
Phylogenetic Relationships and Adaptation in Deep-Sea Mussels: Insights from Mitochondrial Genomes


1
Department of Ocean Science, Hong Kong Branch of the Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou), The Hong Kong University of Science and Technology, Hong Kong 93117, China
2
Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou), Guangzhou 510225, China
3
Department of Biology, Hong Kong Baptist University, Hong Kong 93117, China
*
Authors to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(4), 1900; https://doi.org/10.3390/ijms22041900
Submission received: 14 January 2021
/
Revised: 31 January 2021
/
Accepted: 8 February 2021
/
Published: 14 February 2021
(This article belongs to the Section Molecular Genetics and Genomics)
Abstract
:Mitochondrial genomes (mitogenomes) are an excellent source of information for phylogenetic and evolutionary studies, but their application in marine invertebrates is limited. In the present study, we utilized mitogenomes to elucidate the phylogeny and environmental adaptation in deep-sea mussels (Mytilidae: Bathymodiolinae). We sequenced and assembled seven bathymodioline mitogenomes. A phylogenetic analysis integrating the seven newly assembled and six previously reported bathymodioline mitogenomes revealed that these bathymodiolines are divided into three well-supported clades represented by five Gigantidas species, six Bathymodiolus species, and two “Bathymodiolus” species, respectively. A Common interval Rearrangement Explorer (CREx) analysis revealed a gene order rearrangement in bathymodiolines that is distinct from that in other shallow-water mytilids. The CREx analysis also suggested that reversal, transposition, and tandem duplications with subsequent random gene loss (TDRL) may have been responsible for the evolution of mitochondrial gene orders in bathymodiolines. Moreover, a comparison of the mitogenomes of shallow-water and deep-sea mussels revealed that the latter lineage has experienced relaxed purifying selection, but 16 residues of the atp6, nad4, nad2, cob, nad5, and cox2 genes have underwent positive selection. Overall, this study provides new insights into the phylogenetic relationships and mitogenomic adaptations of deep-sea mussels
1. Introduction
Mytilidae is a family of highly diverse mussels that are widely distributed from freshwater to marine water and from coastal areas to the deep-sea [1,2]. This family is currently divided into seven subfamilies. Although most of these subfamilies are represented by shallow-water species, the subfamily Bathymodiolinae consists of exclusively deep-sea species [2,3]. A previous study has established that Bathymodiolinae split from its shallow-water sister group Modiolinae roughly 110.4 Million years ago (Ma) [4], but given the difficulty in sampling these deep-sea animals, little is known about their biology. Members of Bathymodiolinae, especially those belonging to the genera Bathymodiolus, Gigantidas, Idas, and Tamu, are often conspicuous animals in chemosynthesis-based ecosystems, including hydrothermal vents, cold seeps, and organic falls [5,6]. Despite the lack of phytoplankton-derived food in the deep-sea, these mussels thrive in the special deep-sea habitats due to their symbiosis with bacteria that are dependent on simple organic molecules, such as methane and hydrogen sulfide, as a source of energy [1,4]. Owing to their ecological importance and remarkable biological characteristics, deep-sea mussels have been considered a suitable animal model for studying adaptation and symbiosis [4,7,8,9,10].
The taxonomy of Bathymodiolinae is in disarray because of their high morphological plasticity. Molecular phylogenetic studies have divided deep-sea mussels previously referred to as Bathymodiolus into nine genera, namely, Gigantidas, Bathymodiolus, Adipicola, Benthomodiolus, Idas, Tamu, Terua, Vulcanidas, and “Bathymodiolus” [5,11,12]. “Bathymodiolus”, which is currently represented by only two species (“B.” aduloides and “B.” manusensis), is vastly different from Bathymodiolus sensu stricto, but it has not been given a formal genus name yet [6,12,13].
Previous phylogenetic studies of deep-sea mussels mainly used one or very few gene fragments. Consequently, they provided limited information on the evolutionary history of these animals. In the present study, we examined the phylogenetics and evolution of the deep-sea mussels based on mitochondrial genomes (mitogenomes). Mitogenomes carry useful evolutionary information and have been widely applied in phylogenetic and evolutionary studies of terrestrial and shallow-water organisms, including mussels [14,15,16,17]. Given that the mitochondria play a key role in the energetic metabolism of metazoans, the hypoxia and high hydrostatic pressure environments in cold seeps and hydrothermal vents could exert selective pressure on the evolution of these energy-producing organelles [18,19,20,21]. However, only six Bathymodiolinae mitogenomes are currently available, thereby hindering our understanding of their evolution.
In the present study, we sequenced seven complete Bathymodiolinae mitogenomes and analyzed them together with the previously published mitogenomes to infer their phylogenetic relationships, understand their gene order rearrangement patterns, determine their divergence times, and ascertain the adaptive evolution of protein-coding genes (PCGs).
2. Results
2.1. Genome Organizations and Gene Rearrangement
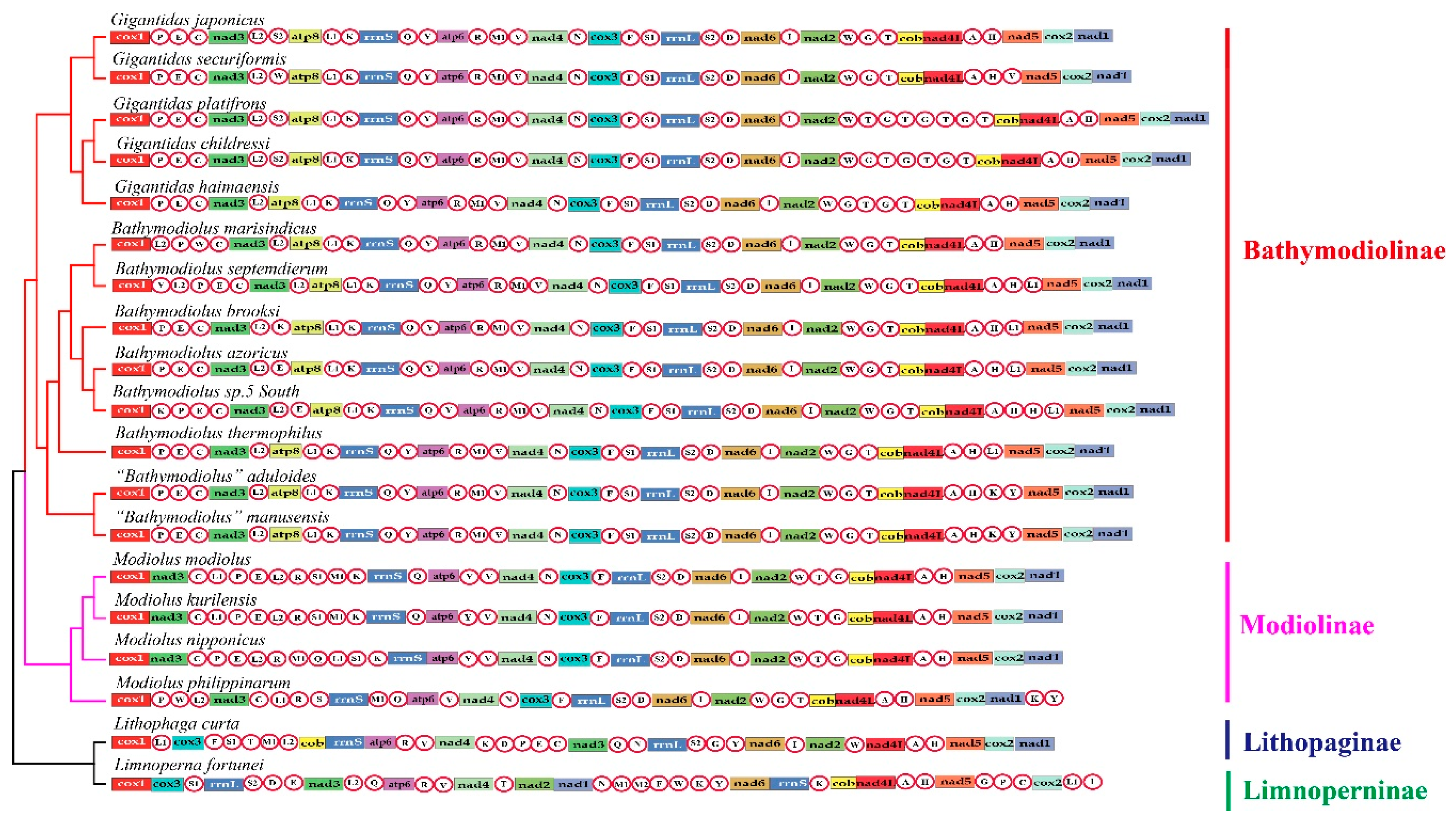
The size of the seven newly assembled mitogenomes ranged from 17,138 bp (B. marisindicus) to 18,376 bp (B. sp. 5 South) (Table 1). They all included 13 PCGs and two rRNA genes. However, the number of tRNA genes varied (23–28), an observation consistent with that reported for other groups of bivalves [22,23]. Within the Gigantidas group, some species had multiple copies of trnT and trnG: G. platifrons had three copies of trnG and four copies of trnT, G. childressi had three copies of trnG and trnT, and G. haimaensis had two copies of trnG and trnT. These copies of trnT and trnG were located between trnW and cob. Within the Bathymodiolus group, all species had two copies of trnL1, except for B. marisindicus. Both species of the “Bathymodiolus” group had two copies of trnK and trnY. A comparison among the bathymodioline mitogenomes revealed that they had a highly conserved gene order, except for the translocation or inversion of some tRNAs (Figure 1). The four Modiolus species and L. curta from shallow seawater and L. fortunei from freshwater lacked the atp8 gene (Figure 1). Regions homologous to the atp8 gene were detected in the mitogenomes of four Modiolus species using the Align by Muscle model implemented in MEGA v.7.0. These regions were located between nad1 and cox1, but the gene structure was incomplete.
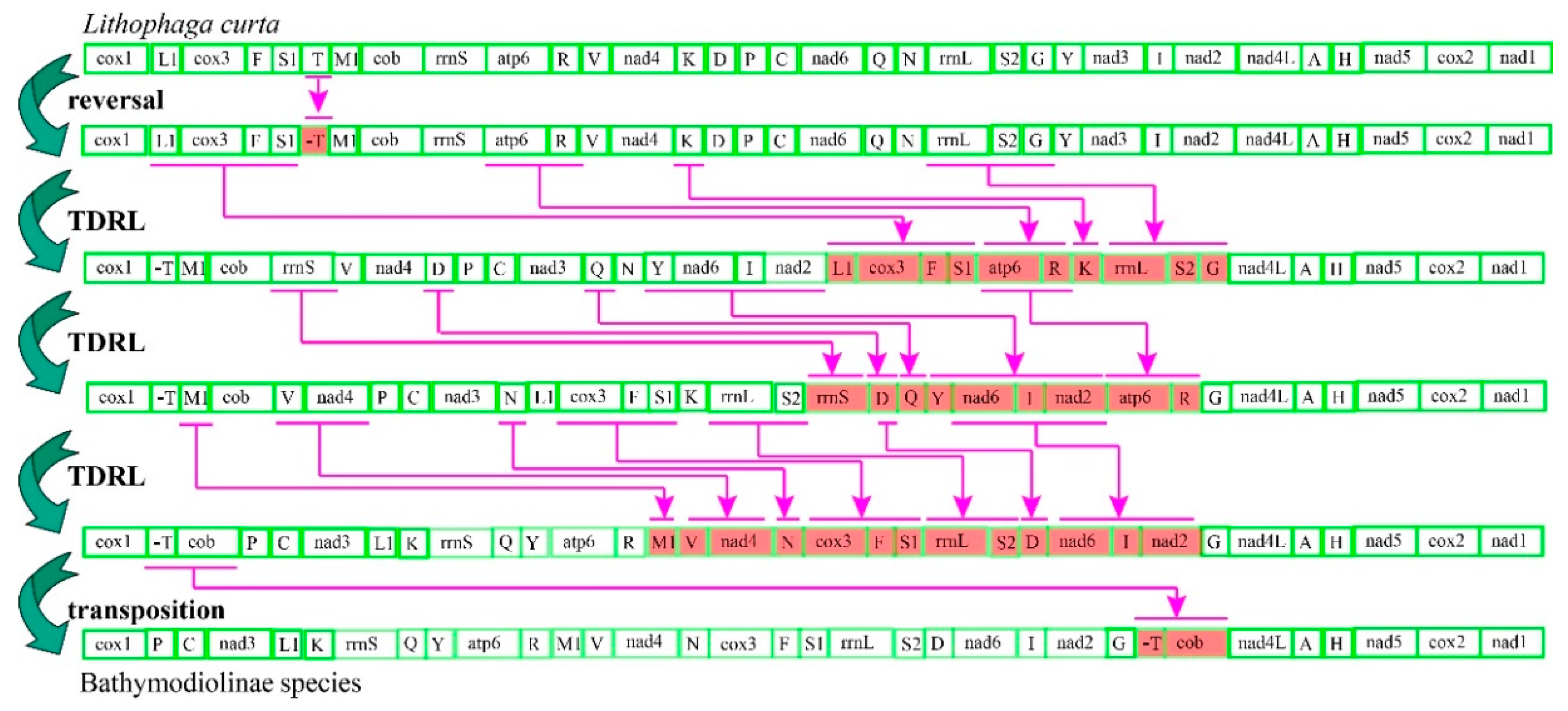
Common interval Rearrangement Explorer (CREx) analysis was performed to determine the likely gene order rearrangement events that occurred during the evolution of deep-sea mussels. After incomplete and duplicated gene constitutions were removed, six unique gene arrangements among the 19 mitogenomes analyzed were detected. All bathymodioline mitogenomes possessed a gene order notably different from that of other mussels. Three gene order rearrangements were detected in the four Modiolus species, indicating that this group has a higher diversity of gene orders than the other groups. We designated the gene order of L. curta as the ancestral Bathymodiolinae gene order for CREx analysis because it is an outgroup taxon that shares a similar gene order with Bathymodiolinae and Modiolinae species [2]. CREx analysis suggested that the distinct gene order of bathymodioline mussels might have evolved from the putative ancestral bathymodioline gene order through five evolutionary steps (Figure 2). These steps included one reversal, one transposition, and three complex tandem duplications with subsequent random gene losses (TDRLs). Aside from the recurrent rearrangements of tRNAs, rearrangements were also observed in the PCGs. Specifically, the gene cluster cox3+trnF+trnS1 moved from between trnL1 and trnT to a position between trnN and rrnL, nad3 moved from between trnY and trnI to a position between trnC and trnL1, and cob moved from between trnM1 and rrnS to between trnT and nad4L. When the tRNA genes were excluded from the comparisons, three conserved gene blocks (A, trnV–nad4–trnN–cox3–trnF; B, rrnL–trnS2–trnD–nad6-I–nad2; C, cob–nad4L–trnA–trnH–nad5–cox2–nad1) were identified in the mitogenomes of Bathymodiolinae and Modiolinae species.
2.2. Phylogenetic Relationships and Divergence Times
A total of 3243 amino acid positions were found in the aligned sequence dataset. The best sequence evolution models identified herein by using PartitionFinder included JTT+G+F for atp6, nad2, nad3, nad4L, nad5, and nad6; LG+G+F for cox2, cox3, and nad1; and MTART+G+F for cob, cox1, and nad4 (Table S1).
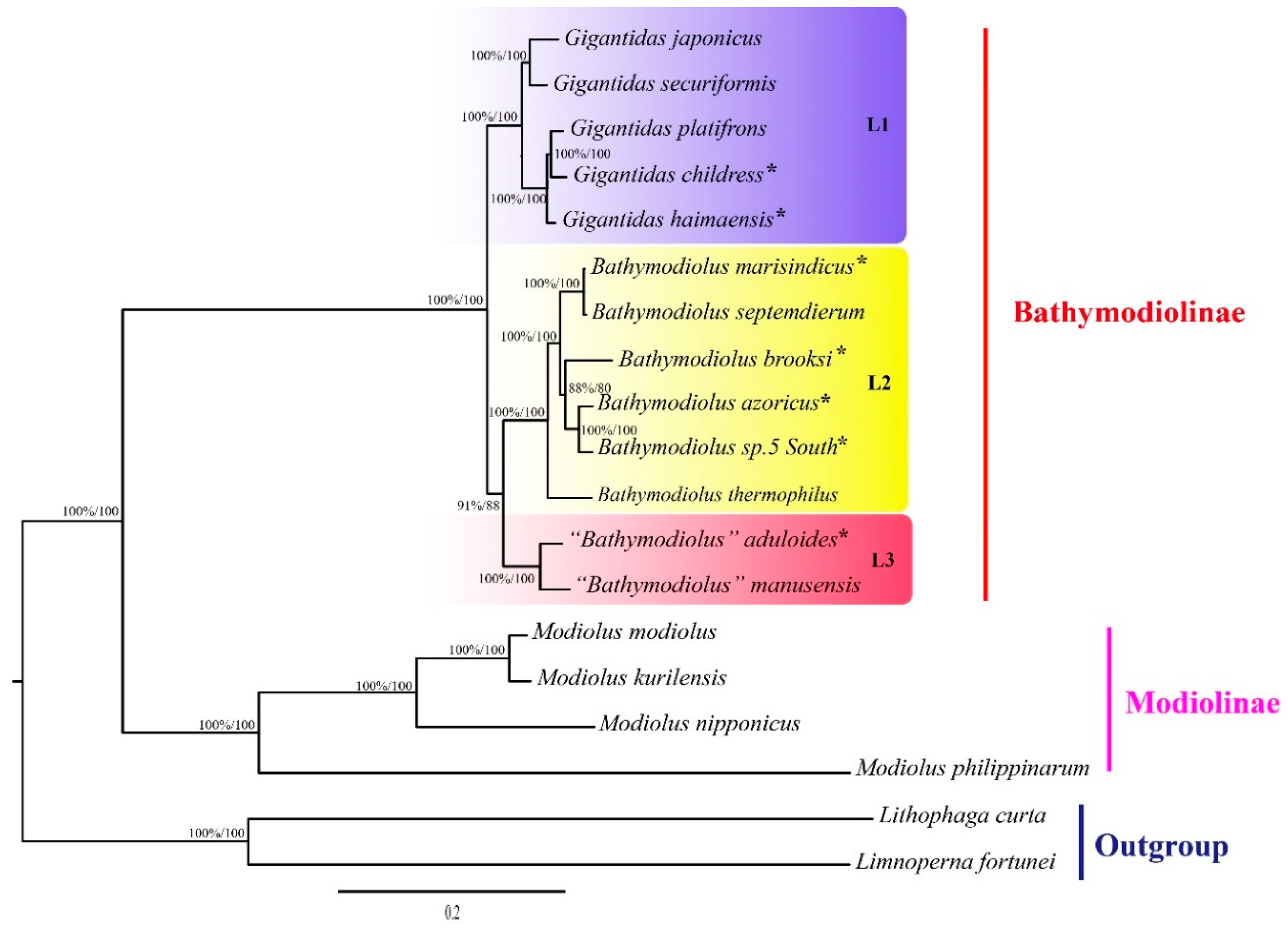
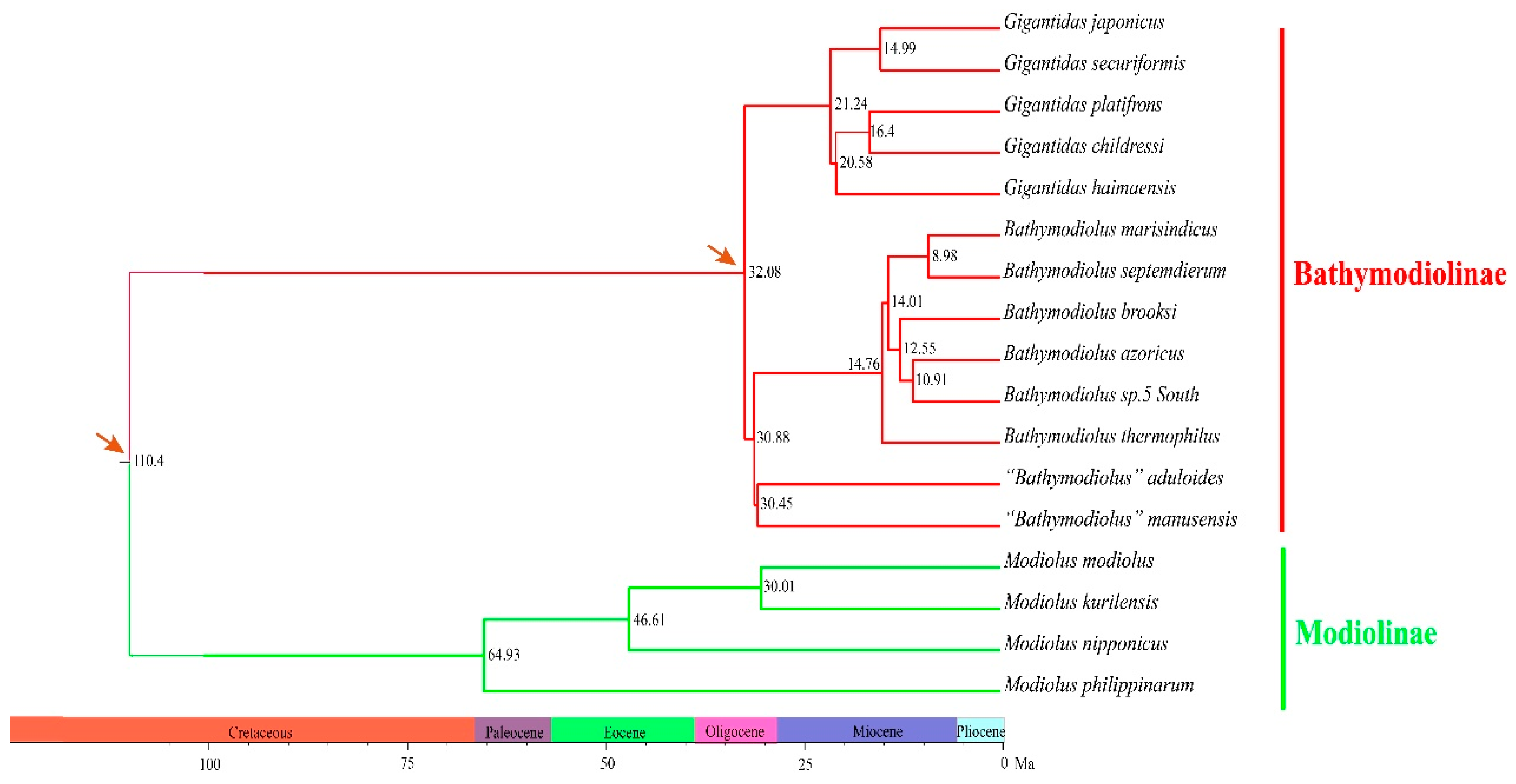
The phylogenetic trees constructed using both the maximum likelihood (ML) and Bayesian inference (BI) methods consistently showed that bathymodioline mussels analyzed were divided into three separate clades (L1, L2, and L3) with high support values (Figure 3). Molecular dating results indicated that the three clades diversified in the last 32.08 Ma (Figure 4). The five Gigantidas species formed the L1 clade, which included species from cold seeps and those that harbor mainly methane-oxidizing symbionts. The Gigantidas clade diverged from the other bathymodiolines, approximately 21.24 Ma. The L2 clade consisted of six Bathymodiolus species, including B. marisindicus, B. septemdierum, and B. azoricus, as well as a species from hydrothermal vents that has not been formally described (Bathymodiolus sp. 5 South), and B. brooksi from cold seeps. Two species from hydrothermal vents, namely, “Bathymodiolus” aduloides, and “Bathymodiolus” manusensis, comprised the L3 clade. These results are consistent with those of a previous study that showed that these two species do not belong to Bathymodiolus [6] but had higher bootstrap values in the tree. The present estimate for the divergence between “Bathymodiolus” mussels and Bathymodiolus mussels was 30.88 Ma (Figure 4).
2.3. Genetic Distance
The Kimura-2-parameter (K2P) distance of the cox1 sequences of the bathymodiolines analyzed herein varied from 0.8% to 18.85%, whereas that for Modiolus species ranged from 12.33% to 33.36% (Table S2). The K2P distance of the combined PCG sequences varied from 1.04% to 23.16% for the bathymodiolines and from 11.39% to 44.82% for the modiolines. Among the bathymodiolines, the smallest genetic distance was between B. marisindicus and B. septemdierum regardless if it was based on cox1 or the combined PCG sequences. Based on the cox1 gene, the highest genetic distances among the three major bathymodioline clades were 17.99% (between the L1 and L2 clades), 16.58% (between the L1 and L3 clades), and 18.85% (between the L2 and L3 clades).
2.4. Elevated Nucleotide Substitution Rates in Deep-Sea Mussels
CodeML from the PAML package was used to evaluate whether positive selection might have contributed to the adaptation of mussels to deep-sea environments. No significant differences were observed between the one- and two-ratio models of ω (Ka/Ks) for all PCGs (Table S3). This result suggested that ω did not change more quickly than expected along the Bathymodiolinae branch. Moreover, the ω values of all mitochondrial PCGs in both models were substantially lower than 1, ranging from 0.00105 to 0.14038 for all PCGs for the deep-sea bathymodiolines and from 0.00239 to 0.06698 for the shallow-water mussels. Nevertheless, compared with shallow-water mussels, the deep-sea mussels had higher ω values for all PCGs, except for cox1.
Branch-site models were employed to detect positively selected sites in the mitogenomes of deep-sea mussels. Sixteen residues located on atp6, nad4, nad2, cob, nad5, and cox2 were detected as positively selected sites along the Bathymodiolinae branch (>95%) (Table 2). These results suggested that the bathymodiolines were affected more heavily than the shallow-water species after they diverged from their common ancestor.
3. Discussion
3.1. General Features of Bathymodiolinae Mitogenomes
Seven complete Bathymodiolinae mitogenomes ranging in length from 17,138 bp (B. marisindicus) to 18,376 bp (B. sp. 5 South) were newly assembled in the present study. This narrow range of genome size was consistent with that of a previous study that reported that the size of the mitogenomes of four other bathymodioline species ranging from 17,069 bp to 18,819 bp [2,24]. The variations in genome size were mainly attributed to the size differences in the control region [2], ranging from 469 bp in B. marisindicus to 1963 bp in G. haimaensis. The arrangement and number of tRNA genes are highly variable among different mussel subfamilies, which is also true for other bivalves [2,22]. Nevertheless, when tRNA genes are not included, the mitogenomes of mussels in Bathymodiolinae and Modiolinae have similar gene contents and gene order arrangements. However, although atp8 is present in most of the sequenced mitogenomes of mussels in other subfamilies of Mytilidae, including all species of deep-sea mussels, this gene is missing in some species of Modiolinae, Lithophaginae, Limnopernimae, and Brachidontinae [2], or has become a pseudogene [25].
3.2. Molecular Phylogeny of Deep-Sea Mussels
The topologies inferred from the amino acid sequences of PCGs via the BI and ML methods were consistent. Results showed that the deep-sea mussels investigated could be divided into three clades (L1, L2, and L3) with high branch support values (Figure 3). These results are consistent with the findings of earlier studies [5,12], which support the classification of deep-sea mussels that species in the L3 clade belong to Nipponiomodiolus and those in the L1 clade belong to Gigantidas [6,12,13]. Nevertheless, the present results do not completely concur with those of previous studies that analyzed a combined dataset of mitochondrial and nuclear gene sequences [3,6]. For instance, the present study placed Gigantidas (L1 clade) as the sister group of (L2 + L3); in contrast, a recent molecular analysis using a combined dataset of mitochondrial and nuclear gene sequences indicates that this genus is closely clustered with the L2 clade [3]. Moreover, B. aduloides and B. manusensis (L3 clade) form a sister group to the L2 clade. This result is consistent with that of our previous works [12,13] but in stark contrast to that of some earlier phylogenetic analyses of few mitochondrial and nuclear datasets that argued that these two species are more closely related to the L1 clade (Gigantidas) [5,26]. Furthermore, the tree topology test rejected the hypothesis that the L3 clade is sister to the L1 clade (Table S4).
CREx analysis revealed that all bathymodioline species analyzed herein had identical gene order arrangements, a remarkable result because the gene orders in many other groups of Mytilidae substantially vary [2]. These deep-sea bathymodiolines are phylogenetically closely related, indicating that their gene orders have not changed since their common ancestor diverged from other lineages of mussels. The K2P genetic distance between the bathymodiolines analyzed herein varied from 0.8% to 18.95% for cox1 and from 1.04% to 23.16% for all PCGs. These results demonstrated that these bathymodiolines (L1 + L2 + L3) are a monophyletic group, consistent with the findings of previous studies [3,13,26]. Unlike Bathymodionae species, which have only one gene order, the Modiolinae species analyzed herein have three gene orders, and their K2P genetic distances varied from 11.39% to 44.82% for PCGs. Nevertheless, the taxonomic distribution of the available samples in this study was biased toward Bathymodiolinae, and several genera of this subfamily were not included in the analysis due to the lack of access to specimens.
DNA barcoding is widely used for species delimitation [27]. Thubaut et al. (2013) used a 2.0% K2P genetic distance as the threshold for species delimitation in bathymodioline mussels [6]. However, among the recognized species, the genetic divergence between B. septemdierum and B. marisindicus is 0.8% for the cox1 gene and 1.04% for all PCGs (Table S2). Therefore, these two species are likely conspecific [26,28].
3.3. Mitochondrial Gene Rearrangement
Mollusks have been utilized as animal models for investigating mitogenome gene order rearrangements [29,30]. In the present study, a novel gene order was observed in the mitogenomes of deep-sea Bathymodiolinea mussels that greatly differed from the gene orders of other mussel groups. Although these deep-sea mussels have an identical mitochondrial gene order, the shallow-water mussels used in this study possess three mitochondrial gene orders, suggesting only one rearrangement following the divergence of bathymodiolines from the other mussels. Given that the mitochondrial gene order in deep-sea Bathymodiolinae species is different from that of shallow-water modioline mussels, including Modiolus and Lithophaga, we speculate that this unique gene order rearrangement pattern might have occurred after the shallow-water mussels invaded the deep-sea. Future studies should examine the mitochondrial gene order of other genera of deep-sea mussels, especially Benthomodiolus species, which usually inhabit sunken woods or whale falls that are considered as transitional habits to the most specialized vent and seep habitats [5,6].
Mitochondrial gene order arrangement involves four gene rearrangement types, namely, transpositions, reverse transpositions, inversions, and TDRL [31,32]. CREx analysis suggested that transposition and TDRL are associated with the evolution of the mitogenomes of deep-sea mussels. These substantial rearrangements indicated that dramatic mitogenome organizations occurred during the invasion of the deep-sea by mussels. This result is consistent with that of other studies of deep-sea species showing their gene orders were also altered, including tRNA and PCG transportation or gene cluster invasion, during their invasion into the deep-sea [33,34,35]. How such gene order rearrangements might be adaptive to deep-sea mussels is unknown. Nevertheless, gene recombination has been suggested to enhance the survival of deep-sea species by offsetting the high mutational rates of mitochondrial DNA [17,36].
3.4. Adaptations to Deep-Sea Environments
As reported by earlier studies, mitochondrial PCGs may experience positive selection in deep-sea animals and thus may help them adjust their metabolism to tolerate the deep-sea conditions [37,38,39]. In the present study, potential positive selection was evaluated in deep-sea mussels by using CodeML in the PAML package. Analyses of branch models showed that the Ka/Ks ratios for all PCGs in both one- and two-ratio models were substantially lower than 1 (Table S3), implying strong purifying selection has driven the evolution of the mitogenomes of these mussels. In addition, the ω values for all PCGs, except for the cox1 gene, in deep-sea mussels were higher than those in sublittoral species, indicating that the mitogenomes of deep-sea mussels underwent a more relaxed purifying selection. Low ω values for mitochondrial PCGs were also reported in other deep-sea animals, including deep-sea vesicomyids [3,21], a giant Bathynomus sp. [40], and deep-sea polynoids [35]. Moreover, previous studies found that positive selection usually occurs within a short period of evolutionary time and acts on only a few sites. Thus, the sparse signals of positive selection are usually overwhelmed by those for continuous purifying selection on most sites in a gene sequence [41,42].
Branch-site models are used to identify possible positively selected sites in deep-sea mussels. Results suggested that 16 residues located in the atp6, nad4, nad2, cob, nad5, and cox2 genes could have experienced positive selection along the branch ancestral to Bathymodiolinae. As a proton pump, the NADH dehydrogenase complex is the largest and foremost enzyme complex of the respiratory chain. The efficiency of proton pumping procedures can be affected by protein mutation, and thus it may be crucial to adaptive evolution [43,44]. In the deep-sea shrimp family Alvinocarididae, the greatest residues of positively selected sites are within nad1-5 [39], a gene also known to be related to deep-sea hydrothermal vent adaptation. The nad3 and nad5 genes in the mitogenome of the deep-sea crab Chaceon granulates also harbor positively selected residues [20]. Similarly, 11 residues are considered positively selected in the nad2 and nad4 genes of the deep-sea sea cucumber Benthodytes marianensis [39]. Cytochrome c oxidase stimulating the terminal reduction of oxygen and with three mitochondrial PCGs (cox1, cox2, and cox3) that encode the catalytic core is an important positive selection target in hypoxia adaptation [45,46]. In deep-sea clams belonging to the family Vesicomyidae, positively selected residues are found in the cox1 and cox3 genes [20]. The ATP synthase Fo subunit 6 or complex V drives the last step of oxidative phosphorylation for electron transport chain. Evidence supporting the adaptive evolution of the atp6 gene has been reported in the mitogenome of Glyptothorax macromaculatus [47], deep-sea fish [19], and deep-sea polynoids [20]. Although cob is a conserved gene, it is crucial to the ability of the mitochondria to generate energy through reversible electron transfer from ubiquinol to cytochrome c along with proton translocation. cob was shown to have undergone positive selection in deep-sea fish [19] and deep-sea clams [21]. Therefore, mitochondrial genes, particularly atp6, nad4, nad2, cob, nad5, and cox2, may help deep-sea mussels to survive and/or thrive under harsh deep-sea conditions.
4. Materials and Methods
4.1. Acquisition of Mitochondrial Genome Sequences
We analyzed the mitogenomes of 19 species of mussels, 13 of which were deep-sea bathymodiolines (Table 1). Six of the mitogenomes were downloaded from GenBank, whereas seven were newly assembled herein. Out of the newly assembled mitogenomes, four were based on DNA sequences downloaded from GenBank (Bathymodiolus sp. 5 South (ERP115508), B. azoricus (ERP105025), B. brooksi (SRP178172), G. childressi (ERP021949)), and three were based on DNA sequences produced herein: “Bathymodiolus” aduloides was collected from the F-site methane seep in the South China Sea (22°06.921′ N, 119°17.131′ E, depth 1122 m), B. marisindicus was gathered from the Longqi hydrothermal vent field in the Southwest Indian Ocean Ridge (37°47′ S 49°39′ E, depth 2800 m), and G. haimaensis was obtained from the Haima Cold Seep in the South China Sea (16°44′ N, 110°29′ E, depth 1390 m). The adductor muscle of an individual of these three species was dissected, and its DNA was extracted using the DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany). DNA library construction and sequencing were conducted by Novogene, Beijing. Sequencing was conducted in paired-end mode on an Illumina platform to produce approximately 5 Gb of reads of 150 bp read length. Whole genome sequencing datasets were assembled using NOVOPlasty v.3.8.3 under default settings [48].
4.2. Genome Sequence Annotation and Gene Arrangement Analysis
The MITOS webserver was used to annotate the mitogenomes assembled herein [49]. The boundaries of 13 PCGs and two ribosomal RNA (rRNA) genes were determined by comparing them with the homologous genes of other bathymodioline species. Transfer RNA (tRNA) genes were predicted using the programs MiTFi [50] in the MITOS pipeline [49], ARWEN v1.2.3.c [51], and tRNAscan-SE v1.21 [52]. The gene order of Lithophaga curta mitogenome was considered as the ancestral gene order of Bathymodiolinae. Pairwise comparisons of mitogenomes were conducted using Common interval Rearrangement Explorer (CREx) [53] to reconstruct the likely gene order rearrangement events (i.e., reversal, transposition, reverse transposition, tandem duplication/random loss (TDRL)) that might have occurred during the evolution of this lineage of mussels.
4.3. Phylogenetic Analyses and Divergence Time Estimation
Out of the 19 mitochondrial genome sequences used for phylogenetic analyses, two were outgroups (L. curta and Limnoperna fortunei). The amino acid sequences of 12 PCGs were allied separately by using MAFFT v.7.407 [54] under defaults settings. The atp8 gene was not included in the analysis because shallow-water mussels lack this gene. Poorly aligned positions were removed using Gblocks v.0.91b [55]. The best-fit substitution models for the dataset and the best partition schemes were determined using PartitionFinder version 2.1.1 (Australian National University, Canberra, ACT, Australia) [56].
Phylogenetic relationships were reconstructed via the Maximum Likelihood (ML) method implemented in RAxML v.8.2.9 [57] and the Bayesian inference (BI) method implemented in MrBayes v.3.2.7a [58]. For the ML analysis, 10,000 replicates were employed. For the BI analysis, the Markov chain Monte Carlo (MCMC) method was applied considering a chain for 10 million generations, and a tree was sampled every 500 generations. The initial 25% of the runs were discarded as burn-in. Alternative tree topologies were assessed via the approximately unbiased test implemented in IQ-TREE v.2.0 [59] with 20,000 bootstrap replicates. After phylogenetic tree construction, the timing of species divergences was estimated via the Bayesian method by using MCMCTree in the PAML package v.4.9h [60]. Two nodes were time-calibrated. According to previous studies [4,7], bathymodioline and shallow-water mussels split about 110.4 Ma, and G. childressi and B. thermophilus diverged between 21.12 and 32.98 Ma. For the MCMC analysis, 100,000 samples were applied, and the first 20% of all samples were discarded as burn-in. An independent rate model (clock = 2), which follows a lognormal distribution, was used for the MCMC search. The phylogenetic tree was visualized in FigTree v.1.4.3 [61].
Genetic distances between the species tested herein were computed using the Kimura-2-parameter (K2P) model (Kimura, 1980) implemented in MEGA v.7.0 [62] for both cox1 and the combined mitochondrial PCGs.
4.4. Positive Selection Analysis
Positive selection in the branches leading to the deep-sea Bathymodiolinae was determined using the branch model and the branch-site model in the PAML package [60]. Selection pressure was determined by applying the overall database of 12 mitochondrial PCGs. The topology of the phylogenetic tree generated in the previous section was utilized in this analysis. For the branch model, the one-ratio model (model = 0, assuming a single ω0 ratio for all branches in the phylogenetic tree) was used to assess ω distribution values (dN/dS ratio), which were taken as the basis for the probability of adaptive evolution of gene sequences. Subsequently, the two-ratio model (model = 2, setting the bathymodiolinae branch as foreground lineages, ω2; setting all other branches as background lineages, ω1) was ran. Furthermore, the one- and two-ratio models were compared to investigate whether the clade of deep-sea mussels is under greater selection pressure than shallow-water mussels. If the two-ratio model showed a significantly higher probability than the one-ratio model and ω2 > 1, then the deep-sea mussels were considered to be under positive selection. Afterward, positively selected sites in the deep-sea Bothymodiolinae lineage (marked as foreground lineage) were determined via a branch-site model. Bayesian posterior probability of the positively selected sites was obtained via Bayes Empirical Bayes (BEB) analysis.
5. Conclusions
In summary, the mitogenomes of various genera of Bathymodiolinae were found to have a conserved gene order, which differs remarkably from the gene orders of shallow-water mussels. Our results suggested that gene order rearrangements in bathymodiolines can be explained by reversal, transposition, and TDRL of an ancestral mitogenome. Finally, multiple mitochondrial genes carry signals of positive selections in some amino acid residues in deep-sea mussels, a condition indicating adaptation to deep-sea environments.
Supplementary Materials
The following are available online at https://www.mdpi.com/1422-0067/22/4/1900/s1, Table S1: The best schemes and best-fit substitution model for phylogenetic tree, Table S2: Pairwise genetic distance (%) between species belonging to the genera Gigantidas, Bathymodiolus, Modiolus, Lithophaga and Limnoperna, Table S3: Models of selection pressure on the mitochondrial protein-coding genes of the deep-sea mussels, Table S4: The result of approximately unbiased (AU) test.
Author Contributions
K.Z. conducted the experiments, analyzed the data, wrote the manuscript, and prepared the figures and tables. J.S. and T.X. collected the samples and discussed the experimental results. P.-Y.Q. and J.-W.Q. conceived and designed the experiments, contributed reagents/materials/analysis tools, supervised the work, and reviewed drafts of the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou) (GML2019ZD0409), the Major of Basic and Applied Basic Research Project of Guangdong Province (2019B030302004-04), the Hong Kong Branch of Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou) (SMSEGL20SC01, L20190005), and the China Ocean Mineral Resources Research and Development Association (DY135-E2-1-03).
Institutional Review Board Statement
The animal study was reviewed and approved by the Ethics Committee of the Hong Kong University of Science and Technology. Written informed consent was obtained from the owners for the participation of their animals in this study.
Informed Consent Statement
Not applicable.
Data Availability Statement
The sequence data have been deposited in the NCBI database with accession numbers MT916740, MT916741, MT916742, MT916743, MT916744, MT916745 and MT916746.
Acknowledgments
We thank the captains and crew members of R/V Xiangyanghong 9, Dayangyihao, and R/V Haiyang 6; the pilots of Jiaolong; and the operation team of ROV Sea Dragon III and Sea Horse.
Conflicts of Interest
The authors declare no competing interests.
References
- Distel, D.L.; Baco, A.R.; Chuang, E.; Morrill, W.; Cavanaugh, C.; Smith, C.R. Marine ecology: Do mussels take wooden steps to deep-sea vents? Nature 2000, 403, 725–726. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kwak, H.; Shin, J.; Kim, S.C.; Kim, T.; Park, J.K. A mitochondrial genome phylogeny of Mytilidae (Bivalvia: Mytilida). Mol. Phylogenet. Evol. 2019, 139, 106533. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, H.; Zhang, H. Phylogeny and evolutionary radiation of the marine mussels (Bivalvia: Mytilidae) based on mitochondrial and nuclear genes. Mol. Phylogenet. Evol. 2018, 126, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zhang, Y.; Xu, T.; Zhang, Y.; Mu, H.; Zhang, Y.; Lan, Y.; Fields, C.J.; Hui, J.H.L.; Zhang, W.; et al. Adaptation to deep-sea chemosynthetic environments as revealed by mussel genomes. Nat. Ecol. Evol. 2017, 1, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Lorion, J.; Kiel, S.; Faure, B.; Kawato, M.; Ho, S.Y.; Marshall, B.; Tsuchida, S.; Miyazaki, J.I.; Fujiwara, Y. Adaptive radiation of chemosymbiotic deep-sea mussels. Proc. R. Soc. B-Biol. Sci. 2013, 280, 20131243. [Google Scholar] [CrossRef] [PubMed]
- Thubaut, J.; Puillandre, N.; Faure, B.; Cruaud, C.; Samadi, S. The contrasted evolutionary fates of deep-sea chemosynthetic mussels (Bivalvia, Bathymodiolinae). Ecol. Evol. 2013, 3, 4748–4766. [Google Scholar] [CrossRef]
- Lorion, J.; Buge, B.; Cruaud, C.; Samadi, S. New insights into diversity and evolution of deep-sea Mytilidae (Mollusca: Bivalvia). Mol. Phylogenet. Evol. 2010, 57, 71–83. [Google Scholar] [CrossRef]
- Johnson, S.; Won, Y.-J.; Harvey, J.; Vrijenhoek, R. A hybrid zone between Bathymodiolus mussel lineages from eastern Pacific hydrothermal vents. BMC Evol. Biol. 2013, 13, 21. [Google Scholar] [CrossRef]
- Bougerol, M.; Boutet, I.; LeGuen, D.; Jollivet, D.; Tanguy, A. Transcriptomic response of the hydrothermal mussel Bathymodiolus azoricus in experimental exposure to heavy metals is modulated by the Pgm genotype and symbiont content. Mar. Genomics 2015, 21, 63–73. [Google Scholar] [CrossRef] [Green Version]
- Bettencourt, R.; Pinheiro, M.; Egas, C.; Gomes, P.; Afonso, M.; Shank, T.; Santos, R. High-throughput sequencing and analysis of the gill tissue transcriptome from the deep-sea hydrothermal vent mussel Bathymodiolus azoricus. BMC Genomics 2010, 11, 559. [Google Scholar] [CrossRef] [Green Version]
- Genio, L.; Kiel, S.; Cunha, M.R.; Grahame, J.; Little, C.T. Shell microstructures of mussels (Bivalvia: Mytilidae: Bathymodiolinae) from deep-sea chemosynthetic sites: Do they have a phylogenetic significance? Deep Sea Res. Part I 2012, 64, 86–103. [Google Scholar] [CrossRef]
- Xu, T.; Feng, D.; Tao, J.; Qiu, J.W. A new species of deep-sea mussel (Bivalvia: Mytilidae: Gigantidas) from the South China Sea: Morphology, phylogenetic position, and gill-associated microbes. Deep Sea Res. Part I 2019, 146, 79–90. [Google Scholar] [CrossRef]
- Samadi, S.; Puillandre, N.; Pante, E.; Boisselier, M.C.; Corbari, L.; Chen, W.J.; Maestrati, P.; Mana, R.; Thubaut, J.; Zuccon, D.; et al. Patchiness of deep-sea communities in Papua New Guinea and potential susceptibility to anthropogenic disturbances illustrated by seep organisms. Mar. Ecol. 2015, 36, 109–132. [Google Scholar] [CrossRef] [Green Version]
- Gvoždík, V.; Moravec, J.; Klütsch, C.; Kotlík, P. Phylogeography of the Middle Eastern tree frogs (Hyla, Hylidae, Amphibia) as inferred from nuclear and mitochondrial DNA variation, with a description of a new species. Mol. Phylogenet. Evol. 2010, 55, 1146–1166. [Google Scholar] [CrossRef]
- Lei, R.; Shore, G.D.; Brenneman, R.A.; Engberg, S.E.; Sitzmann, B.D.; Bailey, C.A.; Kimmel, L.M.; Randriamampionona, R.; Ranaivoarisoa, J.F.; Louis, E.E., Jr. Complete sequence and gene organization of the mitochondrial genome for Hubbard’s sportive lemur (Lepilemur hubbardorum). Gene 2010, 464, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.P.; Xin, Z.Z.; Liu, Y.; Zhang, D.Z.; Wang, Z.F.; Zhang, H.B.; Chai, X.Y.; Zhou, C.L.; Liu, Q.N. The complete mitochondrial genome of Sesarmops sinensis reveals gene rearrangements and phylogenetic relationships in Brachyura. PLoS ONE 2017, 12. [Google Scholar] [CrossRef]
- Li, J.Y.; Song, Z.L.; Yan, G.Y.; He, L.S. The complete mitochondrial genome of the largest amphipod, Alicella gigantea: Insight into its phylogenetic relationships and deep-sea adaptive characters. Int. J. Biol. Macromol. 2019, 141, 570–577. [Google Scholar] [CrossRef]
- Mu, W.; Liu, J.; Zhang, H. Complete mitochondrial genome of Benthodytes marianensis (Holothuroidea: Elasipodida: Psychropotidae): Insight into deep sea adaptation in the sea cucumber. PLoS ONE 2018, 13, e0208051. [Google Scholar] [CrossRef]
- Shen, X.; Pu, Z.; Chen, X.; Murphy, R.W.; Shen, Y. Convergent Evolution of Mitochondrial Genes in Deep-Sea Fishes. Front. Genet 2019, 10, 925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Wu, Y.; Wang, X.; Jiang, W.; Yin, J.; Lin, Q. Comparative analysis of mitochondrial genome of a deep-sea crab Chaceon granulates reveals positive selection and novel genetic features. J. Oceanol. Limnol. 2019, 38, 427–437. [Google Scholar] [CrossRef]
- Yang, M.; Gong, L.; Sui, J.; Li, X. The complete mitochondrial genome of Calyptogena marissinica (Heterodonta: Veneroida: Vesicomyidae): Insight into the deep-sea adaptive evolution of vesicomyids. PLoS ONE 2019, 14, e0217952. [Google Scholar] [CrossRef] [Green Version]
- Boore, J.L.; Medina, M.; Rosenberg, L.A. Complete sequences of the highly rearranged molluscan mitochondrial genomes of the scaphopod Graptacme eborea and the bivalve Mytilus edulis. Mol. Biol. Evol. 2004, 21, 1492–1503. [Google Scholar] [CrossRef] [Green Version]
- Breton, S.; Stewart, D.T.; Hoeh, W.R. Characterization of a mitochondrial ORF from the gender-associated mtDNAs of Mytilus spp. (Bivalvia: Mytilidae): Identification of the “missing” ATPase 8 gene. Mar. Genomics 2010, 3, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, G.; Shimamura, S.; Takaki, Y.; Yokobori, S.I.; Ohara, Y.; Takishita, K.; Maruyama, T.; Fujikura, K.; Yoshida, T. Updated mitochondrial phylogeny of Pteriomorph and Heterodont Bivalvia, including deep-sea chemosymbiotic Bathymodiolus mussels, vesicomyid clams and the thyasirid clam Conchocele cf. bisecta. Mar. Genomics 2017, 31, 43–52. [Google Scholar] [CrossRef]
- Uliano-Silva, M.; Americo, J.A.; Costa, I.; Schomaker-Bastos, A.; de Freitas Rebelo, M.; Prosdocimi, F. The complete mitochondrial genome of the golden mussel Limnoperna fortunei and comparative mitogenomics of Mytilidae. Gene 2016, 577, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, J.I.; de Oliveira Martins, L.; Fujita, Y.; Matsumoto, H.; Fujiwara, Y. Evolutionary process of deep-sea Bathymodiolus mussels. PLoS ONE 2010, 5, e10363. [Google Scholar] [CrossRef] [Green Version]
- Savolainen, V.; Cowan, R.S.; Vogler, A.P.; Roderick, G.K.; Lane, R. Towards writing the encyclopaedia of life: An introduction to DNA barcoding. Philos. Trans. R. Soc. B 2005, 360, 1805–1811. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zhou, Y.; Chen, C.; Kwan, Y.H.; Sun, Y.; Wang, X.; Yang, L.; Zhang, R.; Wei, T.; Yang, Y.; et al. Nearest vent, dearest friend: Biodiversity of Tiancheng vent field reveals cross-ridge similarities in the Indian Ocean. R. Soc. Open. Sci. 2020, 7, 200110. [Google Scholar] [PubMed] [Green Version]
- Akasaki, T.; Nikaido, M.; Tsuchiya, K.; Segawa, S.; Hasegawa, M.; Okada, N. Extensive mitochondrial gene arrangements in coleoid Cephalopoda and their phylogenetic implications. Mol. Phylogenet. Evol. 2006, 38, 648–658. [Google Scholar] [CrossRef]
- Ki, J.S.; Lee, Y.M.; Jung, S.O.; Horiguchi, T.; Cho, H.S.; Lee, J.S. Mitochondrial genome of Thais clavigera (Mollusca: Gastropoda): Affirmation of the conserved, ancestral gene pattern within the mollusks. Mol. Phylogenet. Evol. 2010, 54, 1016–1020. [Google Scholar] [CrossRef]
- Boore, J.L.; Brown, W.M. Big trees from little genomes: Mitochondrial gene order as a phylogenetic tool. Curr. Opin. Genet. Dev. 1998, 8, 668–674. [Google Scholar] [CrossRef]
- Xie, G.L.; Köhler, F.; Huang, X.C.; Wu, R.W.; Zhou, C.H.; Ouyang, S.; Wu, X.P. A novel gene arrangement among the Stylommatophora by the complete mitochondrial genome of the terrestrial slug Meghimatium bilineatum (Gastropoda, Arionoidea). Mol. Phylogenet. Evol. 2019, 135, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Zeng, C.; Yan, G.Y.; He, L.S. Characterization of the mitochondrial genome of an ancient amphipod Halice sp. MT-2017 (Pardaliscidae) from 10,908 m in the Mariana Trench. Sci. Rep. UK 2019, 9, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogan, R.I.; Hopkins, K.; Wheeler, A.J.; Allcock, A.L.; Yesson, C. Novel diversity in mitochondrial genomes of deep-sea Pennatulacea (Cnidaria: Anthozoa: Octocorallia). Mitochondrial DNA A 2019, 30, 764–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Sun, J.; Rouse, G.W.; Wiklund, H.; Pleijel, F.; Watanabe, H.K.; Chen, C.; Qian, P.Y.; Qiu, J.W. Phylogeny, evolution and mitochondrial gene order rearrangement in scale worms (Aphroditiformia, Annelida). Mol. Phylogenet. Evol. 2019, 125, 220–231. [Google Scholar] [CrossRef]
- Brown, W.M.; George, M.; Wilson, A.C. Rapid evolution of animal mitochondrial DNA. Proc. Natl. Acad. Sci. USA 1979, 76, 1967–1971. [Google Scholar] [CrossRef] [Green Version]
- Plazzi, F.; Puccio, G.; Passamonti, M. Burrowers from the past: Mitochondrial signatures of Ordovician bivalve infaunalization. Genome Biol. Evol. 2017, 9, 956–967. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Shi, X.; Sun, L.; Bai, Y.; Zhang, D.; Tang, B. Evolution of mitochondrial energy metabolism genes associated with hydrothermal vent adaption of Alvinocaridid shrimps. Genes Genomics 2017, 39, 1367–1376. [Google Scholar] [CrossRef]
- Hui, M.; Wang, M.; Sha, Z. The complete mitochondrial genome of the alvinocaridid shrimp Shinkaicaris leurokolos (Decapoda, Caridea): Insight into the mitochondrial genetic basis of deep-sea hydrothermal vent adaptation in the shrimp. Comp. Biochem. Physiol. Part D Genomics Proteomics 2018, 25, 42–52. [Google Scholar]
- Shen, Y.; Kou, Q.; Zhong, Z.; Li, X.; He, L.; He, S.; Gan, X. The first complete mitogenome of the South China deep-sea giant isopod Bathynomus sp. (Crustacea: Isopoda: Cirolanidae) allows insights into the early mitogenomic evolution of isopods. Ecol. Evol. 2017, 7, 1869–1881. [Google Scholar] [CrossRef]
- Shen, Y.Y.; Liang, L.; Zhu, Z.H.; Zhou, W.P.; Irwin, D.M.; Zhang, Y.P. Adaptive evolution of energy metabolism genes and the origin of flight in bats. Proc. Natl. Acad. Sci. USA 2010, 107, 8666–8671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Nielsen, R.; Yang, Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol. Biol. Evol. 2005, 22, 2472–2479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ning, T.; Xiao, H.; Li, J.; Hua, S.; Zhang, Y.P. Adaptive evolution of the mitochondrial ND6 gene in the domestic horse. Genet. Mol. Res. 2010, 9, 144–150. [Google Scholar] [CrossRef]
- Yu, L.; Wang, X.; Ting, N.; Zhang, Y. Mitogenomic analysis of Chinese snub-nosed monkeys: Evidence of positive selection in NADH dehydrogenase genes in high-altitude adaptation. Mitochondrion 2011, 11, 497–503. [Google Scholar] [CrossRef]
- Luo, Y.; Gao, W.; Gao, Y.; Tang, S.; Huang, Q.; Tan, X.; Chen, J.; Huang, T. Mitochondrial genome analysis of Ochotona curzoniae and implication of cytochrome c oxidase in hypoxic adaptation. Mitochondrion 2008, 8, 352–357. [Google Scholar] [CrossRef]
- Mahalingam, S.; McClelland, G.B.; Scott, G.R. Evolved changes in the intracellular distribution and physiology of muscle mitochondria in high-altitude native deer mice. J. Physiol. 2017, 595, 4785–4801. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Li, Y.; Ruan, Z.; Bian, C.; You, X.; Yang, J.; Jiang, W.; Shi, Q. The Complete Mitochondrial Genome of Glyptothorax macromaculatus Provides a Well-Resolved Molecular Phylogeny of the Chinese Sisorid Catfishes. Genes 2018, 9, 282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, F.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Jühling, F.; Pütz, J.; Bernt, M.; Donath, A.; Middendorf, M.; Florentz, C.; Stadler, P.F. Improved systematic tRNA gene annotation allows new insights into the evolution of mitochondrial tRNA structures and into the mechanisms of mitochondrial genome rearrangements. Nucleic Acids Res. 2012, 40, 2833–2845. [Google Scholar] [CrossRef] [Green Version]
- Laslett, D.; Canbäck, B. ARWEN: A program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 2008, 24, 172–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Merkle, D.; Ramsch, K.; Fritzsch, G.; Perseke, M.; Bernhard, D.; Schlegel, M.; Stadler, P.F.; Middendorf, M. CREx: Inferring genomic rearrangements based on common intervals. Bioinformatics 2007, 23, 2957–2958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, T.; Yamada, K.D.; Tomii, K.; Katoh, K. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics 2018, 34, 2490–2492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castresana, J. GBLOCKS: Selection of Conserved Blocks from Multiple Alignments for Their Use in Phylogenetic Analysis. Version 0.91 b; EMBL: Heidelberg, Germany, 2002. [Google Scholar]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 46, W537–W545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [PubMed] [Green Version]
- Rambaut, A. FigTree v1.4.3. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 4 June 2020).
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Linearized mitochondrial gene arrangement patterns of the investigated species superimposed on the phylogenetic tree. Gene and genome size are not in scale. The tRNAs are labeled by single-letter abbreviations of the amino acid code.
Figure 1.
Linearized mitochondrial gene arrangement patterns of the investigated species superimposed on the phylogenetic tree. Gene and genome size are not in scale. The tRNAs are labeled by single-letter abbreviations of the amino acid code.

Figure 2.
Putative gene rearrangement events from Lithophaga curta to Bathymodiolinae species. Purple lines and genes are colored to indicate tandem duplication random loss (TDRL) events step by step as identified by Common interval Rearrangement Explorer (CREx) analysis.
Figure 2.
Putative gene rearrangement events from Lithophaga curta to Bathymodiolinae species. Purple lines and genes are colored to indicate tandem duplication random loss (TDRL) events step by step as identified by Common interval Rearrangement Explorer (CREx) analysis.

Figure 3.
Phylogenetic tree of the deep-sea and shallow-water mussels inferred from 12 protein genes by maximum likelihood. Bayesian posterior probabilities/maximum likelihood (ML) bootstrap support values are shown for each node. An asterisk denotes mtDNA sequence newly determined in this study. Scale bar represents nucleotide substitutions per site.
Figure 3.
Phylogenetic tree of the deep-sea and shallow-water mussels inferred from 12 protein genes by maximum likelihood. Bayesian posterior probabilities/maximum likelihood (ML) bootstrap support values are shown for each node. An asterisk denotes mtDNA sequence newly determined in this study. Scale bar represents nucleotide substitutions per site.

Figure 4.
Molecular dating of species divergence events within the Bathymodiolinae and Modiolinae. Calibration points are marked by an arrow.
Figure 4.
Molecular dating of species divergence events within the Bathymodiolinae and Modiolinae. Calibration points are marked by an arrow.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Complete mitochondrial genomes used for phylogenetic analysis in this study.
| Species | Subfamily | bp | Accession No. | Reference |
|---|---|---|---|---|
| Gigantidas japonicus | Bathymodiolinae | 17,510 | AP014560 | Robicheau et al. (2017) |
| Gigantidas platifrons | Bathymodiolinae | 17,653 | AP014561 | Robicheau et al. (2017) |
| Bathmodiolus septemdierm | Bathymodiolinae | 17,069 | AP014562 | Robicheau et al. (2017) |
| Bathymodiolus thermophilus | Bathymodiolinae | 18,819 | MK721544 | Lee et al. (2019) |
| Bathymodiolus securiformis | Bathymodiolinae | 17,199 | NC_039552 | - |
| Bathymodiolus manusensis | Bathymodiolinae | 16,801 | KY270856 | - |
| Bathymodiolus sp. 5 South | Bathymodiolinae | 18,376 | MT916740 | This study |
| Bathymodiolus aduloides | Bathymodiolinae | 17,243 | MT916741 | This study |
| Bathymodiolus azoricus | Bathymodiolinae | 17,598 | MT916742 | This study |
| Bathymodiolus brooksi | Bathymodiolinae | 17,728 | MT916743 | This study |
| Bathymodiolus childressi | Bathymodiolinae | 17,637 | MT916744 | This study |
| Bathymodiolus marisindicus | Bathymodiolinae | 17,138 | MT916745 | This study |
| Gigantidas haimaensis | Bathymodiolinae | 18,283 | MT916746 | This study |
| Modiolus modiolus | Modiolinae | 15,816 | KX821782 | Robicheau et al. (2017) |
| Modiolus kurilensis | Modiolinae | 16,210 | KY242717 | - |
| Modiolus nipponicus | Modiolinae | 15,638 | MK721547 | Lee et al. (2019) |
| Modiolus philippinarum | Modiolinae | 16,389 | KY705073 | Sun et al. (2017) |
| Lithophaga curta | Lithophaginae | 16,580 | MK721546 | Lee et al. (2019) |
| Limnoperna fortunei | Limnoperninae | 18,145 | KP756905 | Uliano-Silva et al. (2016) |
Table 2.
Possible sites under positive selection in the deep-sea mussels.
| Gene | Codon | Amino Acid | BEB Values |
|---|---|---|---|
| atp6 | 50 | L | 0.994 |
| nad4 | 76 | P | 0.992 |
| 234 | K | 0.991 | |
| nad2 | 34 | S | 0.991 |
| 37 | S | 0.995 | |
| 59 | K | 0.991 | |
| 60 | S | 0.994 | |
| 122 | W | 0.997 | |
| 331 | G | 0.992 | |
| cob | 54 | S | 0.991 |
| 317 | N | 0.991 | |
| 351 | L | 0.994 | |
| nad5 | 85 | S | 0.995 |
| 439 | S | 0.997 | |
| 503 | S | 0.991 | |
| cox2 | 95 | K | 0.993 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhang, K.; Sun, J.; Xu, T.; Qiu, J.-W.; Qian, P.-Y. Phylogenetic Relationships and Adaptation in Deep-Sea Mussels: Insights from Mitochondrial Genomes. Int. J. Mol. Sci. 2021, 22, 1900. https://doi.org/10.3390/ijms22041900
AMA Style
Zhang K, Sun J, Xu T, Qiu J-W, Qian P-Y. Phylogenetic Relationships and Adaptation in Deep-Sea Mussels: Insights from Mitochondrial Genomes. International Journal of Molecular Sciences. 2021; 22(4):1900. https://doi.org/10.3390/ijms22041900
Chicago/Turabian StyleZhang, Kai, Jin Sun, Ting Xu, Jian-Wen Qiu, and Pei-Yuan Qian. 2021. "Phylogenetic Relationships and Adaptation in Deep-Sea Mussels: Insights from Mitochondrial Genomes" International Journal of Molecular Sciences 22, no. 4: 1900. https://doi.org/10.3390/ijms22041900
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.