
The New Therapeutic Approaches in the Treatment of Non-Alcoholic Fatty Liver Disease

 , , , ,
, , , ,
Abstract
:1. Introduction
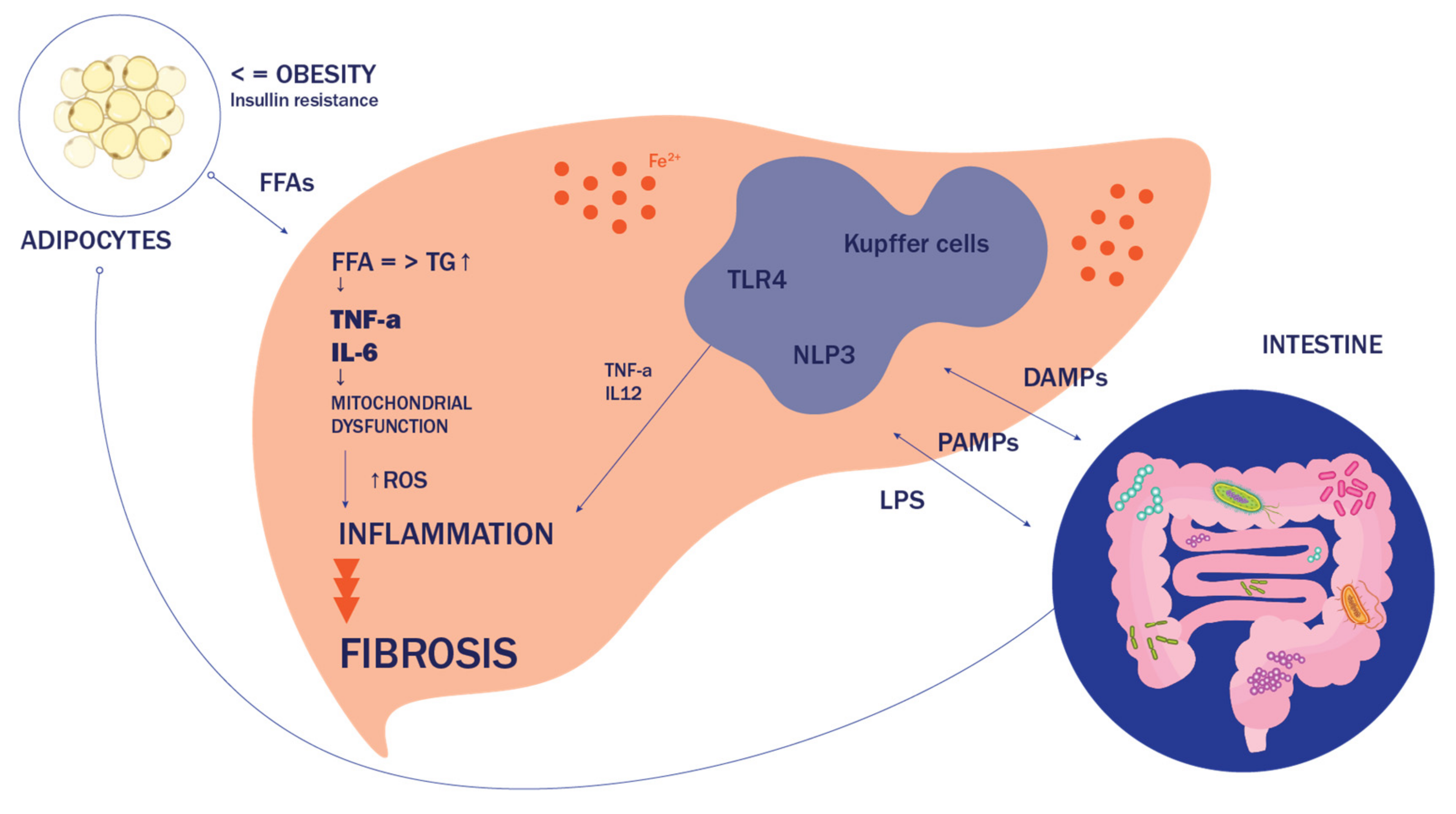
2. Etiopathogenesis of NAFLD
2.1. General Hypothesis
2.2. Lipotoxicity, Organelle Distress and Inflammasome Activation—The Road to NASH
2.3. Does the Answer Lie in the Gut?
2.3.1. Increased Gut Permeability
2.3.2. Disruption of Inflammatory Response and LPS Endotoxemia
2.3.3. Microbiota-Related Alterations of Host Metabolism
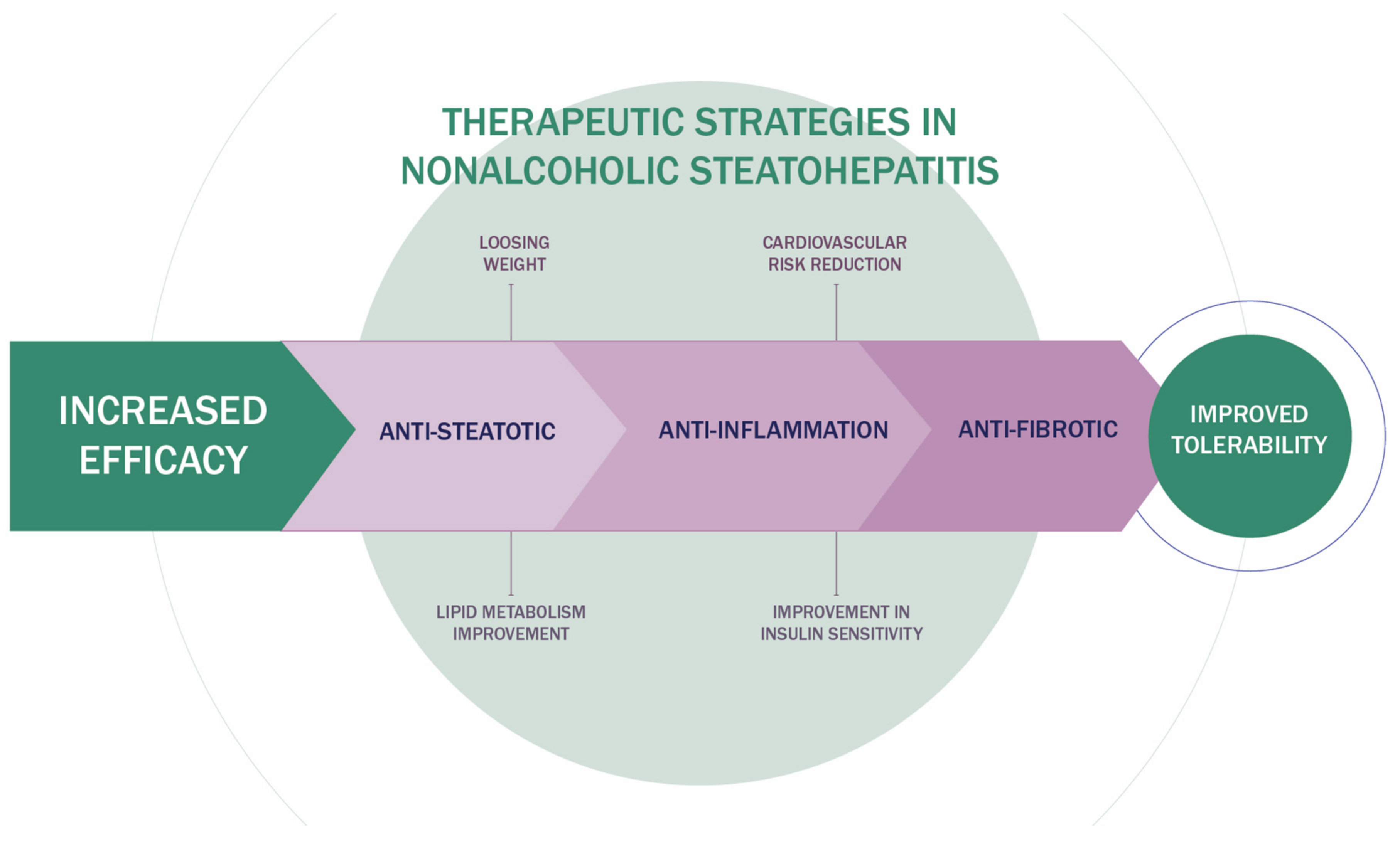
3. Innovative Therapeutic Strategies in NAFLD
3.1. Lifestyle Modification
3.2. Antioxidant
3.3. Farnesoid X Receptor (FXR) Agonists
3.4. Peroxisome Proliferator-Activated Receptor (PPARs) Agonists
3.5. Glucagon-Like Peptide-1 (GLP-1) Agonists
3.6. Sodium/Glucose Transport Protein 2 (SGLT2) Inhibitors
3.7. Probiotics and Synbiotics
3.8. Lipogenesis Inhibitors
3.9. Thyroid Hormone Receptor Beta (TRβ) Agonists
3.10. CCL Receptor Type 2 (CCR2) and Type 5 (CCR5) Antagonists
3.11. Fibroblast Growth Factor-19 (FGF19) Analogue
4. Combination Therapy of NASH
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Younossi, Z.M.; Marchesini, G.; Pinto-Cortez, H.; Petta, S. Epidemiology of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis: Implications for Liver Transplantation. Transplantation 2019, 103, 22–27. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; LaVine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Lim, J.K.; Patton, H.; El-Serag, H.B. AGA Clinical Practice Update on Screening and Surveillance for Hepatocellular Carcinoma in Patients With Nonalcoholic Fatty Liver Disease: Expert Review. Gastroenterology 2020, 158, 1822–1830. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.M.; Golabi, P.; de Avila, L.; Paik, J.M.; Srishord, M.; Fukui, N.; Qiu, Y.; Burns, L.; Afendy, A.; Nader, F. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic review and meta-analysis. J. Hepatol. 2019, 71, 793–801. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL–EASD–EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wong, V.W.-S.; Dufour, J.-F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef]
- Liu, J.; Ayada, I.; Zhang, X.; Wang, L.; Li, Y.; Wen, T.; Ma, Z.; Bruno, M.J.; de Knegt, R.J.; Cao, W.; et al. Estimating Global Prevalence of Metabolic Dysfunction-Associated Fatty Liver Disease in Overweight or Obese Adults. Clin. Gastroenterol. Hepatol. 2021, 21. [Google Scholar] [CrossRef]
- Kim, D.; Kim, W.R. Nonobese Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2017, 15, 474–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, L.; Anstee, Q.M.; Tilg, H.; Targher, G. Non-alcoholic fatty liver disease and its relationship with cardiovascular disease and other extrahepatic diseases. Gut 2017, 66, 1138–1153. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.-L.; Patman, G.L.; Leathart, J.B.S.; Piguet, A.-C.; Burt, A.D.; Dufour, J.-F.; Day, C.P.; Daly, A.K.; Reeves, H.L.; Anstee, Q.M. Carriage of the PNPLA3 rs738409 C >G polymorphism confers an increased risk of non-alcoholic fatty liver disease associated hepatocellular carcinoma. J. Hepatol. 2014, 61, 75–81. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Xiaoli, A.M.; Yang, F. Regulation and Metabolic Significance of De Novo Lipogenesis in Adipose Tissues. Nutrients 2018, 10, 1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godoy-Matos, A.F.; Júnior, W.S.S.; Valerio, C.M. NAFLD as a continuum: From obesity to metabolic syndrome and diabetes. Diabetol. Metab. Syndr. 2020, 12, 60. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M. Toxic AGEs (TAGE) theory: A new concept for preventing the development of diseases related to lifestyle. Diabetol. Metab. Syndr. 2020, 12, 105. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, L.E.; Petta, S.; Fracanzani, A.L.; Nevola, R.; Coppola, C.; Narciso, V.; Rinaldi, L.; Calvaruso, V.; Pafundi, P.C.; Lombardi, R.; et al. Reduced incidence of type 2 diabetes in patients with chronic hepatitis C virus infection cleared by direct-acting antiviral therapy: A prospective study. Diabetes Obes. Metab. 2020, 22, 2408–2416. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, L.E.; Petta, S.; Fracanzani, A.L.; Coppola, C.; Narciso, V.; Nevola, R.; Rinaldi, L.; Calvaruso, V.; Staiano, L.; Di Marco, V.; et al. Impact of hepatitis C virus clearance by direct-acting antiviral treatment on the incidence of major cardiovascular events: A prospective multicentre study. Atherosclerosis 2020, 296, 40–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasso, F.C.; Pafundi, P.C.; Caturano, A.; Galiero, R.; Vetrano, E.; Nevola, R.; Petta, S.; Fracanzani, A.L.; Coppola, C.; Di Marco, V.; et al. Impact of direct acting antivirals (DAAs) on cardiovascular events in HCV cohort with pre-diabetes. Nutr. Metab. Cardiovasc. Dis. 2021, 31, 2345–2353. [Google Scholar] [CrossRef] [PubMed]
- Caturano, A.; Acierno, C.; Nevola, R.; Pafundi, P.C.; Galiero, R.; Rinaldi, L.; Salvatore, T.; Adinolfi, L.E.; Sasso, F.C. Non-Alcoholic Fatty Liver Disease: From Pathogenesis to Clinical Impact. Processes 2021, 9, 135. [Google Scholar] [CrossRef]
- Rinaldi, L.; Pafundi, P.C.; Galiero, R.; Caturano, A.; Morone, M.V.; Silvestri, C.F.; Giordano, M.; Salvatore, T.; Sasso, F.C. Mechanisms of Non-Alcoholic Fatty Liver Disease in the Metabolic Syndrome. A Narrative Review. Antioxidants 2021, 10, 270. [Google Scholar] [CrossRef] [PubMed]
- Masarone, M.; Rosato, V.; Aglitti, A.; Bucci, T.; Caruso, R.; Salvatore, T.; Sasso, F.C.; Tripodi, M.F.; Persico, M. Liver biopsy in type 2 diabetes mellitus: Steatohepatitis represents the sole feature of liver damage. PLoS ONE 2017, 12, e0178473. [Google Scholar] [CrossRef] [PubMed]
- Pierantonelli, I.; Svegliati-Baroni, G. Nonalcoholic Fatty Liver Disease: Basic Pathogenetic Mechanisms in the Progression from NAFLD to NASH. Transplantation 2019, 103, e1–e13. [Google Scholar] [CrossRef] [PubMed]
- Sheka, A.C.; Adeyi, O.; Thompson, J.; Hameed, B.; Crawford, P.A.; Ikramuddin, S. Nonalcoholic Steatohepatitis: A Review. JAMA 2020, 323, 1175–1183. [Google Scholar] [CrossRef]
- Parthasarathy, G.; Revelo, X.; Malhi, H. Pathogenesis of Nonalcoholic Steatohepatitis: An Overview. Hepatol. Commun. 2020, 4, 478–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simões, I.C.M.; Fontes, A.; Pinton, P.; Zischka, H.; Wieckowski, M.R. Mitochondria in non-alcoholic fatty liver disease. Int. J. Biochem. Cell Biol. 2018, 95, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Mota, M.; Banini, B.A.; Cazanave, S.C.; Sanyal, A.J. Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metabolism 2016, 65, 1049–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noureddin, M.; Sanyal, A.J. Pathogenesis of NASH: The Impact of Multiple Pathways. Curr. Hepatol. Rep. 2018, 17, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Bessone, F.; Razori, M.V.; Roma, M.G. Molecular pathways of nonalcoholic fatty liver disease development and progression. Cell. Mol. Life Sci. 2019, 76, 99–128. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caligiuri, A.; Gentilini, A.; Marra, F. Molecular Pathogenesis of NASH. Int. J. Mol. Sci. 2016, 17, 1575. [Google Scholar] [CrossRef] [Green Version]
- Manne, V.; Handa, P.; Kowdley, K.V. Pathophysiology of Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis. Clin. Liver Dis. 2018, 22, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Marra, F.; Svegliati-Baroni, G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J. Hepatol. 2018, 68, 280–295. [Google Scholar] [CrossRef] [PubMed]
- Boutari, C.; Mantzoros, C.S. Adiponectin and leptin in the diagnosis and therapy of NAFLD. Metabolism 2020, 103, 154028. [Google Scholar] [CrossRef]
- Nati, M.; Haddad, D.; Birkenfeld, A.L.; Koch, C.A.; Chavakis, T.; Chatzigeorgiou, A. The role of immune cells in metabolism-related liver inflammation and development of non-alcoholic steatohepatitis (NASH). Rev. Endocr. Metab. Disord. 2016, 17, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Kazankov, K.; Jørgensen, S.M.D.; Thomsen, K.L.; Møller, H.J.; Vilstrup, H.; George, J.; Schuppan, D.; Grønbæk, H. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 145–159. [Google Scholar] [CrossRef]
- Chackelevicius, C.M.; Gambaro, S.E.; Tiribelli, C.; Rosso, N. Th17 involvement in nonalcoholic fatty liver disease progression to non-alcoholic steatohepatitis. World J. Gastroenterol. 2016, 22, 9096–9103. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Debelius, J.; Brenner, D.A.; Karin, M.; Loomba, R.; Schnabl, B.; Knight, R. The gut–liver axis and the intersection with the microbiome. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 397–411. [Google Scholar] [CrossRef]
- Gottlieb, A.; Canbay, A. Why Bile Acids Are So Important in Non-Alcoholic Fatty Liver Disease (NAFLD) Progression. Cells 2019, 8, 1358. [Google Scholar] [CrossRef] [PubMed]
- Cave, M.C.; Clair, H.B.; Hardesty, J.E.; Falkner, K.C.; Feng, W.; Clark, B.J.; Sidey, J.; Shi, H.; Aqel, B.A.; McClain, C.J.; et al. Nuclear receptors and nonalcoholic fatty liver disease. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2016, 1859, 1083–1099. [Google Scholar] [CrossRef] [Green Version]
- Shen, F.; Zheng, R.-D.; Sun, X.-Q.; Ding, W.-J.; Wang, X.-Y.; Fan, J.-G. Gut microbiota dysbiosis in patients with non-alcoholic fatty liver disease. Hepatobiliary Pancreat. Dis. Int. 2017, 16, 375–381. [Google Scholar] [CrossRef]
- Milosevic, I.; Vujovic, A.; Barac, A.; Djelic, M.; Korac, M.; Spurnic, A.R.; Gmizic, I.; Stevanovic, O.; Djordjevic, V.; Lekic, N.; et al. Gut-Liver Axis, Gut Microbiota, and Its Modulation in the Management of Liver Diseases: A Review of the Literature. Int. J. Mol. Sci. 2019, 20, 395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Oliveira, J.M.; Pace, F.L.; Ghetti, F.D.F.; Barbosa, K.V.B.D.; Cesar, D.E.; Chebli, J.M.F.; Ferreira, L.E.V.V.D.C. Non-alcoholic Steatohepatitis: Comparison of Intestinal Microbiota between Different Metabolic Profiles. A Pilot Study. J. Gastrointest. Liver Dis. 2020, 29, 369–376. [Google Scholar] [CrossRef]
- Kolodziejczyk, A.A.; Zheng, D.; Shibolet, O.; Elinav, E. The role of the microbiome in NAFLD and NASH. EMBO Mol. Med. 2019, 11, e9302. [Google Scholar] [CrossRef]
- Magne, F.; Gotteland, M.; Gauthier, L.; Zazueta, A.; Pesoa, S.; Navarrete, P.; Balamurugan, R. The Firmicutes/Bacteroidetes Ratio: A Relevant Marker of Gut Dysbiosis in Obese Patients? Nutrients 2020, 12, 1474. [Google Scholar] [CrossRef] [PubMed]
- Dong, T.S.; Katzka, W.; Lagishetty, V.; Luu, K.; Hauer, M.; Pisegna, J.; Jacobs, J.P. A Microbial Signature Identifies Advanced Fibrosis in Patients with Chronic Liver Disease Mainly Due to NAFLD. Sci. Rep. 2020, 10, 2771. [Google Scholar] [CrossRef] [PubMed]
- Schubert, K.; Olde Damink, S.W.M.; Von Bergen, M.; Schaap, F.G. Interactions between bile salts, gut microbiota, and hepatic innate immunity. Immunol. Rev. 2017, 279, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Odenwald, M.A.; Turner, J.R. The intestinal epithelial barrier: A therapeutic target? Nat. Rev. Gastroenterol. Hepatol. 2016, 14, 9–21. [Google Scholar] [CrossRef]
- Allam-Ndoul, B.; Castonguay-Paradis, S.; Veilleux, A. Gut Microbiota and Intestinal Trans-Epithelial Permeability. Int. J. Mol. Sci. 2020, 21, 6402. [Google Scholar] [CrossRef]
- Chopyk, D.M.; Grakoui, A. Contribution of the Intestinal Microbiome and Gut Barrier to Hepatic Disorders. Gastroenterology 2020, 159, 849–863. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Kolodziejczyk, A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the immune system. Nat. Rev. Immunol. 2017, 17, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Yin, Y.; Li, Z.; Zhang, W. Gut Microbiota-Derived Components and Metabolites in the Progression of Non-Alcoholic Fatty Liver Disease (NAFLD). Nutrients 2019, 11, 1712. [Google Scholar] [CrossRef] [Green Version]
- Hegazy, M.A.; Mogawer, S.M.; Alnaggar, A.R.L.R.; Ghoniem, O.A.; Samie, R.M.A. Serum LPS and CD163 Biomarkers Confirming the Role of Gut Dysbiosis in Overweight Patients with NASH. Diabetes Metab. Syndr. Obes. Targets Ther. 2020, 13, 3861–3872. [Google Scholar] [CrossRef]
- Du Plessis, J.; Korf, H.; Van Pelt, J.; Windmolders, P.; Elst, I.V.; Verrijken, A.; Hubens, G.; Van Gaal, L.; Cassiman, D.; Nevens, F.; et al. Pro-Inflammatory Cytokines but Not Endotoxin-Related Parameters Associate with Disease Severity in Patients with NAFLD. PLoS ONE 2016, 11, e0166048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpino, G.; Del Ben, M.; Pastori, D.; Carnevale, R.; Baratta, F.; Overi, D.; Francis, H.; Cardinale, V.; Onori, P.; Safarikia, S.; et al. Increased Liver Localization of Lipopolysaccharides in Human and Experimental NAFLD. Hepatology 2019, 72, 470–485. [Google Scholar] [CrossRef]
- Ghetti, F.D.F.; Oliveira, D.G.; De Oliveira, J.M.; Ferreira, L.E.V.V.D.C.; Cesar, D.E.; Moreira, A.P.B. Influence of gut microbiota on the development and progression of nonalcoholic steatohepatitis. Eur. J. Nutr. 2017, 57, 861–876. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Xu, C.; Yu, C.; Li, Y. Role of NLRP3 Inflammasome in the Progression of NAFLD to NASH. Can. J. Gastroenterol. Hepatol. 2016, 2016, 6489012. [Google Scholar] [CrossRef] [Green Version]
- Miura, K. Role of gut microbiota and Toll-like receptors in nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 7381–7391. [Google Scholar] [CrossRef]
- Schoeler, M.; Caesar, R. Dietary lipids, gut microbiota and lipid metabolism. Rev. Endocr. Metab. Disord. 2019, 20, 461–472. [Google Scholar] [CrossRef] [Green Version]
- Wahlström, A.; Sayin, S.I.; Marschall, H.-U.; Bäckhed, F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016, 24, 41–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aron-Wisnewsky, J.; Vigliotti, C.; Witjes, J.; Le, P.; Holleboom, A.G.; Verheij, J.; Nieuwdorp, M.; Clément, K. Gut microbiota and human NAFLD: Disentangling microbial signatures from metabolic disorders. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 279–297. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Vitetta, L. Gut Microbiota Metabolites in NAFLD Pathogenesis and Therapeutic Implications. Int. J. Mol. Sci. 2020, 21, 5214. [Google Scholar] [CrossRef] [PubMed]
- Perumpail, B.J.; Li, A.A.; John, N.; Sallam, S.; Shah, N.D.; Kwong, W.; Cholankeril, G.; Kim, D.; Ahmed, A. The Therapeutic Implications of the Gut Microbiome and Probiotics in Patients with NAFLD. Diseases 2019, 7, 27. [Google Scholar] [CrossRef] [Green Version]
- Dufour, J.-F.; Caussy, C.; Loomba, R. Combination therapy for non-alcoholic steatohepatitis: Rationale, opportunities and challenges. Gut 2020, 69, 1877–1884. [Google Scholar] [CrossRef]
- Sumida, Y.; Yoneda, M. Current and future pharmacological therapies for NAFLD/NASH. J. Gastroenterol. 2018, 53, 362–376. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.M.; Corey, K.E.; Lim, J.K. AGA Clinical Practice Update on Lifestyle Modification Using Diet and Exercise to Achieve Weight Loss in the Management of Nonalcoholic Fatty Liver Disease: Expert Review. Gastroenterology 2021, 160, 912–918. [Google Scholar] [CrossRef]
- Anania, C.; Perla, F.M.; Olivero, F.; Pacifico, L.; Chiesa, C. Mediterranean diet and nonalcoholic fatty liver disease. World J. Gastroenterol. 2018, 24, 2083–2094. [Google Scholar] [CrossRef]
- Yang, J.; Fernández-Galilea, M.; Martínez-Fernández, L.; González-Muniesa, P.; Pérez-Chávez, A.; Martínez, J.A.; Moreno-Aliaga, M.J. Oxidative Stress and Non-Alcoholic Fatty Liver Disease: Effects of Omega-3 Fatty Acid Supplementation. Nutrients 2019, 11, 872. [Google Scholar] [CrossRef] [Green Version]
- Berkovic, M.C.; Bilic-Curcic, I.; Mrzljak, A.; Cigrovski, V. NAFLD and Physical Exercise: Ready, Steady, Go! Front. Nutr. 2021, 8, 734859. [Google Scholar] [CrossRef]
- Sumida, Y.; Naito, Y.; Tanaka, S.; Sakai, K.; Inada, Y.; Taketani, H.; Kanemasa, K.; Yasui, K.; Itoh, Y.; Okanoue, T.; et al. Long-term (≥2 year) efficacy of vitamin E for non-alcoholic steatohepatitis. Hepatogastroenterology 2013, 60, 1445–1450. [Google Scholar] [PubMed]
- Younossi, Z.M.; Ratziu, V.; Loomba, R.; Rinella, M.; Anstee, Q.M. Obeticholic acid for the treatment of nonalcoholic steatohepatitis: Interim analysis from a multicentre, randomized, placebo controlled phase 3 trial. Lancet 2019, 394, 2184–2196. [Google Scholar] [CrossRef] [Green Version]
- Ratziu, V.; Sanyal, A.J.; Loomba, R.; Rinella, M.; Harrison, S.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; MacConell, L.; Shringarpure, R.; et al. REGENERATE: Design of a pivotal, randomised, phase 3 study evaluating the safety and efficacy of obeticholic acid in patients with fibrosis due to nonalcoholic steatohepatitis. Contemp. Clin. Trials. 2019, 84, 105803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venetsanaki, V.; Karabouta, Z.; Polyzos, S.A. Farnesoid X nuclear receptor agonists for the treatment of nonalcoholic steatohepatitis. Eur. J. Pharmacol. 2019, 863, 172661. [Google Scholar] [CrossRef] [PubMed]
- Stofan, M.; Guo, G.L. Bile Acids and FXR: Novel Targets for Liver Diseases. Front. Med. 2020, 7, 544. [Google Scholar] [CrossRef]
- Patel, K.; Harrison, S.A.; Elkashab, M.; Trotter, J.F.; Herring, R.; Rojter, S.; Kayali, Z.; Wong, V.W.-S.; Greenbloom, S.; Jayakumar, S.; et al. Cilofexor, a Nonsteroidal FXR Agonist, in Non-Cirrhotic Patients with Nonalcoholic Steatohepatitis: A Phase 2 Randomized Controlled Trial. Hepatology 2020, 72, 58–71. [Google Scholar] [CrossRef]
- Fiorucci, S.; Biagioli, M.; Sepe, V.; Zampella, A.; Distrutti, E. Bile acid modulators for the treatment of nonalcoholic steatohepatitis (NASH). Expert Opin. Investig. Drugs 2020, 29, 623–632. [Google Scholar] [CrossRef]
- Chianelli, D.; Rucker, P.V.; Roland, J.; Tully, D.C.; Nelson, J.; Liu, X.; Bursulaya, B.; Hernandez, E.D.; Wu, J.; Prashad, M.; et al. Nidufexor (LMB763), a Novel FXR Modulator for the Treatment of Nonalcoholic Steatohepatitis. J. Med. Chem. 2020, 63, 3868–3880. [Google Scholar] [CrossRef]
- Boeckmans, J.; Natale, A.; Rombaut, M.; Buyl, K.; Rogiers, V.; De Kock, J.; Vanhaecke, T.; Rodrigues, R.M. Anti-NASH Drug Development Hitches a Lift on PPAR Agonism. Cells 2019, 9, 37. [Google Scholar] [CrossRef] [Green Version]
- Bril, F.; Kalavalapalli, S.; Clark, V.C.; Lomonaco, R.; Soldevila-Pico, C.; Liu, I.-C.; Orsak, B.; Tio, F.; Cusi, K. Response to Pioglitazone in Patients With Nonalcoholic Steatohepatitis With vs Without Type 2 Diabetes. Clin. Gastroenterol. Hepatol. 2018, 16, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, Vitamin E, or Placebo for Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef] [Green Version]
- Cusi, K. Pioglitazone for the treatment of NASH in patients with prediabetes or type 2 diabetes mellitus. Gut 2017, 67, 1371. [Google Scholar] [CrossRef]
- Van Meeteren, M.J.W.; Drenth, J.P.; Tjwa, E.T. Elafibranor: A potential drug for the treatment of nonalcoholic steatohepatitis (NASH). Expert Opin. Investig. Drugs 2020, 29, 117–123. [Google Scholar] [CrossRef]
- Siddiqui, M.S.; Idowu, M.O.; Parmar, D.; Borg, B.B.; Denham, D.; Loo, N.M.; Lazas, D.; Younes, Z.; Sanyal, A.J. A Phase 2 Double Blinded, Randomized Controlled Trial of Saroglitazar in Patients With Nonalcoholic Steatohepatitis. Clin. Gastroenterol. Hepatol. 2020, 19, 2670–2672. [Google Scholar] [CrossRef] [PubMed]
- Sven, M.F.; Pierre, B.; Abdelmalek, M.F.; Anstee, Q.M.; Elisabetta, B.; Vlad, R.; Philippe, H.-M.; Bruno, S.; Jean-Louis, J.; Pierre, B.; et al. A randomised, double-blind, placebo-controlled, multi-centre, dose-range, proof-of-concept, 24-week treatment study of lanifibranor in adult subjects with non-alcoholic steatohepatitis: Design of the NATIVE study. Contemp. Clin. Trials 2020, 98, 106170. [Google Scholar] [CrossRef]
- Petit, J.-M.; Vergès, B. GLP-1 receptor agonists in NAFLD. Diabetes Metab. 2017, 43, 2S28–2S33. [Google Scholar] [CrossRef]
- Petit, J.-M.; Cercueil, J.-P.; Loffroy, R.; Denimal, D.; Bouillet, B.; Fourmont, C.; Chevallier, O.; Duvillard, L.; Vergès, B. Effect of liraglutide therapy on liver fat content in patients with inadequately controlled type 2 diabetes. The Lira-NAFLD study. J. Clin. Endocrinol. Metab. 2016, 102, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Calanna, S.; Cusi, K.; Linder, M.; Okanoue, T.; Ratziu, V.; Sanyal, A.; Sejling, A.-S.; Newsome, P.N. Semaglutide for the treatment of non-alcoholic steatohepatitis: Trial design and comparison of non-invasive biomarkers. Contemp. Clin. Trials 2020, 97, 106174. [Google Scholar] [CrossRef]
- Newsome, P.N.; Buchholtz, K.; Cusi, K.; Linder, M.; Okanoue, T.; Ratziu, V.; Sanyal, A.J.; Sejling, A.-S.; Harrison, S.A. A Placebo-Controlled Trial of Subcutaneous Semaglutide in Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2021, 384, 1113–1124. [Google Scholar] [CrossRef]
- Scheen, A.J. Beneficial effects of SGLT2 inhibitors on fatty liver in type 2 diabetes: A common comorbidity associated with severe complications. Diabetes Metab. 2019, 45, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Latva-Rasku, A.; Honka, M.-J.; Kullberg, J.; Mononen, N.; Lehtimäki, T.; Saltevo, J.; Kirjavainen, A.K.; Saunavaara, V.; Iozzo, P.; Johansson, L.; et al. The SGLT2 Inhibitor Dapagliflozin Reduces Liver Fat but Does Not Affect Tissue Insulin Sensitivity: A Randomized, Double-Blind, Placebo-Controlled Study With 8-Week Treatment in Type 2 Diabetes Patients. Diabetes Care 2019, 42, 931–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aso, Y.; Kato, K.; Sakurai, S.; Kishi, H.; Shimizu, M.; Jojima, T.; Iijima, T.; Maejima, Y.; Shimomura, K.; Usui, I. Impact of dapagliflozin, an SGLT2 inhibitor, on serum levels of soluble dipeptidyl peptidase-4 in patients with type 2 diabetes and non-alcoholic fatty liver disease. Int. J. Clin. Pract. 2019, 73, e13335. [Google Scholar] [CrossRef] [PubMed]
- Kahl, S.; Gancheva, S.; Straßburger, K.; Herder, C.; Machann, J.; Katsuyama, H.; Kabisch, S.; Henkel, E.; Kopf, S.; Lagerpusch, M.; et al. Empagliflozin effectively lowers liver fat content in well-controlled type 2 diabetes: A randomized, double-blind, phase 4, placebo-controlled trial. Diabetes Care 2020, 43, 298–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Li, P.; Liu, Y.; Zhang, Y. Efficacy of Probiotics and Synbiotics in Patients with Nonalcoholic Fatty Liver Disease: A Meta-Analysis. Dig. Dis. Sci. 2019, 64, 3402–3412. [Google Scholar] [CrossRef] [PubMed]
- Alisi, A.; Bedogni, G.; Baviera, G.; Giorgio, V.; Porro, E.; Paris, C.; Giammaria, P.; Reali, L.; Anania, F.; Nobili, V. Randomised clinical trial: The beneficial effects of VSL#3 in obese children with non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther. 2014, 39, 1276–1285. [Google Scholar] [CrossRef] [PubMed]
- Kobyliak, N.; Abenavoli, L.; Mykhalchyshyn, G.; Kononenko, L.; Boccuto, L.; Kyriienko, D.; Dynnyk, O. A Multi-strain Probiotic Reduces the Fatty Liver Index, Cytokines and Aminotransferase levels in NAFLD Patients: Evidence from a Randomized Clinical Trial. J. Gastrointest. Liver Dis. 2018, 27, 41–49. [Google Scholar] [CrossRef] [Green Version]
- Eslamparast, T.; Poustchi, H.; Zamani, F.; Sharafkhah, M.; Malekzadeh, R.; Hekmatdoost, A. Synbiotic supplementation in nonalcoholic fatty liver disease: A randomized, double-blind, placebo-controlled pilot study. Am. J. Clin. Nutr. 2014, 99, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Smits, L.P.; Bouter, K.E.; de Vos, W.M.; Borody, T.; Nieuwdorp, M. Therapeutic Potential of Fecal Microbiota Transplantation. Gastroenterology 2013, 145, 946–953. [Google Scholar] [CrossRef]
- Lechner, S.; Yee, M.; Limketkai, B.N.; Pham, E.A. Fecal Microbiota Transplantation for Chronic Liver Diseases: Current Understanding and Future Direction. Dig. Dis. Sci. 2020, 65, 897–905. [Google Scholar] [CrossRef] [Green Version]
- Issandou, M.; Bouillot, A.; Brusq, J.-M.; Forest, M.-C.; Grillot, D.; Guillard, R.; Martin, S.; Michiels, C.; Sulpice, T.; Daugan, A. Pharmacological inhibition of Stearoyl-CoA Desaturase 1 improves insulin sensitivity in insulin-resistant rat models. Eur. J. Pharmacol. 2009, 618, 28–36. [Google Scholar] [CrossRef]
- Walle, P.; Takkunen, M.; Männistö, V.; Vaittinen, M.; Lankinen, M.; Kärjä, V.; Käkelä, P.; Ågren, J.; Tiainen, M.; Schwab, U.; et al. Fatty acid metabolism is altered in non-alcoholic steatohepatitis independent of obesity. Metabolism 2016, 65, 655–666. [Google Scholar] [CrossRef]
- Fernández-Ramos, D.; Lopitz-Otsoa, F.; Delacruz-Villar, L.; Bilbao, J.; Pagano, M.; Mosca, L.; Bizkarguenaga, M.; Serrano-Macia, M.; Azkargorta, M.; Iruarrizaga-Lejarreta, M.; et al. Arachidyl amido cholanoic acid improves liver glucose and lipid homeostasis in nonalcoholic steatohepatitis via AMPK and mTOR regulation. World J. Gastroenterol. 2020, 26, 5101–5117. [Google Scholar] [CrossRef] [PubMed]
- Tong, L. Acetyl-coenzyme A carboxylase: Crucial metabolic enzyme and attractive target for drug discovery. Cell. Mol. Life Sci. 2005, 62, 1784–1803. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Kayali, Z.; Noureddin, M.; Ruane, P.; Lawitz, E.J.; Bennett, M.; Wang, L.; Harting, E.; Tarrant, J.M.; McColgan, B.J.; et al. GS-0976 Reduces Hepatic Steatosis and Fibrosis Markers in Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2018, 155, 1463–1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, F.; Huffman, M.D.; Macedo, A.F.; Moore, T.H.; Burke, M.; Smith, G.D.; Ward, K.; Ebrahim, S.; Gay, H.C. Statins for the primary prevention of cardiovascular disease. Cochrane Database Syst. Rev. 2013, 2021, CD004816. [Google Scholar] [CrossRef]
- Sinha, R.A.; Bruinstroop, E.; Singh, B.K.; Yen, P.M. Nonalcoholic Fatty Liver Disease and Hypercholesterolemia: Roles of Thyroid Hormones, Metabolites, and Agonists. Thyroid 2019, 29, 1173–1191. [Google Scholar] [CrossRef]
- Harrison, S.; Bashir, M.R.; Guy, C.D.; Zhou, R.; Moylan, C.A.; Frias, J.P.; Alkhouri, N.; Bansal, M.B.; Baum, S.; Neuschwander-Tetri, B.A.; et al. Resmetirom (MGL-3196) for the treatment of non-alcoholic steatohepatitis: A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2019, 394, 2012–2024. [Google Scholar] [CrossRef]
- Sinha, R.A.; Bruinstroop, E.; Singh, B.K.; Yen, P.M. Thyroid Hormones and Thyromimetics: A New Approach to Nonalcoholic Steatohepatitis? Hepatology 2020, 72, 770–771. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Sanyal, A.; Harrison, S.A.; Wong, V.W.S.; Francque, S.; Goodman, Z.; Aithal, G.P.; Kowdley, K.V.; Seyedkazemi, S.; Fischer, L.; et al. Cenicriviroc Treatment for Adults with Nonalcoholic Steatohepatitis and Fibrosis: Final Analysis of the Phase 2b CENTAUR Study. Hepatology 2020, 72, 892–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tacke, F. Cenicriviroc for the treatment of non-alcoholic steatohepatitis and liver fibrosis. Expert Opin. Investig. Drugs 2018, 27, 301–311. [Google Scholar] [CrossRef]
- Harrison, S.A.; Neff, G.; Guy, C.D.; Bashir, M.R.; Paredes, A.H.; Frias, J.P.; Younes, Z.; Trotter, J.F.; Gunn, N.T.; Moussa, S.E.; et al. Efficacy and Safety of Aldafermin, an Engineered FGF19 Analog, in a Randomized, Double-Blind, Placebo-Controlled Trial of Patients with Nonalcoholic Steatohepatitis. Gastroenterology 2021, 160, 219–231. [Google Scholar] [CrossRef]
- Loomba, R.; Ling, L.; Dinh, D.M.; DePaoli, A.M.; Lieu, H.D.; Harrison, S.A.; Sanyal, A.J. The Commensal Microbe V eillonella as a Marker for Response to an FGF19 Analog in NASH. Hepatology 2021, 73, 126–143. [Google Scholar] [CrossRef]
- Pedrosa, M.; Seyedkazemi, S.; Francque, S.; Sanyal, A.; Rinella, M.; Charlton, M.; Loomba, R.; Ratziu, V.; Kochuparampil, J.; Fischer, L.; et al. A randomized, double-blind, multicenter, phase 2b study to evaluate the safety and efficacy of a combination of tropifexor and cenicriviroc in patients with nonalcoholic steatohepatitis and liver fibrosis: Study design of the TANDEM trial. Contemp. Clin. Trials 2020, 88, 105889. [Google Scholar] [CrossRef] [Green Version]
- Fiorucci, S.; Biagioli, M.; Baldoni, M.; Ricci, P.; Sepe, V.; Zampella, A.; Distrutti, E. The identification of farnesoid X receptor modulators as treatment options for nonalcoholic fatty liver disease. Expert Opin. Drug Discov. 2021, 16, 1193–1208. [Google Scholar] [CrossRef]
- Bril, F.; Biernacki, D.M.; Kalavalapalli, S.; Lomonaco, R.; Subbarayan, S.K.; Lai, J.; Tio, F.; Suman, A.; Orsak, B.K.; Hecht, J.; et al. Role of Vitamin E for Nonalcoholic Steatohepatitis in Patients With Type 2 Diabetes: A Randomized Controlled Trial. Diabetes Care 2019, 42, 1481–1488. [Google Scholar] [CrossRef]
- Dougherty, J.A.; Guirguis, E.; Thornby, K.-A. A Systematic Review of Newer Antidiabetic Agents in the Treatment of Nonalcoholic Fatty Liver Disease. Ann. Pharmacother. 2020, 55, 65–79. [Google Scholar] [CrossRef]
- Pockros, P.J.; Fuchs, M.; Freilich, B.; Schiff, E.; Kohli, A.; Lawitz, E.J.; Hellstern, P.A.; Owens-Grillo, J.; Van Biene, C.; Shringarpure, R.; et al. Control: A randomized phase 2 study of obeticholic acid and atorvastatin on lipoproteins in nonalcoholic steatohepatitis patients. Liver Int. 2019, 39, 2082–2093. [Google Scholar] [CrossRef]
- Alkhouri, N.; Lawitz, E.; Noureddin, M.; DeFronzo, R.; Shulman, G.I. GS-0976 (Firsocostat): An investigational liver-directed acetyl-CoA carboxylase (ACC) inhibitor for the treatment of non-alcoholic steatohepatitis (NASH). Expert Opin. Investig. Drugs 2020, 29, 135–141. [Google Scholar] [CrossRef]
- Zein, C.O.; Yerian, L.M.; Gogate, P.; Lopez, R.; Kirwan, J.P.; Feldstein, A.E.; McCullough, A.J. Pentoxifylline improves nonalcoholic steatohepatitis: A randomized placebo-controlled trial. Hepatology 2011, 54, 1610–1619. [Google Scholar] [CrossRef]
- Li, Y.; Liu, L.; Wang, B.; Wang, J.; Chen, D. Metformin in non-alcoholic fatty liver disease: A systematic review and meta-analysis. Biomed. Rep. 2013, 1, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Ćulafić, M.; Vezmar-Kovačević, S.; Dopsaj, V.; Oluic, B.; Bidzic, N. Pentoxifylline with metformin treatment improves biochemical parameters in patients with nonalcoholic steatohepatitis. J. Med. Biochem. 2020, 39, 290–298. [Google Scholar] [CrossRef]
- Jeong, S.W. Nonalcoholic Fatty Liver Disease: A Drug Revolution Is Coming. Diabetes Metab. J. 2020, 44, 640–657. [Google Scholar] [CrossRef]
- Li, K.; Zhang, K.; Wang, H.; Wu, Y.; Chen, N.; Chen, J.; Qiu, C.; Cai, P.; Li, M.; Liang, X.; et al. Hrd1-mediated ACLY ubiquitination alleviate NAFLD in db/db mice. Metabolism 2020, 114, 154349. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Pathway | Mechanism of Action | Drug |
|---|---|---|
| Metabolism | Farnesoid X receptor(FXR) agonists | Obeticholic acid (OCA) |
| Tropifexor | ||
| Cilofexor | ||
| Nidufexor | ||
| Peroxisome proliferator-activated receptor (PPARs) agonists | Pioglitazone | |
| Elafibranor | ||
| Saroglitazar | ||
| Lanifibranor | ||
| Acetyl-CoA carboxylase inhibition | Aramchol | |
| Firsocostat | ||
| Glucagon-like peptide-1(GLP-1) agonists | Liraglutide | |
| Semaglutide | ||
| Thyroid hormone receptor beta (TRβ) agonists | Resmetirom | |
| Fibroblast growth factor-19 (FGF19) analogue | Aldafermin | |
| Sodium/glucose transport protein 2 (SGLT2) inhibitors | Dapagliflozin | |
| Empagliflozin | ||
| Inflammation | CCL receptor type 2 (CCR2) and type 5 (CCR5) antagonists | Cenicriviroc |
| Gut- liver axis | Probiotics, symbiotics | VSL3 Multi-strain probiotic (14 probiotic bacteria genera Bifidobacterium, Lactobacillus, Lactococcus, Propionibacterium) Synbiotic (7 probiotic strains Lactobacillus casei, L. bulgaricus, Lactobacillus rhamnosus, Lactobacillus acidophilus, Bifidobacterium breve, B. longum, and S. thermophilus) |
| Cell death | Antioxidant | Vitamin E |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Filipovic, B.; Lukic, S.; Mijac, D.; Marjanovic-Haljilji, M.; Vojnovic, M.; Bogdanovic, J.; Glisic, T.; Filipovic, N.; Al Kiswani, J.; Djokovic, A.; et al. The New Therapeutic Approaches in the Treatment of Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2021, 22, 13219. https://doi.org/10.3390/ijms222413219
Filipovic B, Lukic S, Mijac D, Marjanovic-Haljilji M, Vojnovic M, Bogdanovic J, Glisic T, Filipovic N, Al Kiswani J, Djokovic A, et al. The New Therapeutic Approaches in the Treatment of Non-Alcoholic Fatty Liver Disease. International Journal of Molecular Sciences. 2021; 22(24):13219. https://doi.org/10.3390/ijms222413219
Chicago/Turabian StyleFilipovic, Branka, Snezana Lukic, Dragana Mijac, Marija Marjanovic-Haljilji, Marko Vojnovic, Jelena Bogdanovic, Tijana Glisic, Natasa Filipovic, Jamal Al Kiswani, Aleksandra Djokovic, and et al. 2021. "The New Therapeutic Approaches in the Treatment of Non-Alcoholic Fatty Liver Disease" International Journal of Molecular Sciences 22, no. 24: 13219. https://doi.org/10.3390/ijms222413219