1343–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonelli, R.; McIntyre, A.; Camerin, C.; Walters, Z.S.; Di Leo, K.; Selfe, J.; Purgato, S.; Missiaglia, E.; Tortori, A.; Renshaw, J.; et al. Antitumor activity of sustained N-Myc reduction in rhabdomyosarcomas and transcriptional block by antigene therapy. Clin. Cancer Res. 2012, 18, 796–807. [Google Scholar] [CrossRef] [PubMed]
- AHN, E.H.; Lee, M.B.; Seo, D.J.; Lee, J.; Kim, Y.; Gupta, K. Sphingosine induces apoptosis and down-regulation of MYCN in PAX3-FOXO1-positive alveolar rhabdomyosarcoma cells irrespective of TP53 mutation. Anticancer Res. 2018, 38, 71–76. [Google Scholar] [PubMed]
- Luo, X.; Liu, Y.; Kubicek, S.; Myllyharju, J.; Tumber, A.; Ng, S.; Che, K.H.; Podoll, J.; Heightman, T.D.; Oppermann, U.; et al. A selective inhibitor and probe of the cellular functions of Jumonji C domain-containing histone demethylases. J. Am. Chem. Soc. 2011, 133, 9451–9456. [Google Scholar] [CrossRef] [PubMed]
- Hernlund, E.; Ihrlund, L.S.; Khan, O.; Ates, Y.O.; Linder, S.; Panaretakis, T.; Shoshan, M.C. Potentiation of chemotherapeutic drugs by energy metabolism inhibitors 2-Deoxyglucose and etomoxir. Int. J. Cancer 2008, 123, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Dagher, R.; Long, L.M.; Read, E.J.; Leitman, S.F.; Carter, C.S.; Tsokos, M.; Goletz, T.J.; Avila, N.; Berzofsky, J.A.; Helman, L.J.; et al. Pilot trial of tumor-specific peptide vaccination and continuous infusion Interleukin-2 in patients with recurrent ewing sarcoma and alveolar rhabdomyosarcoma: An inter-institute NIH study. Med. Pediatr. Oncol. 2002, 38, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Rodeberg, D.A.; Nuss, R.A.; Heppelmann, C.J.; Celis, E. Lack of effective T-Lymphocyte response to the PAX3/FKHR translocation area in alveolar rhabdomyosarcoma. Cancer Immunol. Immunother. 2005, 54, 526–534. [Google Scholar] [CrossRef] [PubMed]
- van den Broeke, L.T.; Pendleton, C.D.; Mackall, C.; Helman, L.J.; Berzofsky, J.A. Identification and epitope enhancement of a PAX-FKHR fusion protein breakpoint epitope in alveolar rhabdomyosarcoma cells created by a tumorigenic chromosomal translocation inducing CTL capable of lysing human tumors. Cancer Res. 2006, 66, 1818–1823. [Google Scholar] [CrossRef] [PubMed]
- Mackall, C.L.; Rhee, E.H.; Read, E.J.; Khuu, H.M.; Leitman, S.F.; Bernstein, D.; Tesso, M.; Long, L.M.; Grindler, D.; Merino, M.; et al. A pilot study of consolidative immunotherapy in patients with high-risk pediatric sarcomas. Clin. Cancer Res. 2008, 14, 4850–4858. [Google Scholar] [CrossRef] [PubMed]
- Mayeenuddin, L.H.; Yu, Y.; Kang, Z.; Helman, L.J.; Cao, L. Insulin-like growth factor 1 receptor antibody induces rhabdomyosarcoma cell death via a process involving AKT and Bcl-xL. Oncogene 2010, 29, 6367–6377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pappo, A.S.; Patel, S.R.; Crowley, J.; Reinke, D.K.; Kuenkele, K.-P.; Chawla, S.P.; Toner, G.C.; Maki, R.G.; Meyers, P.A.; Chugh, R.; et al. R1507, a monoclonal antibody to the insulin-like growth factor 1 receptor, in patients with recurrent or refractory ewing sarcoma family of tumors: results of a phase II sarcoma alliance for research through collaboration study. J. Clin. Oncol. 2011, 29, 4541–4547. [Google Scholar] [CrossRef] [PubMed]
- Vela, M. Anti CXCR4 antibody combined with activated and expanded natural killer cells for sarcoma immunotherapy. J. Clin. Oncol. 2018, 36 (Suppl. S15), 11541. [Google Scholar] [CrossRef]
- Fesnak, A.D.; June, C.H.; Levine, B.L. Engineered T Cells: The promise and challenges of cancer immunotherapy. Nat. Rev. Cancer 2016, 16, 566–581. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Wolchok, J.D.; Chan, T.A.; Mellman, I.; Palucka, K.; Banchereau, J.; Rosenberg, S.A.; Dane Wittrup, K. Immunotherapy: The path to win the war on cancer? Cell 2015, 161, 185–186. [Google Scholar] [PubMed]
- Porter, D.L.; Hwang, W.-T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in children and young adults with B-Cell lymphoblastic leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Kebriaei, P.; Singh, H.; Huls, M.H.; Figliola, M.J.; Bassett, R.; Olivares, S.; Jena, B.; Dawson, M.J.; Kumaresan, P.R.; Su, S.; et al. Phase I trials using sleeping beauty to generate CD19-specific CAR T cells. J. Clin. Investig. 2016, 126, 3363–3376. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Frey, N.V.; Grupp, S.A.; Maude, S.L. CAR T cell therapy in acute Lymphoblastic Leukemia and potential for chronic lymphocytic leukemia. Curr. Treat. Options Oncol. 2016, 17, 28. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Riddell, S.R. Chimeric Antigen Receptor T Cell Therapy: Challenges to bench-to-bedside efficacy. J. Immunol. 2018, 200, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Newick, K.; O’Brien, S.; Moon, E.; Albelda, S.M. CAR T cell therapy for solid tumors. Annu. Rev. Med. 2017, 68, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Yong, C.S.M.; Dardalhon, V.; Devaud, C.; Taylor, N.; Darcy, P.K.; Kershaw, M.H. CAR T-cell therapy of solid tumors. Immunol. Cell Biol. 2017, 95, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Skarzynski, M.; Shivaprasad, N.; Subramanian, B.; Azorsa, D.; Zhu, Z.; Dimitrov, D.; Khan, J. Abstract 693: Antibody-based targeting of the cell surface receptor tyrosine kinase FGFR4 in rhabdomyosarcoma and other cancers. Cancer Res. 2017, 77 (Suppl. S13), 693. [Google Scholar] [CrossRef]
- Huang, X.; Park, H.; Greene, J.; Pao, J.; Mulvey, E.; Zhou, S.X.; Albert, C.M.; Moy, F.; Sachdev, D.; Yee, D.; et al. IGF1R- and ROR1-specific CAR T cells as a potential therapy for high risk sarcomas. PLoS ONE 2015, 10, e0133152. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, C. Identification of a new class of PAX3-FKHR target promoters: A role of the Pax3 paired box DNA binding domain. Oncogene 2006, 26, 1595–1605. [Google Scholar] [CrossRef] [PubMed]
- Phelps, M.; Yang, H.; Chen, E. Targeted ablation of essential oncogenes in rhabdomyosarcoma with CRISPR/Cas9 gene therapy. Cancer Res. 2017, 77 (Suppl. S13), 5096. [Google Scholar] [CrossRef]
- Baquir, B.; Hancock, R.E.W. Exosomes, your body’s answer to immune health. Ann. Transl. Med. 2017, 5, 81. [Google Scholar] [CrossRef] [PubMed]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.M.; Nguyen, Q.H.; Singh, M.; Razorenova, O.V. Approaches to identifying synthetic lethal interactions in cancer. Yale J. Biol. Med. 2015, 88, 145–155. [Google Scholar] [PubMed]
- Scholl, C.; Fröhling, S.; Dunn, I.F.; Schinzel, A.C.; Barbie, D.A.; Kim, S.Y.; Silver, S.J.; Tamayo, P.; Wadlow, R.C.; Ramaswamy, S.; et al. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell 2009, 137, 821–834. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; McMillan, E.; Kim, H.S.; Venkateswaran, N.; Makkar, G.; Rodriguez-Canales, J.; Villalobos, P.; Neggers, J.E.; Mendiratta, S.; Wei, S.; et al. XPO1-dependent nuclear export is a druggable vulnerability in KRAS-mutant lung cancer. Nature 2016, 538, 114–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Zhang, X.A.; Li, X.; Xie, W.; Huang, S. MYC-mediated synthetic lethality for treating tumors. Curr. Cancer Drug Targets 2015, 15, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Gillet, J.P.; Varma, S.; Gottesman, M.M. The clinical relevance of cancer cell lines. J. Natl. Cancer Inst. 2013, 105, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Wei, X.; Lin, S.; Qin, L.; Cheng, L.; Li, P. Current status and perspectives of patient-derived Xenograft models in cancer research. J. Hematol. Oncol. 2017, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, R.M. Patient-derived orthotopic Xenografts: Better mimic of metastasis than subcutaneous Xenografts. Nat. Rev. Cancer 2015, 15, 451–452. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Protein domain structures of the wild-type PAX3, FOXO1 and PAX3-FOXO1 fusion proteins. The numbers indicate the corresponding amino acids present on each protein. Abbreviations: PD, Paired box domain; OP, octapeptide motif; HD, homeobox domain; P-TAD, PAX3 transactivation domain; FH, forkhead domain; KD, KIX-binding domain; F-TAD, FOXO1 transactivation domain; NLS, nuclear localization signal; NES, nuclear export signal.
Figure 1.
Protein domain structures of the wild-type PAX3, FOXO1 and PAX3-FOXO1 fusion proteins. The numbers indicate the corresponding amino acids present on each protein. Abbreviations: PD, Paired box domain; OP, octapeptide motif; HD, homeobox domain; P-TAD, PAX3 transactivation domain; FH, forkhead domain; KD, KIX-binding domain; F-TAD, FOXO1 transactivation domain; NLS, nuclear localization signal; NES, nuclear export signal.

Figure 2.
Targetable regulations of PAX3-FOXO1 activity. GSK3β, CK2, CDK4 and PLK1 phosphorylate PAX3-FOXO1 at various serine residues. The acetyltransferase enzyme P/CAF acetylates PAX3-FOXO1 at K426/K429 lysine residues. BRD4 acts as co-transcriptional activator of PAX3-FOXO1 at its DNA target sites. The mechanism by which HDAC regulates the mRNA expression level of PAX3-FOXO1 remains unknown. These chromatin modifiers, which modulate expression of PAX3-FOXO1 target genes, can be targeted by small molecule inhibitors.
Figure 2.
Targetable regulations of PAX3-FOXO1 activity. GSK3β, CK2, CDK4 and PLK1 phosphorylate PAX3-FOXO1 at various serine residues. The acetyltransferase enzyme P/CAF acetylates PAX3-FOXO1 at K426/K429 lysine residues. BRD4 acts as co-transcriptional activator of PAX3-FOXO1 at its DNA target sites. The mechanism by which HDAC regulates the mRNA expression level of PAX3-FOXO1 remains unknown. These chromatin modifiers, which modulate expression of PAX3-FOXO1 target genes, can be targeted by small molecule inhibitors.


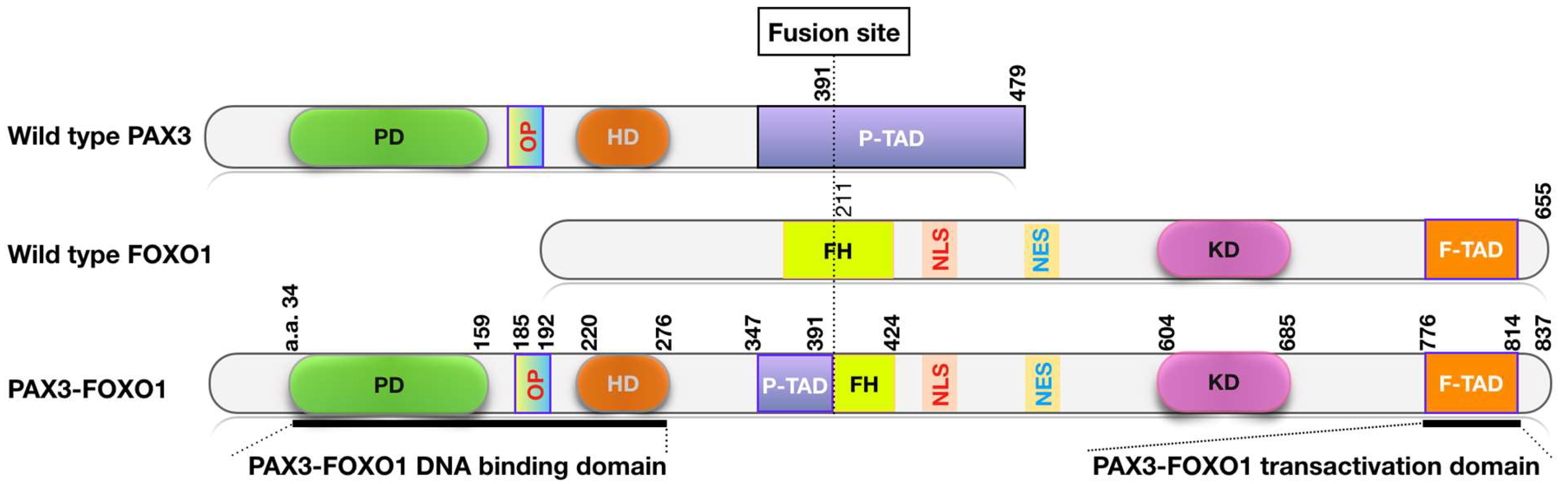{kind=link}
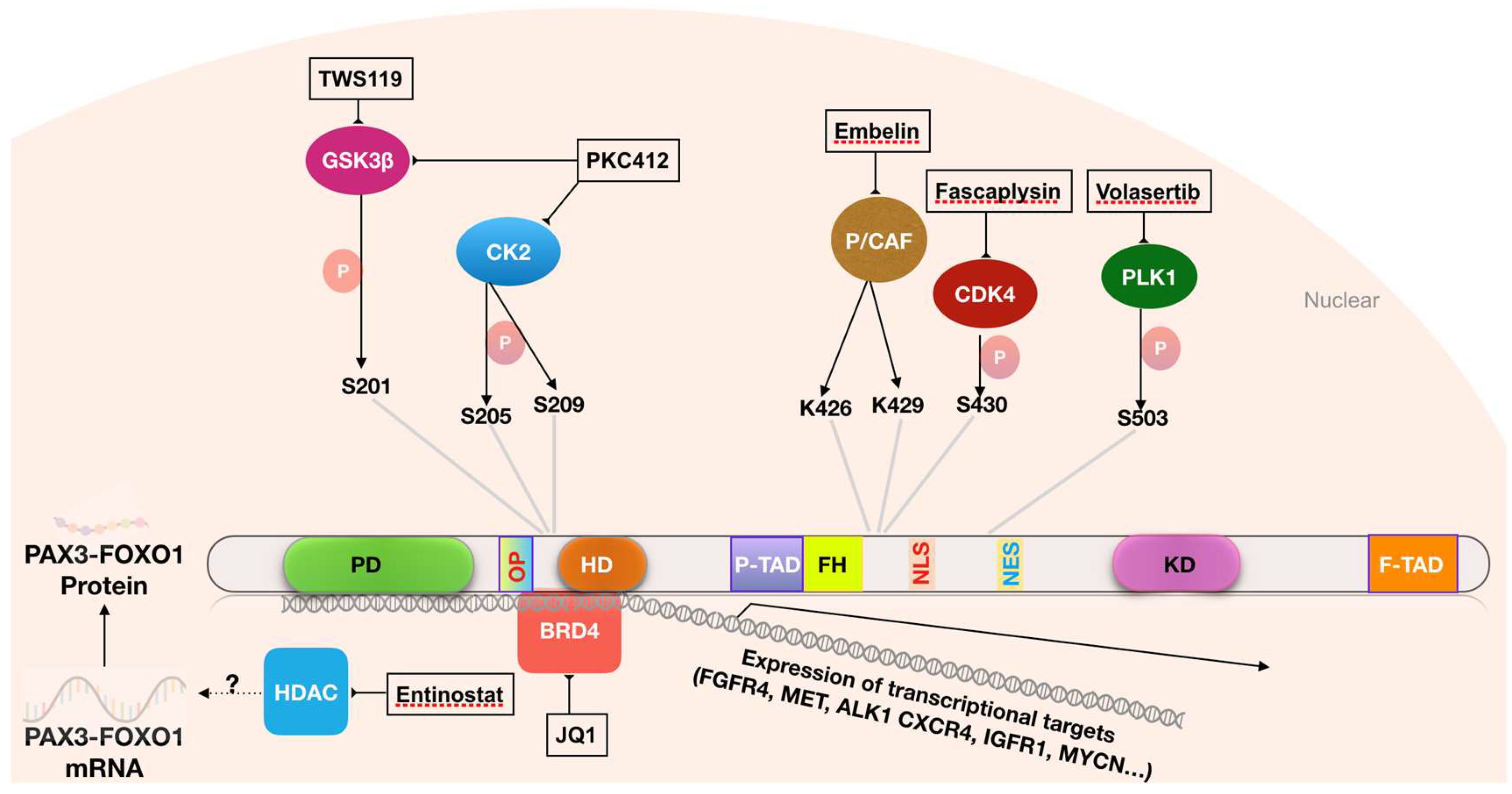{kind=link}
Table 1.
RMS subtypes and their major associated genetic alterations.
| Histologic Pattern | Fusion Status | Frequency | Associated Genetic Changes | Outcome |
|---|---|---|---|---|
| ARMS | FP | ~20% | ● PAX3/7-FOXO1 fusions [16,41,42] ● Amplification [43,44]: 2p24 (MYCN) 12q13-14 (CDK4) 13q31-32 (MIR17HG) | Poor |
| FN | ~5% | ● Point mutations [17]: RAS genes TP53 PIK3CA FGFR4 NF1 BCOR FBXW7 ● Loss of heterozygosity [17]: 11p15.5 9p21.3 (CDKN2A) ● Amplification [17]: 12q15 (MDM2) ● Chromosome copy number gains [45,46]: 2, 8 and 12 | Good | |
| ERMS | ~75% |
Table 2.
List of small molecules reported to inhibit PAX3-FOXO1 activity.
| Small Molecules | Relevant Molecular Targets | References |
|---|---|---|
| 1. Kinase inhibitors | ||
| TWS119 | GSK3 | [78] |
| SB212763 | GSK3 | [78] |
| TDZD-8 | GSK3 | [78] |
| Fascaplysin | CDK4 | [79] |
| PKC412 | Multiple kinases including CK2 and GSK3 | [19] |
| BI-2536 | PLK1 | [80] |
| BI-6727 (volasertib) | PLK1 | [80] |
| Thapsigargin | Ca2+ATPases | [84] |
| 2. Inhibitors of chromatin modifying complexes | ||
| JQ1 | BRD4 and BET proteins | [85] |
| OTX015 | BRD4 and BET proteins | [85] |
| iBET151 | BRD4 and BET proteins | [85] |
| iBET726 | BRD4 and BET proteins | [85] |
| iBET762 | BRD4 and BET proteins | [85] |
| Embelin | P/CAF | [86] |
| Entinostat | HDAC | [87] |
| SAHA | HDAC | [87] |
| 3. Unknown inhibitors | ||
| Fenretinide | Unknown | [88] |
Table 3.
PAX3-FOXO1 downstream targets and specific agents that could be potentially explored for treatment of FP RMS.
Table 3.
PAX3-FOXO1 downstream targets and specific agents that could be potentially explored for treatment of FP RMS.
| Genes | Targeting Agents | References | Clinical Trials #1 |
|---|---|---|---|
| 1. FGFR4 | Ponatinib | [124] | |
| PD173074 | [109,125] | ||
| FGF401 | NCT02325739 (Phase II) | ||
| FGFR4 MAB #2 | [126] | ||
| FGFR4 CAR T | [127] | ||
| 2. ALK1 | Crizotinib | [116] | NCT02034981 (II) |
| LDK378 | [128,129] | NCT01742286 (I) | |
| 3. IGF1R | Ganitumab #2 | [130] | NCT03041701(II) |
| Dalotuzumab #2 | [131] | NCT00694356 (I) | |
| R1507 | [132] | NCT00642941 (II) | |
| Picropodophyllin | [133] | ||
| 4. PDGFRα | Sorafenib | [134,135] | NCT01502410 (II) |
| Dasatinib | [119] | NCT00464620 (II) | |
| Sunitinib | [134] | NCT00474994 (II) | |
| Axitinib | [136] | NCT01140737 (II) | |
| Pazopanib | [137,138] | NCT01956669 (II) | |
| Olaratumab #2 | [139] | NCT01185964 (II) | |
| 5. MET | Crizotinib | [116] | NCT02034981 (II) |
| Tivantinib | [140] | ||
| 6. CXCR4 | MAB CF172 #2 | [141] | |
| 7. MYCN | PNA-MYCN | [142] | |
| Sphingosine | [143] | ||
| 8. JARID2 | JHDM inhibitor | [144] | |
| 9. CB1 | AM251 | [69] | |
| HU210 | [122] | ||
| THC | [122] | ||
| 10. CPT1A | Etomoxir | [145] |
#1 Clinical trials that recruit RMS patients. #2 MAB, mab: monoclonal antibody.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Nguyen, T.H.; Barr, F.G. Therapeutic Approaches Targeting PAX3-FOXO1 and Its Regulatory and Transcriptional Pathways in Rhabdomyosarcoma. Molecules 2018, 23, 2798. https://doi.org/10.3390/molecules23112798
AMA Style
Nguyen TH, Barr FG. Therapeutic Approaches Targeting PAX3-FOXO1 and Its Regulatory and Transcriptional Pathways in Rhabdomyosarcoma. Molecules. 2018; 23(11):2798. https://doi.org/10.3390/molecules23112798
Chicago/Turabian StyleNguyen, Thanh Hung, and Frederic G. Barr. 2018. "Therapeutic Approaches Targeting PAX3-FOXO1 and Its Regulatory and Transcriptional Pathways in Rhabdomyosarcoma" Molecules 23, no. 11: 2798. https://doi.org/10.3390/molecules23112798