
Troponin Variants in Congenital Myopathies: How They Affect Skeletal Muscle Mechanics


Abstract
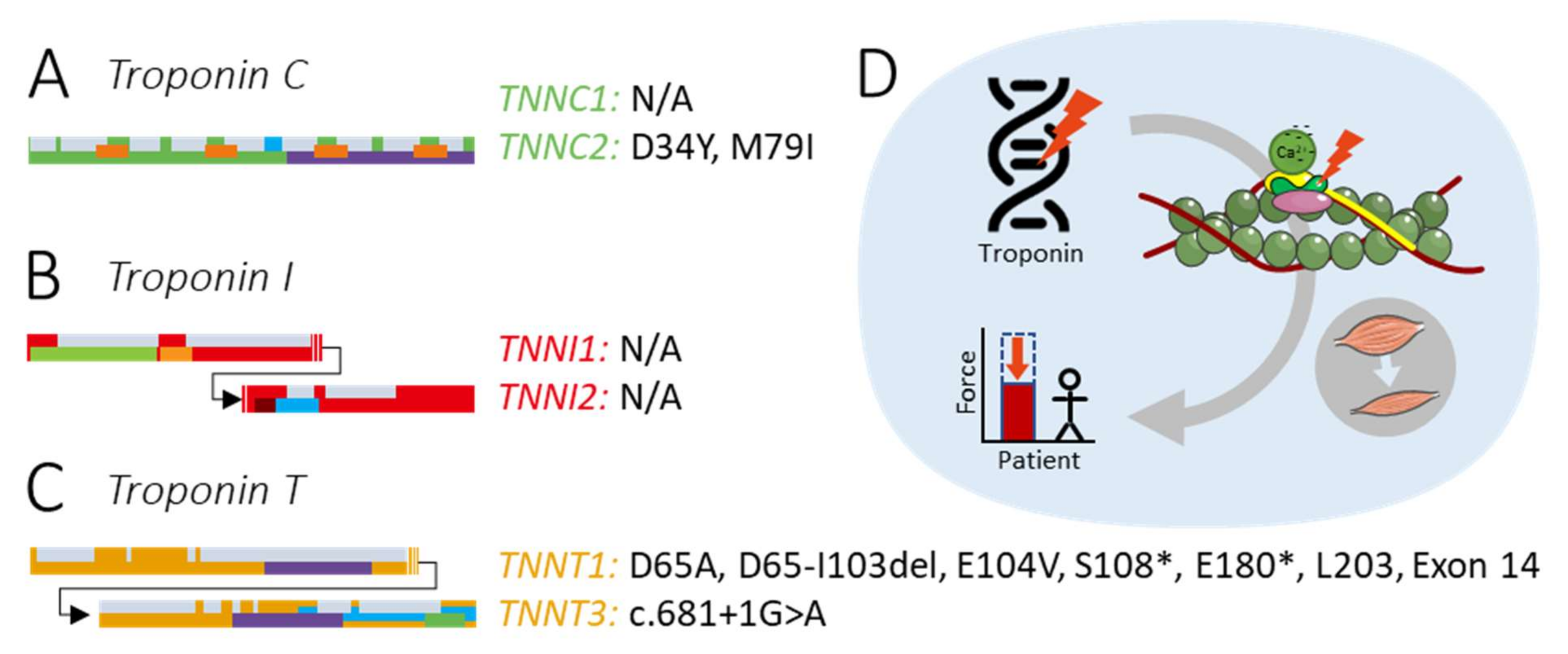
:1. Introduction
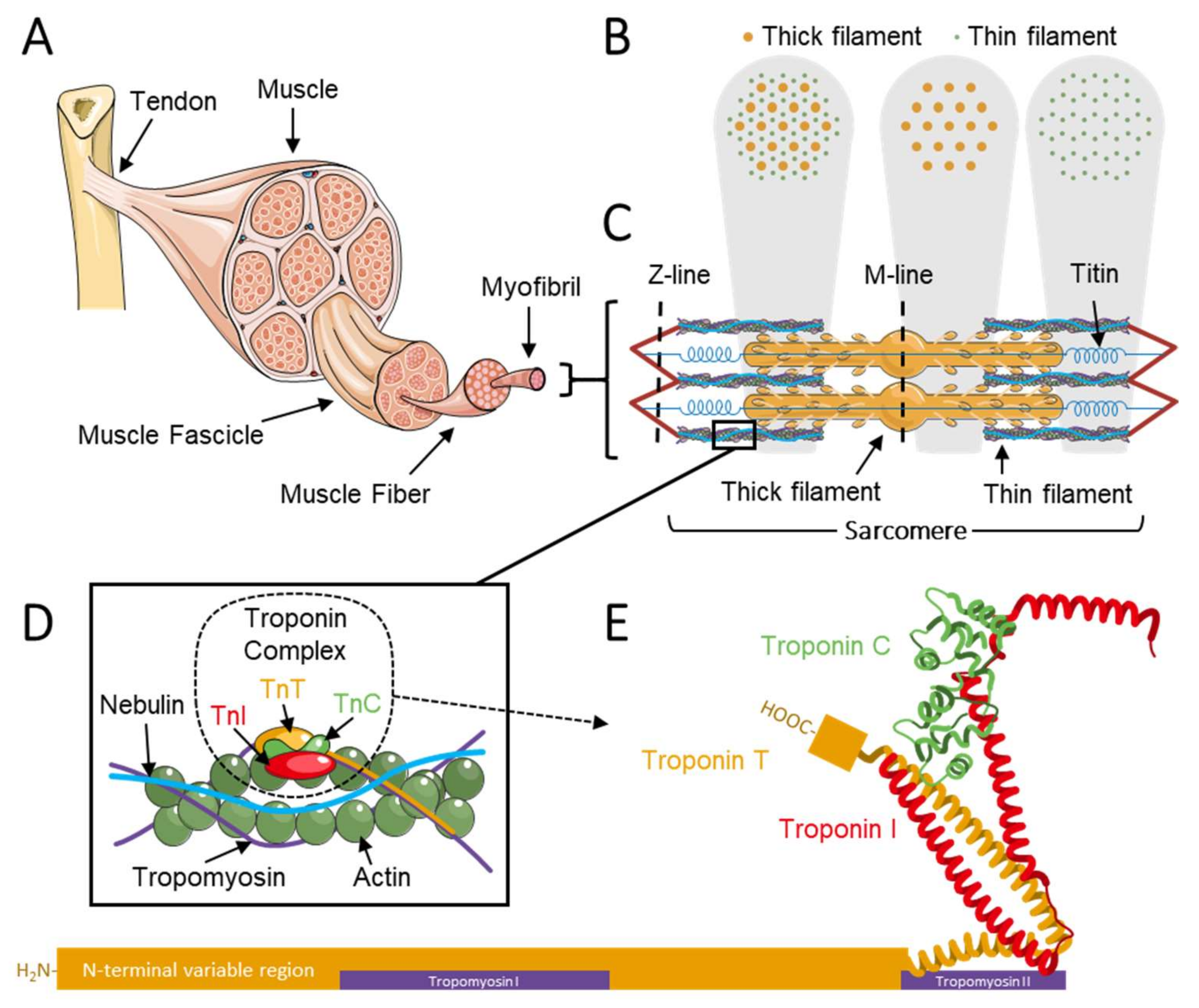
1.1. The Troponin Complex Regulates Muscle Contraction
1.2. Troponin in Congenital Myopathies
2. The (Patho)Physiology of Troponin C
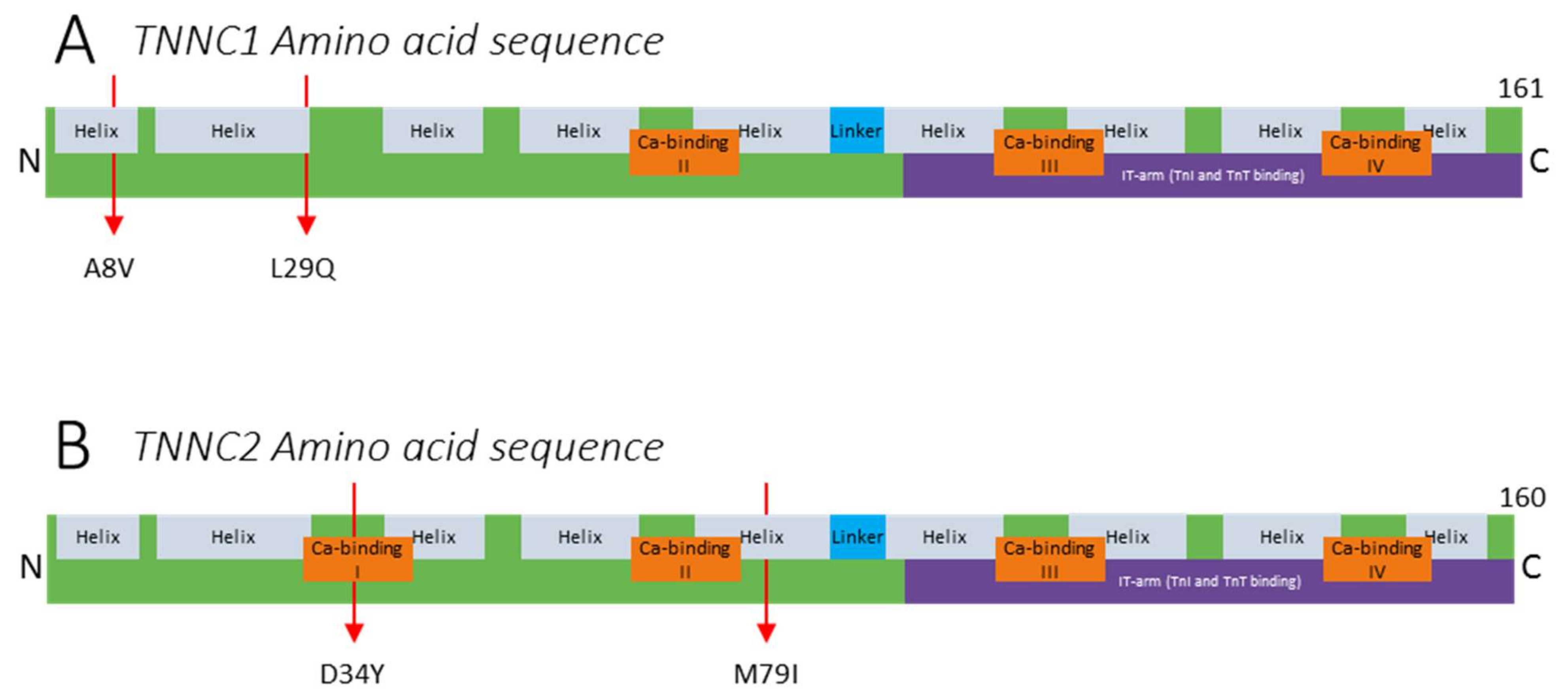
2.1. Variants in TNNC1
2.2. Variants in TNNC2
3. The (Patho)Physiology of Troponin I
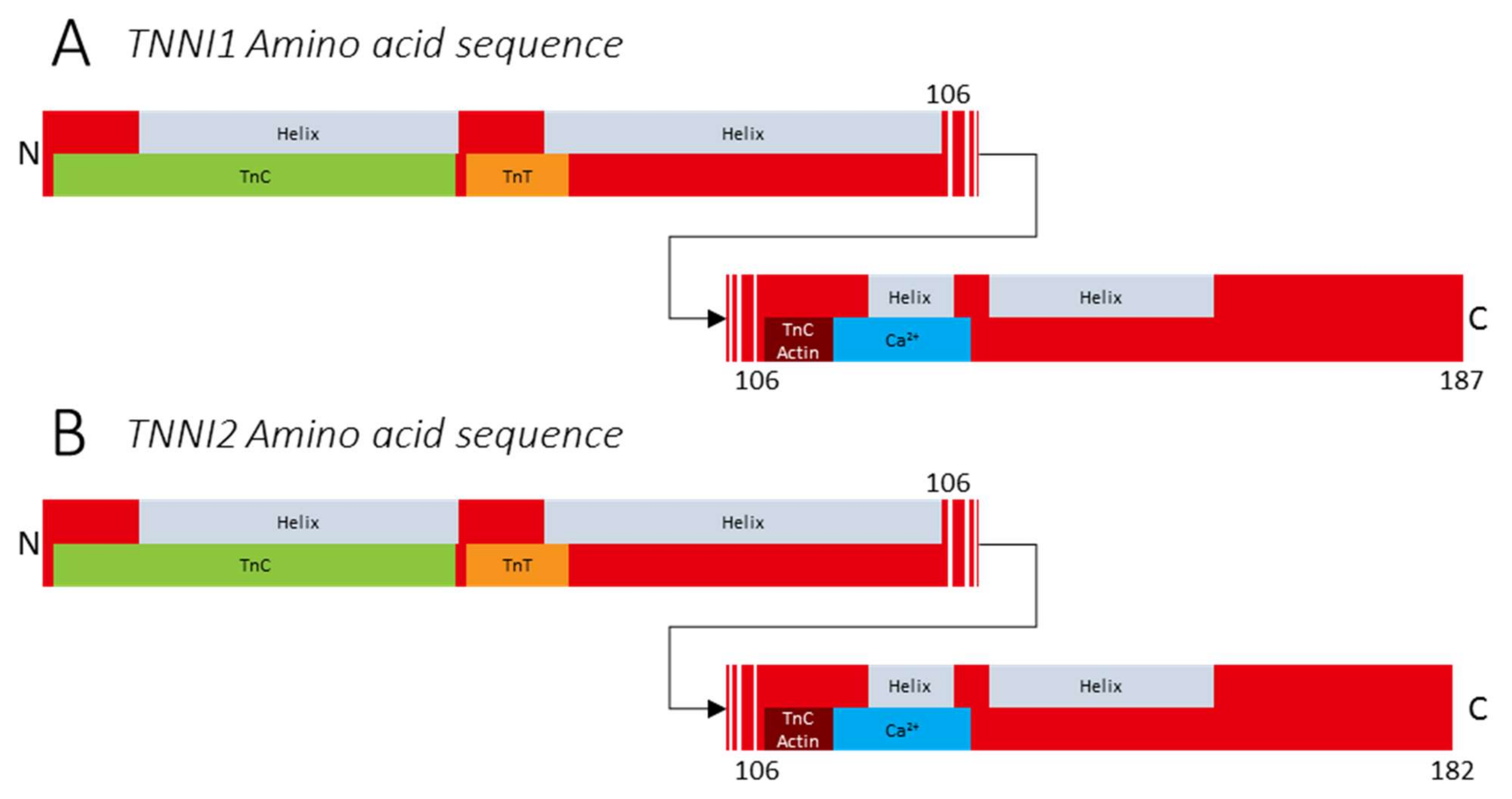
3.1. Variants in TNNI1
3.2. Variants in TNNI2
4. The (Patho)Physiology of Troponin T
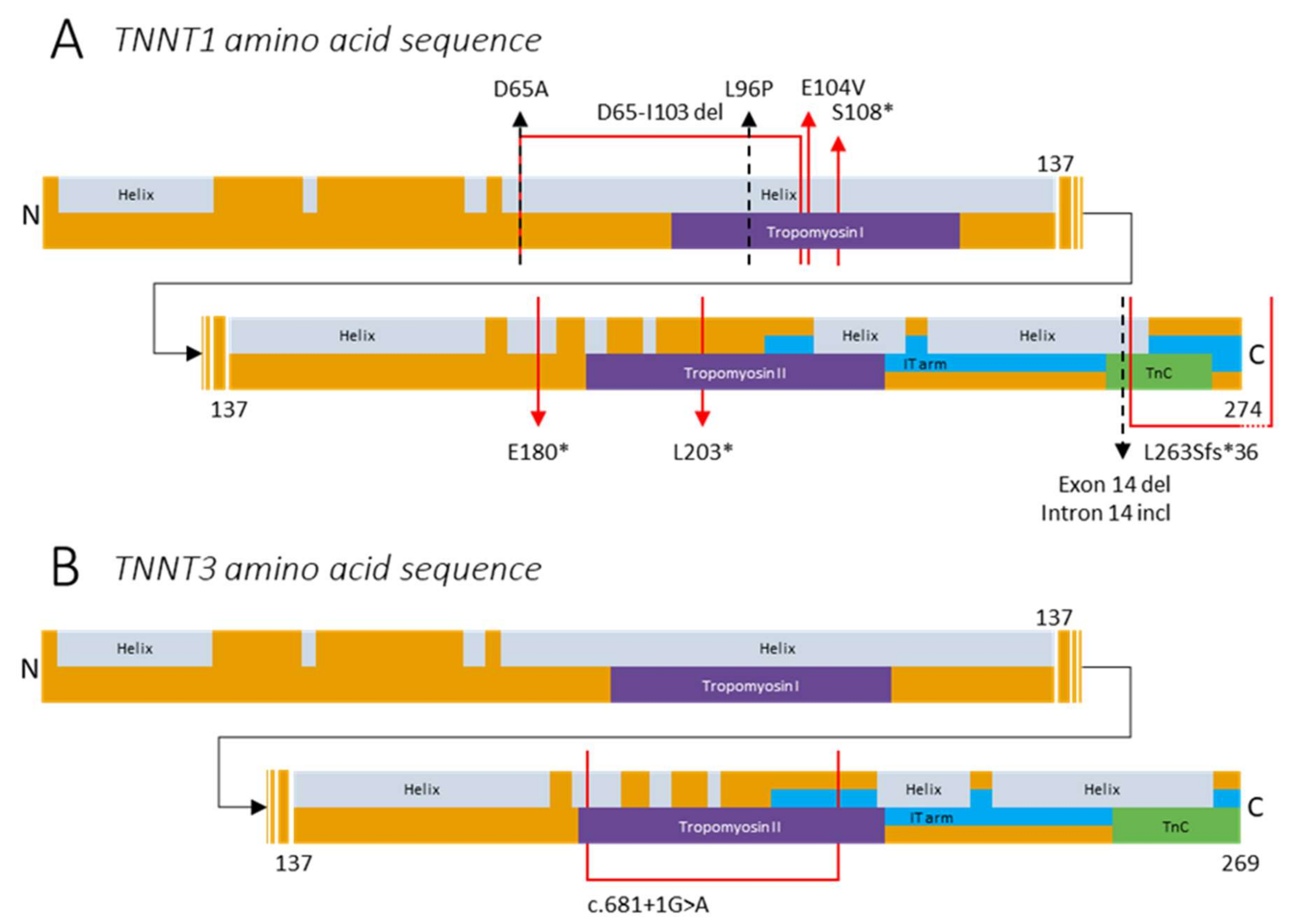
4.1. Variants in TNNT1
4.2. Variants in TNNT3
5. Therapeutic Strategies
5.1. Troponin Activators
5.2. AAV Virus
5.3. Myosin Modulation
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Szczesna, D.; Potter, J.D. The role of troponin in the Ca2+-regulation of skeletal muscle contraction. Mol. Interact. Actin 2002, 36, 171–190. [Google Scholar] [CrossRef]
- Kuo, I.; Ehrlich, B.E. Signaling in muscle contraction. Cold Spring Harb. Perspect. Biol. 2015, 7, a006023. [Google Scholar] [CrossRef] [PubMed]
- Zot, A.S.; Potter, J.D. Structural aspects of troponin-tropomyosin regulation of skeletal muscle contraction. Annu. Rev. Biophys. Biophys. Chem. 1987, 16, 535–559. [Google Scholar] [CrossRef]
- Murakami, U.; Uchida, K. Contents of myofibrillar proteins in cardiac, skeletal, and smooth muscles. J. Biochem. 1985, 98, 187–197. [Google Scholar] [CrossRef]
- Yates, L.D.; Greaser, M.L.; Huxley, H. Quatitative determination of myosin and actin in rabbit skeletal muscle. J. Mol. Biol. 1983, 168, 123–141. [Google Scholar] [CrossRef]
- Gomes, A.V.; Potter, J.D.; Szczesna-Cordary, D. The role of troponins in muscle contraction. IUBMB Life 2002, 54, 323–333. [Google Scholar] [CrossRef]
- Solaro, R.J. Troponin C—Troponin I interactions and molecular signalling in cardiac myofilaments. Mol. Subcell. Cardiol. 1995, 382, 109–115. [Google Scholar] [CrossRef]
- Chandra, M.; Montgomery, D.E.; Kim, J.J.; Solaro, R. The N-terminal region of troponin T is essential for the maximal activation of rat cardiac myofilaments. J. Mol. Cell. Cardiol. 1999, 31, 867–880. [Google Scholar] [CrossRef] [PubMed]
- Grabarek, Z. Structural basis for diversity of the EF-hand calcium-binding proteins. J. Mol. Biol. 2006, 359, 509–525. [Google Scholar] [CrossRef]
- Marston, S.; Zamora, J.E. Troponin structure and function: A view of recent progress. J. Muscle Res. Cell Motil. 2019, 41, 71–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geeves, M.A.; Holmes, K.C. Structural mechanism of muscle contraction. Annu. Rev. Biochem. 1999, 68, 687–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Au, Y. The muscle ultrastructure: A structural perspective of the sarcomere. Cell. Mol. Life Sci. 2004, 61, 3016–3033. [Google Scholar] [CrossRef]
- Cooper, G.M. Actin, myosin, and cell movement. In The Cell: A Molecular Approach, 2nd ed.; Cooper, G.M., Ed.; American Society of Microbiology: Washington, DC, USA, 2000. Available online: http://www.ncbi.nlm.nih.gov/books/NBK9961/ (accessed on 4 June 2020).
- Farah, C.S.; Reinach, F.C. The troponin complex and regulation of muscle contraction. FASEB J. 1995, 9, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Reggiani, C. Molecular diversity of myofibrillar proteins: Gene regulation and functional significance. Physiol. Rev. 1996, 76, 371–423. [Google Scholar] [CrossRef]
- Wilkinson, J.M. Troponin C from rabbit slow skeletal and cardiac muscle is the product of a single gene. JBIC J. Biol. Inorg. Chem. 1980, 103, 179–188. [Google Scholar] [CrossRef]
- Schreier, T.; Kedes, L.; Gahlmann, R. Cloning, structural analysis, and expression of the human slow twitch skeletal muscle/cardiac troponin C gene. J. Biol. Chem. 1990, 265, 21247–21253. [Google Scholar] [CrossRef]
- Gahlmann, R.; Kedes, L. Cloning, structural analysis, and expression of the human fast twitch skeletal muscle troponin C gene. J. Biol. Chem. 1990, 265, 12520–12528. [Google Scholar] [CrossRef]
- Sheng, J.-J.; Jin, J.-P. TNNI1, TNNI2 and TNNI3: Evolution, regulation, and protein structure-function relationships. Gene 2015, 576, 385–394. [Google Scholar] [CrossRef] [Green Version]
- Wei, B.; Jin, J.-P. TNNT1, TNNT2, and TNNT3: Isoform genes, regulation, and structure–function relationships. Gene 2016, 582, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Staron, R.S. Human skeletal muscle fiber types: Delineation, development, and distribution. Can. J. Appl. Physiol. 1997, 22, 307–327. [Google Scholar] [CrossRef]
- Demer, J.L. The orbital pulley system: A revolution in concepts of orbital anatomy. Ann. N. Y. Acad. Sci. 2002, 956, 17–32. [Google Scholar] [CrossRef]
- Lennerstrand, G. Strabismus and eye muscle function. Acta Ophthalmol. Scand. 2007, 85, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Katrukha, I.A.; Gusev, N.B. Enigmas of cardiac troponin T phosphorylation. J. Mol. Cell. Cardiol. 2013, 65, 156–158. [Google Scholar] [CrossRef] [PubMed]
- Li, M.X.; Hwang, P.M. Structure and function of cardiac troponin C (TNNC1): Implications for heart failure, cardiomyopathies, and troponin modulating drugs. Gene 2015, 571, 153–166. [Google Scholar] [CrossRef] [Green Version]
- Ryan, M.M.; Schnell, C.; Strickland, C.D.; Shield, L.K.; Morgan, G.; Iannaccone, S.T.; Laing, N.G.; Beggs, A.H.; North, K.N. Nemaline myopathy: A clinical study of 143 cases. Ann. Neurol. 2001, 50, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Shieh, P.B. Muscular dystrophies and other genetic myopathies. Neurol. Clin. 2013, 31, 1009–1029. [Google Scholar] [CrossRef]
- North, K.N.; Wang, C.H.; Clarke, N.; Jungbluth, H.; Vainzof, M.; Dowling, J.J.; Amburgey, K.; Quijano-Roy, S.; Beggs, A.; Sewry, C.; et al. Approach to the diagnosis of congenital myopathies. Neuromuscul. Disord. 2014, 24, 97–116. [Google Scholar] [CrossRef] [Green Version]
- De Winter, J.M.; Ottenheijm, C.A. Sarcomere dysfunction in nemaline myopathy. J. Neuromuscul. Dis. 2017, 4, 99–113. [Google Scholar] [CrossRef] [Green Version]
- Jungbluth, H.; Treves, S.; Zorzato, F.; Sarkozy, A.; Ochala, J.; Sewry, C.; Phadke, R.; Gautel, M.; Muntoni, F. Congenital myopathies: Disorders of excitation-contraction coupling and muscle contraction. Nat. Rev. Neurol. 2018, 14, 151–167. [Google Scholar] [CrossRef]
- Colombo, I.; Scoto, M.; Manzur, A.Y.; Robb, S.A.; Maggi, L.; Gowda, V.; Cullup, T.; Yau, M.; Phadke, R.; Sewry, C.; et al. Congenital myopathies: Natural history of a large pediatric cohort. Neurology 2014, 84, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Clarkson, E.; Costa, C.F.; Machesky, L.M. Congenital myopathies: Diseases of the actin cytoskeleton. J. Pathol. 2004, 204, 407–417. [Google Scholar] [CrossRef]
- Cassandrini, D.; Rodolico, C.; Trovato, R.; Rubegni, A.; Lenzi, S.; Fiorillo, C.; Baldacci, J.; Minetti, C.; Astrea, G.; Bruno, C.; et al. Congenital myopathies: Clinical phenotypes and new diagnostic tools. Ital. J. Pediatr. 2017, 43, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Tubridy, N.; Fontaine, B.; Eymard, B. Congenital myopathies and congenital muscular dystrophies. Curr. Opin. Neurol. 2001, 14, 575–582. [Google Scholar] [CrossRef]
- Winter, J.M.D.; Joureau, B.; Lee, E.-J.; Kiss, B.; Yuen, M.; Gupta, V.A.; Pappas, C.; Gregorio, C.C.; Stienen, G.; Edvardson, S.; et al. Mutation-specific effects on thin filament length in thin filament myopathy. Ann. Neurol. 2016, 79, 959–969. [Google Scholar] [CrossRef] [Green Version]
- Ochala, J. Thin filament proteins mutations associated with skeletal myopathies: Defective regulation of muscle contraction. J. Mol. Med. 2008, 86, 1197–1204. [Google Scholar] [CrossRef]
- Lewit-Bentley, A.; Réty, S. EF-hand calcium-binding proteins. Curr. Opin. Struct. Biol. 2000, 10, 637–643. [Google Scholar] [CrossRef]
- Gifford, J.L.; Walsh, M.P.; Vogel, H.J. Structures and metal-ion-binding properties of the Ca2+-binding helix-loop-helix EF-hand motifs. Biochem. J. 2007, 405, 199–221. [Google Scholar] [CrossRef] [PubMed]
- Dyer, E.C.; Jacques, A.M.; Hoskins, A.C.; Ward, D.G.; Gallon, C.E.; Messer, A.E.; Kaski, J.P.; Burch, M.; Kentish, J.C.; Marston, S.B. Functional analysis of a unique troponin C mutation, GLY159ASP, that causes familial dilated cardiomyopathy, studied in explanted heart muscle. Circ. Heart Fail. 2009, 2, 456–464. [Google Scholar] [CrossRef] [Green Version]
- Putkey, J.A.; Sweeney, H.L.; Campbell, S.T. Site-directed mutation of the trigger calcium-binding sites in cardiac troponin C. J. Biol. Chem. 1989, 264, 12370–12378. [Google Scholar] [CrossRef]
- Kobayashi, T.; Jin, L.; De Tombe, P.P. Cardiac thin filament regulation. Pflügers Arch.-Eur. J. Physiol. 2008, 457, 37–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moss, R.L.; Lauer, M.R.; Giulian, G.G.; Greaser, M.L. Altered Ca2+ dependence of tension development in skinned skeletal muscle fibers following modification of troponin by partial substitution with cardiac troponin C. J. Biol. Chem. 1986, 261, 6096–6099. [Google Scholar] [CrossRef]
- Gulati, J.; Scordilis, S.; Babu, A. Effect of troponin C on the cooperativity in Ca2+ activation of cardiac muscle. FEBS Lett. 1988, 236, 441–444. [Google Scholar] [CrossRef] [Green Version]
- Babu, A.; Lehman, W.; Gulati, J. Characterization of the Ca2+-switch in skeletal and cardiac muscles. FEBS Lett. 1989, 251, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Morimoto, S. The effect of Mg2+ on the Ca2+ binding to troponin C in rabbit fast skeletal myofibrils. Biochim. Biophys. Acta (BBA) Gen. Subj. 1991, 1073, 336–340. [Google Scholar] [CrossRef]
- Landstrom, A.; Parvatiyar, M.; Pinto, J.R.; Marquardt, M.L.; Bos, J.M.; Tester, D.J.; Ommen, S.R.; Potter, J.D.; Ackerman, M.J. Molecular and functional characterization of novel hypertrophic cardiomyopathy susceptibility mutations in TNNC1-encoded troponin C. J. Mol. Cell. Cardiol. 2008, 45, 281–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, J.R.; Parvatiyar, M.; Jones, M.A.; Liang, J.; Ackerman, M.J.; Potter, J.D. A functional and structural study of troponin C mutations related to hypertrophic cardiomyopathy. J. Biol. Chem. 2009, 284, 19090–19100. [Google Scholar] [CrossRef] [Green Version]
- Schmidtmann, A.; Lindow, C.; Villard, S.; Heuser, A.; Mügge, A.; Geßner, R.; Granier, C.; Jaquet, K. Cardiac troponin C-L29Q, related to hypertrophic cardiomyopathy, hinders the transduction of the protein kinase A dependent phosphorylation signal from cardiac troponin I to C. FEBS J. 2005, 272, 6087–6097. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, B.; Schmidt-Traub, H.; Perrot, A.; Osterziel, K.J.; Geßner, R. First mutation in cardiac troponin C, L29Q, in a patient with hypertrophic cardiomyopathy. Hum. Mutat. 2001, 17, 524. [Google Scholar] [CrossRef]
- Van de Locht, M.; Donkervoort, S.; de Winter, J.M.; Conijn, S.; Begthel, L.; Kusters, B.; Mohassel, P.; Hu, Y.; Medne, L.; Quinn, C.; et al. Pathogenic variants in TNNC2 cause congenital myopathy due to an impaired force response to calcium. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Schiaffino, S.; Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filatov, V.L.; Katrukha, A.G.; Bulargina, T.V.; Gusev, N.B. Troponin: Structure, properties, and mechanism of functioning. Biochemistry 1999, 64, 969–985. [Google Scholar]
- Jiang, M.; Zhao, X.; Han, W.; Bian, C.; Li, X.; Wang, G.; Ao, Y.; Li, Y.; Yi, D.; Zhe, Y.; et al. A novel deletion in TNNI2 causes distal arthrogryposis in a large Chinese family with marked variability of expression. Qual. Life Res. 2006, 120, 238–242. [Google Scholar] [CrossRef]
- Talbot, J.; Hodges, R. Comparative studies on the inhibitory region of selected species of troponin-I. The use of synthetic peptide analogs to probe structure-function relationships. J. Biol. Chem. 1981, 256, 12374–12378. [Google Scholar] [CrossRef]
- Mak, A.S.; Golosinska, K.; Smillie, L.B. Induction of nonpolymerizable tropomyosin binding to F-actin by troponin and its components. J. Biol. Chem. 1983, 258, 14330–14334. [Google Scholar] [CrossRef]
- Zhu, L.; Lyons, G.E.; Juhasz, O.; Joya, J.E.; Hardeman, E.C.; Wade, R. Developmental regulation of troponin I isoform genes in striated muscles of transgenic mice. Dev. Biol. 1995, 169, 487–503. [Google Scholar] [CrossRef] [Green Version]
- Sung, S.S.; Brassington, A.-M.E.; Krakowiak, P.A.; Carey, J.C.; Jorde, L.B.; Bamshad, M. Mutations in TNNT3 cause multiple congenital contractures: A second locus for distal arthrogryposis type 2B. Am. J. Hum. Genet. 2003, 73, 212–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimber, E.; Tajsharghi, H.; Kroksmark, A.-K.; Oldfors, A.; Tulinius, M. A mutation in the fast skeletal muscle troponin I gene causes myopathy and distal arthrogryposis. Neurology 2006, 67, 597–601. [Google Scholar] [CrossRef]
- Bamshad, M.; Jorde, L.B.; Carey, J.C. A revised and extended classification of the distal arthrogryposes. Am. J. Med. Genet. 1996, 65, 277–281. [Google Scholar] [CrossRef]
- Bamshad, M.; Van Heest, A.; Pleasure, D. Arthrogryposis: A review and update. J. Bone Jt. Surg. Am. Vol. 2009, 91, 40–46. [Google Scholar] [CrossRef] [Green Version]
- Clarke, N.F. Skeletal muscle disease due to mutations in tropomyosin, troponin and cofilin. In The Sarcomere and Skeletal Muscle Disease; Laing, N.G., Ed.; Springer: New York, NY, USA, 2008; Volume 642, pp. 40–54. [Google Scholar]
- Zhao, N.; Jiang, M.; Han, W.; Bian, C.; Li, X.; Huang, F.; Kong, Q.; Li, J. A novel mutation in TNNT3 associated with Sheldon–Hall syndrome in a Chinese family with vertical talus. Eur. J. Med. Genet. 2011, 54, 351–353. [Google Scholar] [CrossRef]
- Sandaradura, S.A.; Bournazos, A.; Mallawaarachchi, A.; Cummings, B.B.; Waddell, L.B.; Jones, K.J.; Troedson, C.; Sudarsanam, A.; Nash, B.; Peters, G.B.; et al. Nemaline myopathy and distal arthrogryposis associated with an autosomal recessive TNNT3 splice variant. Hum. Mutat. 2018, 39, 383–388. [Google Scholar] [CrossRef] [Green Version]
- Kowalczyk, B.; Felus, J. Arthrogryposis: An update on clinical aspects, etiology, and treatment strategies. Arch. Med. Sci. 2016, 1, 10–24. [Google Scholar] [CrossRef] [Green Version]
- Sung, S.S.; Brassington, A.-M.E.; Grannatt, K.; Rutherford, A.; Whitby, F.G.; Krakowiak, P.A.; Jorde, L.B.; Carey, J.C.; Bamshad, M. Mutations in genes encoding fast-twitch contractile proteins cause distal arthrogryposis syndromes. Am. J. Hum. Genet. 2003, 72, 681–690. [Google Scholar] [CrossRef] [Green Version]
- Robinson, P.; Lipscomb, S.; Preston, L.C.; Altin, E.; Watkins, H.; Ashley, C.C.; Redwood, C.S. Mutations in fast skeletal troponin I, troponin T, and β-tropomyosin that cause distal arthrogryposis all increase contractile function. FASEB J. 2006, 21, 896–905. [Google Scholar] [CrossRef]
- Zhu, X.; Wang, F.; Zhao, Y.; Yang, P.; Chen, J.; Sun, H.; Liu, L.; Li, W.; Pan, L.; Guo, Y.; et al. A gain-of-function mutation in TNNI2 impeded bone development through increasing HIF3A Expression in DA2B Mice. PLoS Genet. 2014, 10, e1004589. [Google Scholar] [CrossRef] [PubMed]
- Moses, M.A.; Wiederschain, D.; Wu, I.; Fernandez, C.A.; Ghazizadeh, V.; Lane, W.S.; Flynn, E.; Sytkowski, A.; Tao, T.; Langer, R. Troponin I is present in human cartilage and inhibits angiogenesis. Proc. Natl. Acad. Sci. USA 1999, 96, 2645–2650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Shen, P.Y.; Wu, G.; Chen, X.-Z. Polycystin-2 interacts with troponin I, an angiogenesis inhibitor. Biochemistry 2003, 42, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Pearlstone, J.R.; Carpenter, M.R.; Johnson, P.; Smillie, L.B. Amino-acid sequence of tropomyosin-binding component of rabbit skeletal muscle troponin. Proc. Natl. Acad. Sci. USA 1976, 73, 1902–1906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, J.J.; Kelley, R.I.; Crawford, T.O.; Morton, D.H.; Agarwala, R.; Koch, T.; Schäffer, A.A.; Francomano, C.A.; Biesecker, L.G. A novel nemaline myopathy in the Amish caused by a mutation in troponin T1. Am. J. Hum. Genet. 2000, 67, 814–821. [Google Scholar] [CrossRef] [Green Version]
- Van Der Pol, W.L.; Leijenaar, J.F.; Spliet, W.G.M.; Lavrijsen, S.W.; Jansen, N.J.G.; Braun, K.P.J.; Mulder, M.; Timmers-Raaijmakers, B.; Ratsma, K.; Dooijes, D.; et al. Nemaline myopathy caused byTNNT1 mutations in a Dutch pedigree. Mol. Genet. Genom. Med. 2013, 2, 134–137. [Google Scholar] [CrossRef]
- Fox, M.D.; Carson, V.J.; Feng, H.-Z.; Lawlor, M.W.; Gray, J.T.; Brigatti, K.W.; Jin, J.-P.; Strauss, K.A. TNNT1 nemaline myopathy: Natural history and therapeutic frontier. Hum. Mol. Genet. 2018, 27, 3272–3282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barton, P.J.R.; Townsend, P.J.; Brand, N.J.; Yacoub, M.H. Localization of the fast skeletal muscle troponin I gene (TNNI2) to 11p15.5: Genes for troponin I and T are organized in pairs. Ann. Hum. Genet. 1997, 61, 519–523. [Google Scholar] [CrossRef]
- Toyota, N.; Shimada, Y. Isoform variants of troponin in skeletal and cardiac muscle cells cultured with and without nerves. Cell 1983, 33, 297–304. [Google Scholar] [CrossRef]
- Cooper, T.A.; Ordahl, C.P. A single cardiac troponin T gene generates embryonic and adult isoforms via developmentally regulated alternate splicing. J. Biol. Chem. 1985, 260, 11140–11148. [Google Scholar] [CrossRef]
- Anderson, P.A.; Malouf, N.N.; Oakeley, A.E.; Pagani, E.D.; Allen, P.D. Troponin T isoform expression in humans. A comparison among normal and failing adult heart, fetal heart, and adult and fetal skeletal muscle. Circ. Res. 1991, 69, 1226–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, S.V. Troponin T: Genetics, properties and function. J. Muscle Res. Cell Motil. 1998, 19, 575–602. [Google Scholar] [CrossRef]
- Ohtsuki, I. Molecular arrangement of troponin-T in the thin filament. J. Biochem. 1979, 86, 491–497. [Google Scholar] [CrossRef]
- Takeda, S.; Yamashita, A.; Maeda, K.; Maéda, Y. Structure of the core domain of human cardiac troponin in the Ca2+-saturated form. Nature 2003, 424, 35–41. [Google Scholar] [CrossRef]
- Potter, J.D.; Sheng, Z.; Pan, B.-S.; Zhao, J. A direct regulatory role for troponin T and a dual role for troponin C in the Ca2+ regulation of muscle contraction. J. Biol. Chem. 1995, 270, 2557–2562. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.-P.; Zhang, Z.; Bautista, J.A. Isoform diversity, regulation, and functional adaptation of troponin and calponin. Crit. Rev. Eukaryot. Gene Expr. 2008, 18, 93–124. [Google Scholar] [CrossRef]
- Biesiadecki, B.J.; Elder, B.D.; Yu, Z.-B.; Jin, J.-P. Cardiac troponin T variants produced by aberrant splicing of multiple exons in animals with high instances of dilated cardiomyopathy. J. Biol. Chem. 2002, 277, 50275–50285. [Google Scholar] [CrossRef] [Green Version]
- Feng, H.-Z.; Biesiadecki, B.J.; Yu, Z.-B.; Hossain, M.M.; Jin, J.-P. Restricted N-terminal truncation of cardiac troponin T: A novel mechanism for functional adaptation to energetic crisis. J. Physiol. 2008, 586, 3537–3550. [Google Scholar] [CrossRef]
- Wei, B.; Jin, J.-P. Troponin T isoforms and posttranscriptional modifications: Evolution, regulation and function. Arch. Biochem. Biophys. 2011, 505, 144–154. [Google Scholar] [CrossRef] [Green Version]
- Oki, K.; Wei, B.; Feng, H.; Jin, J. The loss of slow skeletal muscle isoform of troponin T in spindle intrafusal fibres explains the pathophysiology of Amish nemaline myopathy. J. Physiol. 2019, 597, 3999–4012. [Google Scholar] [CrossRef]
- Jin, J.-P.; Brotto, M.A.; Hossain, M.M.; Huang, Q.-Q.; Brotto, L.S.; Nosek, T.; Morton, D.H.; Crawford, T.O. Truncation by Glu180 nonsense mutation results in complete loss of slow skeletal muscle troponin T in a lethal nemaline myopathy. J. Biol. Chem. 2003, 278, 26159–26165. [Google Scholar] [CrossRef] [Green Version]
- Abdulhaq, U.N.; Daana, M.; Dor, T.; Fellig, Y.; Eylon, S.; Schuelke, M.; Shaag, A.; Elpeleg, O.; Edvardson, S. Nemaline body myopathy caused by a novel mutation in troponin T1 (TNNT1). Muscle Nerve 2015, 53, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Konersman, C.G.; Freyermuth, F.; Winder, T.L.; Lawlor, M.W.; Lagier-Tourenne, C.; Patel, S.B. Novel autosomal dominant TNNT1 mutation causing nemaline myopathy. Mol. Genet. Genom. Med. 2017, 5, 678–691. [Google Scholar] [CrossRef] [PubMed]
- Sewry, C.A.; Laitila, J.M.; Wallgren-Pettersson, C. Nemaline myopathies: A current view. J. Muscle Res. Cell Motil. 2019, 40, 111–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maquat, L. When cells stop making sense: Effects of nonsense codons on RNA metabolism in vertebrate cells. RNA 1995, 1, 453–465. [Google Scholar]
- Jha, P.K.; Leavis, P.C.; Sarkar, S. Interaction of deletion mutants of troponins I and T: COOH-terminal truncation of troponin T abolishes troponin I binding and reduces Ca2+ sensitivity of the reconstituted regulatory system. Biochemistry 1996, 35, 16573–16580. [Google Scholar] [CrossRef]
- Amarasinghe, C.; Hossain, M.M.; Jin, J.-P. Functional basis of three new recessive mutations of slow skeletal muscle troponin T found in non-Amish TNNT1 nemaline myopathies. Biochemistry 2016, 55, 4560–4567. [Google Scholar] [CrossRef] [PubMed]
- Marra, J.D.; Engelstad, K.E.; Ankala, A.; Tanji, K.; Dastgir, J.; De Vivo, D.C.; Coffee, B.; Chiriboga, C.A. Identification of a novel nemaline myopathy-causing mutation in the troponin T1 (TNNT1) gene: A case outside of the old order Amish. Muscle Nerve 2015, 51, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Fattahi, Z.; Kalhor, Z.; Fadaee, M.; Vazehan, R.; Parsimehr, E.; Abolhassani, A.; Beheshtian, M.; Zamani, G.; Nafissi, S.; Nilipour, Y.; et al. Improved diagnostic yield of neuromuscular disorders applying clinical exome sequencing in patients arising from a consanguineous population. Clin. Genet. 2016, 91, 386–402. [Google Scholar] [CrossRef] [PubMed]
- Spiro, A.J.; Kennedy, C. Hereditary occurrence of nemaline myopathy. Arch. Neurol. 1965, 13, 155–159. [Google Scholar] [CrossRef]
- Gonatas, N.K.; Shy, G.M.; Godfrey, E.H. Nemaline myopathy. N. Engl. J. Med. 1966, 274, 535–539. [Google Scholar] [CrossRef]
- Fattori, F.; D’Amico, A.; Verardo, M.; Bellacchio, E.; Brizzi, T.; Fiumara, A.; Rodolico, C.; Van De Locht, M.; Ottenheijm, C.; Bertini, E. EP.126 congenital fiber type disproportion related to novel autosomal dominant mutation in TNNT1. Neuromuscul. Disord. 2019, 29, S204. [Google Scholar] [CrossRef]
- Pellerin, D.; Aykanat, A.; Ellezam, B.; Troiano, E.C.; Karamchandani, J.; Dicaire, M.; Petitclerc, M.; Robertson, R.; Allard-Chamard, X.; Brunet, D.; et al. Novel recessive TNNT1 congenital core-rod myopathy in French Canadians. Ann. Neurol. 2020, 87, 568–583. [Google Scholar] [CrossRef]
- Petrucci, A.; Primiano, G.; Savarese, M.; Sancricca, C.; Udd, B.; Servidei, S. Novel TNNT1 mutation and mild nemaline myopathy phenotype in an Italian patient. Neuromuscul. Disord. 2021, 31, 532–538. [Google Scholar] [CrossRef]
- Beck, A.E.; McMillin, M.J.; Gildersleeve, H.I.; Kezele, P.R.; Shively, K.M.; Carey, J.C.; Regnier, M.; Bamshad, M.J. Spectrum of mutations that cause distal arthrogryposis types 1 and 2B. Am. J. Med. Genet. Part A 2013, 161, 550–555. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.H.; Dowling, J.J.; North, K.; Schroth, M.K.; Sejersen, T.; Shapiro, F.; Bellini, J.; Weiss, H.; Guillet, M.; Amburgey, K.; et al. Consensus statement on standard of care for congenital myopathies. J. Child. Neurol. 2012, 27, 363–382. [Google Scholar] [CrossRef] [Green Version]
- Hwee, D.T.; Kennedy, A.; Ryans, J.; Russell, A.J.; Jia, Z.; Hinken, A.C.; Morgans, D.J.; Malik, F.I.; Jasper, J.R. Fast skeletal muscle troponin activator tirasemtiv increases muscle function and performance in the B6SJL-SOD1G93A ALS mouse model. PLoS ONE 2014, 9, e96921. [Google Scholar] [CrossRef]
- De Winter, J.M.; Buck, D.; Hidalgo, C.; Jasper, J.R.; Malik, F.I.; Clarke, N.F.; Stienen, G.; Lawlor, M.W.; Beggs, A.; Ottenheijm, C.A.C.; et al. Troponin activator augments muscle force in nemaline myopathy patients with nebulin mutations. J. Med. Genet. 2013, 50, 383–392. [Google Scholar] [CrossRef]
- Russell, A.J.; Hartman, J.J.; Hinken, A.C.; Muci, A.R.; Kawas, R.; Driscoll, L.; Godinez, G.; Lee, K.H.; Marquez, D.; Browne, W.F.; et al. Activation of fast skeletal muscle troponin as a potential therapeutic approach for treating neuromuscular diseases. Nat. Med. 2012, 18, 452–455. [Google Scholar] [CrossRef] [Green Version]
- Edes, I.; Kiss, E.; Kitada, Y.; Powers, F.M.; Papp, J.G.; Kranias, E.G.; Solaro, R.J. Effects of levosimendan, a cardiotonic agent targeted to troponin C, on cardiac function and on phosphorylation and Ca2+ sensitivity of cardiac myofibrils and sarcoplasmic reticulum in guinea pig heart. Circ. Res. 1995, 77, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Nieminen, M.S.; Fruhwald, S.; Heunks, L.M.A.; Suominen, P.K.; Gordon, A.; Kivikko, M.; Pollesello, P. Levosimendan: Current data, clinical use and future development. Heart Lung Vessel. 2013, 5, 227–245. [Google Scholar]
- Doorduin, J.; Sinderby, C.A.; Beck, J.E.; Stegeman, D.F.; Van Hees, H.W.H.; Van Der Hoeven, J.G.; Heunks, L.M.A. The calcium sensitizer levosimendan improves human diaphragm function. Am. J. Respir. Crit. Care Med. 2012, 185, 90–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Hees, H.W.H.; Dekhuijzen, P.N.R.; Heunks, L.M.A. Levosimendan enhances force generation of diaphragm muscle from patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2009, 179, 41–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Hees, H.; Acuña, G.A.; Linkels, M.; Dekhuijzen, P.; Heunks, L. Levosimendan improves calcium sensitivity of diaphragm muscle fibres from a rat model of heart failure. Br. J. Pharmacol. 2011, 162, 566–573. [Google Scholar] [CrossRef] [Green Version]
- Roesthuis, L.; Van Der Hoeven, H.; Sinderby, C.; Frenzel, T.; Ottenheijm, C.; Brochard, L.; Doorduin, J.; Heunks, L. Effects of levosimendan on respiratory muscle function in patients weaning from mechanical ventilation. Intensive Care Med. 2019, 45, 1372–1381. [Google Scholar] [CrossRef] [Green Version]
- Donkervoort, S.; Papadaki, M.; de Winter, J.M.; Neu, M.B.; Kirschner, J.; Bolduc, V.; Yang, M.L.; Gibbons, M.A.; Hu, Y.; Do, J.D.; et al. TPM3 deletions cause a hypercontractile congenital muscle stiffness phenotype. Ann. Neurol. 2015, 78, 982–994. [Google Scholar] [CrossRef] [Green Version]
- De Winter, J.M.; Gineste, C.; Minardi, E.; Brocca, L.; Rossi, M.; Borsboom, T.; Beggs, A.H.; Bernard, M.; Bendahan, D.; Hwee, D.T.; et al. Acute and chronic tirasemtiv treatment improves in vivo and in vitro muscle performance in actin-based nemaline myopathy mice. Hum. Mol. Genet. 2021, 30, 1305–1320. [Google Scholar] [CrossRef] [PubMed]
- Shefner, J.M.; Cudkowicz, M.E.; Hardiman, O.; Cockroft, B.M.; Lee, J.H.; Malik, F.I.; Meng, L.; Rudnicki, S.A.; Wolff, A.A.; Andrews, J.A.; et al. A phase III trial of tirasemtiv as a potential treatment for amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2019, 20, 584–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, J.A.; Miller, T.M.; Vijayakumar, V.; Stoltz, R.; James, J.K.; Meng, L.; Wolff, A.A.; Malik, F.I. CK-2127107 amplifies skeletal muscle response to nerve activation in humans. Muscle Nerve 2017, 57, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Shefner, J.M.; Andrews, J.A.; Genge, A.; Jackson, C.; Lechtzin, N.; Miller, T.M.; Cockroft, B.M.; Meng, L.; Wei, J.; Wolff, A.A.; et al. A phase 2, double-blind, randomized, dose-ranging trial of reldesemtiv in patients with ALS. Amyotroph. Lateral Scler. Front. Degener. 2020, 22, 287–299. [Google Scholar] [CrossRef]
- Mendell, L.M. The size principle: A rule describing the recruitment of motoneurons. J. Neurophysiol. 2005, 93, 3024–3026. [Google Scholar] [CrossRef] [Green Version]
- Gregory, C.M.; Bickel, C.S. Recruitment patterns in human skeletal muscle during electrical stimulation. Phys. Ther. 2005, 85, 358–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Samulski, R.J. Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 2020, 21, 255–272. [Google Scholar] [CrossRef] [PubMed]
- Salabarria, S.; Nair, J.; Clement, N.; Smith, B.; Raben, N.; Fuller, D.; Byrne, B.; Corti, M. Advancements in AAV-mediated gene therapy for Pompe disease. J. Neuromuscul. Dis. 2020, 7, 15–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, T.; Ferran, B.; Tsukahara, Y.; Shang, Q.; Desai, S.; Fedoce, A.; Pimentel, D.R.; Luptak, I.; Adachi, T.; Ido, Y.; et al. Production of adeno-associated virus vectors for in vitro and in vivo applications. Sci. Rep. 2019, 9, 13601. [Google Scholar] [CrossRef] [Green Version]
- Keeler, A.M.; Flotte, T.R. Recombinant adeno-associated virus gene therapy in light of Luxturna (and Zolgensma and Glybera): Where are we, and how did we get here? Annu. Rev. Virol. 2019, 6, 601–621. [Google Scholar] [CrossRef]
- Smith, B.K.; Collins, S.W.; Conlon, T.J.; Mah, C.S.; Lawson, L.A.; Martin, A.D.; Fuller, D.D.; Cleaver, B.D.; Clément, N.; Phillips, D.; et al. Phase I/II Trial of adeno-associated virus–mediated alpha-glucosidase gene therapy to the diaphragm for chronic respiratory failure in Pompe Disease: Initial safety and ventilatory outcomes. Hum. Gene Ther. 2013, 24, 630–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindqvist, J.; Levy, Y.; Pati-Alam, A.; Hardeman, E.C.; Gregorevic, P.; Ochala, J. Modulating myosin restores muscle function in a mouse model of nemaline myopathy. Ann. Neurol. 2016, 79, 717–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, F.I.; Hartman, J.J.; Elias, K.A.; Morgan, B.P.; Rodriguez, H.; Brejc, K.; Anderson, R.L.; Sueoka, S.H.; Lee, K.H.; Finer, J.T.; et al. Cardiac myosin activation: A potential therapeutic approach for systolic heart failure. Science 2011, 331, 1439–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindqvist, J.; Lee, E.-J.; Karimi, E.; Kolb, J.; Granzier, H. Omecamtiv mecarbil lowers the contractile deficit in a mouse model of nebulin-based nemaline myopathy. PLoS ONE 2019, 14, e0224467. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Protein(s) | Abbreviation | Molecular Weight | Associated with |
|---|---|---|---|---|
| TNNC1 | Slow skeletal troponin C Cardiac troponin C | ssTnC | 18.4 kDa | Cardiomyopathy |
| TNNC2 | Fast skeletal troponin C | fsTnC | 18.1 kDa | Congenital myopathy |
| TNNI1 | Slow skeletal troponin I | ssTnI | 21.7 kDa | N/A |
| TNNI2 | Fast skeletal troponin I | fsTnI | 24.0 kDa | Distal arthrogryposis |
| TNNI3 | Cardiac troponin I | cTnI | 21.3 kDa | Cardiomyopathy |
| TNNT1 | Slow skeletal troponin T | ssTnT | 30.1 kDa | Nemaline myopathy |
| TNNT2 | Cardiac troponin T | cTnT | 35.9 kDa | Cardiomyopathy |
| TNNT3 | Fast skeletal troponin T | fsTnT | 31.8 kDa | Nemaline myopathy |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van de Locht, M.; Borsboom, T.C.; Winter, J.M.; Ottenheijm, C.A.C. Troponin Variants in Congenital Myopathies: How They Affect Skeletal Muscle Mechanics. Int. J. Mol. Sci. 2021, 22, 9187. https://doi.org/10.3390/ijms22179187
van de Locht M, Borsboom TC, Winter JM, Ottenheijm CAC. Troponin Variants in Congenital Myopathies: How They Affect Skeletal Muscle Mechanics. International Journal of Molecular Sciences. 2021; 22(17):9187. https://doi.org/10.3390/ijms22179187
Chicago/Turabian Stylevan de Locht, Martijn, Tamara C. Borsboom, Josine M. Winter, and Coen A. C. Ottenheijm. 2021. "Troponin Variants in Congenital Myopathies: How They Affect Skeletal Muscle Mechanics" International Journal of Molecular Sciences 22, no. 17: 9187. https://doi.org/10.3390/ijms22179187