
New Tricholomalides D–G from the Mushroom Tricholoma ustaloides Grown in an Italian Beech Wood
by
 , and
, and
Gianluca Gilardoni
1 ,
,
Francesca Negri
2,
Paola Vita Finzi
2,
Faiq H. S. Hussain
3 and
and
Giovanni Vidari
2,3,* 1
Departamento de Química, Universidad Técnica Particular de Loja (UTPL), Loja 110107, Ecuador
2
Dipartimento di Chimica, Università degli Studi di Pavia, Via Taramelli 10, 27100 Pavia, Italy
3
Department of Medical Analysis, Faculty of Applied Science, Tishk International University, Erbil 44001, Iraq
*
Author to whom correspondence should be addressed.
Molecules 2023, 28(21), 7446; https://doi.org/10.3390/molecules28217446
Submission received: 22 September 2023
/
Revised: 25 October 2023
/
Accepted: 3 November 2023
/
Published: 6 November 2023
(This article belongs to the Special Issue Terpenoid Natural Products: Discovery, Biological Evaluation, and Structural Modification)
Abstract
:Four novel seconeodolastane diterpenoids, named tricholomalides D–G, were isolated, together with the known tricholomalide C, from the fruiting bodies of Tricholoma ustaloides Romagn., a species belonging to the large Tricholoma genus of higher mushrooms (Basidiomycota, family Tricholomataceae). They were isolated through multiple chromatographic separations, and the structures, including the absolute configuration, were established through a detailed analysis of MS, NMR, and CD spectral data and comparison with related compounds reported in the literature, which has been thoroughly revised.
1. Introduction
More than 250 species are included in the genus Tricholoma (Fr.) Staude [1], which is the largest in the family Tricholomataceae of the order Agaricales (Basidiomycota). The species have a worldwide distribution and are mainly found in temperate and subtropical zones in both the southern and northern hemispheres [2], from Australia and China to North America and Europe [3,4,5,6,7,8].
The use of molecular methods based on nuclear rDNA internal transcribed spacer ITS1-5.8S-ITS2 (ITS) sequences is becoming more and more important in phylogenetic studies of higher mushrooms [7,8,9], accompanying or even substituting the traditional studies of fungal morphological characters. Thus, it has often been demonstrated that mushrooms growing in different habitats/countries but classified as the same species based on morphological characters, must instead be placed in distinct clades/subclades or even different taxa [8,10,11]. In this context, the distinct chemical contents determined through phytochemical analysis have often confirmed the possible existence of different varieties or taxa [12].
Our interest in the chemical analysis of Tricholoma species grown in Italy was thus motivated by taxonomical reasons, as the morphological characters alone make it difficult to differentiate some species, e.g., those included in the section Genuina [7]. However, not less important was the fact that the fruiting bodies of most Tricholoma produce a wide variety of secondary metabolites, exhibiting unusual/rare structures and important biological activities, including cytotoxic and antimicrobial properties, neurite outgrowth stimulation effects, acetylcholinesterase inhibitory activity, etc. [13].
Continuing our studies on Tricholoma species grown in Italy, our attention was recently drawn to the phytochemical study of Tricholoma ustaloides Romagn. (Figure 1A), which has been included by Heilmann-Clausen and Christensen in the critical section Genuina [7]. This mushroom, which is considered inedible in Europe, is generally rare in Italy, where it typically grows from late summer to late autumn in association with oak, chestnut, hornbeam, and beech trees [4,5] (Figure 1B).
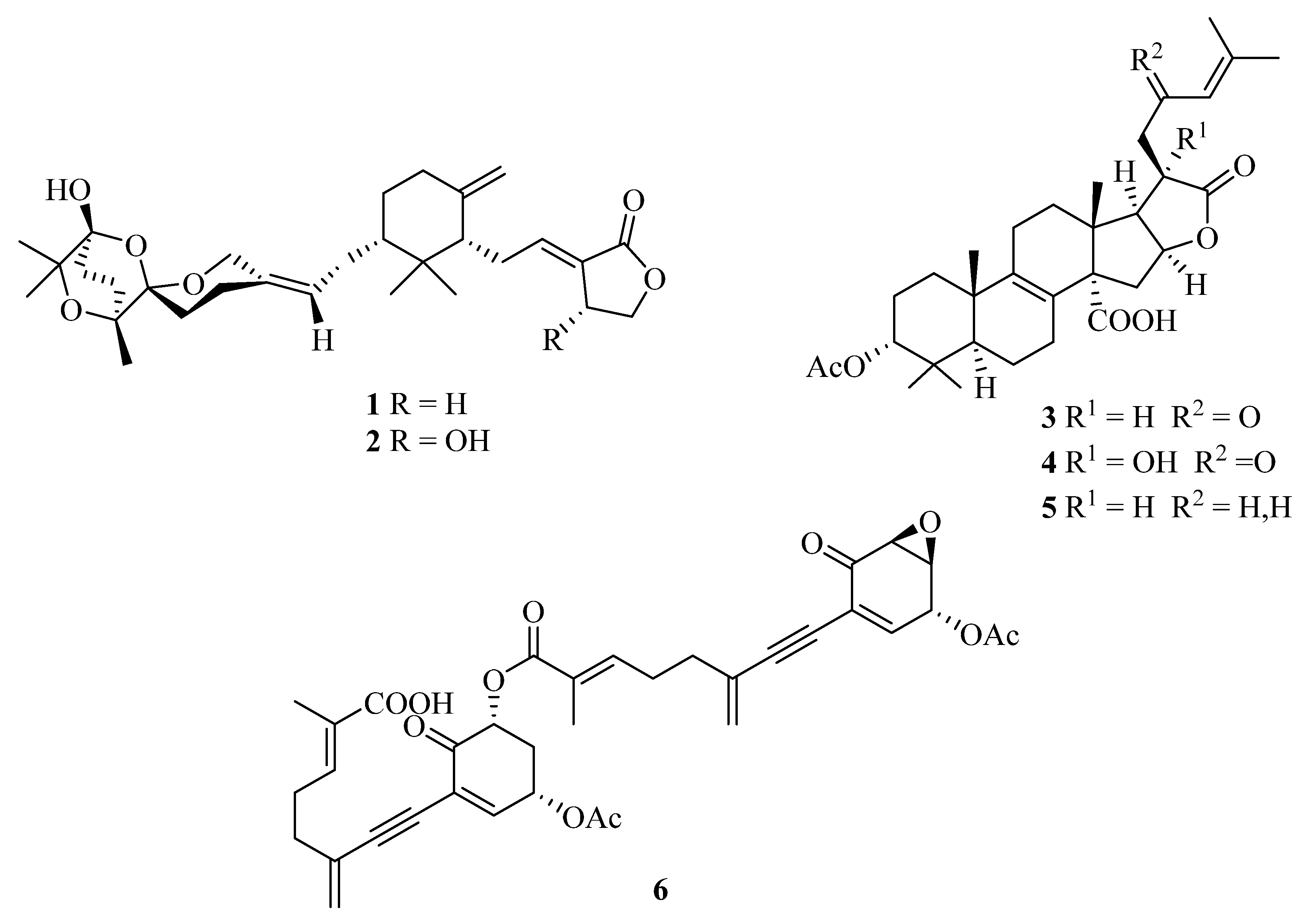
In our initial phytochemical investigation of an aqueous methanol sub-extract of an EtOAc extract of T. ustaloides fruiting bodies, we isolated two rare C-30 terpenoids, saponaceolides J (1) and F (2), three cyclic lactone-containing lanostane triterpenoids, tricholidic acid (3), the new tricholidic acids B (4) and C (5), and the rare tricholomenyn C (6) (Figure 2). In addition, mixtures of undetermined triglycerides and free fatty acids, together with five unidentified diterpenoids (A1–A5), were isolated [14].
In this paper we report the structures of compounds A1–A5, which were established through interpretation of IR, MS, 1D, and 2D NMR spectral data. The assignment of the absolute configuration (AC) to these compounds was based on the interpretation of electronic circular dichroism (ECD) spectra. In general, determination of the absolute configuration of natural products using ECD compares the spectrum of a new compound having an unknown AC to those of analogous compounds of known ACs [15]. However, AC determination by predicting the sign of one or more bands in the ECD spectrum using empirical, semiempirical, or nonempirical rules is often an option [15,16]. Another widely used option is to compare calculated and experimental ECD spectra [15].
The assignment of stereostructures to the compounds A1–A5 heavily depended on those of related diterpenoids previously isolated from other Tricholoma species. Since there are several discrepancies in the literature about the structures of the latter compounds [13], especially about their ACs, the discussion of the structures of A1–A5 is preceded by a short critical review of the existing literature.
2. Diterpenoids Isolated from Tricholoma Fruiting Bodies: A Short Critical Review
Diterpenoids are a class of terpenes formally composed of four isoprene units, that are biosynthesized by plants, animals, and fungi, including higher mushrooms (Basidiomycota) [17], via the HMG-CoA reductase pathway, with geranylgeranyl pyrophosphate being a primary intermediate [18]. Only a few examples of diterpenoids have been isolated so far from fruiting bodies of Tricholoma [13].
2.1. Trichoaurantianolides and Tricholomalides
All the diterpenoids isolated from fruiting bodies of Tricholoma species have a rare rearranged terpenoid skeleton, which has been named seconeodolastane in accordance with the proposed biosynthetic pathway (vide infra, Scheme 1). They belong to two diastereomeric families, the trichoaurantianolides and the tricholomalides, respectively, depending on the stereochemistry of the OH group at the C-8 position of the seconeodolastane skeleton (vide infra).
The first group of diterpenoids includes trichoaurantianolides A–D (7–10), which were isolated from wild fruiting bodies of T. aurantium (Schaeff.: Fr.) Ricken collected in Italy [19,20]; trichaurantin 8 (synonym of trichoaurantianolide B), which was isolated from the fruiting bodies of T. aurantium and T. fracticum (Britz.) Kreis. (syn. T. batschii Gulden) collected in Germany [21]; and acetyl trichaurantin (11), which was isolated from T. aurantium [21].
Structures 7–11 were established using 2D NMR data and using single-crystal X-ray analysis in the case of alcohol 8 [20,21]. Moreover, the absolute configurations of trichoaurantianolides B (8), C (9), and D (10) (Figure 3) were firmly established using anomalous X-ray diffraction and chemical interconversions [20,21], and enantioselective total synthesis [22]. Thus, the absolute stereochemistry of trichoaurantianolide C (9) was identical to the one initially assigned to this compound on the basis of the weak Cotton effect (CE) observed at 300 nm (Δε = +0.14) [20]. In fact, based on an empirical rule [16], the positive sign of the CE of compound 9, for which the molecular modeling indicated a “twisted” conformation of the cyclopentanone ring, corresponded to the signs of the octants occupied by the carbons of the ring (see structure A in Figure 4). Instead, misapplication of the octant rule [16] to the positive CE of trichoaurantianolide C resulted in the erroneous assignment of the enantiomeric configuration ent-9 (see structure ent-A in Figure 4) [23].
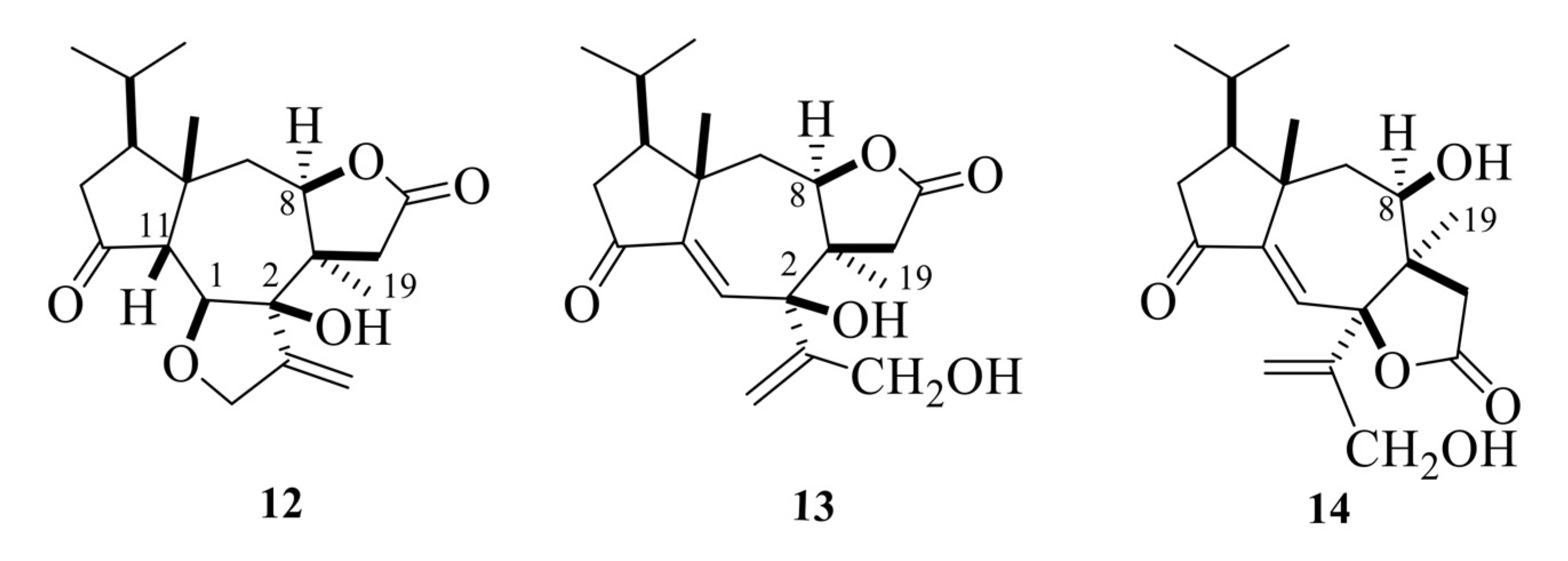
Tricholomalides A–C were isolated from the methanol extract of fresh fruiting bodies of an undetermined species of Tricholoma collected in Japan [23]. Noteworthily, these diterpenoids significantly induced neurite outgrowth in rat pheochromocytoma cells at concentrations of 100 μM [23]. The stereostructures of tricholomalides A–C, which were similar to those of trichoaurantianolides 7–11, were definitely established as 12–14 (Figure 5) using total synthesis and single-crystal X-ray diffraction. Thus, the stereochemistry at C-2 in tricholomalides A (12) and B (13) [24] was established to be opposite to the one originally assigned to these compounds based on spectral data only [23]. Moreover, H-8 was cis to H3-19, while it was trans in compounds 7–11.
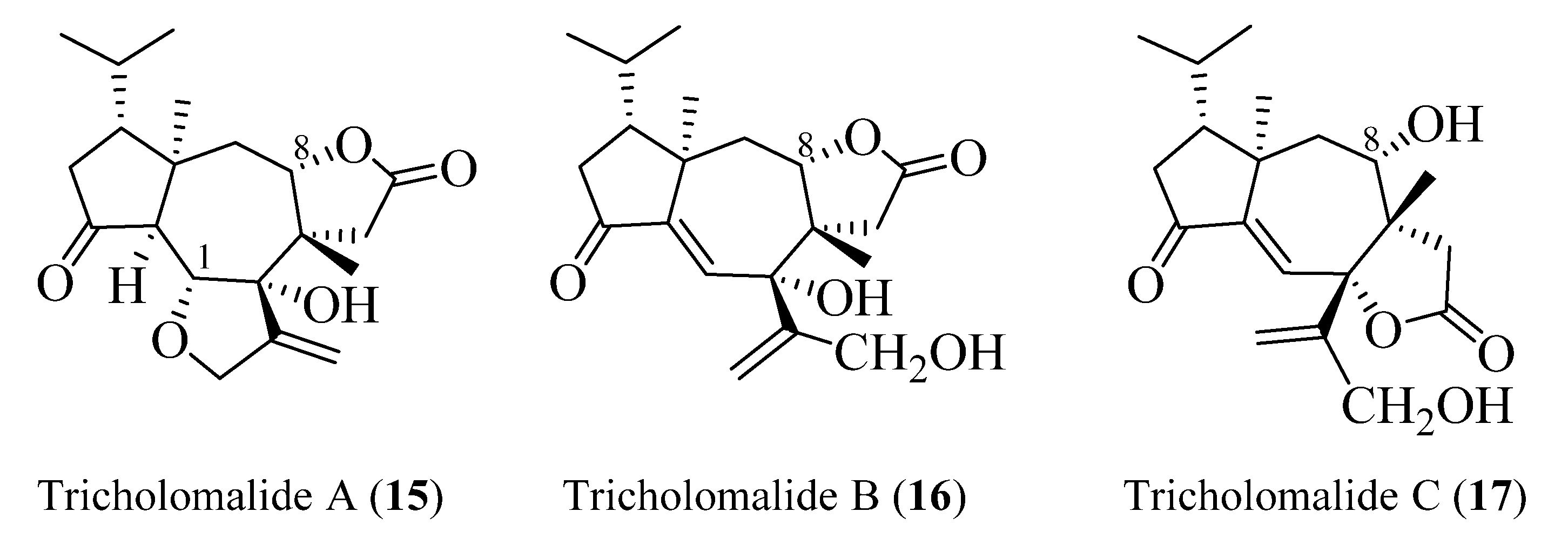
The absolute configuration of tricholomalides A–C has not yet been determined unambiguously. However, biosynthetic considerations (vide infra, Scheme 1) and interpretation of the CD data applying the empirical rule of twisted cyclopentanones [16], instead of the octant rule used in the paper reporting the isolation of tricholomalides [23], have strongly suggested that the configuration is identical to that of trichoaurantianolides, thus inverting the original assignment [23]. In fact, like trichoaurantianolide C (9) [20], tricholomalide A (15) exhibited a positive Cotton effect at 302 nm (Δε + 0.31) [23], indicating a similar “positively twisted” conformation of the cyclopentanone ring (see the discussion above). In addition, tricholomalide B (16) was converted to 15 during storage at 4 °C in DMSO [23], and the CD spectra of the cisoid α,β-unsaturated ketones 16 and 17 revealed CEs with the same signs for the corresponding peaks at 250 (Δε − 0.57) and 336 nm (Δε + 0.53) for 16 and at 234 (Δε − 0.22) and 344 nm (Δε + 0.16) for 17 [23].
In summary, the absolute stereochemistry depicted in Figure 6 is proposed for tricholomalides A–C (15–17).
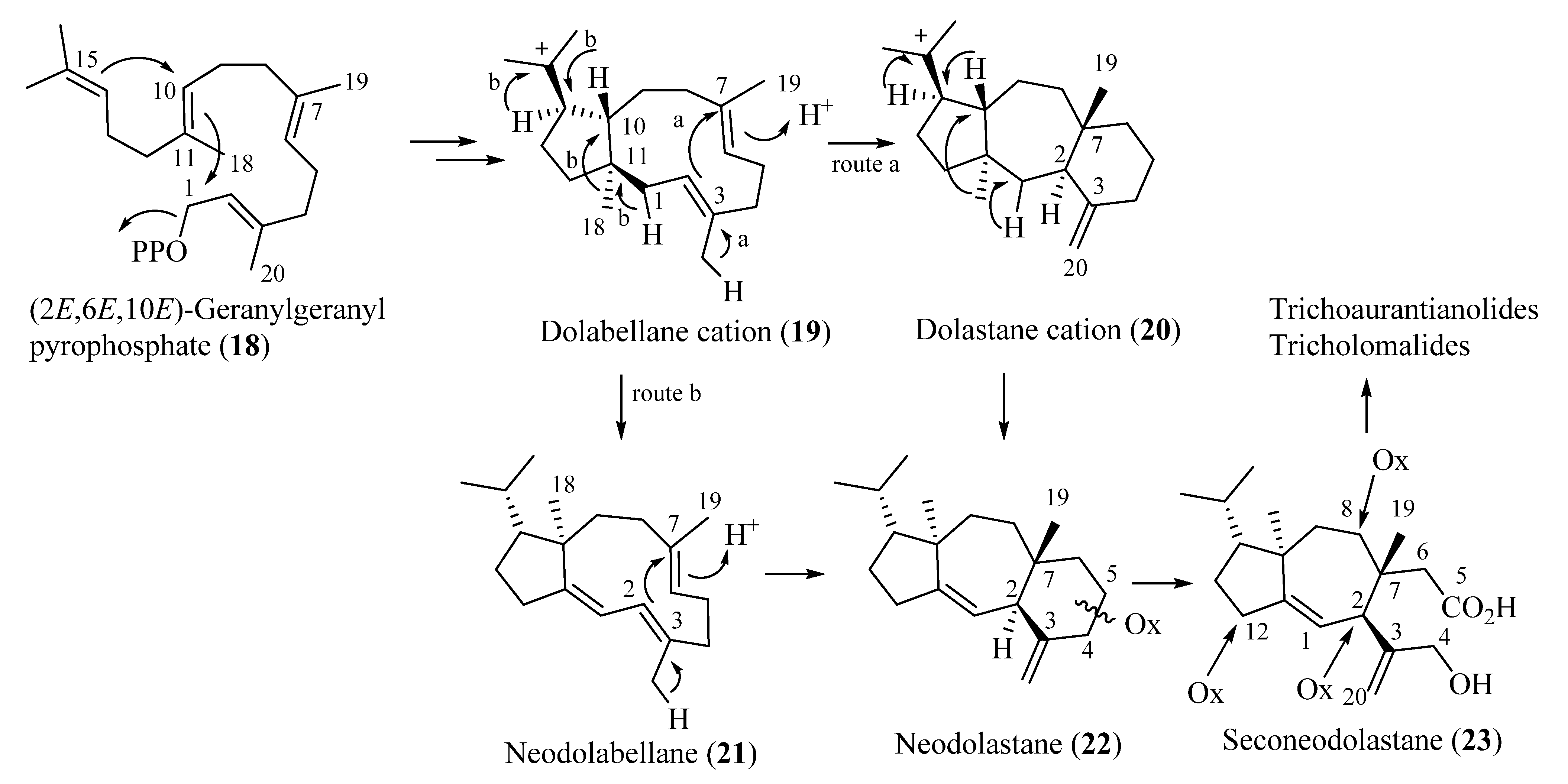
Biosynthetically, trichoaurantianolides and tricholomalides should originate from an enzymatically controlled stereoselective cyclization of geranylgeranyl pyrophosphate (18) producing nonracemic dolabellane cation 19. This intermediate would be the precursor of the dolastane (20), neodolabellane (21), and neodolastane (guanacastane) (22) skeletons through a series of stereospecific transannular cyclizations and Wagner–Meerwein migrations (Scheme 1). The seconeodolastane skeleton (23) of trichoaurantianolides and tricholomalides would finally result from oxidation and cleavage of the six-membered ring in 22 (Scheme 1), while oxidative decoration of the structures at C-2, C-8, and C-12 would likely occur in the latest steps of the biosynthetic path to trichoaurantianolides and tricholomalides. It is interesting to note that diterpenoids originating from a common biogenetic pathway in algae, fungi, liverworts, and higher plants are enantiomeric to the corresponding metabolites isolated from species of marine invertebrates [13,25].
3. Results and Discussion
3.1. Tricholomalides from Tricholoma Ustaloides
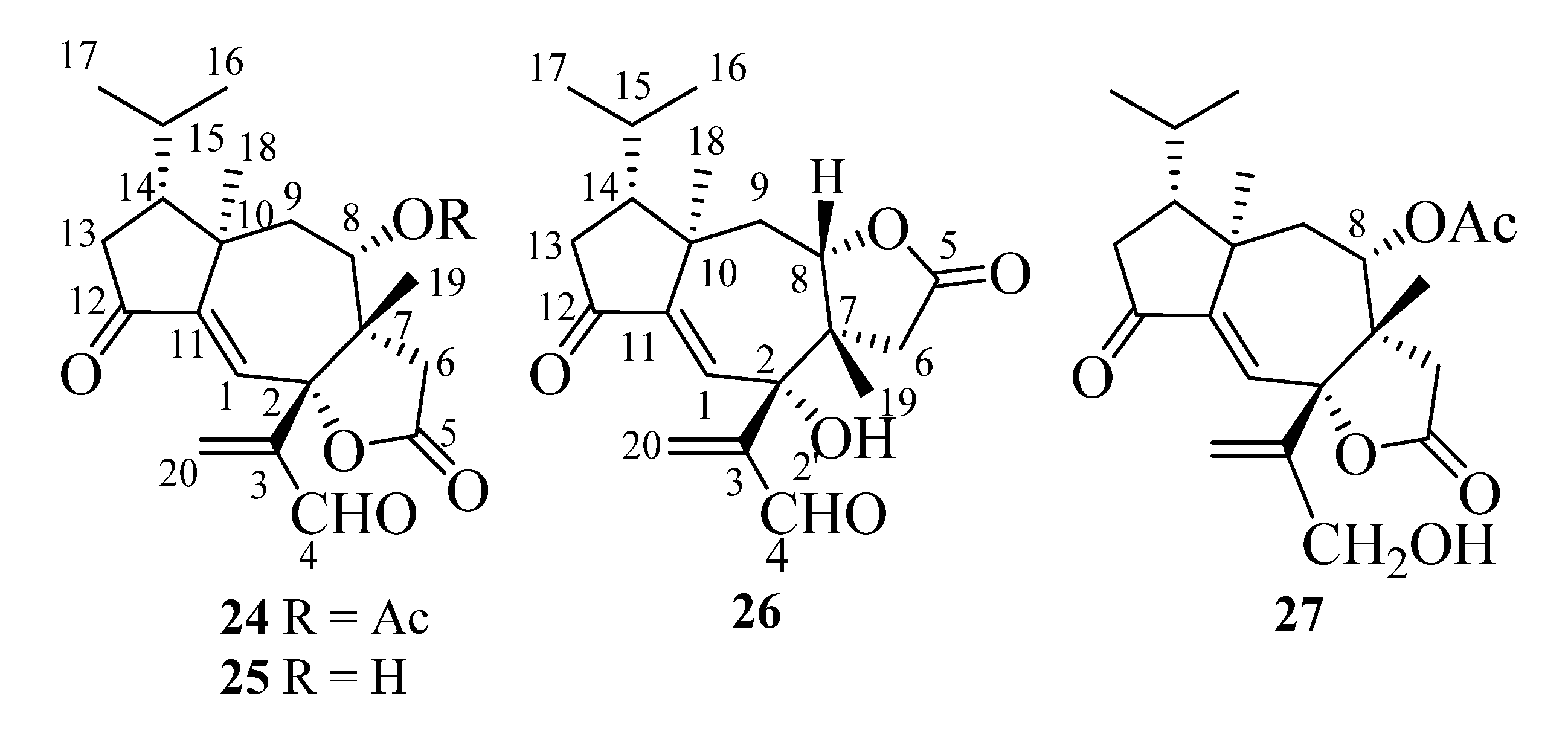
In the following paragraphs, we will describe the determination of the structures of compounds A1–A5, whose isolation from T. ustaloides fruiting bodies was reported in a recent paper [14]. A1–A4 were new diterpenoids, named tricholomalides D–G (24–27 in Figure 7), respectively, while A5 was identified as the known tricholomalide C (17) [23]. At first, we will discuss the structures of tricholomalides D (24), E (25), and G (27), which showed a γ-lactone ring cis-fused at C-2 and C-7 with the central cycloheptene ring of the seconeodolastane skeleton. Subsequently, we will describe the structure of tricholomalide F (26), in which the γ-lactone ring is cis-fused with the cycloheptene ring at C-7 and C-8.
3.1.1. Tricholomalides C, D, E, and G
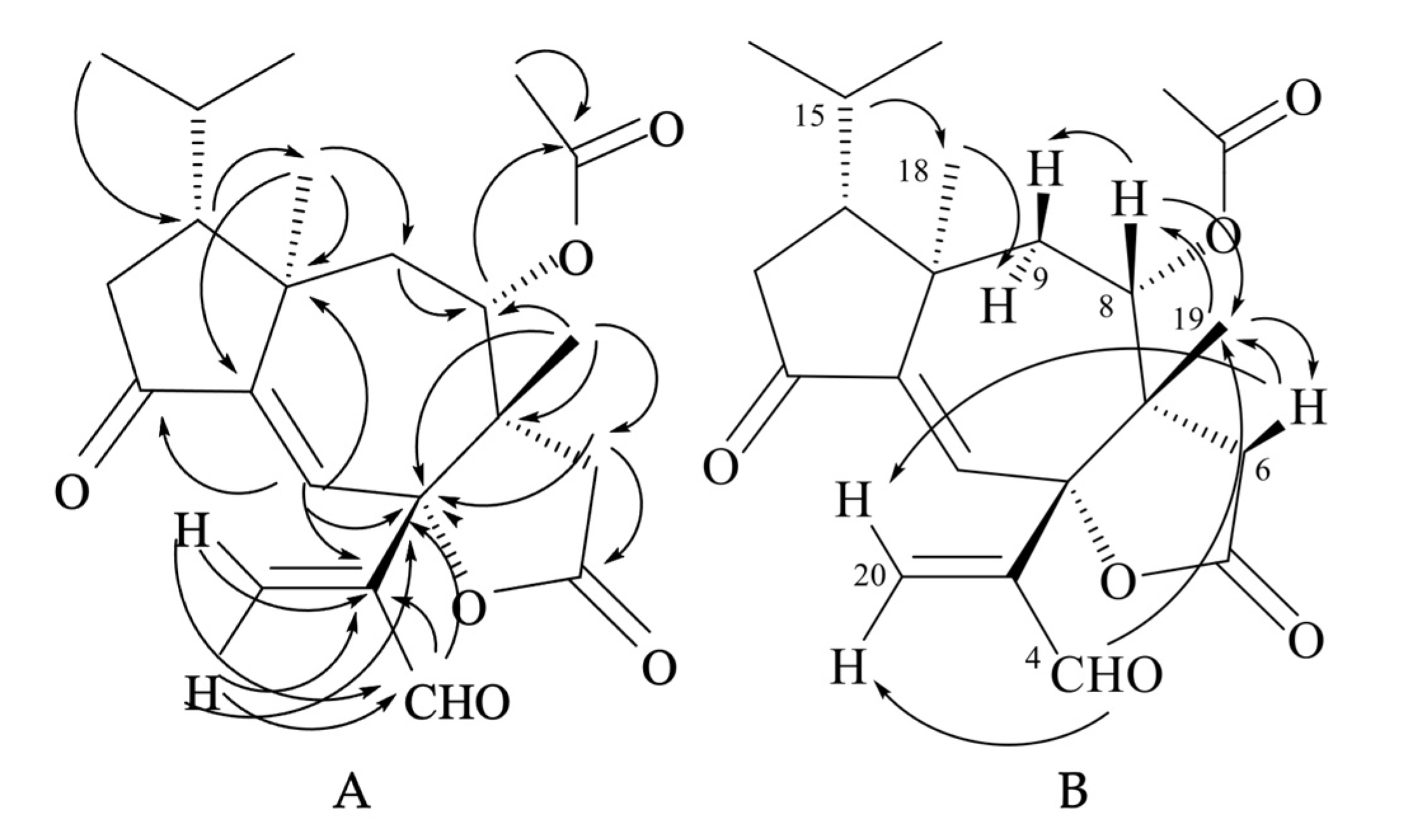
The molecular formula of tricholomalide D (24) was established as C22H28O6 by EIMS (Figure S9 in the Supplementary Materials), elemental analysis, and 1H and 13C NMR spectra (Table S1 in the Supplementary Materials). It indicated nine degrees of unsaturation. The oxygen-containing functionalities in the molecule were established as one cyclopentanone (C-12), one formyl (C-4), one γ-lactone (C-5), and one acetoxy (C-21) group from the IR bands (1783, 1736, 1699 cm−1) and the characteristic chemical shifts (δC 205.0, 191.8, 175.2, and 169.4 ppm, respectively) of the corresponding carbonyl carbons in the 13C NMR spectrum (Figures S7 and S8 in the Supplementary Materials).
The NMR spectra of compound 24 (Figures S5 and S7 and Table S1 in the Supplementary Materials) showed signals for one trisubstituted (δC 125.9 (d, C-1) and 151.3 (s, C-11); δH 6.63 (1H, s, H-1)) and one exo-methylene double bond (δC 138.4 (t, C-20) and 148.6 (s, C-3); δH 6.41 (1H, br s, HZ-20) and 6.62 (1H, br s, HE-20)). HMBC correlations (Figure 8A) indicated that the two olefins were α,β-conjugated to the ketone (C-12) and the aldehyde (C-4) groups, respectively, while they were bound to each other through an oxygenated quaternary carbon placed in the lactone ring (δC 87.4 (s, C-2)). The remaining two unsaturations of tricholomalide D (24) were then assigned to two rings that formed a tricyclic carbon skeleton together with the γ-lactone. The 1H NMR spectrum of 24 (Figure S5 in the Supplementary Materials) showed two methyl singlets at δH 0.98 (H3-19) and 1.12 (H3-18) that were attached to quaternary carbons C-7 (δC 49.8 s) and C-10 (δC 44.7 s), respectively, and two methyl doublets of an isopropyl group at δH 0.95 (J = 6.5 Hz, H3-17) and 1.02 (J = 6.5 Hz, H3-16). The remaining proton and carbon atoms of compound 24 were assigned to three separate moieties, I–III based on 1H-1H COSY data (Figure S6 in the Supplementary Materials) and selective homonuclear decoupling experiments. The moiety I linked C-13 to C-16 and included the H-15 (δH 1.75–1.85, 1H, m) to H3-17 linkage. The spin system of the unit II was an isolated AB quartet (δH 2.29 and 2.88, 2H, JAB = 18.5 Hz), which was assigned to an isolated methylene (H2-6) attached to the γ-lactone carbonyl group (δC 175.2, s, C-5) by HMBC correlations (Figure 8A). The moiety III gave rise to a distorted AMX system which includes a methylene (H2-9; δH 1.79 (1H, dd, 15.0, 11.5) and δH 2.15 (1H, dd, 15.0, 2.0)) and a methine hydrogen (H-8; δH 4.72 (1H, ddd, 11.5, 2.0, 1.0)). This proton was geminal to the acetoxy carbonyl carbon (δC 169.4, s, C-21) via an HMBC cross peak with C-21 (Figure 8A). Assembling these partial structures using the two- and three-bond C-H connectivity data from HMBC correlations (Figure 8A), we unambiguously assigned structure 24 to tricholomalide D. The structure of the ring system was determined by (i) the bonds around the angular methyl (H3-19) attached to C-7 (δC 49.8 s), which linked C-6 (δC 35.3 t) and C-9 (δC 37.7 t) to C-19 (δC 22.4 q) via C-7 and C-8 (δC 74.4 d); (ii) the linkages from H3-19, H2-20, and H-4 (δH 9.70 (1H, s)) to C-2 (δC 87.4 s) and those from H-1 (δH 6.63 (1H, s)) to C-2, thanks to which the acrolein moiety was connected to C-2 and the arrangement of the substituents of the γ-lactone ring was defined; (iii) the connectivities from H3-18 to C-10 (δC 44.7 s), C-9 and C-11 (δC 151.3 s), and H-1 to C-10, of which C-12 and C-2 indicated the presence of a cycloheptene ring bearing an angular methyl (H3-18) on an allylic sp3 quaternary carbon (C-10); (iv) the linkage of the isopropyl group to C-18 (δC 21.4 q) via C-14 (δC 47.2 d), which defined the placement of the moiety I to form a cyclopentanone ring. These data and 1D NOE correlations (Figure 8B), H-15/H3-18; H3-18/H-9α; H-8/H-9β and H-8/H3-19; H3-19/H-6β, H-4/H3-19, and H-6β/H-20E (Figure 8B), suggested that tricholomalide D (24) was the C-8 epimer of trichoaurantianolide A (7) [19]. Thus, the 8-acetoxy group was trans to H3-19 in 24, while it was cis in 7 [19]. This finding was confirmed by the significantly different chemical shift and coupling constants of H-8 in 24 (δH 4.72; J8β-9β = 2.0 and J8β-9α = 11.5 Hz) and in 7 (δH 5.15; J8α-9β = 9.5 and J8α-9α = 1.5 Hz) [19].
The molecular formula of tricholomalide E (25), C20H26O5, was established from the EIMS (Figure S15 in the Supplementary Materials) and the 1H and 13C NMR spectra (Figures S11–S14 in the Supplementary Materials). The NMR spectral data (Table S1 in the Supplementary Materials) clearly indicated that it was the deacetyl derivative of tricholomalide D (24). In fact, the signals of an acetyl group were missing in the 1H and 13C NMR spectra of 25 (Figures S11 and S13 in the Supplementary Materials), while the signals of H-8 (δH 3.63, br d, 11.5) and C-8 (δC 71.6 d) in the 1H and 13C NMR spectra, respectively, of alcohol 25 moved upfield by about 1 and 3 ppm, respectively, in comparison with the corresponding signals of the acetate 24 (vide supra). Finally, the structure of 25 was confirmed with standard acetylation with Ac2O/Py, which afforded a product that was identical with acetate 24.
Tricholomalide G (27), with the molecular formula C20H26O5 from the EIMS (Figure S27 in the Supplementary Materials) and the 1H and 13C NMR spectra (Figures S23–S26 in the Supplementary Materials), was identified as the dihydroderivative of aldehyde 24 through the following significant differences between the NMR spectra of the two compounds (Figures S23–S26 and Table S1 in the Supplementary Materials): (i) the presence of two diastereotopic protons (H2-4) at δH 4.22 (1H, dd, 13.0, 1.1) and 4.27 (1H, br d, 13) in the 1H NMR spectrum of 27, assignable to an allylic CH2OH group, which replaced the signal H-4 (δH 9.70 (1H, s)) of aldehyde 24; (ii) the upfield shift of the H2-20 protons (δH 5.32 (1H, s) and δH 5.67 (1H, t, 1.1)) and the C-20 carbon (δC 118.3, t), respectively, in the 1H and 13C NMR spectra of 27, compared with the corresponding signals of the βCH2 group of the acrolein moiety in 24. Finally, the structure of compound 27 was confirmed through the oxidation of the allylic alcohol with PDC (pyridinium dichromate), which afforded a product indistinguishable from aldehyde 24.
The NMR spectral data (Figures S1–S3 in the Supplementary Materials) and the signs of the CD maxima of compound A5 (Figure S4 in the Supplementary Materials) were identical with the data reported in the literature for tricholomalide C (17) [23]. Moreover, selective oxidation of A5 (≡17) with PDC gave a product indistinguishable from aldehyde 25.
3.1.2. Tricholomalide F
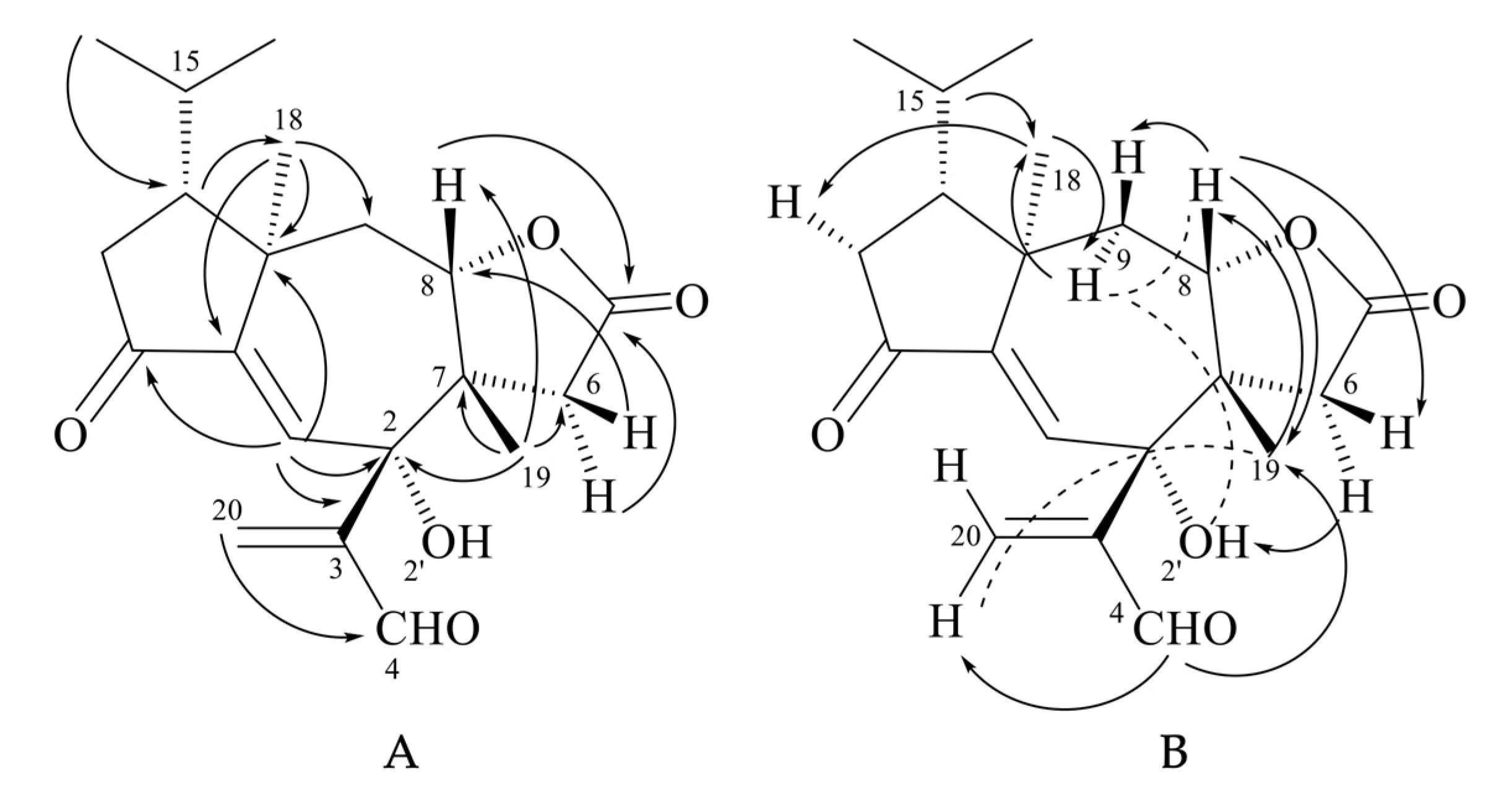
Tricholomalide F (26) possessed the same molecular formula as tricholomalide E (25), C20H26O5, as inferred from the EIMS spectrum (Figure S21 in the Supplementary Materials) and the 1H and 13C NMR spectral data (Figures S17–S20 and Table S2 in the Supplementary Materials). These data resembled those reported for tricholomalide B (16) [23], except for the presence of a formyl group (H4 (δH 9.55, 1H, s) and a C-4 carbonyl group (δC 196.3, d)) bound to C-3 (δC 148.6 s) in 26 which replaced the allylic CH2OH group occurring in compound 16 [23]. The presence of a free tertiary OH group attached to C-2 of 26 was revealed by the singlet at δC 76.1 in the 13C NMR spectrum (Figure S19 in the Supplementary Materials), while the signals of H-8 (δH 3.92, 1H, dd, 12.8, 2.5) and C-8 (δC 85.0, d) in the NMR spectra of 26 (Figures S17 and S19 in the Supplementary Materials) indicated the ring closure of the γ-lactone unit on C-8.
The two- and three-bond HMBC correlations (Figure 9A) were fully consistent with the gross structure of 26, while the relative stereochemistry of the stereogenic carbons in 26 was established through selective 1D NOE experiments (Figure 9B). In fact, the correlations of H-15 (δH 1.75–1.92 (1H, m)) with H3-18 (δH 1.20 (3H, s)) and between H3-18 and H-9α (δH 2.79 (1H, dd, 14.0, 12.8)) indicated that the isopropyl group, H3-18, and H-9α were oriented on the same side of the tricyclic structure, while the interactions of H-8 with H-9β (δH 2.23 (1H, dd, 14.0, 2.5)) and H3-19 (δH 0.97 (3H, br s)) and H-4 with H3-19 (Figure 9B) revealed that they were placed on the opposite sides of H3-18 and H-9α. The resulting stereochemistry of compound 26 was further corroborated by the J = 12.8 Hz of H-8β with H-9α and by the NOE interactions of H-8β with H-6β (δH 1.86 (1H, d, 17.0)) and H-6α (δH 3.24 (1H, dd, 17.0, 1.0)) with 2-OH (δH 4.92 (1H, d, 1.0)). Interestingly, H-1 (δH 6.97 (1H, d, 1.0)) was coupled with 2-OH by a JW coupling of 1 Hz, which indicated a rather rigid orientation of the tertiary hydroxy group.
In summary, tricholomalides D–G (24–27) exhibited a relative stereochemistry identical to that of tricholomalides B (16) and C (17), while they were epimers of trichoaurantianolides at C-8.
3.1.3. Absolute Configuration of Tricholomalides D–G
The absolute configuration of tricholomalides D–G (24–27) shown in Figure 7 was established on the basis of chemical interconversions, biosynthetic considerations (see above), and CD data (Figures S10, S16, S22 and S28 in the Supplementary Materials). The cisoid α,β-unsaturated cyclopentanone system occurring in 24–27 is also a characteristic feature of the structures of tricholomalides B (16) and C (17), as well as of trichoaurantianolides 7, 8, and 11, whose configuration was thoroughly discussed in a previous Section 2.1. All the CD spectra of these compounds revealed Cotton effects with positive signs for the respective peaks in the region of 300–350 nm (Figures S10, S16, S22 and S28 in the Supplementary Materials), which were thus closely associated with similar conformational and dissymmetric substituent effects on the cisoid α,β-unsaturated ketone unit. Therefore, tricholomalides D–G (24–27) were assigned the same absolute configuration as tricholomalides B (16) and C (17) (Figure 6 and Figure 7), having a stereochemistry at C-8 that was opposite to that determined for trichoaurantianolides 7, 8, and 11 (Figure 3). This finding is not unexpected, as the configurations at C-7, C-10, and C-14 arise from stereospecific transannular cyclizations and Meerwein–Wagner rearrangements in the first steps of the common biosynthetic path leading to the tricholomalides and trichoaurantianolides (Scheme 1). Instead, the hydroxylation at C-8 likely involved a neodolastane or seconeodolastane derivative at a late step of the biosynthetic path, and it was controlled by enzymes with opposite diastereoselectivities for tricholomalides and trichoaurantianolides, respectively.
4. Materials and Methods
4.1. General Experimental Techniques and Procedures
Preparative chromatographic separations were carried out on open columns at atmospheric pressure (CC). The columns were manually packed with silica gel (Merck Kieselgel 60, 40–63 μm, Rahway, NJ, USA) or reversed-phase C18 (Merck LiChroprep RP-18, 25–40 μm) purchased from Sigma-Aldrich (St. Louis, MO, USA). Thin-layer chromatographic (TLC) analyses were conducted over glass-supported silica gel 60 (0.25 mm; GF254, Merck) or RP-18 (F254s, Merck) plates (Sigma-Aldrich). Spots on TLC plates were initially visualized under UV light (254 and 366 nm); subsequently, they were sprayed with a 0.5% solution of vanillin in H2SO4/ethanol 4:1 and finally heated with a hot gun until reaching maximum color development. Semipreparative medium-pressure liquid chromatographic (MPLC) separations were performed with an Isolera instrument (Biotage, Uppsala, Sweden) equipped with silica gel and RP-18 reversed-phase cartridges and a dual-wavelength UV detector. Reagent-grade solvents, purchased from Carlo Erba (Milan, Italy) or from Aldrich, were used for extraction and chromatographic separations. Optical rotation was conducted using PerkinElmer 241 polarimeter (Walthman, MA, USA); CD spectra were obtained using Jasco J-1500 CD spectrometer (Tokyo, Japan) in MeOH. IR spectra were obtained using PerkinElmer Paragon 100 PC FT-IR spectrometer (Walthman, MA, USA) on KBr disks. NMR spectra were obtained using Bruker AV300 and 400 spectrometers at 300 and 400 MHz (1H), respectively, and at 75.47 and 100 MHz (13C), respectively, operating at 22 °C (Billerica, MA, USA). 1H NMR and 13C NMR chemical shifts are relative to signals of residual CHCl3 (δH 7.25, singlet) and 13CDCl3 δC (77.0, central line of a triplet) in CDCl3 (Sigma-Aldrich, Steinheim, Germany); coupling constants (J) were measured in Hz; multiplicity (=number of attached hydrogens) of each C-atom was determined using DEPT experiments; COSY, DEPT, and HSQC spectra and NOE effects were recorded using standard pulse sequences. EIMS and DCI-MS (NH3) spectra were obtained using Finnigan-MAT 822 mass spectrometer.
4.2. Fungal Material
The fruiting bodies of Tricholoma ustaloides Romagn. were collected in a beech wood at the end of September—beginning of October 2021 and identified by Alfredo Gatti, as reported in the inaugural study of this mushroom [14]. A sample specimen (accession code: TU001) was deposited at the Department of Chemistry, University of Pavia, Italy.
4.3. Extraction and Isolation
Extraction of the fresh fruiting bodies (990 g) with EtOAc, evaporation of the extract, and partition of the resulting residue between MeOH-H2O at 90:10 and hexane were described in a previous paper [14]. The residue (2.8 g) from the aqueous methanol layer was subjected to multiple separations using semipreparative MPLC silica gel and RP-18 columns to afford compounds A1–A5, named tricholomalides D (24, 51 mg, 0.0051% on fresh fruiting bodies), E (25, 18.6 mg, 0.0019%), F (26, 5.5 mg, 0.0006%), G (27, 15.2 mg, 0.0015%), and C (17, 13.6 mg, 0.0014%), respectively [14].
4.3.1. Tricholomalide C (17)
Colorless, sticky oil; [α]D22 + 32.9 (c 10.1 mg/mL, CH2Cl2); Rf 0.77 (RP18 TLC; MeOH-H2O, 6:1); CD (MeOH) Δε236 −0.61, Δε345 +0.51; lH NMR (300 MHz, CDCl3) δH 0.98 (3H, d, 6.5, H3-17), 1.07 (3H, d, 6.5, H3-16), 1.14 (3H, s, H3-19), 1.16 (3H, s, H3-18), 1.68-1.89 (2H, m, H-14 and H-15), 1.84 (1H, dd, 15.5 and 11.5, Hα-9), 2.11 (1H, dd, 15.5 and 2.0, Hβ-9), 2.14 (1H, dd, 18.5 and 12.5, Hα-13), 2.37 (1H, dd, 18.0 and 1.0, Hβ-6), 2.47 (1H, dd, 18.5 and 8.0, Hβ-13), 2.84 (1H, d, 18.0, Hα-6), 3.48 (1H, ddd, 11.5, 2.0, and 1.0, H-8), 4.21 (2H, br s, H2-4), 5.30 (1H, s, HZ-20), 5.63 (IH, s, HE-20), 6.62 (1H, s, H-1); 13C NMR (75.5 MHz, CDCl3) δC 21.2 (q, C-18), 21.7 (q, C-17), 21.8 (q, C-19), 24.0 (q, C-16), 28.5 (d, C-15), 34.2 (t, C-6), 39.6 (t, C-9), 41.2 (t, C-13), 44.6 (s, C-10), 49.0 (d, C-14), 50.2 (s, C-7), 63.2 (t, C-4), 72.2 (d, C-8), 88.9 (s, C-2), 117.7 (t, C-20), 128.4 (d, C-1), 148.0 (s, C-3), 151.4 (s, C-11), 176.8 (s, C-5), 205.4 (C-12); IR (film): ῡmax 3425, 2969, 1758, 1726, 1647, 1461, 1246, 1199, 1020, 907 cm−1; EIMS m/z (relative intensity) 348 [M]+ (C20H28O5)+ (6), 333 [M-CH3]+ (46), 263 (100), 175 (21), 167 (59), 139 (23), 109 (25), 97 (39), 83 (21), 69 (50), 55 (36), 41 (60). Anal. Calcd for C20H28O5: C, 68.94; H, 8.10. Found: C, 69.15; H, 8.18.
4.3.2. Tricholomalide D (24)
Colorless, sticky oil; [α]D22 + 31.2 (c 10.9 mg/mL, CH2Cl2); Rf 0.53 (silica gel TLC; CH2Cl2-Me2CO, 30:1); CD (MeOH) Δε220 −0.45, Δε246 +0.34, Δε350 +0.11; 1H and 13C NMR spectral data, see Table S1 in the Supplementary Materials; IR (film): ῡmax 2968, 1783, 1736, 1699, 1648, 1367, 1241, 1027, 990, 961 cm−1; EIMS m/z (relative intensity) 388 [M]+ (C22H28O6)+ (54), 359 [M-CHO]+ (11), 346 [M-CH2CO]+ (18), 328 [M-AcOH]+ (34), 313 (12), 285 (45), 269 (21), 263 (33), 178 (23), 163 (48), 133 (37), 106 (78), 95 (33), 83 (42), 69 (35), 58 (69), 43 (100). Anal. Calcd for C22H28O6: C, 66.02; H, 7.27. Found: C, 66.15; H, 7.44.
4.3.3. Tricholomalide E (25)
Colorless, sticky oil; [α]D22 + 1.2 (c 5.2 mg/mL, CH2Cl2); Rf 0.67 (RP18 TLC; MeOH-H2O, 80:20); CD (MeOH) Δε225 −2.28, Δε246 +0.11, Δε272 −0.68, Δε350 +0.46; 1H and 13C NMR spectral data, see Table S1 in the Supplementary Materials; IR (film): ῡmax 3474, 2929, 1780, 1723, 1696, 1647, 1461, 1375, 1246, 1024, 962 cm−1; EIMS m/z (relative intensity) 346 [M]+ (C20H26O5)+ (31), 287 (40), 263 (58), 231 (22), 203 (22), 191 (36), 173 (25), 167 (82), 163 (51), 151 (37), 133 (39), 121 (30), 109 (32), 91 (40), 83 (61), 69 (68), 55 (68), 43 (100). Anal. Calcd for C20H26O5: C, 69.34; H, 7.57. Found: C, 69.75; H, 7.78.
4.3.4. Tricholomalide F (26)
Colorless, sticky oil; [α]D22 + 63.9 (c 1.8 mg/mL, CH2Cl2); Rf 0.57 (RP18 TLC; MeOH-H2O, 80:20); CD (MeOH) Δε255 +0.47, Δε275 −0.05, Δε340 +0.20; 1H and 13C NMR spectral data, see Table S2 in the Supplementary Materials; IR (film): ῡmax 3561, 3416, 2941, 2876, 1783, 1765, 1725, 1638, 1461, 1371, 1242, 1051, 976 cm−1; EIMS m/z (relative intensity) 346 [M]+ (C20H26O5)+ (30), 331 [M-CH3]+ (9), 317 [M-CHO]+ (33), 303 (35), 287 (23), 263 (99), 235 (46), 203 (29), 191 (61), 175 (27), 163 (61), 149 (43), 135 (48), 121 (33), 109 (38), 97 (47), 83 (58), 69 (84), 55 (88), 43 (70), 41 (100). Anal. Calcd for C20H26O5: C, 69.34; H, 7.57. Found: C, 69.65; H, 7.73.
4.3.5. Tricholomalide G (27)
Colorless, sticky oil; [α]D22 + 55.2 (c 4.8 mg/mL, CH2Cl2); Rf 0.57 (RP18 TLC; MeOH-H2O, 80:20); CD (MeOH) Δε250 +0.43, Δε345 +0.18; 1H and 13C NMR spectral data, see Table S1 in the Supplementary Materials; IR (film): ῡmax 3449, 3060, 2960, 2929, 1772, 1731, 1644, 1463, 1370, 1241, 1020, 963, 938, 735 cm−1; EIMS m/z (relative intensity) 390 [M]+ (C22H30O6)+ (9), 375 [M-CH3]+ (54), 315 [M-AcOH]+ (15), 263 (45), 191 (17), 181 (24), 167 (32), 135 (15), 109 (16), 95 (24), 69 (39), 55 (27), 43 (100), 41 (44). Anal. Calcd for C22H30O6: C, 67.67; H, 7.74. Found: C, 67.81; H, 7.62.
4.4. Acetylation of Tricholomalide E (25) to Tricholomalide D (24)
Tricholomalide E (25) (3 mg, 8.7 μmol) in CH2Cl2 (1 mL) was added to pyridine (50 μL), followed by excess Ac2O (50 μL). The mixture was stirred for 2 h at 22 °C, quenched with MeOH (0.5 mL), and evaporated. The residue was filtered through a short pad of silica gel (300 mg). Elution with CH2Cl2-MeOH at 99:1 gave a product (3.3 mg, 99%) identical ([α]D22, NMR data) to tricholomalide D (24).
4.5. Oxidation of Tricholomalide G (27) to Tricholomalide D (24) and Tricholomalide C (17) to Tricholomalide E (25)
Freshly prepared PDC (9 mg, 24 μmol) [26] was added in one portion to tricholomalide G (27) (3 mg, 7.7 μmol) dissolved in CH2Cl2 (0.5 mL). The mixture was stirred for 6 h at 22 °C; subsequently, it was diluted with abundant Et2O. The organic layer was washed with aqueous NaHCO3, dried (Na2SO4), and evaporated. The residue was filtered through a short pad of silica gel (300 mg). Elution with CH2Cl2-MeOH at 99:1 gave a product (2.3 mg, 77%) identical ([α]D22, NMR data) to tricholomalide D (24).
Isolated tricholomalide C (17) was converted to tricholomalide E (25) in 75% yield using the procedure described above.
5. Conclusions
The isolation of four novel seconeodolastane diterpenoids, tricholomalides D–G, from T. ustaloides has confirmed that the fruiting bodies of Tricholoma mushrooms are rich sources of new compounds, most of which have unique chemical structures. The rare seconeodolastane skeleton of tricholomalides seems to be a characteristic feature of the diterpenoids present in the fruiting bodies of Tricholoma; however, epimeric derivatives, e.g., compounds 7 and 24, are produced by different species, and this finding may be chemotaxonomically significant. So far, besides being isolated from fruiting bodies of the genus Tricholoma, seconeodolastane diterpenoids have only been isolated from a mycelial culture of Lepista sordida [27]. Interestingly, the genera Tricholoma and Lepista both belong to the family Tricholomataceae (Agaricales).
In the future, we will extend our investigations to the chemical contents of other Tricholoma species grown in the Italian woods, and, regarding tricholomalides C–G, we intend to study their biological activities using in vitro tests.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/molecules28217446/s1: 1H NMR graphical spectra of tricholomalides C–G; COSY graphical spectra of tricholomalides D–G; 13C NMR graphical spectra of tricholomalides C–G; DEPT 13C NMR graphical spectra of tricholomalides C–G; EIMS graphical spectra of tricholomalides D–G; CD graphical spectra of tricholomalides C–G; tabulated NMR spectral data of tricholomalides D–G.
Author Contributions
Conceptualization, G.V. and F.H.S.H.; investigation, F.N. and G.G.; writing—original draft preparation, F.N. and G.V.; writing—review and editing, G.V.; supervision, G.V. and P.V.F. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Raw data are available from one of the authors (G.V.).
Acknowledgments
We are grateful to the Universidad Técnica Particular de Loja (UTPL) for supporting open access publication. We warmly thank Alfredo Gatti (Gruppo Micologico Vogherese) for the mushroom identification and the wood picture in Figure 1, and Teresio Restelli (Gruppo Micologico Pavese) for the mushroom picture in Figure 1.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Staude, F. Die Schwämme Mitteldeutschlands, insbesondere des Herzogthums Coburg; Dietz: Hannover, Germany, 1857. [Google Scholar]
- Tedersoo, L.; May, T.W.; Smith, M.E. Ectomycorrhizal lifestyle in fungi: Global diversity, distribution, and evolution of phylogenetic lineages. Mycorrhiza 2010, 20, 217–263. [Google Scholar] [CrossRef]
- Bessette, A.E.; Bessette, A.R.; Roody, W.C.; Trudell, S.A. Tricholomas of North America: A Mushroom Field Guide; University of Texas Press: Austin, TX, USA, 2013; p. 1. [Google Scholar]
- Bon, M. Champignons de France et d’Europe Occidentale; Flammarion: Paris, France, 2004. [Google Scholar]
- Riva, A. Tricholoma (Fr.) Staude. Fungi Europaei; Candusso Editrice: Origgio, Italy, 2003; Volume 3, ISBN 88-901057-1-2A. [Google Scholar]
- Christensen, M.; Heilmann-Clausen, J. The Genus Tricholoma. Fungi of Northern Europe; Svampetrik: Tilst, Denmark, 2013; Volume 4. [Google Scholar]
- Heilmann-Clausen, J.; Christensen, M.; Frøslev, T.G.; Kjøller, R. Taxonomy of Tricholoma in northern Europe based on ITS sequence data and morphological characters. Persoonia 2017, 38, 38–57. [Google Scholar] [CrossRef]
- Reschke, K.; Popa, F.; Yang, Z.L.; Kost, G. Diversity and taxonomy of Tricholoma species from Yunnan, China, and notes on species from Europe and North America. Mycologia 2018, 110, 1081–1109. [Google Scholar] [CrossRef]
- Cui, Y.Y.; Ding, X.X.; Kost, G.; Yang, Z.L. Tricholoma sect. Tricholoma (Tricholomataceae) from China: Molecular phylogeny and taxonomy. Mycol. Prog. 2022, 21, 35. [Google Scholar] [CrossRef]
- Yang, Z.L.; Ding, X.-X.; Kost, G.; Rexer, K.-H. New species in the Tricholoma pardinum complex from Eastern Himalaya. Phytotaxa 2017, 305, 1–10. [Google Scholar] [CrossRef]
- Aoki, W.; Endo, N.; Ushijima, S.; Nagai, H.; Ito, T.; Fukuda, M.; Yamada, A. Taxonomic revision of the Japanese Tricholoma ustale and closely related species based on molecular phylogenetic and morphological data. Mycoscience 2021, 62, MYC548. [Google Scholar] [CrossRef]
- Clericuzio, M.; Hussain, F.H.S.; Amin, H.I.M.; Salis, A.; Damonte, G.; Pavela, R.; Vidari, G. New acetylenic metabolites from the toxic mushroom Tricholoma pardinum. Nat. Prod. Res. 2021, 35, 5081–5088. [Google Scholar] [CrossRef]
- Clericuzio, M.; Mellerio, G.G.; Vita Finzi, P.; Vidari, G. Secondary metabolites isolated from Tricholoma species (Basidiomycota, Tricholomatacee): A review. Nat. Prod. Commun. 2018, 13, 1213–1224. [Google Scholar]
- Gilardoni, G.; Negri, F.; Vita Finzi, P.; Hussain, F.H.S.; Vidari, G. New tricholidic acid triterpenoids from the mushroom Tricholoma ustaloides collected in an Italian beech wood. Molecules 2023, 28, 3864. [Google Scholar] [CrossRef]
- Nugroho, A.E.; Morita, H. Circular dichroism calculation for natural products. J. Nat. Med. 2014, 68, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kirk, D.N. The chiroptical properties of carbonyl compounds. Tetrahedron 1986, 42, 777–818. [Google Scholar] [CrossRef]
- Zhang, F.-L.; Feng, T. Diterpenes specially produced by fungi: Structures, biological activities, and biosynthesis (2010–2020). J. Fungi 2022, 8, 244. [Google Scholar] [CrossRef]
- Breitmaier, E. Diterpenes. In Terpenes: Flavors, Fragrances, Pharmaca, Pheromones; Wiley-VCH: Berlin, Germany, 2006; Chapter 4; pp. 52–81. [Google Scholar] [CrossRef]
- Gamba Invernizzi, A.; Vidari, G.; Vita Finzi, P. Trichoaurantianolide A, a new diterpene with an unprecedented carbon skeleton from Tricholoma aurantium. Tetrahedron Lett. 1995, 36, 1905–1908. [Google Scholar] [CrossRef]
- Benevelli, F.; Carugo, O.; Gamba-Invernizzi, A.; Vidari, G. The structures of trichoaurantianolides B, C and D, Novel Diterpenes from Tricholoma aurantium. Tetrahedron Lett. 1995, 36, 3035–3038. [Google Scholar] [CrossRef]
- Knops, L.; Nieger, M.; Steffan, B.; Steglich, W. Novel diterpenoids from the toadstools T. aurantium and T. fracticum (Agaricales). Liebigs Ann. 1995, 1995, 77–80. [Google Scholar] [CrossRef]
- Williams, D.R.; Gladen, P.T.; Pinchman, J.R. Total synthesis of neodolastane trichoaurantanolides C and D. J. Org. Chem. 2015, 80, 5474–5493. [Google Scholar] [CrossRef]
- Tsukamoto, S.; Macabalang, A.D.; Nakatani, K.; Obara, Y.; Nakahata, N.; Ohta, T. Tricholomalides A-C, new neurotrophic diterpenes from the mushroom Tricholoma sp. J. Nat. Prod. 2003, 66, 1578–1581. [Google Scholar] [CrossRef]
- Wang, Z.; Min, S.-J.; Danishefsky, S.J. Total synthesis and structural revision of (±)-tricholomalides A and B. J. Am. Chem. Soc. 2009, 131, 10848–10849. [Google Scholar] [CrossRef]
- Hiersemann, M.; Helmboldt, H. Recent Progress in the total synthesis of dolabellane and dolastane diterpenes. Top. Curr. Chem. 2005, 243, 73–136. [Google Scholar]
- Corey, E.J.; Schmidt, G. Useful procedures for the oxidation of alcohols involving pyridinium dichromate in aprotic media. Tetrahedron Lett. 1979, 20, 399–402. [Google Scholar] [CrossRef]
- Mazur, X.; Becker, U.; Anke, T.; Sterner, O. Two new bioactive diterpenes from Lepista sordida. Phytochemistry 1996, 43, 405–407. [Google Scholar] [CrossRef]
Figure 1.
Specimens of Tricholoma ustaloides (A) collected in a beech wood (B) in Northern Apennines near Pavia, Italy (photos provided by Teresio Restelli and Alfredo Gatti).
Figure 1.
Specimens of Tricholoma ustaloides (A) collected in a beech wood (B) in Northern Apennines near Pavia, Italy (photos provided by Teresio Restelli and Alfredo Gatti).

Figure 2.
Structures of saponaceolides J (1) and F (2), tricholidic acid (3), tricholidic acids B (4) and C (5), and tricholomenyn C (6) isolated from Tricholoma ustaloides.
Figure 2.
Structures of saponaceolides J (1) and F (2), tricholidic acid (3), tricholidic acids B (4) and C (5), and tricholomenyn C (6) isolated from Tricholoma ustaloides.

Figure 3.
Structures of trichoaurantianolides A–D (7–10) and acetyl trichaurantin (11).

Figure 4.
A: octan projections of the “twisted” conformation of the cyclopentanone ring in trichoaurantianolide C (9); B: octan projection of ent-9.
Figure 4.
A: octan projections of the “twisted” conformation of the cyclopentanone ring in trichoaurantianolide C (9); B: octan projection of ent-9.


{kind=link}
{kind=link}
{kind=link}
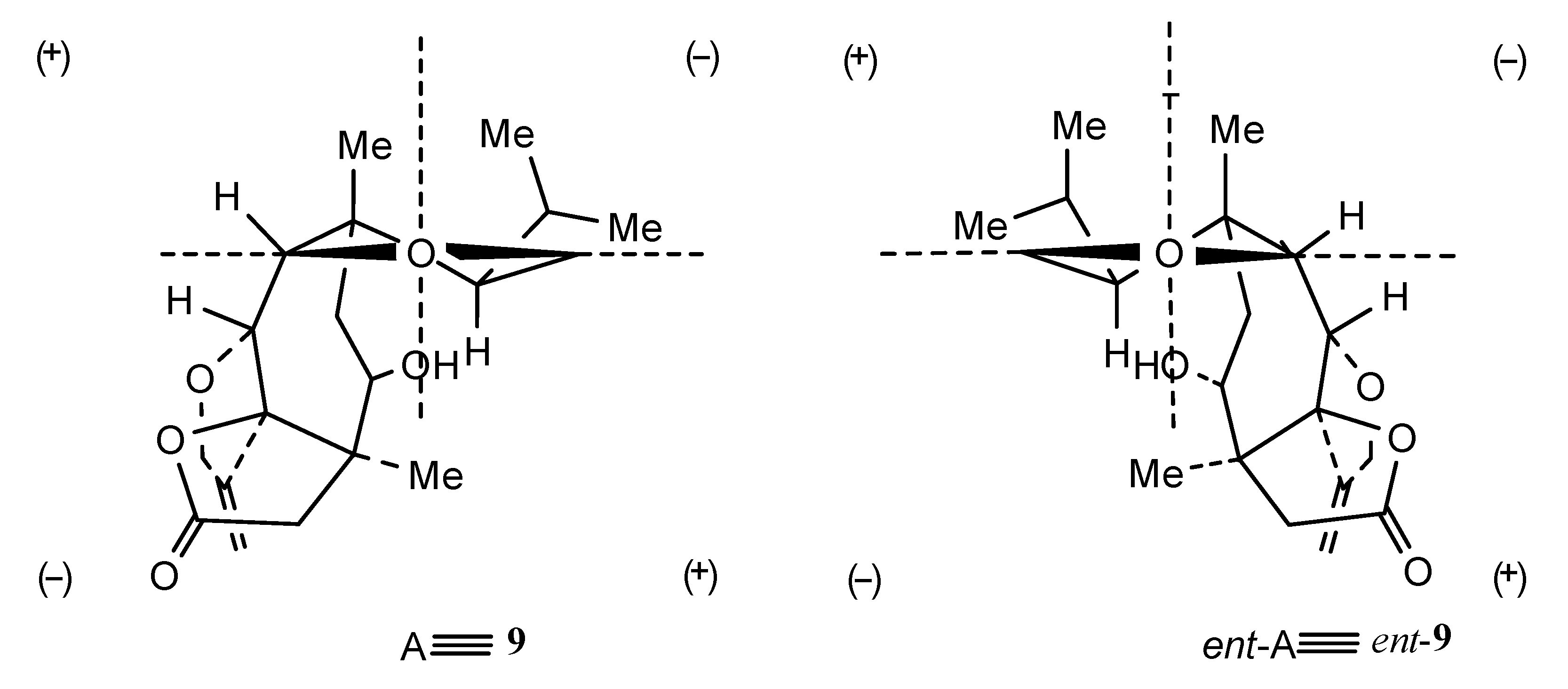{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 6.
Absolute configurations proposed for tricholomalides A–C.

Scheme 1.
Proposed biosynthetic pathways of trichoaurantianolides and tricholomalides.

Figure 7.
Structures of tricholomalides D–G (24–27).

Figure 8.
HMBC (H → C) correlations (A) and 1D NOE correlations (H → H) (B) observed for tricholomalide D (24).
Figure 8.
HMBC (H → C) correlations (A) and 1D NOE correlations (H → H) (B) observed for tricholomalide D (24).

Figure 9.
(A) HMBC (H → C) correlations; (B) 1D NOE correlations (H → H) observed for tricholomalide F (26); ![Molecules 28 07446 i001]() strong NOE effect;
strong NOE effect; ![Molecules 28 07446 i002]() weak NOE effect.
weak NOE effect.
 strong NOE effect;
strong NOE effect;  weak NOE effect.
weak NOE effect.
Figure 9.
(A) HMBC (H → C) correlations; (B) 1D NOE correlations (H → H) observed for tricholomalide F (26); ![Molecules 28 07446 i001]() strong NOE effect;
strong NOE effect; ![Molecules 28 07446 i002]() weak NOE effect.
weak NOE effect.
strong NOE effect; weak NOE effect.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Gilardoni, G.; Negri, F.; Vita Finzi, P.; Hussain, F.H.S.; Vidari, G. New Tricholomalides D–G from the Mushroom Tricholoma ustaloides Grown in an Italian Beech Wood. Molecules 2023, 28, 7446. https://doi.org/10.3390/molecules28217446
AMA Style
Gilardoni G, Negri F, Vita Finzi P, Hussain FHS, Vidari G. New Tricholomalides D–G from the Mushroom Tricholoma ustaloides Grown in an Italian Beech Wood. Molecules. 2023; 28(21):7446. https://doi.org/10.3390/molecules28217446
Chicago/Turabian StyleGilardoni, Gianluca, Francesca Negri, Paola Vita Finzi, Faiq H. S. Hussain, and Giovanni Vidari. 2023. "New Tricholomalides D–G from the Mushroom Tricholoma ustaloides Grown in an Italian Beech Wood" Molecules 28, no. 21: 7446. https://doi.org/10.3390/molecules28217446