(Phospho)proteomic Profiling of Microsatellite Unstable CRC Cells Reveals Alterations in Nuclear Signaling and Cholesterol Metabolism Caused by Frameshift Mutation of NMD Regulator UPF3A

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. Identification of NMD-Related Genes Harboring cMNRs
2.2. cMNR Frameshift Mutations in NMD-Associated Genes
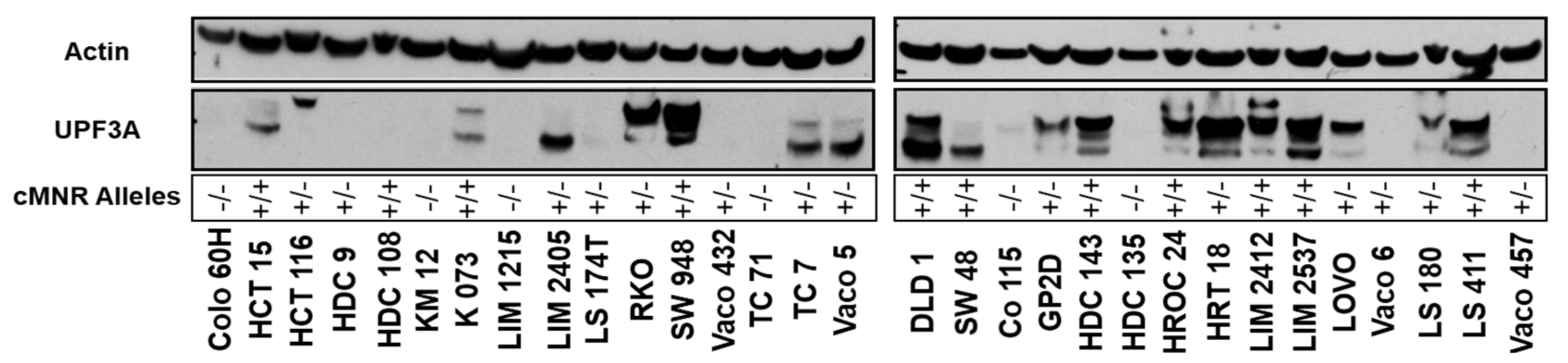
2.3. UPF3A Protein Expression in MSI Colorectal Cancer Cell Lines
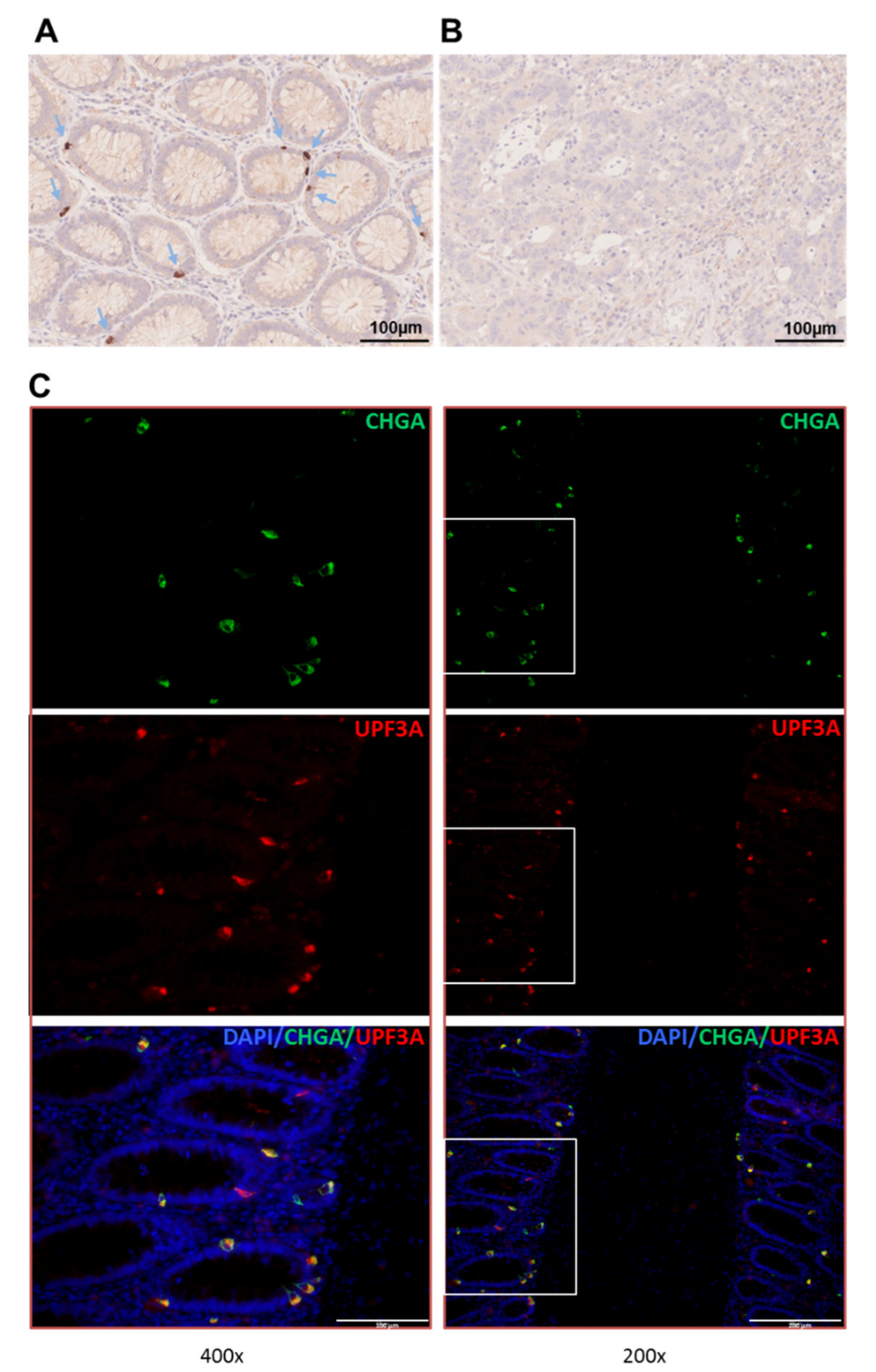
2.4. Expression of UPF3A in Normal Colon Epithelium and Colorectal Tumors
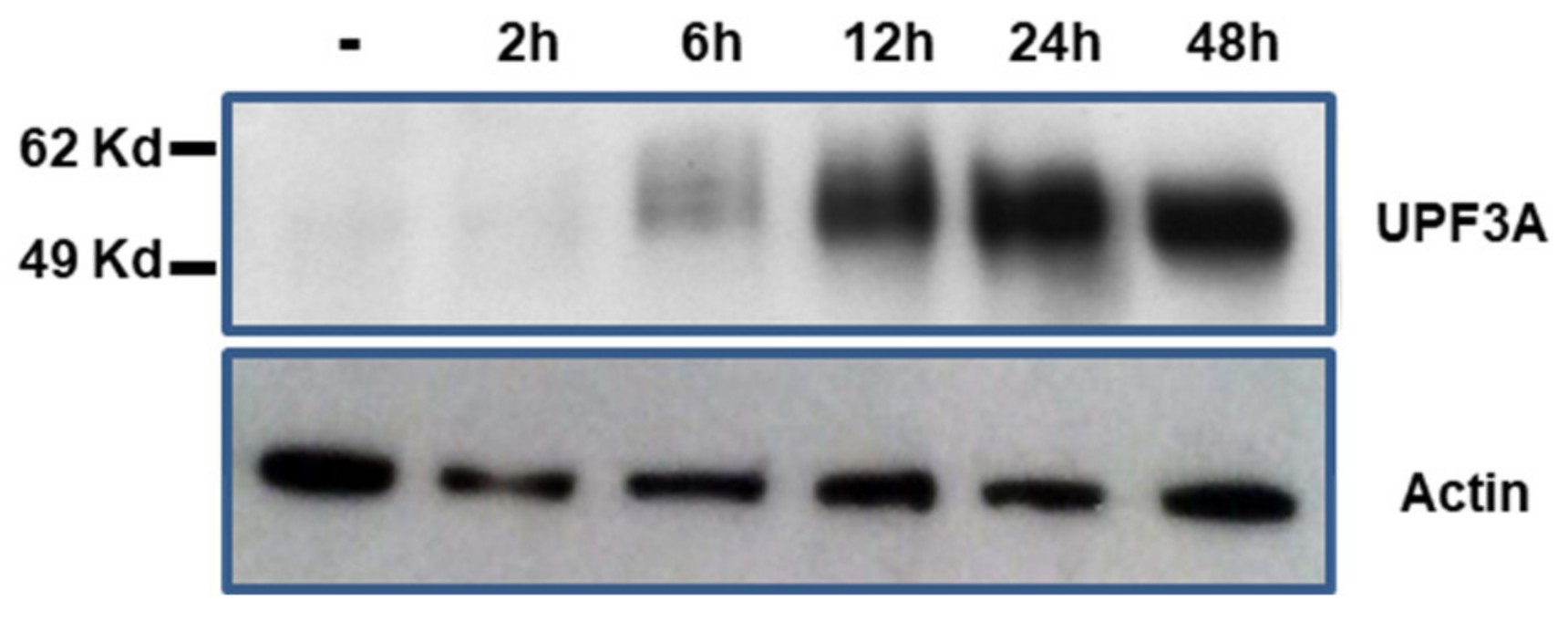
2.5. Characterization of KM12-UPF3A Model Cell Line
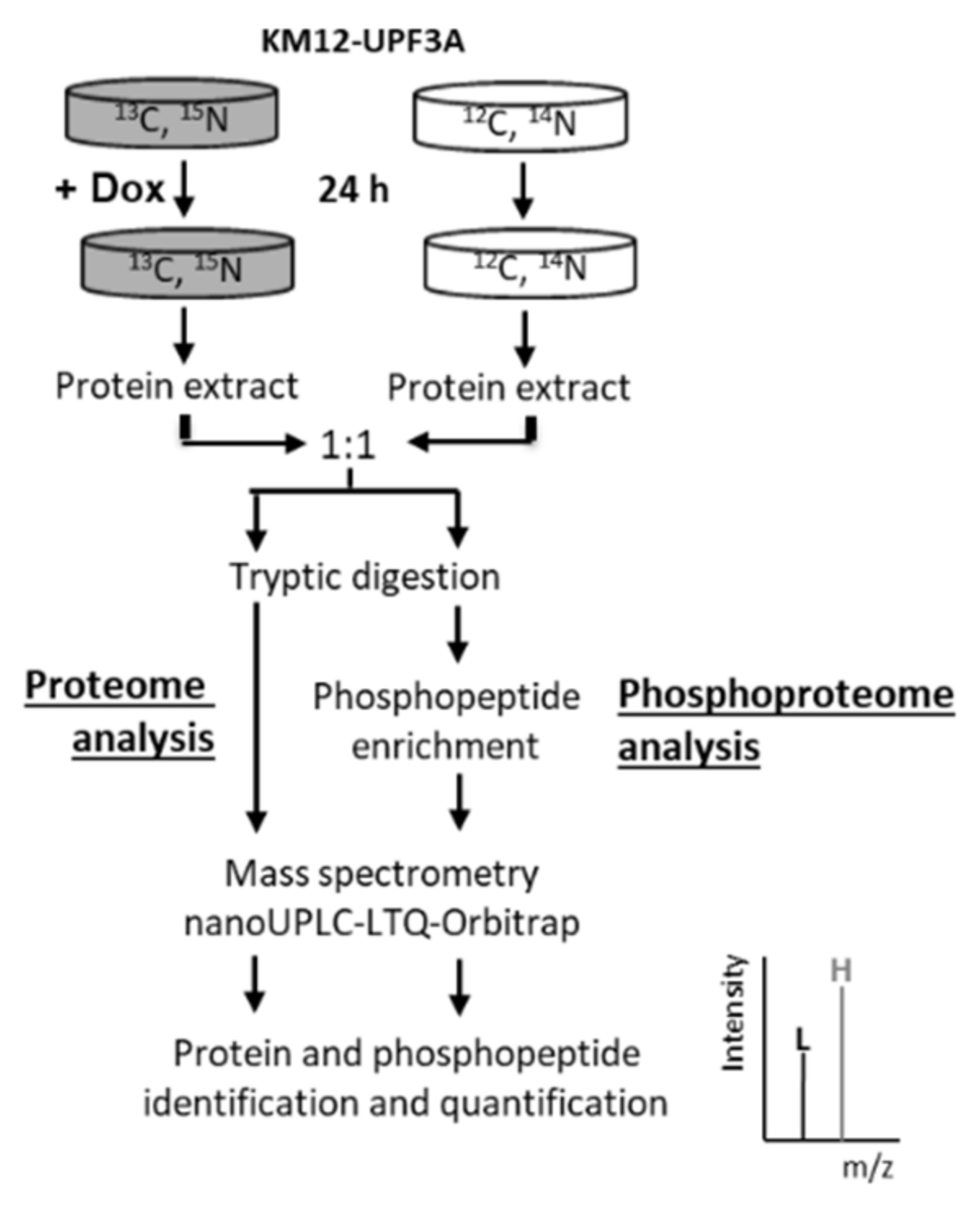
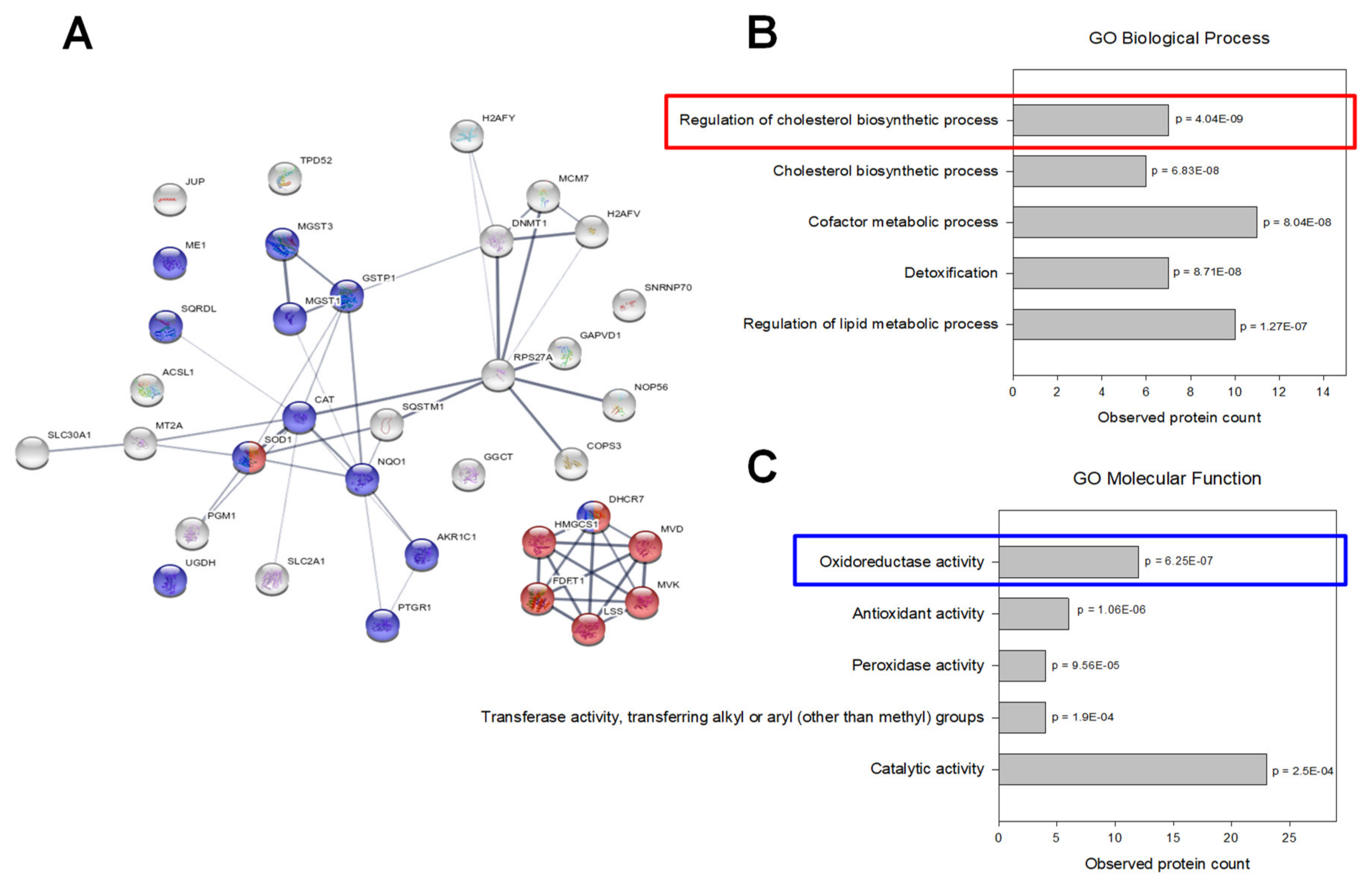
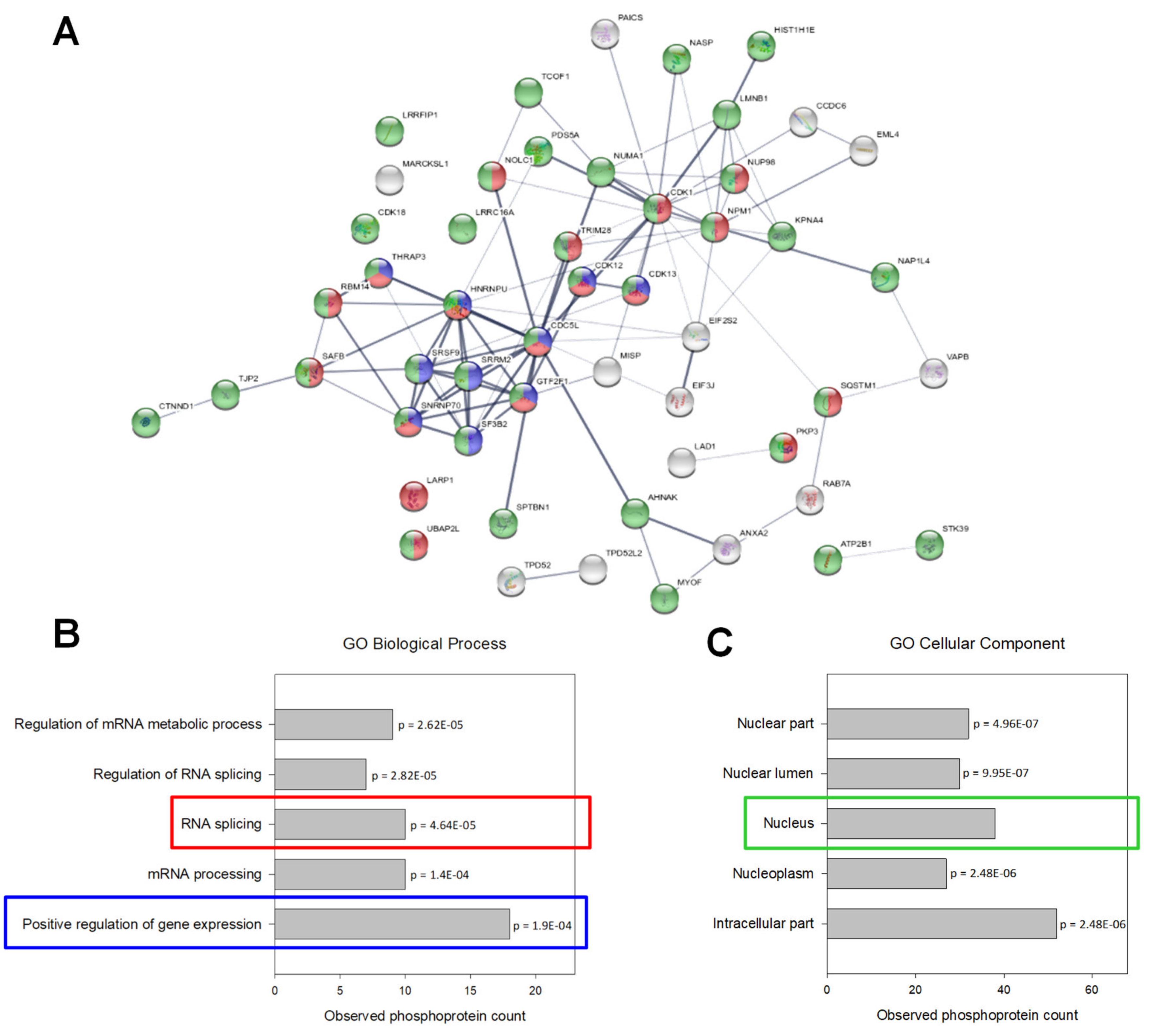
2.6. UPF3A Induces Proteomic and Phosphoproteomic Changes
3. Discussion
4. Materials and Methods
4.1. Database Analyses
4.2. Cancer Cell Lines and Human Tissue
4.3. Nucleic Acid Isolation, Analysis, and RT-PCR
4.4. Generation of a Dox-Inducible UPF3A Expression Plasmid
4.5. Stably Transfected Cells
4.6. Coding Microsatellite Frameshift Mutation Analysis
4.7. Western Blot Analysis
4.8. Immunohistochemistry
4.9. Immunofluorescence Staining and Imaging
4.10. SILAC Labeling and Protein Extraction
4.11. Tryptic Digestion
4.12. Phosphopeptide Enrichment
4.13. LC-MS/MS
4.14. Protein and Phosphopeptide Identification and Quantification
4.15. Data Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| cMNR | Coding mononucleotide repeat |
| CRC | Colorectal cancer |
| Dox | Doxycycline |
| dUPF3A | UPF3A-deficient |
| FTMS | Fourier-transform mass spectrometry |
| MMR | DNA mismatch repair |
| MS | Mass spectrometry |
| MSI | Microsatellite instability |
| MSS | Microsatellite stability |
| NMD | Nonsense-mediated RNA decay |
| PPI | Protein-protein interaction |
| PTC | Premature termination codon |
| pUPF3A | UPF3A-proficient |
| SILAC | Stable isotope labeling with amino acids in cell culture |
| UPLC | Ultra high-performance liquid chromatography |
| WT | Wild type |
References
- Buckowitz, A.; Knaebel, H.; Benner, A.; Bläker, H.; Gebert, J.; Kienle, P.; von Knebel Doeberitz, M.; Kloor, M. Microsatellite instability in colorectal cancer is associated with local lymphocyte infiltration and low frequency of distant metastases. Br. J. Cancer 2005, 92, 1746–1753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, H.T.; Smyrk, T.; Lynch, J.F. Molecular genetics and clinical-pathology features of hereditary nonpolyposis colorectal carcinoma (Lynch syndrome). Oncology 1998, 55, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Popat, S.; Hubner, R.; Houlston, R. Systematic review of microsatellite instability and colorectal cancer prognosis. J. Clin. Oncol. 2005, 23, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Woerner, S.M.; Gebert, J.; Yuan, Y.P.; Sutter, C.; Ridder, R.; Bork, P.; von Knebel Doeberitz, M. Systematic identification of genes with coding microsatellites mutated in DNA mismatch repair-deficient cancer cells. Int. J. Cancer 2001, 93, 12–19. [Google Scholar] [CrossRef]
- Mori, Y.; Yin, J.; Rashid, A.; Leggett, B.A.; Young, J.; Simms, L.; Kuehl, P.M.; Langenberg, P.; Meltzer, S.J.; Stine, O.C. Instabilotyping: Comprehensive identification of frameshift mutations caused by coding region microsatellite instability. Cancer Res. 2001, 61, 6046–6049. [Google Scholar]
- Duval, A.; Gayet, J.; Zhou, X.-P.; Iacopetta, B.; Thomas, G.; Hamelin, R. Frequent frameshift mutations of the TCF-4 gene in colorectal cancers with microsatellite instability. Cancer Res. 1999, 59, 4213–4215. [Google Scholar]
- Holbrook, J.A.; Neu-Yilik, G.; Hentze, M.W.; Kulozik, A.E. Nonsense-mediated decay approaches the clinic. Nat. Genet. 2004, 36, 801–808. [Google Scholar] [CrossRef]
- Isken, O.; Maquat, L.E. The multiple lives of NMD factors: Balancing roles in gene and genome regulation. Nat. Rev. Genet. 2008, 9, 699–712. [Google Scholar] [CrossRef] [Green Version]
- Kurosaki, T.; Popp, M.W.; Maquat, L.E. Quality and quantity control of gene expression by nonsense-mediated mRNA decay. Nat. Rev. Mol. Cell Biol. 2019, 20, 406–420. [Google Scholar] [CrossRef]
- Kunz, J.B.; Neu-Yilik, G.; Hentze, M.W.; Kulozik, A.E.; Gehring, N.H. Functions of hUpf3a and hUpf3b in nonsense-mediated mRNA decay and translation. RNA 2006, 12, 1015–1022. [Google Scholar] [CrossRef] [Green Version]
- Neu-Yilik, G.; Raimondeau, E.; Eliseev, B.; Yeramala, L.; Amthor, B.; Deniaud, A.; Huard, K.; Kerschgens, K.; Hentze, M.W.; Schaffitzel, C. Dual function of UPF3B in early and late translation termination. EMBO J. 2017, 36, 2968–2986. [Google Scholar] [CrossRef]
- Shum, E.Y.; Jones, S.H.; Shao, A.; Dumdie, J.; Krause, M.D.; Chan, W.-K.; Lou, C.-H.; Espinoza, J.L.; Song, H.-W.; Phan, M.H. The antagonistic gene paralogs Upf3a and Upf3b govern nonsense-mediated RNA decay. Cell 2016, 165, 382–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasif, S.; Contu, L.; Mühlemann, O. Beyond quality control: The role of nonsense-mediated mRNA decay (NMD) in regulating gene expression. Semin. Cell Dev. Biol. 2018, 75, 78–87. [Google Scholar] [CrossRef]
- Hall, G.; Thein, S. Nonsense codon mutations in the terminal exon of the beta-globin gene are not associated with a reduction in beta-mRNA accumulation: A mechanism for the phenotype of dominant beta-thalassemia. Blood 1994, 83, 2031–2037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarpey, P.S.; Raymond, F.L.; Nguyen, L.S.; Rodriguez, J.; Hackett, A.; Vandeleur, L.; Smith, R.; Shoubridge, C.; Edkins, S.; Stevens, C. Mutations in UPF3B, a member of the nonsense-mediated mRNA decay complex, cause syndromic and nonsyndromic mental retardation. Nat. Genet. 2007, 39, 1127–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Addington, A.; Gauthier, J.; Piton, A.; Hamdan, F.; Raymond, A.; Gogtay, N.; Miller, R.; Tossell, J.; Bakalar, J.; Germain, G. A novel frameshift mutation in UPF3B identified in brothers affected with childhood onset schizophrenia and autism spectrum disorders. Mol. Psychiatry 2011, 16, 238–239. [Google Scholar] [CrossRef]
- Chan, W.-K.; Bhalla, A.D.; Le Hir, H.; Nguyen, L.S.; Huang, L.; Gecz, J.; Wilkinson, M.F. A UPF3-mediated regulatory switch that maintains RNA surveillance. Nat. Struct. Mol. Biol. 2009, 16, 747–753. [Google Scholar] [CrossRef]
- Laumonnier, F.; Shoubridge, C.; Antar, C.; Nguyen, L.; Van Esch, H.; Kleefstra, T.; Briault, S.; Fryns, J.; Hamel, B.; Chelly, J. Mutations of the UPF3B gene, which encodes a protein widely expressed in neurons, are associated with nonspecific mental retardation with or without autism. Mol. Psychiatry 2010, 15, 767–776. [Google Scholar] [CrossRef] [Green Version]
- El-Bchiri, J.; Buhard, O.; Penard-Lacronique, V.; Thomas, G.; Hamelin, R.; Duval, A. Differential nonsense mediated decay of mutated mRNAs in mismatch repair deficient colorectal cancers. Human Mol. Genet. 2005, 14, 2435–2442. [Google Scholar] [CrossRef] [Green Version]
- El-Bchiri, J.; Guilloux, A.; Dartigues, P.; Loire, E.; Mercier, D.; Buhard, O.; Sobhani, I.; de la Grange, P.; Auboeuf, D.; Praz, F.; et al. Nonsense-Mediated mRNA Decay Impacts MSI-Driven Carcinogenesis and Anti-Tumor Immunity in Colorectal Cancers. PLoS ONE 2008, 3, e2583. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Karam, R.; Zhou, Y.; Su, F.; Ji, Y.; Li, G.; Xu, G.; Lu, L.; Wang, C.; Song, M. The UPF1 RNA surveillance gene is commonly mutated in pancreatic adenosquamous carcinoma. Nat. Med. 2014, 20, 596–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, L.; Li, C.; Guo, T.; Wang, H.; Ma, W.; Yuan, Y.; Liu, Q.; Ye, Q.; Liu, Z. The human RNA surveillance factor UPF1 regulates tumorigenesis by targeting Smad7 in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2016, 35, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popp, M.W.; Maquat, L.E. Nonsense-mediated mRNA decay and cancer. Curr. Opin. Genet. Dev. 2018, 48, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Woerner, S.M.; Benner, A.; Sutter, C.; Schiller, M.; Yuan, Y.P.; Keller, G.; Bork, P.; Doeberitz, M.V.K.; Gebert, J.F. Pathogenesis of DNA repair-deficient cancers: A statistical meta-analysis of putative Real Common Target genes. Oncogene 2003, 22, 2226–2235. [Google Scholar] [CrossRef] [Green Version]
- Su, A.I.; Wiltshire, T.; Batalov, S.; Lapp, H.; Ching, K.A.; Block, D.; Zhang, J.; Soden, R.; Hayakawa, M.; Kreiman, G.; et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. USA 2004, 101, 6062–6067. [Google Scholar] [CrossRef] [Green Version]
- Safran, M.; Dalah, I.; Alexander, J.; Rosen, N.; Iny Stein, T.; Shmoish, M.; Nativ, N.; Bahir, I.; Doniger, T.; Krug, H.; et al. GeneCards Version 3: The human gene integrator. Database 2010, 2010. [Google Scholar] [CrossRef]
- Woerner, S.M.; Yuan, Y.P.; Benner, A.; Korff, S.; von Knebel Doeberitz, M.; Bork, P. SelTarbase, a database of human mononucleotide-microsatellite mutations and their potential impact to tumorigenesis and immunology. Nucleic Acids Res. 2010, 38, 682–689. [Google Scholar] [CrossRef] [Green Version]
- Kloor, M.; Michel, S.; von Knebel Doeberitz, M. Immune evasion of microsatellite unstable colorectal cancers. Int. J. Cancer 2010, 127, 1001–1010. [Google Scholar] [CrossRef]
- Serin, G.; Gersappe, A.; Black, J.D.; Aronoff, R.; Maquat, L.E. Identification and Characterization of Human Orthologues to Saccharomyces cerevisiae Upf2 Protein and Upf3 Protein (Caenorhabditis elegans SMG-4). Mol. Cell. Biol. 2001, 21, 209–223. [Google Scholar] [CrossRef] [Green Version]
- Lykke-Andersen, J.; Shu, M.-D.; Steitz, J.A. Human Upf Proteins Target an mRNA for Nonsense-Mediated Decay When Bound Downstream of a Termination Codon. Cell 2000, 103, 1121–1131. [Google Scholar] [CrossRef] [Green Version]
- Gehring, N.H.; Neu-Yilik, G.; Schell, T.; Hentze, M.W.; Kulozik, A.E. Y14 and hUpf3b Form an NMD-Activating Complex. Mol. Cell 2003, 11, 939–949. [Google Scholar] [CrossRef]
- Jaffrey, S.R.; Wilkinson, M.F. Nonsense-mediated RNA decay in the brain: Emerging modulator of neural development and disease. Nat. Rev. Neurosci. 2018, 19, 715–728. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.S.; Wilkinson, M.F.; Gecz, J. Nonsense-mediated mRNA decay: Inter-individual variability and human disease. Neurosci. Biobehav. Rev. 2014, 46, 175–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamelgarn, M.; Chen, J.; Kuang, L.; Jin, H.; Kasarskis, E.J.; Zhu, H. ALS mutations of FUS suppress protein translation and disrupt the regulation of nonsense-mediated decay. Proc. Natl. Acad. Sci. USA 2018, 115, E11904–E11913. [Google Scholar] [CrossRef] [Green Version]
- Bohórquez, D.V.; Shahid, R.A.; Erdmann, A.; Kreger, A.M.; Wang, Y.; Calakos, N.; Wang, F.; Liddle, R.A. Neuroepithelial circuit formed by innervation of sensory enteroendocrine cells. J. Clin. Investig. 2015, 125, 782–786. [Google Scholar] [CrossRef] [Green Version]
- Siperstein, M.D. Role of cholesterogenesis and isoprenoid synthesis in DNA replication and cell growth. J. Lipid Res. 1984, 25, 1462–1468. [Google Scholar]
- Rao, K. The significance of the cholesterol biosynthetic pathway in cell growth and carcinogenesis. Anticancer Res. 1995, 15, 309–314. [Google Scholar]
- Coleman, P.S.; Lavietes, B.B.; Sabine, J.R. Membrane cholesterol, tumorigenesis, and the biochemical phenotype of neoplasia. Crit. Rev. Biochem. 1981, 11, 341–393. [Google Scholar] [CrossRef]
- Bathaie, S.Z.; Ashrafi, M.; Azizian, M.; Tamanoi, F. Mevalonate pathway and human cancers. Curr. Mol. Pharmacol. 2017, 10, 77–85. [Google Scholar] [PubMed]
- Gorin, A.; Gabitova, L.; Astsaturov, I. Regulation of cholesterol biosynthesis and cancer signaling. Curr. Opin. Pharmacol. 2012, 12, 710–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribas, V.; García-Ruiz, C.; Fernández-Checa, J.C. Mitochondria, cholesterol and cancer cell metabolism. Clin. Trans. Med. 2016, 5, 1–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Wang, L.; Jia, R. Role of de novo cholesterol synthesis enzymes in cancer. J. Cancer 2020, 11, 1761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuzu, O.F.; Noory, M.A.; Robertson, G.P. The Role of Cholesterol in Cancer. Cancer Res. 2016, 76, 2063–2070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, X.; Cheng, C.; Tan, Z.; Li, N.; Tang, M.; Yang, L.; Cao, Y. Emerging roles of lipid metabolism in cancer metastasis. Mol. Cancer 2017, 16, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caruso, M.G.; Notarnicola, M.; Bifulco, M.; Laezza, C.; Guerra, V.; Altomare, D.; Memeo, V.; Lorusso, D.; Demma, I.; Di Leo, A. Increased Farnesyltranferase Activity in Human Colorectal Cancer: Relationship with Clinicopathological Features and K-ras Mutation. Scand. J. Gastroenterol. 2003, 38, 80–85. [Google Scholar] [CrossRef]
- Caruso, M.G.; Notarnicola, M.; Santillo, M.; Cavallini, A.; Di, A.L. Enhanced 3-hydroxy-3-methyl-glutaryl coenzyme A reductase activity in human colorectal cancer not expressing low density lipoprotein receptor. Anticancer Res. 1999, 19, 451–454. [Google Scholar]
- Maione, F.; Oliaro-Bosso, S.; Meda, C.; Di Nicolantonio, F.; Bussolino, F.; Balliano, G.; Viola, F.; Giraudo, E. The cholesterol biosynthesis enzyme oxidosqualene cyclase is a new target to impair tumour angiogenesis and metastasis dissemination. Sci. Rep. 2015, 5, 9054. [Google Scholar] [CrossRef] [Green Version]
- Messa, C.; Notarnicola, M.; Russo, F.; Cavallini, A.; Pallottini, V.; Trentalance, A.; Bifulco, M.; Laezza, C.; Gabriella Caruso, M. Estrogenic regulation of cholesterol biosynthesis and cell growth in DLD-1 human colon cancer cells. Scand. J. Gastroenterol. 2005, 40, 1454–1461. [Google Scholar] [CrossRef]
- Notarnicola, M.; Messa, C.; Cavallini, A.; Bifulco, M.; Tecce, M.F.; Eletto, D.; Di Leo, A.; Montemurro, S.; Laezza, C.; Caruso, M.G. Higher farnesyl diphosphate synthase activity in human colorectal cancer inhibition of cellular apoptosis. Oncology 2004, 67, 351–358. [Google Scholar] [CrossRef]
- Bardou, M.; Barkun, A.; Martel, M. Effect of statin therapy on colorectal cancer. Gut 2010, 59, 1572–1585. [Google Scholar] [CrossRef] [Green Version]
- Murai, T. Cholesterol lowering: Role in cancer prevention and treatment. Biol. Chem. 2015, 396, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schackmann, R.C.; Tenhagen, M.; van de Ven, R.A.; Derksen, P.W. p120-catenin in cancer–mechanisms, models and opportunities for intervention. J. Cell Sci. 2013, 126, 3515–3525. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Yu, T.; Zhang, N.; Chen, J.; Zhang, P.; Li, S.; Luo, L.; Cui, Z.; Qin, Y.; Liu, F. Nuclear E-cadherin acetylation promotes colorectal tumorigenesis via enhancing β-catenin activity. Mol. Cancer Res. 2019, 17, 655–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhlen, M.; Oksvold, P.; Fagerberg, L.; Lundberg, E.; Jonasson, K.; Forsberg, M.; Zwahlen, M.; Kampf, C.; Wester, K.; Hober, S. Towards a knowledge-based human protein atlas. Nat. Biotechnol. 2010, 28, 1248–1250. [Google Scholar] [CrossRef] [PubMed]
- Kilpinen, S.; Autio, R.; Ojala, K.; Iljin, K.; Bucher, E.; Sara, H.; Pisto, T.; Saarela, M.; Skotheim, R.I.; Björkman, M. Systematic bioinformatic analysis of expression levels of 17,330 human genes across 9783 samples from 175 types of healthy and pathological tissues. Genome Biol. 2008, 9, R139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woerner, S.M.; Kloor, M.; Schwitalle, Y.; Youmans, H.; Doeberitz, M.V.K.; Gebert, J.; Dihlmann, S. The putative tumor suppressor AIM2 is frequently affected by different genetic alterations in microsatellite unstable colon cancers. Genes Chromosomes Cancer 2007, 46, 1080–1089. [Google Scholar] [CrossRef]
- Boland, C.R.; Thibodeau, S.N.; Hamilton, S.R.; Sidransky, D.; Eshleman, J.R.; Burt, R.W.; Meltzer, S.J.; Rodriguez-Bigas, M.A.; Fodde, R.; Ranzani, G.N.; et al. A National Cancer Institute Workshop on Microsatellite Instability for Cancer Detection and Familial Predisposition: Development of International Criteria for the Determination of Microsatellite Instability in Colorectal Cancer. Cancer Res. 1998, 58, 5248–5257. [Google Scholar]
- Findeisen, P.; Kloor, M.; Merx, S.; Sutter, C.; Woerner, S.M.; Dostmann, N.; Benner, A.; Dondog, B.; Pawlita, M.; Dippold, W.; et al. T25 Repeat in the 3′ Untranslated Region of the CASP2 Gene: A Sensitive and Specific Marker for Microsatellite Instability in Colorectal Cancer. Cancer Res. 2005, 65, 8072–8078. [Google Scholar] [CrossRef] [Green Version]
- Martinez, E.; Palhan, V.B.; Tjernberg, A.; Lymar, E.S.; Gamper, A.M.; Kundu, T.K.; Chait, B.T.; Roeder, R.G. Human STAGA Complex Is a Chromatin-Acetylating Transcription Coactivator That Interacts with Pre-mRNA Splicing and DNA Damage-Binding Factors in vivo. Mol. Cell. Biol. 2001, 21, 6782–6795. [Google Scholar] [CrossRef] [Green Version]
- Morikawa, K.; Walker, S.M.; Nakajima, M.; Pathak, S.; Jessup, J.M.; Fidler, I.J. Influence of Organ Environment on the Growth, Selection, and Metastasis of Human Colon Carcinoma Cells in Nude Mice. Cancer Res. 1988, 48, 6863–6871. [Google Scholar]
- Welman, A.; Barraclough, J.; Dive, C. Generation of cells expressing improved doxycycline-regulated reverse transcriptional transactivator rtTA2S-M2. Nat. Protoc. 2006, 1, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Gossen, M.; Bujard, H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. USA 1992, 89, 5547–5551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [PubMed]
- Michalak, M.; Warnken, U.; Schnölzer, M.; Gabius, H.J.; Kopitz, J. Detection of malignancy-associated phosphoproteome changes in human colorectal cancer induced by cell surface binding of growth-inhibitory galectin-4. IUBMB Life 2019, 71, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Kloor, M.; Sutter, C.; Wentzensen, N.; Cremer, F.W.; Buckowitz, A.; Keller, M.; von Knebel Doeberitz, M.; Gebert, J. A large MSH2 Alu insertion mutation causes HNPCC in a German kindred. Human Genet. 2004, 115, 432–438. [Google Scholar] [CrossRef]
- Katzenmaier, E.; Stark, H.-J.; Gebert, J.; Kopitz, J. Galectin-12 colocalizes with splicing factor-rich speckles and shuttles between the nucleus and cytoplasm in colon cancer cells. J. Mol. Biochem. 2018, 7, 28–40. [Google Scholar]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Michalak, M.; Warnken, U.; Andre, S.; Schnolzer, M.; Gabius, H.-J.; Kopitz, J. Detection of proteome changes in human colon cancer induced by cell surface binding of growth-inhibitory human galectin-4 using quantitative SILAC-based proteomics. J. Proteome Res. 2016, 15, 4412–4422. [Google Scholar] [CrossRef]
- Lößner, C.; Warnken, U.; Pscherer, A.; Schnölzer, M. Preventing arginine-to-proline conversion in a cell-line-independent manner during cell cultivation under stable isotope labeling by amino acids in cell culture (SILAC) conditions. Anal. Biochem. 2011, 412, 123–125. [Google Scholar] [CrossRef]
- Wessel, D.M.; Flügge, U. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 1984, 138, 141–143. [Google Scholar] [CrossRef]
- Rappsilber, J.; Mann, M.; Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007, 2, 1896. [Google Scholar] [CrossRef] [PubMed]
- Fricke, F.; Michalak, M.; Warnken, U.; Hausser, I.; Schnölzer, M.; Kopitz, J.; Gebert, J. SILAC-Based Quantification of TGFBR2-Regulated Protein Expression in Extracellular Vesicles of Microsatellite Unstable Colorectal Cancers. Int. J. Mol. Sci. 2019, 20, 4162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized ppb-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- Stohr, G.; Andreas, T. Quantitative LC-MS of Proteins. In Protein and Peptide Analysis by LC-MS: Experimental Strategies; Letzel, T., Ed.; Royal Society of Chemistry: London, UK, 2011. [Google Scholar]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P. STRING v11: Protein—Protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hugo ID | Role in NMD | cMNR | Position a | Mutated |
|---|---|---|---|---|
| SMG1 | UPF1 kinase | T7 | 1339 | 2/18 (11%) |
| T7 | 9018 | 0/19 | ||
| A7 | 10694 | 0/19 | ||
| SMG5 | Promotes UPF1 dephosphorylation | C7 | 12 | 0/19 |
| SMG7 | Promotes UPF1 dephosphorylation | A9 | 2273 | 3/19 (15%) |
| UPF2 | Binds UPF3A/B, recruits UPF1 | A7 | 355 | 0/16 |
| UPF3A | Binds EJC, recruits UPF2 | A7 | 486 b | 0/19 |
| A9 | 790 b | 18/23 (78%) | ||
| UPF3B | Binds EJC, recruits UPF2 | T7 | 244 | 0/19 |
| Tissue | Colorectal | Endometrial | Gastric | Urothelial | ||
|---|---|---|---|---|---|---|
| Stage | Cancer | Adenomas | Cell Lines | Cancer | Cancer | Cancer |
| Mut | 62/101 (61%) a 51/78 (65%) b | 15/27 (55%) | 21/31 (67%) a | 3/13 (23%) | 5/13 (38%) a | 1/11 (9%) |
| Biall. Mut | – | – | 6/31 (19%) | – | – | – |
| Proteins | Total # |
|---|---|
| Identified and quantified | 1298 |
| Regulated * | 35 |
| Phosphosites | |
| Identified and quantified | 2248 |
| Unknown § | 27 |
| Corrected for protein expression | 779 |
| Regulated sites (phosphoproteins) | 85 (52) |
| Known regulation § | 7 |
| Protein Name | Gene Name | Ratio pUPF3A/dUPF3A |
|---|---|---|
| GTPase-activating protein and VPS9 domain-containing protein 1 | GAPVD1 | 2.19 |
| DNA (cytosine-5)-methyltransferase 1 | DNMT1 | 1.88 |
| Tumor protein D52 | TPD52 | 1.70 |
| COP9 signalosome complex subunit 3 | COPS3 | 1.65 |
| U1 small nuclear ribonucleoprotein 70 kDa | SNRNP70 | 1.64 |
| DNA replication licensing factor MCM7 | MCM7 | 1.49 |
| Nucleolar protein 56 | NOP56 | −1.51 |
| Phosphoglucomutase-1 | PGM1 | −1.51 |
| Superoxide dismutase [Cu-Zn] | SOD1 | −1.53 |
| NAD(P)H dehydrogenase [quinone] 1 | NQO1 | −1.56 |
| Microsomal glutathione S-transferase 1 | MGST1 | −1.56 |
| Core histone macro-H2A.1 | H2AFY | −1.60 |
| UDP-glucose 6-dehydrogenase | UGDH | −1.61 |
| Sulfide:quinone oxidoreductase. mitochondrial | SQRDL | −1.67 |
| Glutathione S-transferase P | GSTP1 | −1.67 |
| Hydroxymethylglutaryl-CoA synthase. cytoplasmic | HMGCS1 | −1.71 |
| Mevalonate kinase | MVK | −1.74 |
| NADP-dependent malic enzyme | ME1 | −1.75 |
| Solute carrier family 2. facilitated glucose transporter member 1 | SLC2A1 | −1.80 |
| Ubiquitin-40S ribosomal protein S27a | RPS27A | −1.80 |
| Catalase | CAT | −1.80 |
| Microsomal glutathione S-transferase 3 | MGST3 | −1.80 |
| Diphosphomevalonate decarboxylase | MVD | −1.87 |
| Lanosterol synthase | LSS | −1.88 |
| Gamma-glutamylcyclotransferase | GGCT | −1.90 |
| 7-dehydrocholesterol reductase | DHCR7 | −1.96 |
| Long-chain-fatty-acid--CoA ligase 1 | ACSL1 | −1.97 |
| Histone H2A.V | H2AFV | −2.00 |
| Zinc transporter 1 | SLC30A1 | −2.03 |
| Prostaglandin reductase 1 | PTGR1 | −2.11 |
| Squalene synthase | FDFT1 | −2.52 |
| Aldo-keto reductase family 1 member C1; | AKR1C1 | −2.90 |
| Sequestosome-1 | SQSTM1 | −3.37 |
| Junction plakoglobin | JUP | −4.41 |
| Metallothionein-2 | MT2A | −5.88 |
| Protein Name | Gene Name | Phosphosite | Ratio pUPF3A/dUPF3A |
|---|---|---|---|
| Lamin-B1 * | LMNB1 | T20, S23 | 2.39 |
| Nucleophosmin | NPM1 | S254 | 2.35 |
| Catenin delta-1 § | CTNND1 | S349 | 2.15 |
| Cell division cycle 5-like protein | CDC5L | S303 | 2.05 |
| Multifunctional protein ADE2; Phosphoribosylaminoimidazole-succinocarboxamide synthase | PAICS | S27 | 2.01 |
| Neuroblast differentiation-associated protein AHNAK | AHNAK | S5763 | 1.96 |
| General transcription factor IIF subunit 1 | GTF2F1 | S224 | 1.95 |
| Importin subunit alpha-3 | KPNA4 | S60 | 1.88 |
| MARCKS-related protein | MARCKSL1 | S104 | 1.84 |
| Sister chromatid cohesion protein PDS5 homolog A | PDS5A | S1206 | 1.77 |
| Serine/arginine repetitive matrix protein 2 | SRRM2 | S377 | −2.27 |
| Tumor protein D52 | TPD52 | S223 | −2.30 |
| Tumor protein D54 | TPD52L2 | S19 | −2.71 |
| Serine/arginine repetitive matrix protein 2 * | SRRM2 | S876 | −2.90 |
| RNA-binding protein 14 | RBM14 | S618 | −3.20 |
| Serine/arginine-rich splicing factor 9 | SRSF9 | S211, S216 | −3.41 |
| Nuclear mitotic apparatus protein 1 | NUMA1 | S1969 | −3.43 |
| Catenin delta-1 | CTNND1 | S230 | −3.58 |
| Catenin delta-1 § | CTNND1 | S346, S349, S352 | −3.74 |
| Ras-related protein Rab-7a | RAB7A | S72 | −5.34 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michalak, M.; Katzenmaier, E.-M.; Roeckel, N.; Woerner, S.M.; Fuchs, V.; Warnken, U.; Yuan, Y.P.; Bork, P.; Neu-Yilik, G.; Kulozik, A.; et al. (Phospho)proteomic Profiling of Microsatellite Unstable CRC Cells Reveals Alterations in Nuclear Signaling and Cholesterol Metabolism Caused by Frameshift Mutation of NMD Regulator UPF3A. Int. J. Mol. Sci. 2020, 21, 5234. https://doi.org/10.3390/ijms21155234
Michalak M, Katzenmaier E-M, Roeckel N, Woerner SM, Fuchs V, Warnken U, Yuan YP, Bork P, Neu-Yilik G, Kulozik A, et al. (Phospho)proteomic Profiling of Microsatellite Unstable CRC Cells Reveals Alterations in Nuclear Signaling and Cholesterol Metabolism Caused by Frameshift Mutation of NMD Regulator UPF3A. International Journal of Molecular Sciences. 2020; 21(15):5234. https://doi.org/10.3390/ijms21155234
Chicago/Turabian StyleMichalak, Malwina, Eva-Maria Katzenmaier, Nina Roeckel, Stefan M. Woerner, Vera Fuchs, Uwe Warnken, Yan P. Yuan, Peer Bork, Gabriele Neu-Yilik, Andreas Kulozik, and et al. 2020. "(Phospho)proteomic Profiling of Microsatellite Unstable CRC Cells Reveals Alterations in Nuclear Signaling and Cholesterol Metabolism Caused by Frameshift Mutation of NMD Regulator UPF3A" International Journal of Molecular Sciences 21, no. 15: 5234. https://doi.org/10.3390/ijms21155234