
Nephritis-Associated Plasmin Receptor (NAPlr): An Essential Inducer of C3-Dominant Glomerular Injury and a Potential Key Diagnostic Biomarker of Infection-Related Glomerulonephritis (IRGN)

Abstract
:1. Introduction
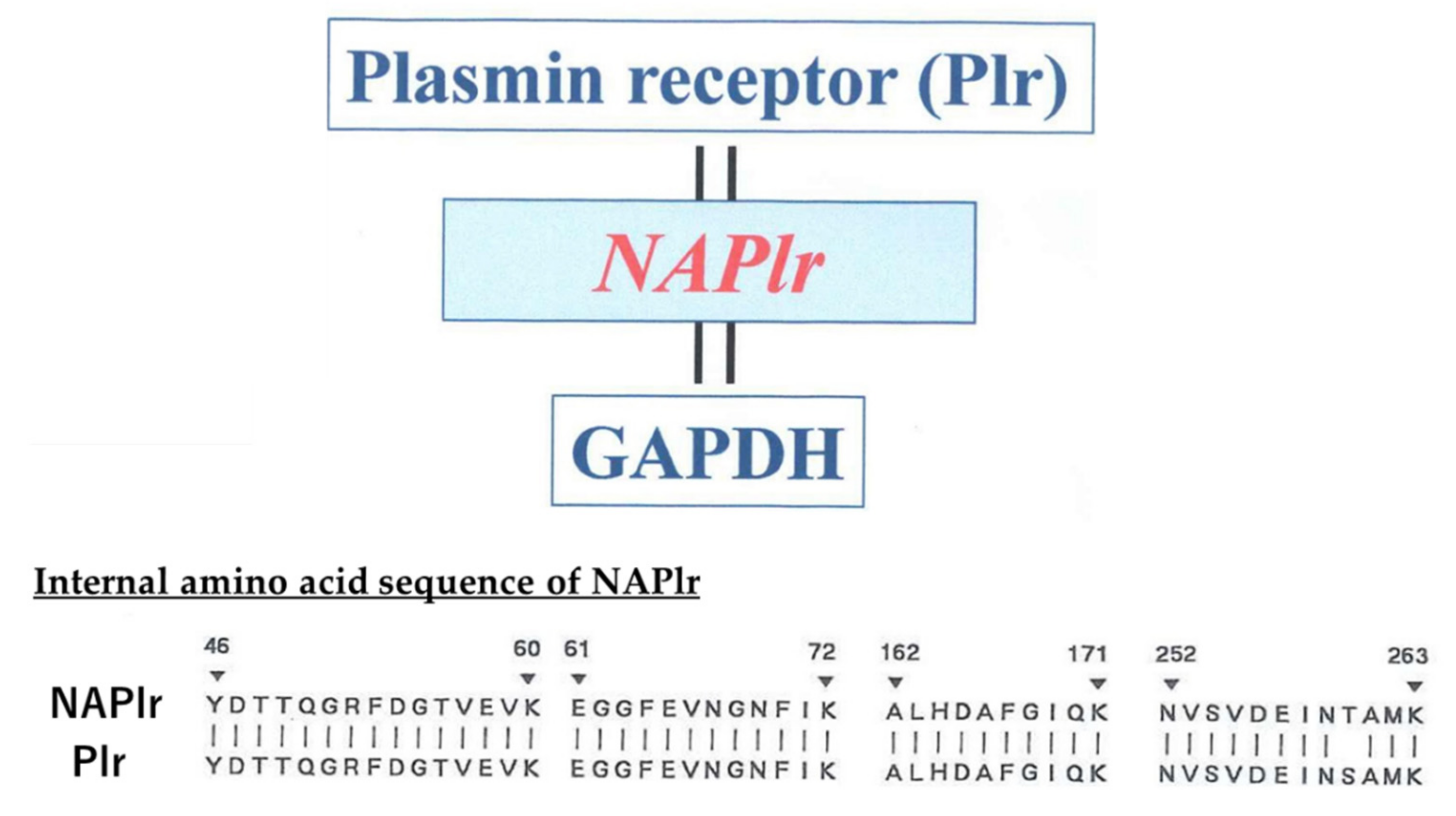
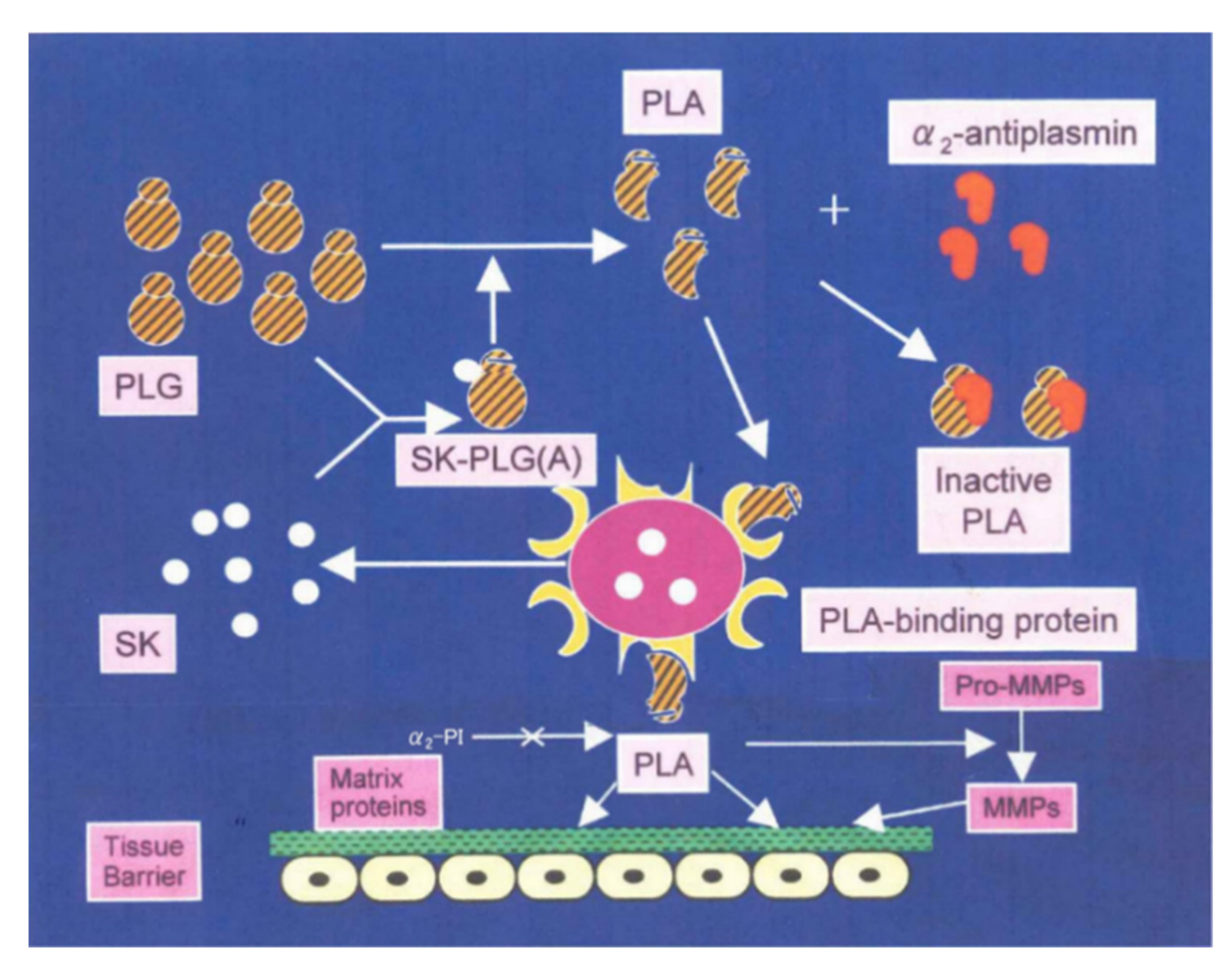
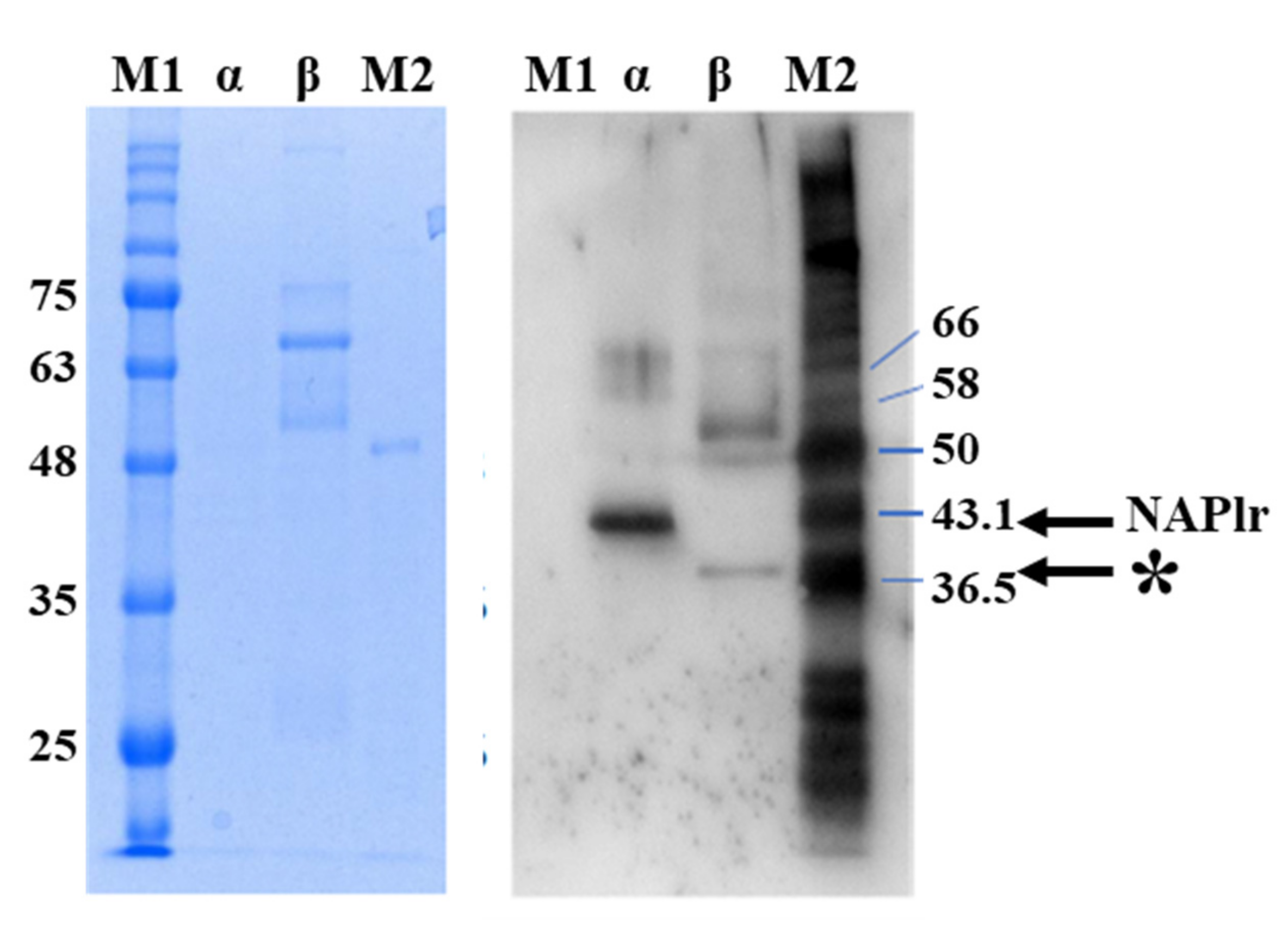
2. Isolation and Characterization of NAPlr
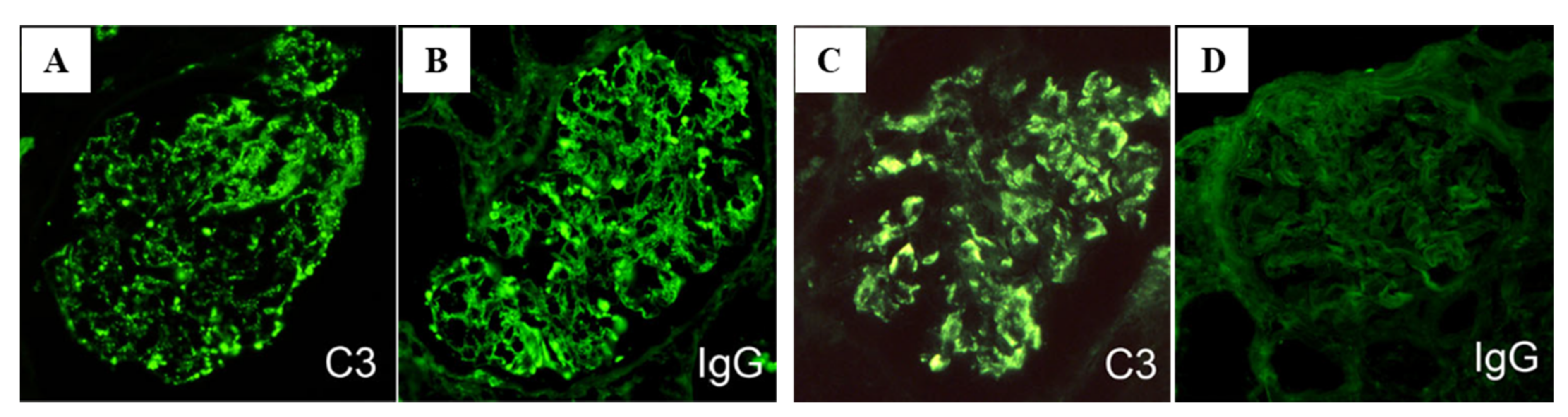
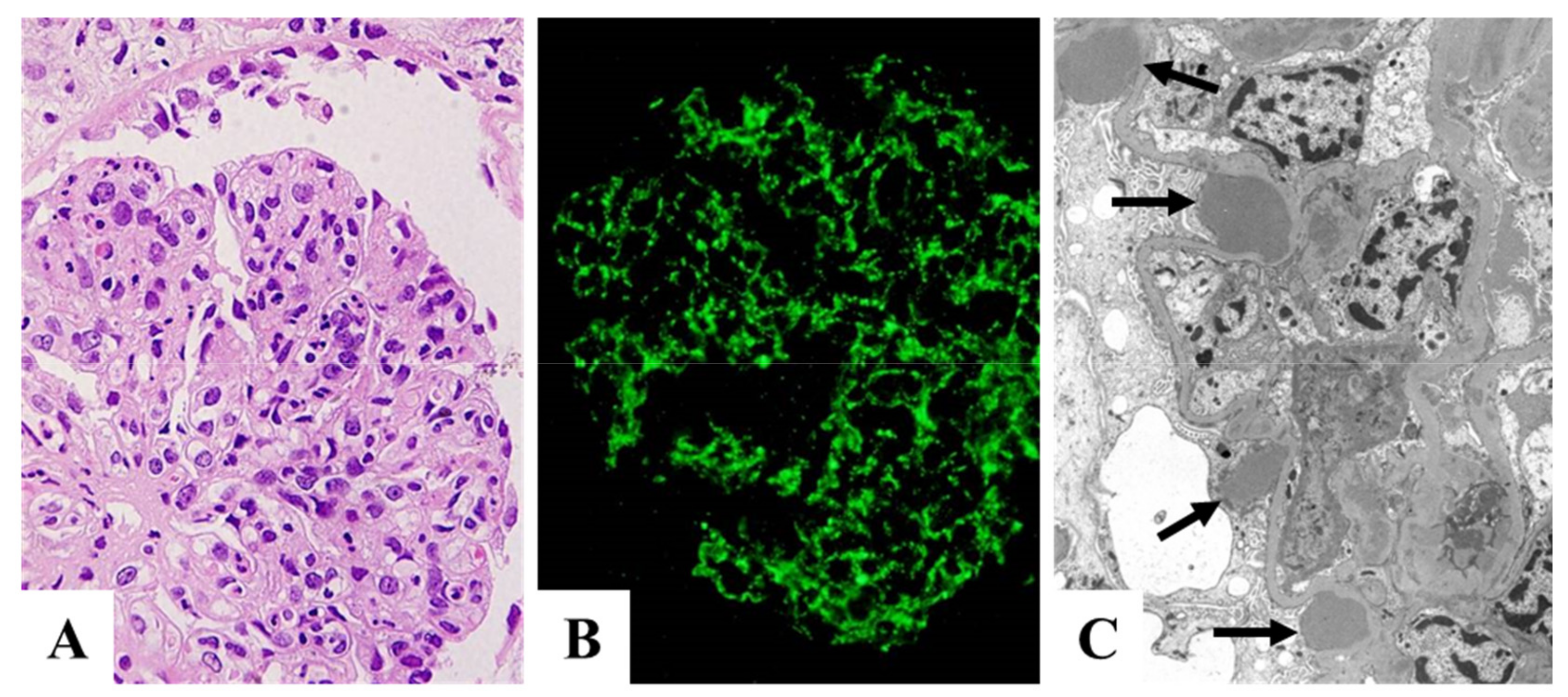
3. Clinical and Histological Characteristics of PSAGN in Relation to Complements, ICs, and NAPlr
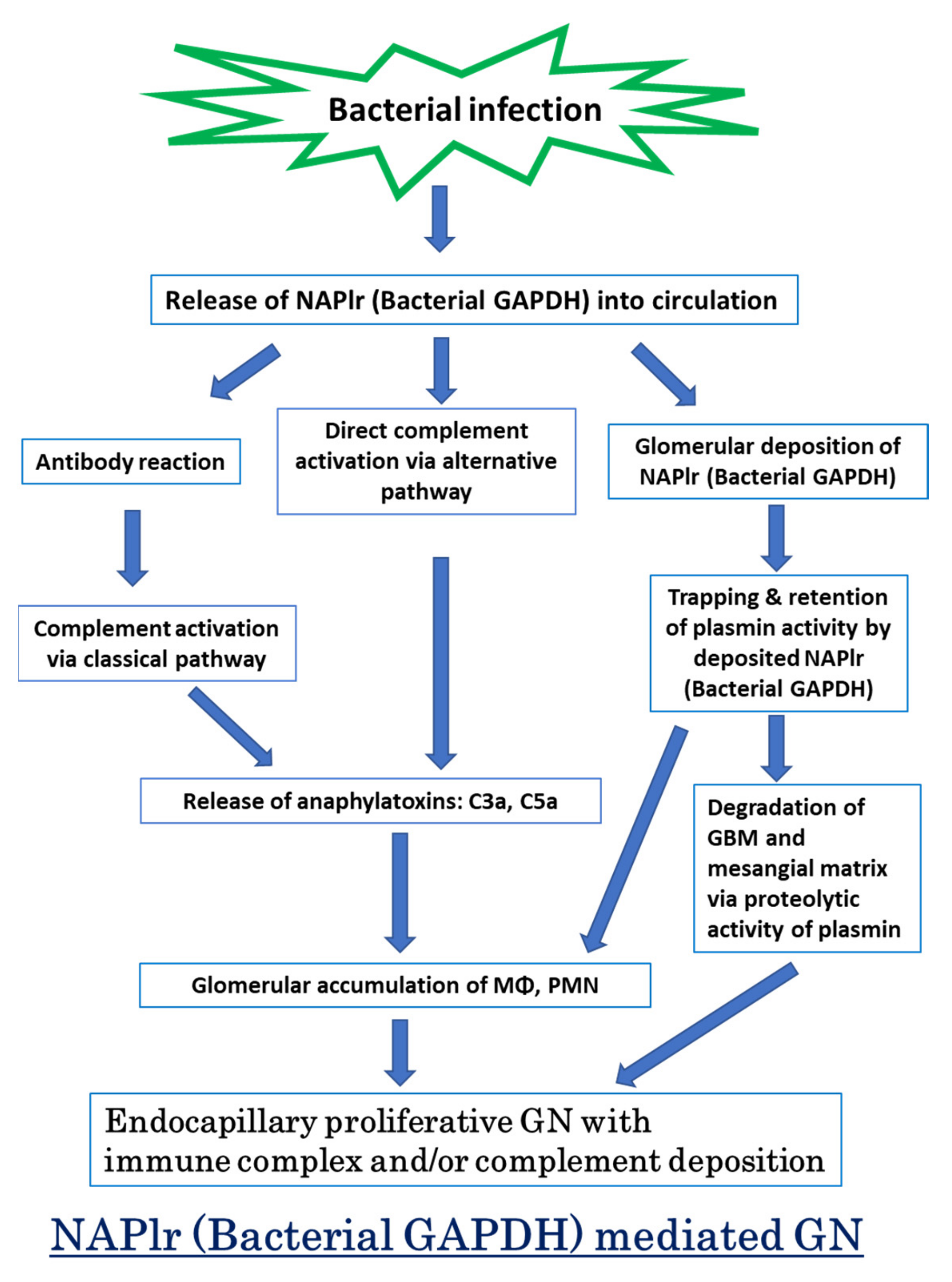
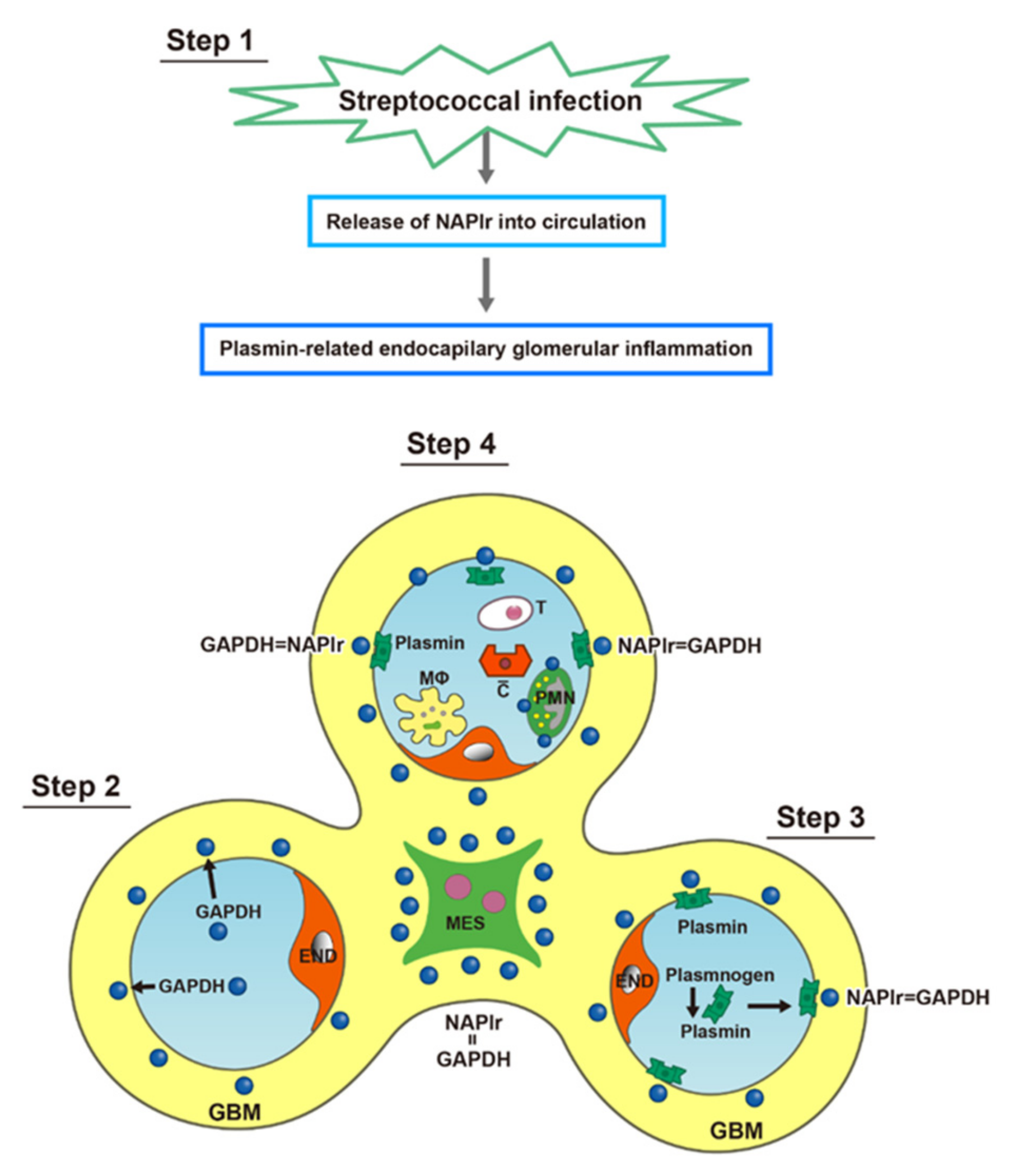
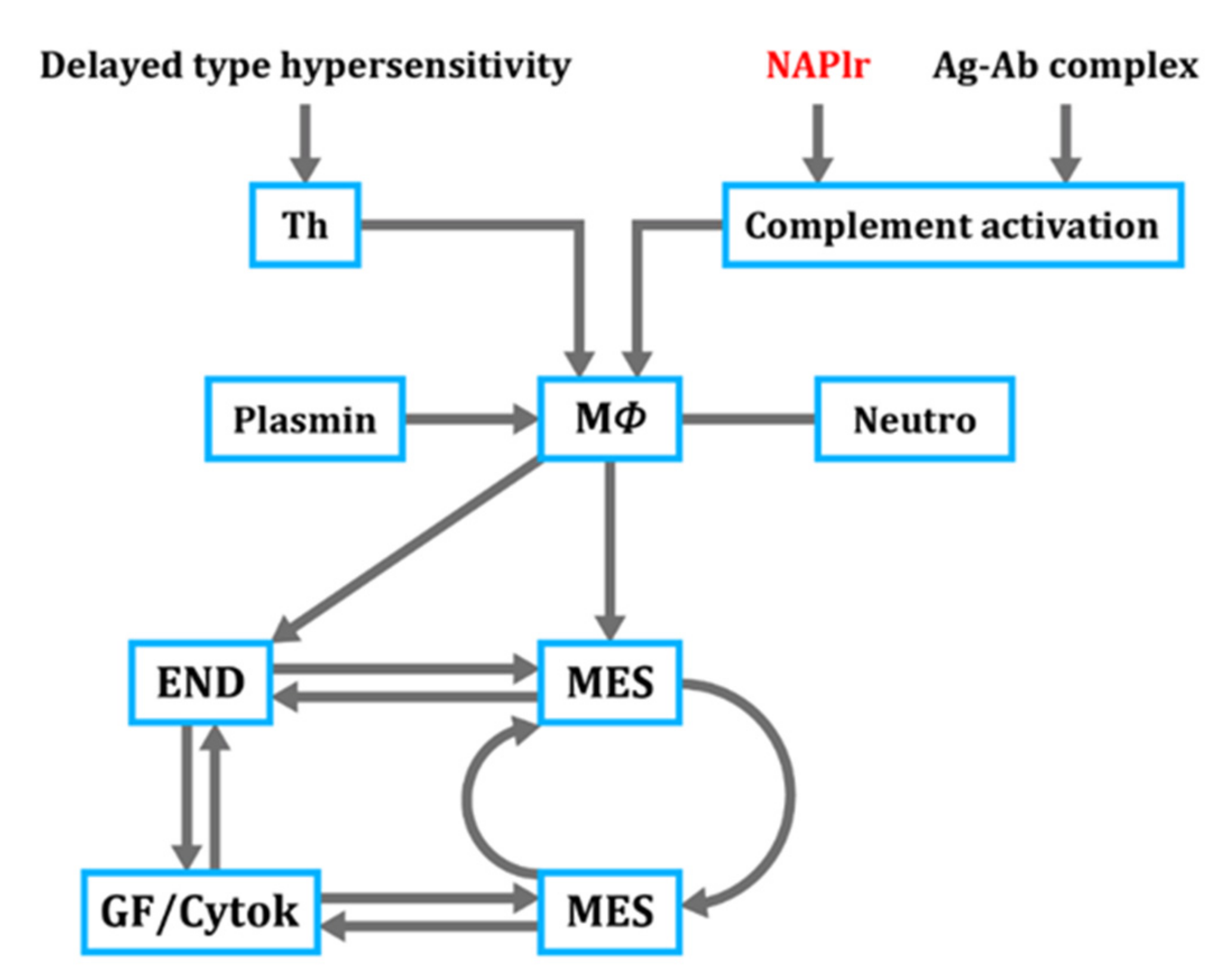
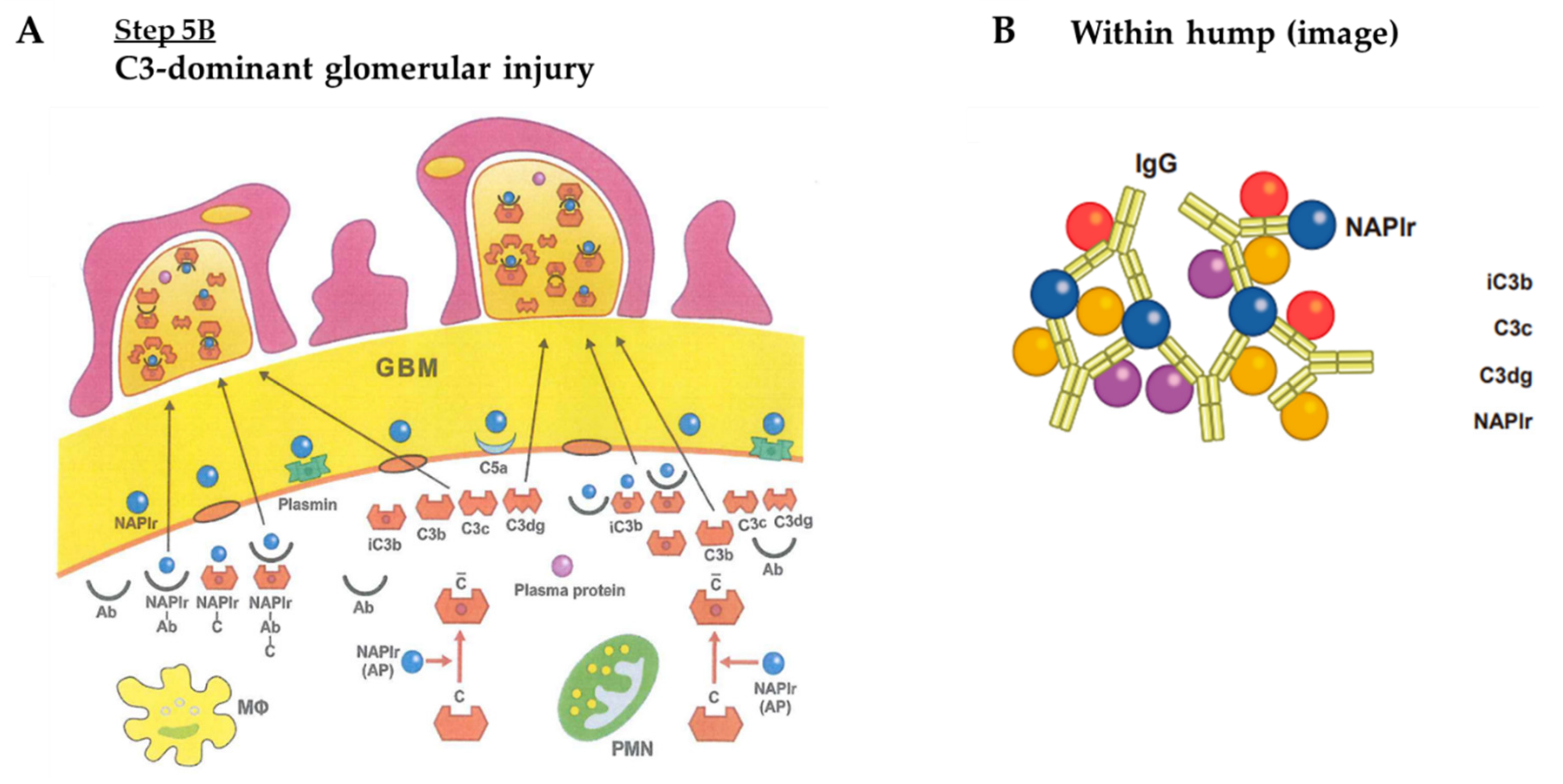
4. Proposed Mechanism for PSAGN
- (1)
- Complement profile in PSAGN
- (2)
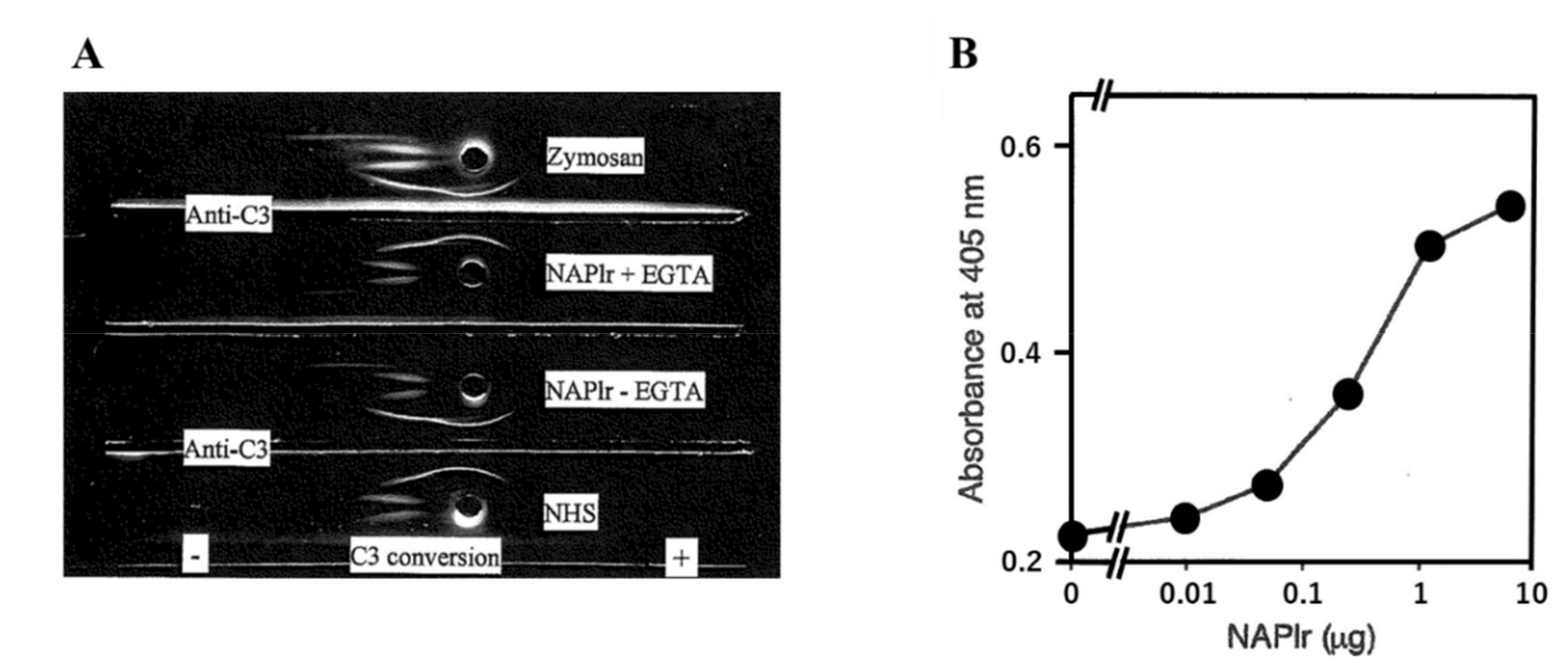
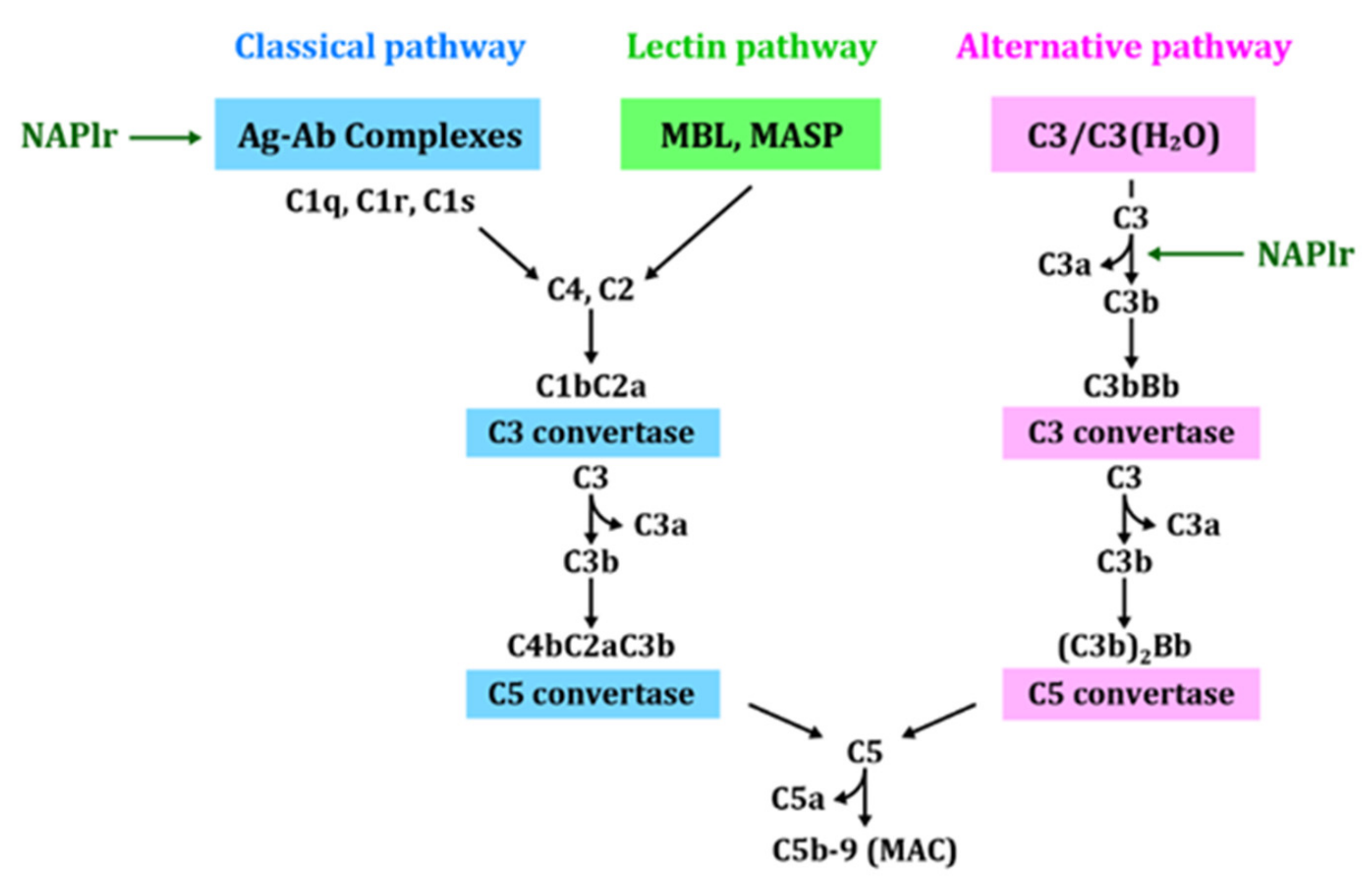
- Complement activation by NAPlr
- (3)
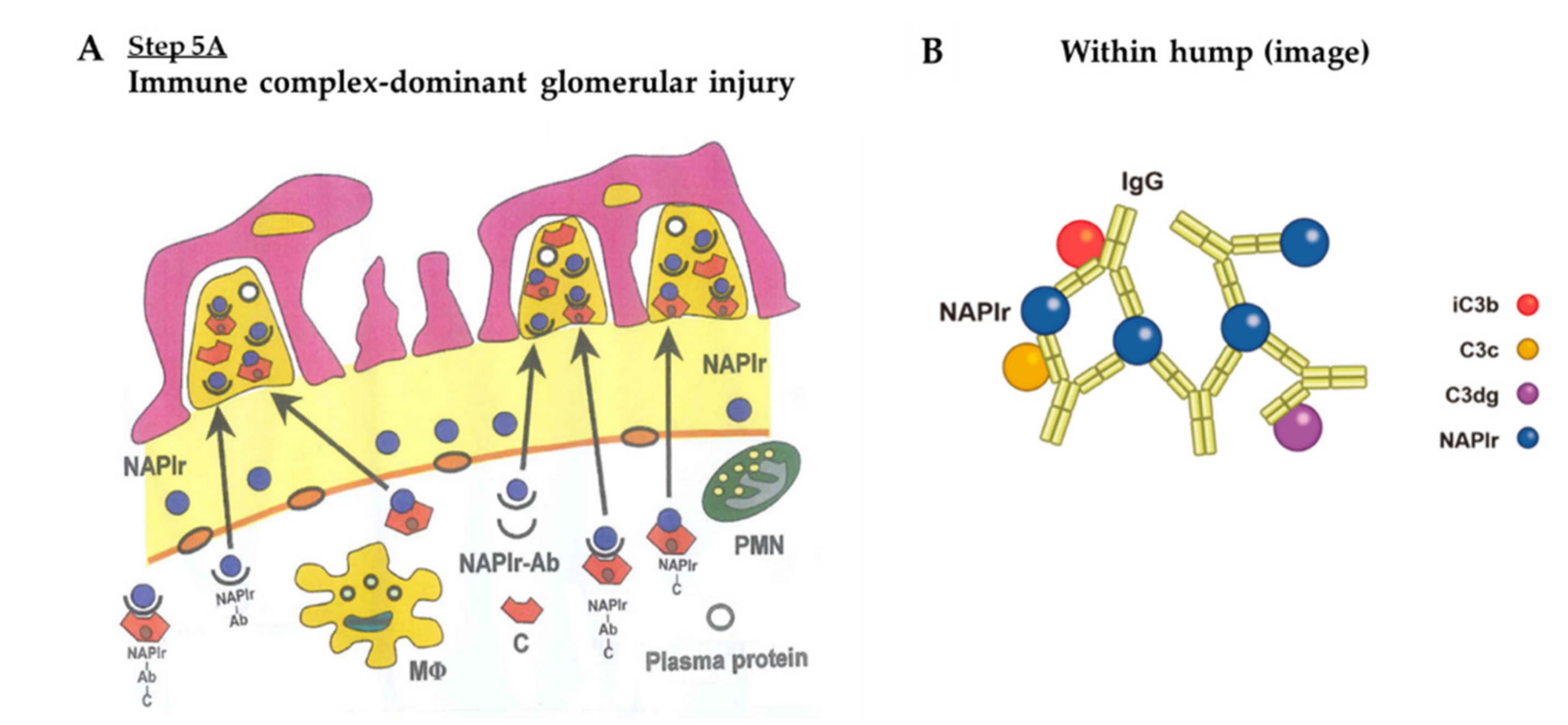
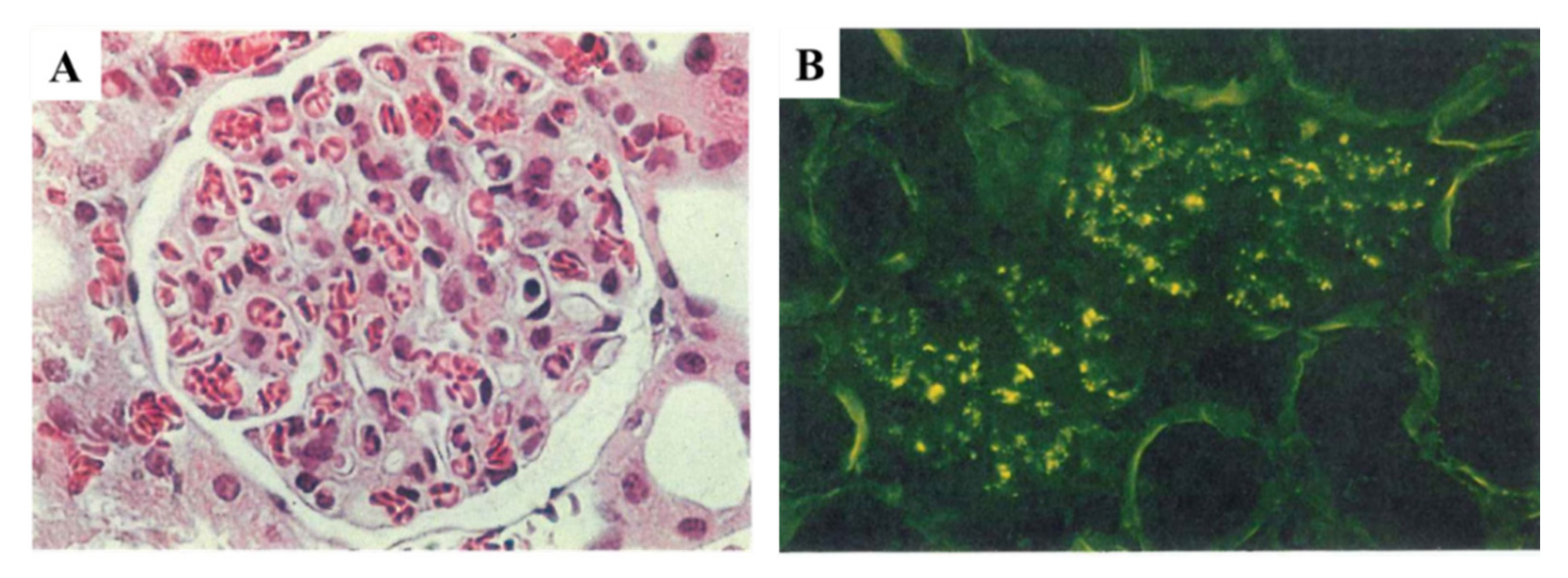
- C3-dominant glomerular injury

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC-Dominant GI | C3-Dominant GI | ||
|---|---|---|---|
| Major difference | C3 with IgG | C3 without IgG | |
| NAPlr deposition | +~++ | +~++ | |
| Plasmin activation | +~++ | +~++ | |
| Complement activation | |||
| Alternative pathway | + | ++ | |
| Classical pathway | + | + | |
| Serum complement | |||
| CH50 | ↓ | ↓ | |
| C3 | ↓ | ↓ | |
| C4 | → | → | |
| Underlying complement dysregulation | unknown | unknown | |
| C3 Nephritic factor | + ? | + ? | [68] |
| Anti-factor B antibody | ++ ? | ++ ? | [67] |
| Pathology | |||
| LM | |||
| Endocapillary GN | ++ | ++ | |
| Exudative change | ++ | ++ | |
| IF | |||
| C3 | ++~+++ | ++~+++ | |
| IgG | +~++ | − | |
| C4 | +~− | − | |
| EM | |||
| Hump | ++ | ++ | |
| Circulating IC | ++ | ++ | [63] |
| Circulating NAPlr antibody | ++ | ++ | [2] |
5. NAPlr as a Key Diagnostic Biomarker of IRGN
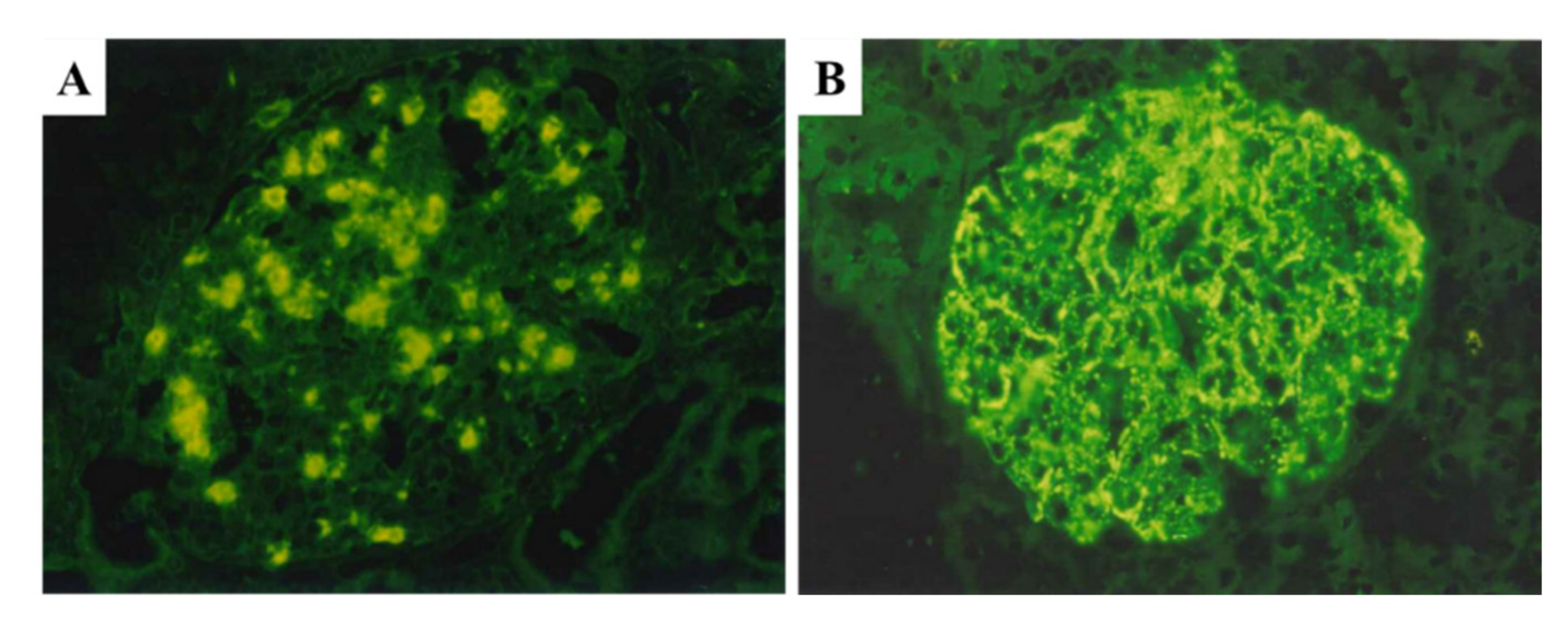
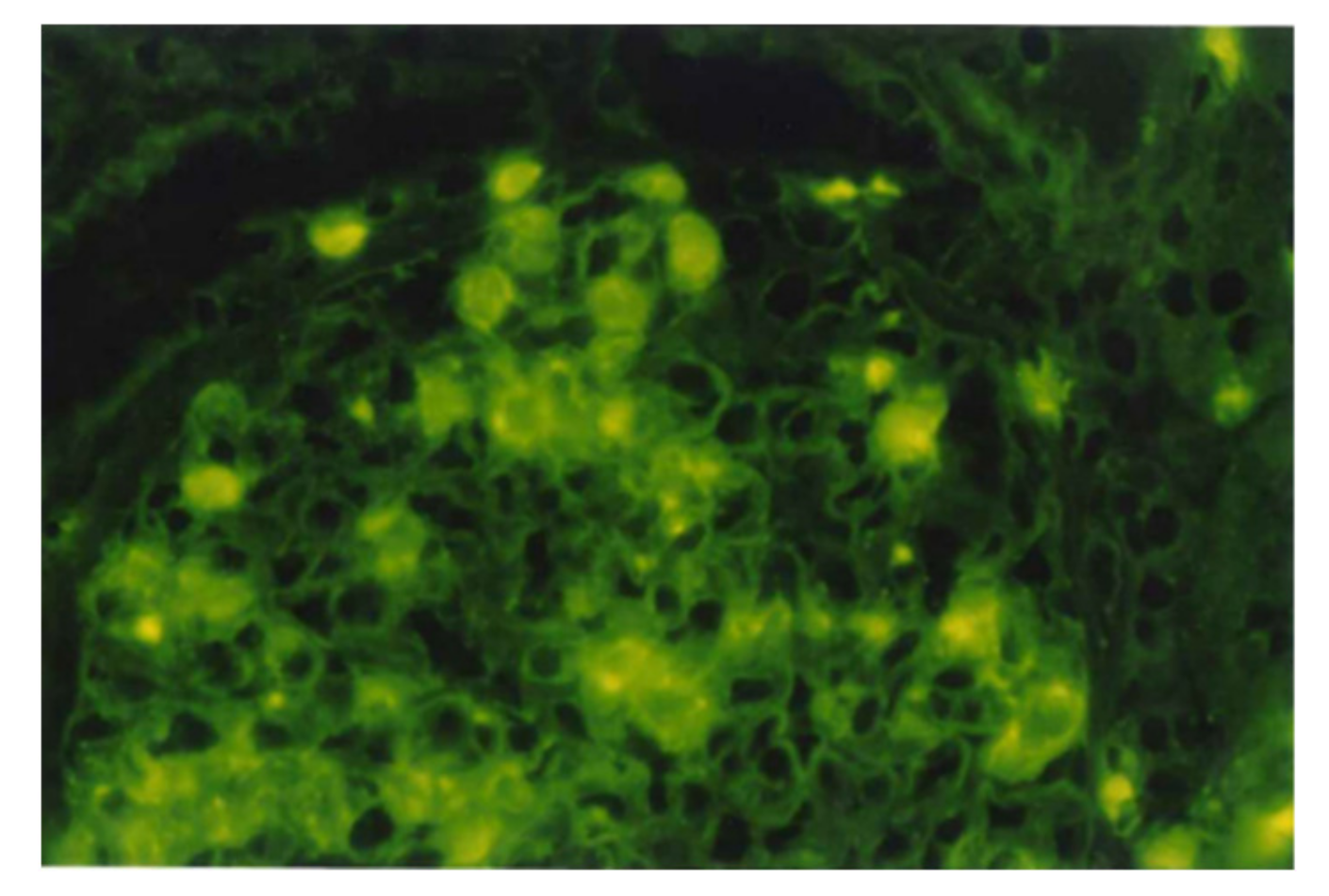
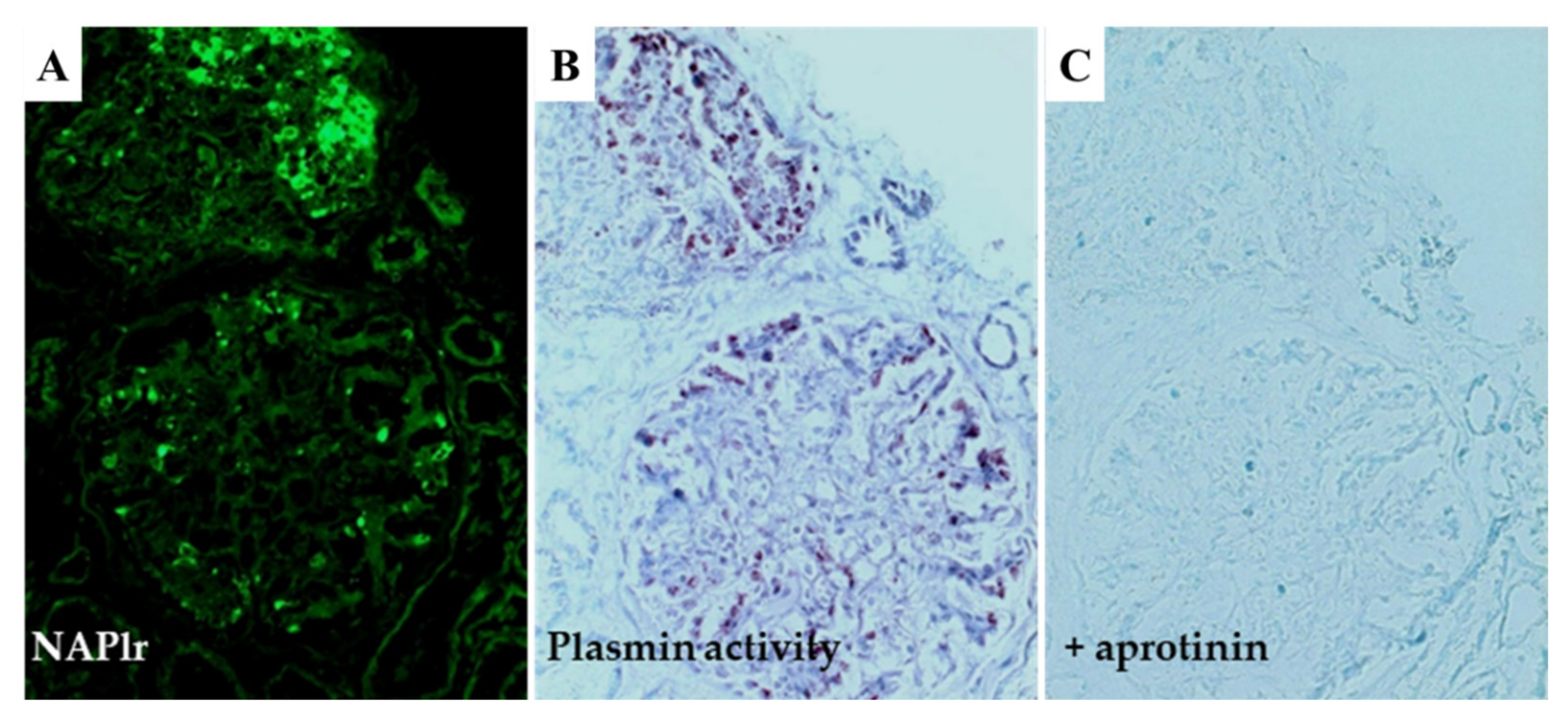
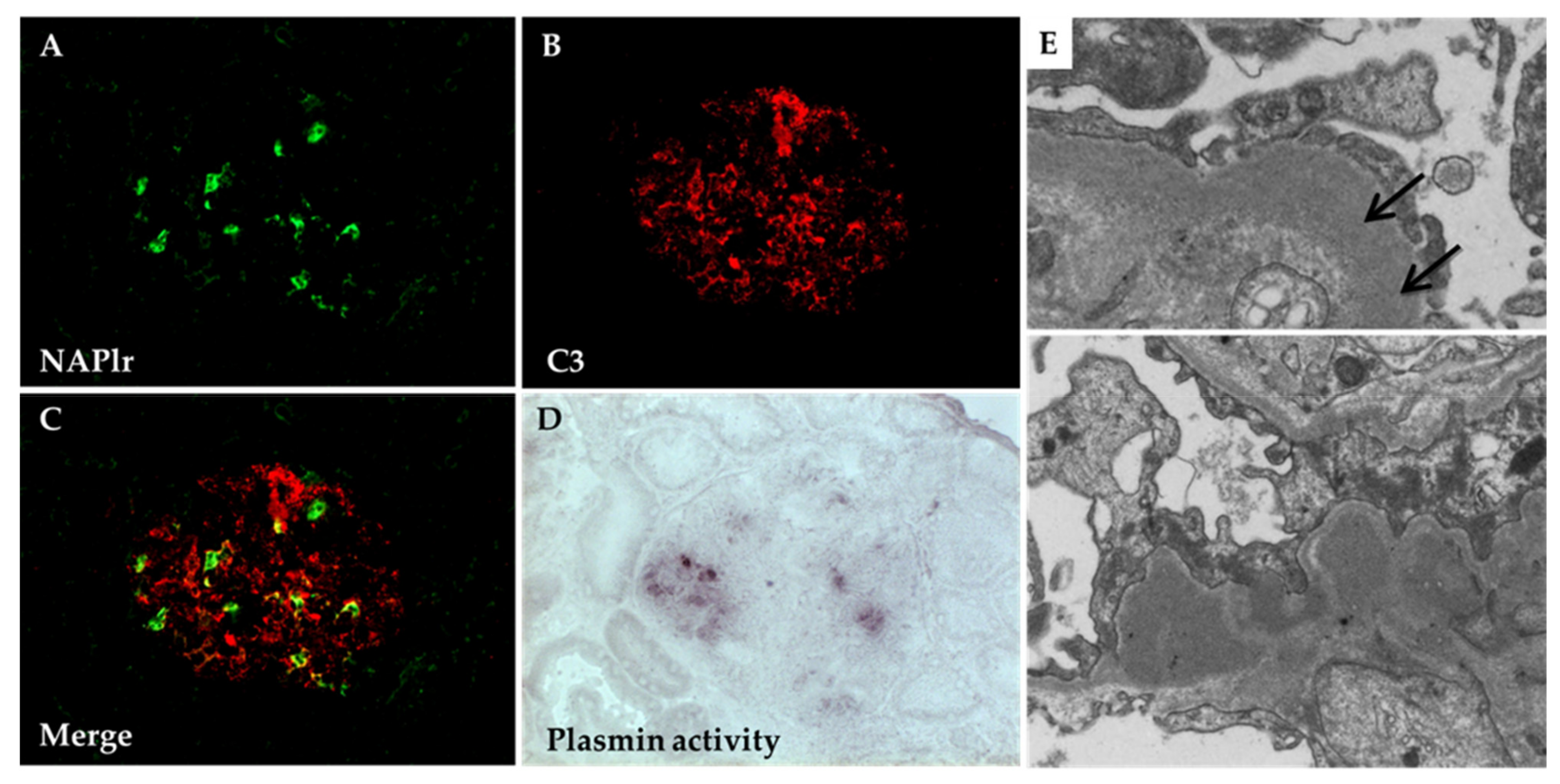
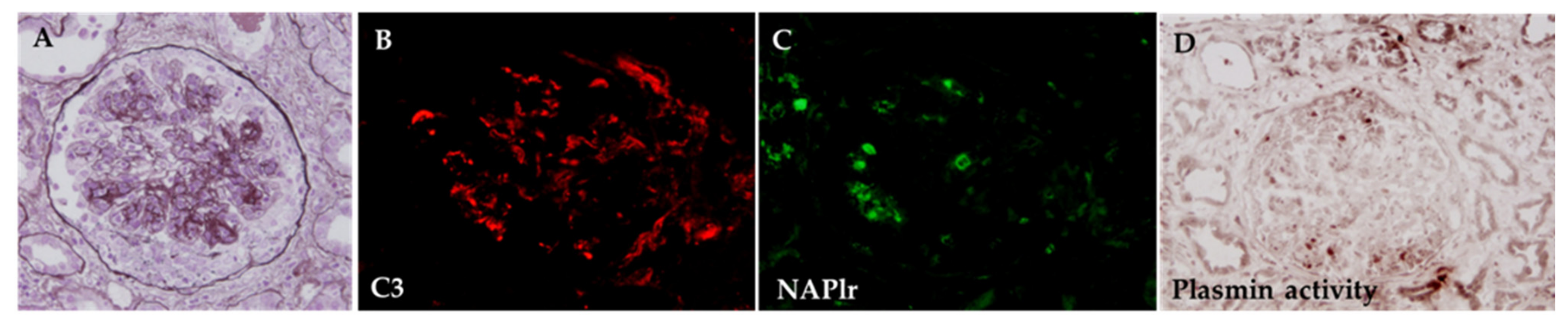
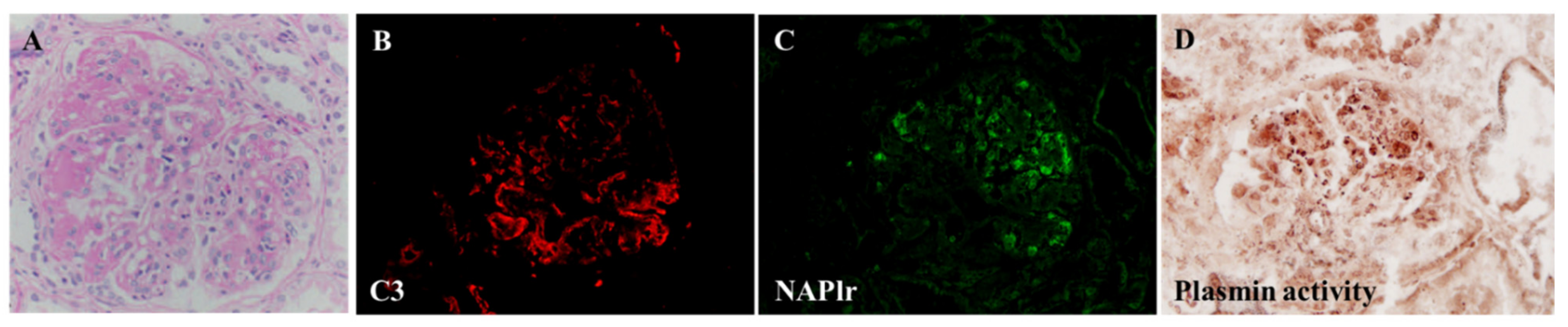
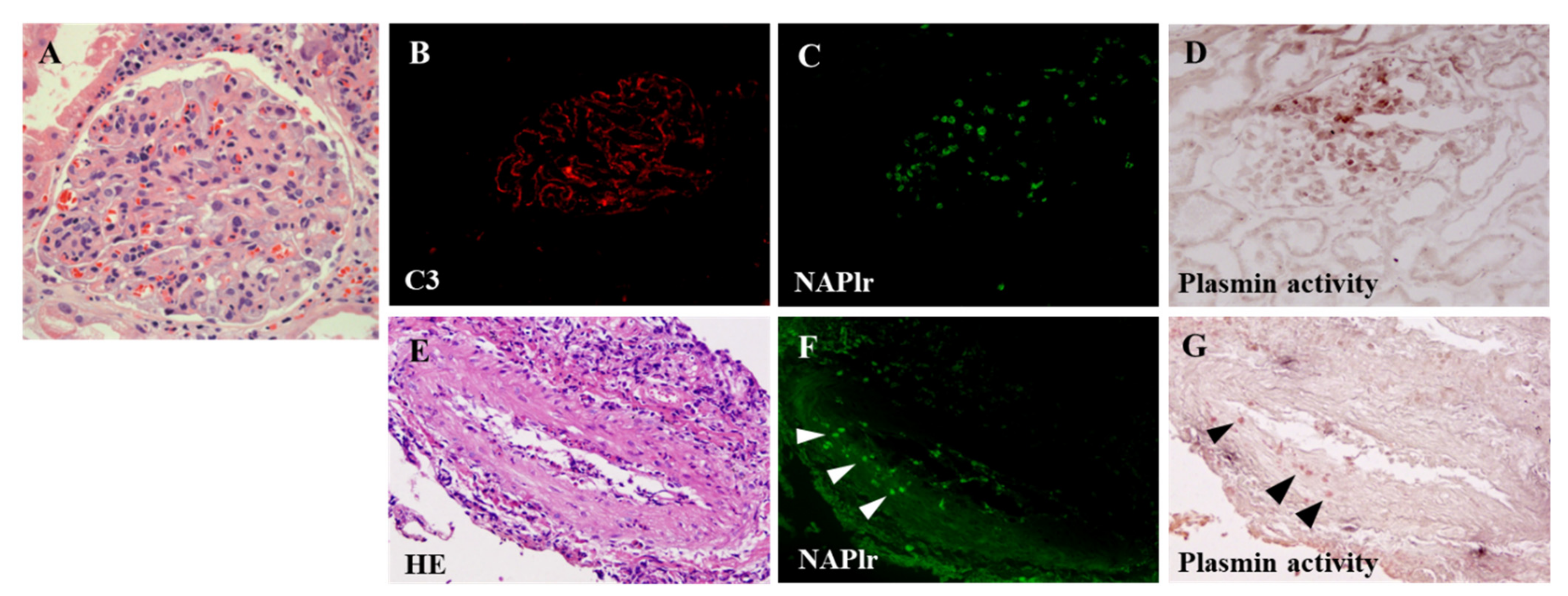
5.1. Glomerular NAPlr Deposition and Related Plasmin Activity in Streptococcal Infection-Related Nephritis
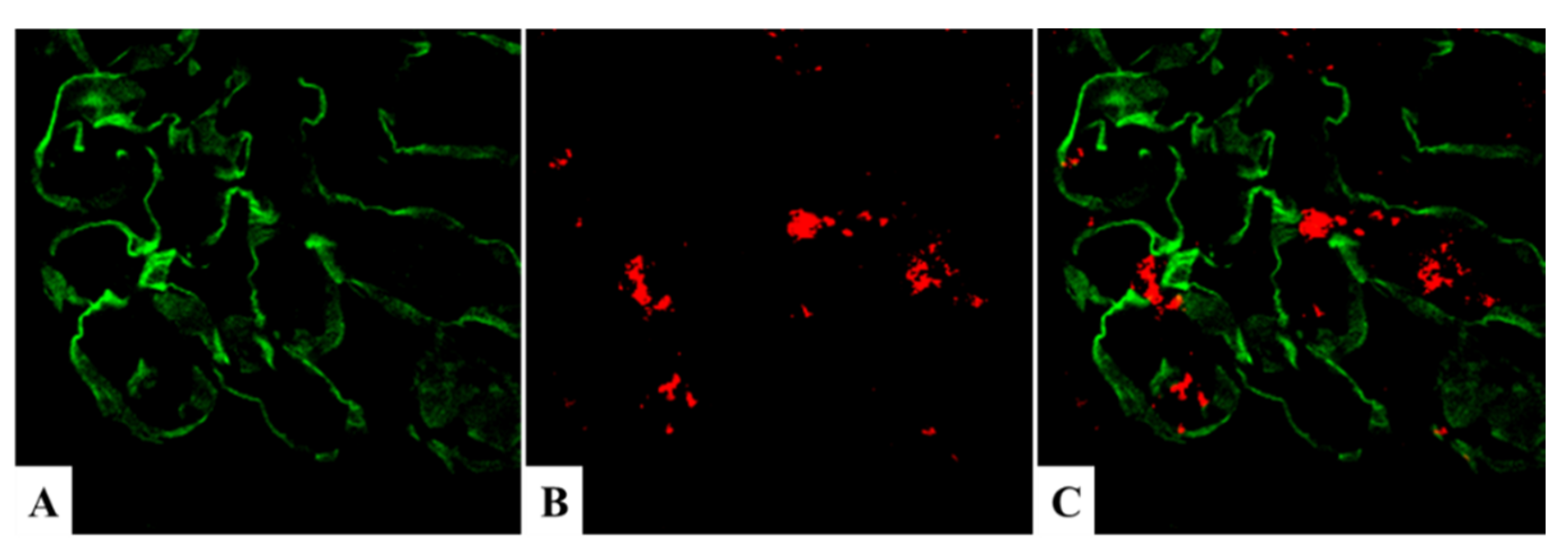
5.2. Positive Staining for NAPlr/Plasmin Activity in Non-Streptococcal IRGN
5.3. Plasmin-Binding Capacity of GAPDH in Non-Streptococcal Bacteria
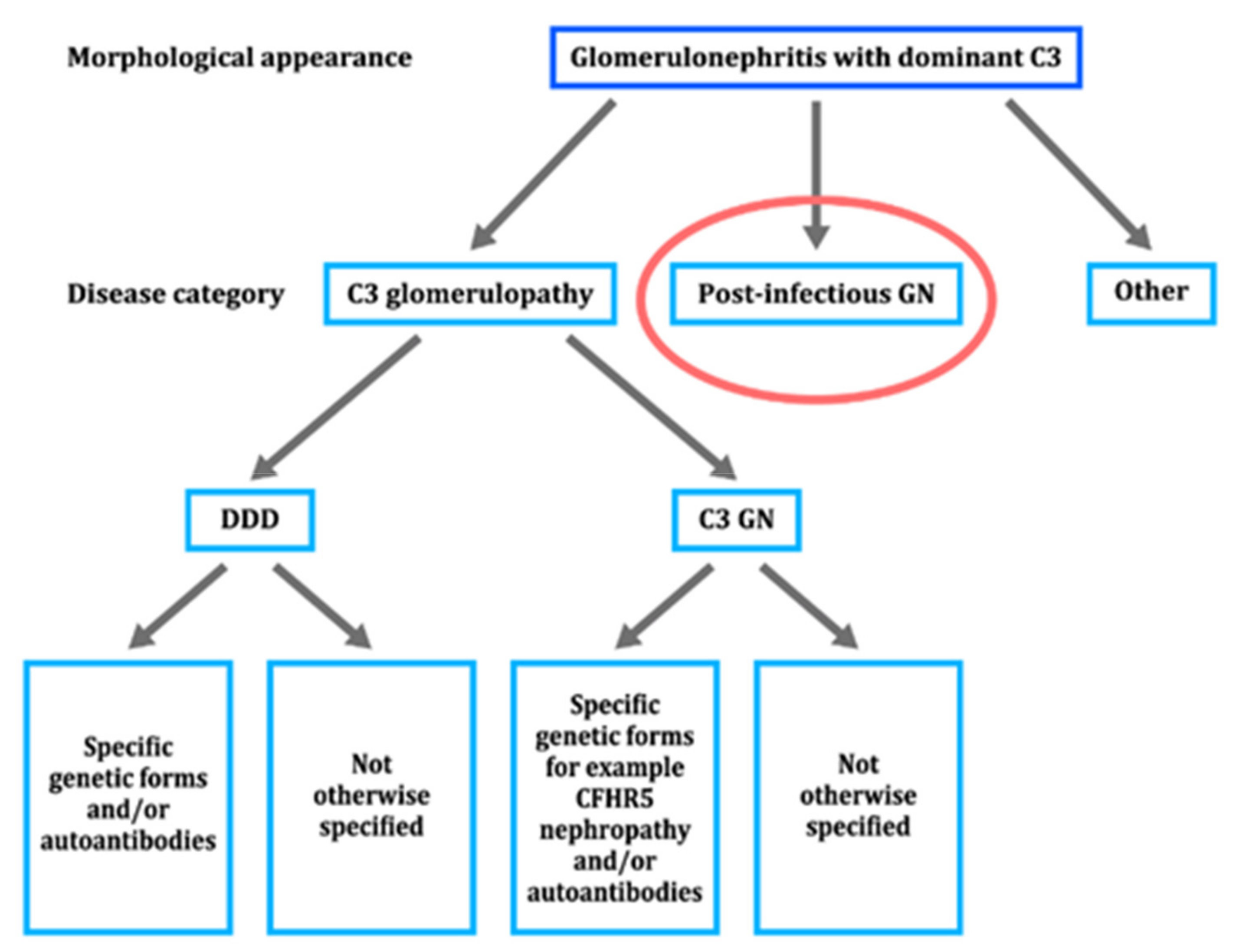
5.4. NAPlr as a General Diagnostic Biomarker of IRGN
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Yamakami, K.; Yoshizawa, N.; Wakabayashi, K.; Takeuchi, A.; Tadakuma, T.; Boyle, M.D. The potential role for nephritis-associated plasmin receptor in acute poststreptococcal glomerulonephritis. Methods 2000, 21, 185–197. [Google Scholar] [CrossRef]
- Yoshizawa, N.; Yamakami, K.; Fujino, M.; Oda, T.; Tamura, K.; Matsumoto, K.; Sugisaki, T.; Boyle, M.D. Nephritis-associated plasmin receptor and acute poststreptococcal glomerulonephritis: Characterization of the antigen and associated immune response. J. Am. Soc. Nephrol. 2004, 15, 1785–1793. [Google Scholar] [CrossRef] [PubMed]
- Couser, W.G.; Johnson, R.J. Post-infecitous glomerulonephritis. In Immunologic Renal Diseases; Neilson, E.G., Couser, W.G., Eds.; Lippincott-Raven: Philadelphia, PA, USA, 1997; pp. 915–943. [Google Scholar]
- Nadasdy, T.; Silva, F.G. Acute postinfectious glomerulonephritis and glomerulonephritis complicating persistent bacterial infection. In Heptinstall’s Pathology of the Kidney, 6th ed.; Jennette, J.C., Olson, J.L., Schwartz, M.M., Silva, F.G., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; pp. 321–396. [Google Scholar]
- Nasr, S.H.; Fidler, M.E.; Valeri, A.M.; Cornell, L.D.; Sethi, S.; Zoller, A.; Stokes, M.B.; Markowitz, G.S.; D’Agati, V.D. Postinfectious glomerulonephritis in the elderly. J. Am. Soc. Nephrol. 2011, 22, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Oda, T.; Yoshizawa, N.; Yamakami, K.; Tamura, K.; Kuroki, A.; Sugisaki, T.; Sawanobori, E.; Higashida, K.; Ohtomo, Y.; Hotta, O.; et al. Localization of nephritis-associated plasmin receptor in acute poststreptococcal glomerulonephritis. Hum. Pathol. 2010, 41, 1276–1285. [Google Scholar] [CrossRef] [PubMed]
- Treser, G.; Semar, M.; McVicar, M.; Franklin, M.; Ty, A.; Sagel, I.; Lange, K. Antigenic streptococcal components in acute glomerulonephritis. Science 1969, 163, 676–677. [Google Scholar] [CrossRef]
- Winram, S.B.; Lottenberg, R. The plasmin-binding protein Plr of group A streptococci is identified as glyceraldehyde-3-phosphate dehydrogenase. Microbiology 1996, 142, 2311–2320. [Google Scholar] [CrossRef] [PubMed]
- Lottenberg, R.; Broder, C.C.; Boyle, M.D. Identification of a specific receptor for plasmin on a group A streptococcus. Infect. Immun. 1987, 55, 1914–1918. [Google Scholar] [CrossRef] [PubMed]
- Lottenberg, R.; DesJardin, L.E.; Wang, H.; Boyle, M.D. Streptokinase-producing streptococci grown in human plasma acquire unregulated cell-associated plasmin activity. J. Infect. Dis. 1992, 166, 436–440. [Google Scholar] [CrossRef]
- Oda, T.; Yamakami, K.; Omasu, F.; Suzuki, S.; Miura, S.; Sugisaki, T.; Yoshizawa, N. Glomerular plasmin-like activity in relation to nephritis-associated plasmin receptor in acute poststreptococcal glomerulonephritis. J. Am. Soc. Nephrol. 2005, 16, 247–254. [Google Scholar] [CrossRef]
- Oda, T.; Yoshizawa, N.; Yamakami, K.; Sakurai, Y.; Takechi, H.; Yamamoto, K.; Oshima, N.; Kumagai, H. The role of nephritis-associated plasmin receptor (NAPlr) in glomerulonephritis associated with streptococcal infection. J. Biomed. Biotechnol. 2012, 2012, 417675. [Google Scholar] [CrossRef]
- Vogt, A.; Batsford, S.; Rodriguez-Iturbe, B.; Garcia, R. Cationic antigens in poststreptococcal glomerulonephritis. Clin. Nephrol. 1983, 20, 271–279. [Google Scholar]
- Rodriguez-Iturbe, B. Autoimmunity in Acute Poststreptococcal GN: A Neglected Aspect of the Disease. J. Am. Soc. Nephrol. 2021, 32, 534–542. [Google Scholar] [CrossRef]
- Fish, A.J.; Herdman, R.C.; Michael, A.F.; Pickering, R.J.; Good, R.A. Epidemic acute glomerulonephritis associated with type 49 streptococcal pyoderma. II. Correlative study of light, immunofluorescent and electron microscopic findings. Am. J. Med. 1970, 48, 28–39. [Google Scholar] [CrossRef]
- Westberg, N.G.; Naff, G.B.; Boyer, J.T.; Michael, A.F. Glomerular deposition of properdin in acute and chronic glomerulonephritis with hypocomplementemia. J. Clin. Investig. 1971, 50, 642–649. [Google Scholar] [CrossRef]
- Verroust, P.J.; Wilson, C.B.; Cooper, N.R.; Edgington, T.S.; Dixon, F.J. Glomerular complement components in human glomerulonephritis. J. Clin. Investig. 1974, 53, 77–84. [Google Scholar] [CrossRef]
- Kluthe, R.; Vogt, A.; Batsford, S.R. Glomerulonephritis: International Conference on Pathogenesis, Pathology and Treatment, Freiburg, Germany, 18–19th June 1976; Thieme: New York, NY, USA, 1976. [Google Scholar]
- Wyatt, R.J.; McAdams, A.J.; Forristal, J.; Snyder, J.; West, C.D. Glomerular deposition of complement-control proteins in acute and chronic glomerulonephritis. Kidney Int. 1979, 16, 505–512. [Google Scholar] [CrossRef]
- Sorger, K.; Gessler, U.; Hubner, F.K.; Kohler, H.; Schulz, W.; Stuhlinger, W.; Thoenes, G.H.; Thoenes, W. Subtypes of acute postinfectious glomerulonephritis. Synopsis of clinical and pathological features. Clin. Nephrol. 1982, 17, 114–128. [Google Scholar]
- Madaio, M.P.; Harrington, J.T. Current concepts. The diagnosis of acute glomerulonephritis. N. Engl. J. Med. 1983, 309, 1299–1302. [Google Scholar] [CrossRef]
- Al-Ghaithi, B.; Chanchlani, R.; Riedl, M.; Thorner, P.; Licht, C. C3 Glomerulopathy and post-infectious glomerulonephritis define a disease spectrum. Pediatr. Nephrol. 2016, 31, 2079–2086. [Google Scholar] [CrossRef]
- Oda, T. Acute glomerulonephritis syndrome (AGS). In Shin Rinsho Naikagaku (PRACTICE OF INTERNAL MEDICINE), 10th ed; Yazaki, Y., Ed.; Igaku-Shoin Ltd.: Tokyo, Japan, 2020; pp. 1181–1183. [Google Scholar]
- Yoshizawa, N. Acute glomerulonephritis. Intern. Med. 2000, 39, 687–694. [Google Scholar] [CrossRef]
- Plesner, T.; Ploug, M.; Ellis, V.; Ronne, E.; Hoyer-Hansen, G.; Wittrup, M.; Pedersen, T.L.; Tscherning, T.; Dano, K.; Hansen, N.E. The receptor for urokinase-type plasminogen activator and urokinase is translocated from two distinct intracellular compartments to the plasma membrane on stimulation of human neutrophils. Blood 1994, 83, 808–815. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Song, Y.P.; Boel, G.; Kochar, J.; Pancholi, V. Group A streptococcal surface GAPDH, SDH, recognizes uPAR/CD87 as its receptor on the human pharyngeal cell and mediates bacterial adherence to host cells. J. Mol. Biol. 2005, 350, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Boyle, M.D.; Lottenberg, R. Plasminogen activation by invasive human pathogens. Thromb. Haemost. 1997, 77, 001–010. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Ploplis, V.A.; Castellino, F.J. Bacterial plasminogen receptors utilize host plasminogen system for effective invasion and dissemination. J. Biomed. Biotechnol. 2012, 2012, 482096. [Google Scholar] [CrossRef]
- Pancholi, V.; Fischetti, V.A. A major surface protein on group A streptococci is a glyceraldehyde-3-phosphate-dehydrogenase with multiple binding activity. J. Exp. Med. 1992, 176, 415–426. [Google Scholar] [CrossRef]
- Uchida, T.; Oda, T. Glomerular Deposition of Nephritis-Associated Plasmin Receptor (NAPlr) and Related Plasmin Activity: Key Diagnostic Biomarkers of Bacterial Infection-related Glomerulonephritis. Int. J. Mol. Sci. 2020, 21, 2595. [Google Scholar] [CrossRef]
- Vassalli, J.D.; Sappino, A.P.; Belin, D. The plasminogen activator/plasmin system. J. Clin. Investig. 1991, 88, 1067–1072. [Google Scholar] [CrossRef]
- Mignatti, P.; Rifkin, D.B. Biology and biochemistry of proteinases in tumor invasion. Physiol. Rev. 1993, 73, 161–195. [Google Scholar] [CrossRef]
- Montrucchio, G.; Lupia, E.; De Martino, A.; Silvestro, L.; Savu, S.R.; Cacace, G.; De Filippi, P.G.; Emanuelli, G.; Camussi, G. Plasmin promotes an endothelium-dependent adhesion of neutrophils. Involvement of platelet activating factor and P-selectin. Circulation 1996, 93, 2152–2160. [Google Scholar] [CrossRef]
- Burysek, L.; Syrovets, T.; Simmet, T. The serine protease plasmin triggers expression of MCP-1 and CD40 in human primary monocytes via activation of p38 MAPK and janus kinase (JAK)/STAT signaling pathways. J. Biol. Chem. 2002, 277, 33509–33517. [Google Scholar] [CrossRef]
- Syrovets, T.; Simmet, T. Novel aspects and new roles for the serine protease plasmin. Cell Mol. Life Sci. 2004, 61, 873–885. [Google Scholar] [CrossRef]
- Yoshizawa, N.; Oda, T.; Oshikawa, Y.; Akashi, Y.; Suzuki, Y.; Shimizu, J.; Shimizu, E.; Niwa, H.; Treser, G. Cell-mediated immune response in acute poststreptococcal glomerulonephritis. Nihon Jinzo Gakkai Shi 1994, 36, 322–330. [Google Scholar]
- Oda, T.; Yoshizawa, N.; Takeuchi, A.; Nakabayashi, I.; Nishiyama, J.; Ishida, A.; Tazawa, K.; Murayama, M.; Hotta, O.; Taguma, Y. Glomerular proliferating cell kinetics in acute post-streptococcal glomerulonephritis (APSGN). J. Pathol. 1997, 183, 359–368. [Google Scholar] [CrossRef]
- Platt, J.L.; Grant, B.W.; Eddy, A.A.; Michael, A.F. Immune cell populations in cutaneous delayed-type hypersensitivity. J. Exp. Med. 1983, 158, 1227–1242. [Google Scholar] [CrossRef]
- Arai, M.; Mii, A.; Kashiwagi, T.; Shimizu, A.; Sakai, Y. The severity of glomerular endothelial cell injury is associated with infiltrating macrophage heterogeneity in endocapillary proliferative glomerulonephritis. Sci. Rep. 2021, 11, 13339. [Google Scholar] [CrossRef]
- Yoshizawa, N.; Treser, G.; Sagel, I.; Ty, A.; Ahmed, U.; Lange, K. Demonstration of antigenic sites in glomeruli of patients with acute poststreptococcal glomerulonephritis by immunofluorescein and immunoferritin technics. Am. J. Pathol. 1973, 70, 131–150. [Google Scholar]
- Seegal, B.C.; Andres, G.A.; Hsu, K.C.; Zabriskie, J.B. Studies on the Pathogenesis of Acute and Progressive Glomerulonephritis in Man by Immunofluorescein and Immunoferritin Techniques. Fed. Proc. 1965, 24, 100–108. [Google Scholar]
- Andres, G.A.; Accinni, L.; Hsu, K.C.; Zabriskie, J.B.; Seegal, B.C. Electron microscopic studies of human glomerulonephritis with ferritin-conjugated antibody. Localization of antigen-antibody complexes in glomerular structures of patients with acute glomerulonephritis. J. Exp. Med. 1966, 123, 399–412. [Google Scholar] [CrossRef]
- Batsford, S.R.; Mezzano, S.; Mihatsch, M.; Schiltz, E.; Rodriguez-Iturbe, B. Is the nephritogenic antigen in post-streptococcal glomerulonephritis pyrogenic exotoxin B (SPE B) or GAPDH? Kidney Int. 2005, 68, 1120–1129. [Google Scholar] [CrossRef]
- Oshima, S.; Yoshizawa, N. Role of the membrane attack complex (MAC) in acute poststreptococcal glomerulonephritis. Nihon Jinzo Gakkai Shi 1988, 30, 811–819. [Google Scholar]
- Yoshizawa, N.; Oshima, S.; Takeuchi, A.; Kondo, S.; Oda, T.; Shimizu, J.; Nishiyama, J.; Ishida, A.; Nakabayashi, I.; Tazawa, K.; et al. Experimental acute glomerulonephritis induced in the rabbit with a specific streptococcal antigen. Clin. Exp. Immunol. 1997, 107, 61–67. [Google Scholar] [CrossRef]
- Yoshizawa, N.; Oshima, S.; Sagel, I.; Shimizu, J.; Treser, G. Role of a streptococcal antigen in the pathogenesis of acute poststreptococcal glomerulonephritis. Characterization of the antigen and a proposed mechanism for the disease. J. Immunol. 1992, 148, 3110–3116. [Google Scholar]
- Satoskar, A.A.; Silva, F.G. Acute postinfectious glomerulonephritis and glomerulonephritis caused by persistent bacterial infection. In Heptinstall’s Pathology of the Kidney, 7th ed.; Jennette, J.C., Olson, J.L., Silva, F.G., D’Agati, V.D., Eds.; Wolters Kluwer: Philadelphia, PA, USA, 2015; pp. 367–436. [Google Scholar]
- Pickering, M.C.; D’Agati, V.D.; Nester, C.M.; Smith, R.J.; Haas, M.; Appel, G.B.; Alpers, C.E.; Bajema, I.M.; Bedrosian, C.; Braun, M.; et al. C3 glomerulopathy: Consensus report. Kidney Int. 2013, 84, 1079–1089. [Google Scholar] [CrossRef]
- Sethi, S.; Gamez, J.D.; Vrana, J.A.; Theis, J.D.; Bergen, H.R., 3rd; Zipfel, P.F.; Dogan, A.; Smith, R.J. Glomeruli of Dense Deposit Disease contain components of the alternative and terminal complement pathway. Kidney Int. 2009, 75, 952–960. [Google Scholar] [CrossRef]
- Ito, N.; Ohashi, R.; Nagata, M. C3 glomerulopathy and current dilemmas. Clin. Exp. Nephrol. 2017, 21, 541–551. [Google Scholar] [CrossRef]
- Ravindran, A.; Fervenza, F.C.; Smith, R.J.H.; Sethi, S. C3 glomerulopathy associated with monoclonal Ig is a distinct subtype. Kidney Int. 2018, 94, 178–186. [Google Scholar] [CrossRef]
- Fakhouri, F.; Le Quintrec, M.; Fremeaux-Bacchi, V. Practical management of C3 glomerulopathy and Ig-mediated MPGN: Facts and uncertainties. Kidney Int. 2020, 98, 1135–1148. [Google Scholar] [CrossRef]
- Levine, A.P.; Chan, M.M.Y.; Sadeghi-Alavijeh, O.; Wong, E.K.S.; Cook, H.T.; Ashford, S.; Carss, K.; Christian, M.T.; Hall, M.; Harris, C.L.; et al. Large-Scale Whole-Genome Sequencing Reveals the Genetic Architecture of Primary Membranoproliferative GN and C3 Glomerulopathy. J. Am. Soc. Nephrol. 2020, 31, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Chauvet, S.; Roumenina, L.T.; Aucouturier, P.; Marinozzi, M.C.; Dragon-Durey, M.A.; Karras, A.; Delmas, Y.; Le Quintrec, M.; Guerrot, D.; Jourde-Chiche, N.; et al. Both Monoclonal and Polyclonal Immunoglobulin Contingents Mediate Complement Activation in Monoclonal Gammopathy Associated-C3 Glomerulopathy. Front. Immunol. 2018, 9, 2260. [Google Scholar] [CrossRef] [PubMed]
- Sethi, S.; Vrana, J.A.; Fervenza, F.C.; Theis, J.D.; Sethi, A.; Kurtin, P.J.; Zhang, Y.; Smith, R.J.H. Characterization of C3 in C3 glomerulopathy. Nephrol. Dial. Transplant. 2017, 32, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Bomback, A.S.; Santoriello, D.; Avasare, R.S.; Regunathan-Shenk, R.; Canetta, P.A.; Ahn, W.; Radhakrishnan, J.; Marasa, M.; Rosenstiel, P.E.; Herlitz, L.C.; et al. C3 glomerulonephritis and dense deposit disease share a similar disease course in a large United States cohort of patients with C3 glomerulopathy. Kidney Int. 2018, 93, 977–985. [Google Scholar] [CrossRef]
- Kaartinen, K.; Safa, A.; Kotha, S.; Ratti, G.; Meri, S. Complement dysregulation in glomerulonephritis. Semin. Immunol. 2019, 45, 101331. [Google Scholar] [CrossRef]
- Lemaire, M.; Noone, D.; Lapeyraque, A.L.; Licht, C.; Fremeaux-Bacchi, V. Inherited Kidney Complement Diseases. Clin. J. Am. Soc. Nephrol. 2021, 16, 942–956. [Google Scholar] [CrossRef]
- Cook, H.T.; Pickering, M.C. C3 glomerulopathies, including dense deposit disease. In Heptinstall’s Pathology of the Kidney, 7th ed.; Jennette, J.C., Olson, J.L., Silva, F.G., D’Agati, V.D., Eds.; Wolters Kluwer: Philadelphia, PA, USA, 2015; pp. 341–366. [Google Scholar]
- Takahashi, M.; Takahashi, S.; Hirose, S. Solubilization of antigen-antibody complexes: A new function of complement as a regulator of immune reactions. Prog. Allergy 1980, 27, 134–166. [Google Scholar]
- Gadd, K.J.; Reid, K.B. The binding of complement component C3 to antibody-antigen aggregates after activation of the alternative pathway in human serum. Biochem. J. 1981, 195, 471–480. [Google Scholar] [CrossRef]
- Takata, Y.; Tamura, N.; Fujita, T. Interaction of C3 with antigen-antibody complexes in the process of solubilization of immune precipitates. J. Immunol. 1984, 132, 2531–2537. [Google Scholar]
- Yoshizawa, N.; Treser, G.; McClung, J.A.; Sagel, I.; Takahashi, K. Circulating immune complexes in patients with uncomplicated group A streptococcal pharyngitis and patients with acute poststreptococcal glomerulonephritis. Am. J. Nephrol. 1983, 3, 23–29. [Google Scholar] [CrossRef]
- Messias, N.C.; Walker, P.D.; Larsen, C.P. Paraffin immunofluorescence in the renal pathology laboratory: More than a salvage technique. Mod. Pathol. 2015, 28, 854–860. [Google Scholar] [CrossRef]
- Tauber, J.W.; Polley, M.J.; Zabriskie, J.B. Nonspecific complement activation by streptococcal structures. II. Properdin-independent initiation of the alternate pathway. J. Exp. Med. 1976, 143, 1352–1366. [Google Scholar] [CrossRef] [Green Version]
- Vernon, K.A.; Goicoechea de Jorge, E.; Hall, A.E.; Fremeaux-Bacchi, V.; Aitman, T.J.; Cook, H.T.; Hangartner, R.; Koziell, A.; Pickering, M.C. Acute presentation and persistent glomerulonephritis following streptococcal infection in a patient with heterozygous complement factor H-related protein 5 deficiency. Am. J. Kidney Dis. 2012, 60, 121–125. [Google Scholar] [CrossRef]
- Chauvet, S.; Berthaud, R.; Devriese, M.; Mignotet, M.; Vieira Martins, P.; Robe-Rybkine, T.; Miteva, M.A.; Gyulkhandanyan, A.; Ryckewaert, A.; Louillet, F.; et al. Anti-Factor B Antibodies and Acute Postinfectious GN in Children. J. Am. Soc. Nephrol. 2020, 31, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Sethi, S.; Fervenza, F.C.; Zhang, Y.; Zand, L.; Meyer, N.C.; Borsa, N.; Nasr, S.H.; Smith, R.J. Atypical postinfectious glomerulonephritis is associated with abnormalities in the alternative pathway of complement. Kidney Int. 2013, 83, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Nasr, S.H.; Radhakrishnan, J.; D’Agati, V.D. Bacterial infection-related glomerulonephritis in adults. Kidney Int. 2013, 83, 792–803. [Google Scholar] [CrossRef] [PubMed]
- Sawanobori, E.; Umino, A.; Kanai, H.; Matsushita, K.; Iwasa, S.; Kitamura, H.; Oda, T.; Yoshizawa, N.; Sugita, K.; Higashida, K. A prolonged course of Group A streptococcus-associated nephritis: A mild case of dense deposit disease (DDD)? Clin. Nephrol. 2009, 71, 703–707. [Google Scholar] [CrossRef]
- Suga, K.; Kondo, S.; Matsuura, S.; Kinoshita, Y.; Kitano, E.; Hatanaka, M.; Kitamura, H.; Hidaka, Y.; Oda, T.; Kagami, S. A case of dense deposit disease associated with a group A streptococcal infection without the involvement of C3NeF or complement factor H deficiency. Pediatr. Nephrol. 2010, 25, 1547–1550. [Google Scholar] [CrossRef]
- Okabe, M.; Tsuboi, N.; Yokoo, T.; Miyazaki, Y.; Utsunomiya, Y.; Hosoya, T. A case of idiopathic membranoproliferative glomerulonephritis with a transient glomerular deposition of nephritis-associated plasmin receptor antigen. Clin. Exp. Nephrol. 2012, 16, 337–341. [Google Scholar] [CrossRef]
- Iseri, K.; Iyoda, M.; Yamamoto, Y.; Kobayashi, N.; Oda, T.; Yamaguchi, Y.; Shibata, T. Streptococcal Infection-related Nephritis (SIRN) Manifesting Membranoproliferative Glomerulonephritis Type I. Intern. Med. 2016, 55, 647–650. [Google Scholar] [CrossRef]
- Kohatsu, K.; Suzuki, T.; Yazawa, M.; Yahagi, K.; Ichikawa, D.; Koike, J.; Oda, T.; Shibagaki, Y. Granulomatosis with Polyangiitis Induced by Infection. Kidney Int. Rep. 2019, 4, 341–345. [Google Scholar] [CrossRef]
- Yano, K.; Suzuki, H.; Oda, T.; Ueda, Y.; Tsukamoto, T.; Muso, E. Crescentic poststreptococcal acute glomerulonephritis accompanied by small vessel vasculitis: Case report of an elderly male. BMC Nephrol. 2019, 20, 471. [Google Scholar] [CrossRef]
- Kikuchi, Y.; Yoshizawa, N.; Oda, T.; Imakiire, T.; Suzuki, S.; Miura, S. Streptococcal origin of a case of Henoch-Schoenlein purpura nephritis. Clin. Nephrol. 2006, 65, 124–128. [Google Scholar] [CrossRef]
- Odaka, J.; Kanai, T.; Ito, T.; Saito, T.; Aoyagi, J.; Betsui, H.; Oda, T.; Ueda, Y.; Yamagata, T. A case of post-pneumococcal acute glomerulonephritis with glomerular depositions of nephritis-associated plasmin receptor. CEN Case Rep. 2015, 4, 112–116. [Google Scholar] [CrossRef]
- Komaru, Y.; Ishioka, K.; Oda, T.; Ohtake, T.; Kobayashi, S. Nephritis-associated plasmin receptor (NAPlr) positive glomerulonephritis caused by Aggregatibacter actinomycetemcomitans bacteremia: A case report. Clin. Nephrol. 2018, 90, 155–160. [Google Scholar] [CrossRef]
- Hirano, D.; Oda, T.; Ito, A.; Yamada, A.; Kakegawa, D.; Miwa, S.; Umeda, C.; Takemasa, Y.; Tokunaga, A.; Wajima, T.; et al. Glyceraldehyde-3-phosphate dehydrogenase of Mycoplasma pneumoniae induces infection-related glomerulonephritis. Clin. Nephrol. 2019, 92, 263–272. [Google Scholar] [CrossRef]
- Grundel, A.; Pfeiffer, M.; Jacobs, E.; Dumke, R. Network of Surface-Displayed Glycolytic Enzymes in Mycoplasma pneumoniae and Their Interactions with Human Plasminogen. Infect. Immun. 2015, 84, 666–676. [Google Scholar] [CrossRef]
- Lottenberg, R.; Broder, C.C.; Boyle, M.D.; Kain, S.J.; Schroeder, B.L.; Curtiss, R., 3rd. Cloning, sequence analysis, and expression in Escherichia coli of a streptococcal plasmin receptor. J. Bacteriol. 1992, 174, 5204–5210. [Google Scholar] [CrossRef]
- Winram, S.B.; Lottenberg, R. Site-directed mutagenesis of streptococcal plasmin receptor protein (Plr) identifies the C-terminal Lys334 as essential for plasmin binding, but mutation of the plr gene does not reduce plasmin binding to group A streptococci. Microbiology 1998, 144, 2025–2035. [Google Scholar] [CrossRef]
- Takehara, E.; Mandai, S.; Shikuma, S.; Akita, W.; Chiga, M.; Mori, T.; Oda, T.; Kuwahara, M.; Uchida, S. Post-infectious Proliferative Glomerulonephritis with Monoclonal Immunoglobulin G Deposits Associated with Complement Factor H Mutation. Intern. Med. 2017, 56, 811–817. [Google Scholar] [CrossRef]
- Okabe, M.; Takamura, T.; Tajiri, A.; Tsuboi, N.; Ishikawa, M.; Ogura, M.; Ohashi, R.; Oda, T.; Yokoo, T. A case of infection-related glomerulonephritis with massive eosinophilic infiltration. Clin. Nephrol. 2018, 90, 142–147. [Google Scholar] [CrossRef]
- Asano, M.; Oda, T.; Mizuno, M. A case of C3 glomerulopathy with nephritis-associated plasmin receptor positivity without a history of streptococcal infection. CEN Case Rep. 2021, 11, 259–264. [Google Scholar] [CrossRef]
- Komaki, K.; Shiotsu, Y.; Adachi, H.; Urata, N.; Hara, M.; Nakayama, M.; Kusaba, T.; Masuzawa, N.; Konishi, E.; Oda, T.; et al. Nephritis-associated plasmin receptor (NAPlr)-positive glomerulonephritis in a case of ANCA-negative small vessel vasculitis. CEN Case Rep. 2022, 11, 90–96. [Google Scholar] [CrossRef]
- Onishi, A.; Mizumoto, A.; Mitsumoto, K.; Katsunuma, R.; Shingu, T.; Oda, T.; Uzu, T. A man with immunoglobulin A nephropathy complicated by infection-related glomerulonephritis with glomerular depositions of nephritis-associated plasmin receptor. CEN Case Rep. 2021, 10, 490–493. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Suzuki, T.; Watanabe, S.; Nakata, M.; Ichikawa, D.; Koike, J.; Oda, T.; Suzuki, H.; Suzuki, Y.; Shibagaki, Y. Galactose-deficient IgA1 and nephritis-associated plasmin receptors as markers for IgA-dominant infection-related glomerulonephritis: A case report. Medicine 2021, 100, e24460. [Google Scholar] [CrossRef] [PubMed]
- Noda, S.; Mandai, S.; Oda, T.; Shinoto, T.; Sato, H.; Sato, K.; Hirokawa, K.; Noda, Y.; Uchida, S. Asymptomatic sinusitis as an origin of infection-related glomerulonephritis manifesting steroid-resistant nephrotic syndrome: A case report. Medicine 2020, 99, e20572. [Google Scholar] [CrossRef] [PubMed]
- Nasr, S.H.; Galgano, S.J.; Markowitz, G.S.; Stokes, M.B.; D’Agati, V.D. Immunofluorescence on pronase-digested paraffin sections: A valuable salvage technique for renal biopsies. Kidney Int. 2006, 70, 2148–2151. [Google Scholar] [CrossRef] [PubMed]
- Nasr, S.H.; Fidler, M.E.; Said, S.M. Paraffin Immunofluorescence: A Valuable Ancillary Technique in Renal Pathology. Kidney Int. Rep. 2018, 3, 1260–1266. [Google Scholar] [CrossRef]
- Nasr, S.H.; Fidler, M.E.; Said, S.M.; Koepplin, J.W.; Altamirano-Alonso, J.M.; Leung, N. Immunofluorescence staining for immunoglobulin heavy chain/light chain on kidney biopsies is a valuable ancillary technique for the diagnosis of monoclonal gammopathy-associated kidney diseases. Kidney Int. 2021, 100, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Larsen, C.P.; Messias, N.C.; Walker, P.D.; Fidler, M.E.; Cornell, L.D.; Hernandez, L.H.; Alexander, M.P.; Sethi, S.; Nasr, S.H. Membranoproliferative glomerulonephritis with masked monotypic immunoglobulin deposits. Kidney Int. 2015, 88, 867–873. [Google Scholar] [CrossRef] [PubMed]
- Larsen, C.P.; Ambuzs, J.M.; Bonsib, S.M.; Boils, C.L.; Cossey, L.N.; Messias, N.C.; Silva, F.G.; Wang, Y.H.; Gokden, N.; Walker, P.D. Membranous-like glomerulopathy with masked IgG kappa deposits. Kidney Int. 2014, 86, 154–161. [Google Scholar] [CrossRef]
- Howlader, A.; Thajudeen, B.; Sussman, A.N.; Bracamonte, E.; Krahl, L.; Nasr, S.H. Proliferative Glomerulonephritis with Masked Monoclonal Deposits Responsive to Myeloma Therapy. Kidney Int. Rep. 2017, 2, 1233–1237. [Google Scholar] [CrossRef] [Green Version]
- DiFranza, L.T.; Markowitz, G.S.; D’Agati, V.D.; Santoriello, D. Atypical Infection-Related Glomerulonephritis With “Masked” IgG-Kappa Crystalline Hump-Like Deposits. Kidney Int. Rep. 2021, 6, 228–233. [Google Scholar] [CrossRef]
- Yu, X.J.; Hu, N.; Wang, S.X.; Zhou, F.D.; Zhao, M.H. Membranoproliferative glomerulonephritis with deposition of monoclonal IgG evolved from polyclonal IgG: A case report with two consecutive renal biopsies. BMC Nephrol. 2019, 20, 275. [Google Scholar] [CrossRef]
- Johnson, C.K.; Zuniga, S.C.; Dhawale, T.; Zhang, Y.; Smith, R.J.H.; Blosser, C.D. Monoclonal Gammopathy of Renal Significance Causes C3 Glomerulonephritis Via Monoclonal IgG Kappa Inhibition of Complement Factor H. Kidney Int. Rep. 2021, 6, 2505–2509. [Google Scholar] [CrossRef]
- Sethi, S.; Fervenza, F.C.; Zhang, Y.; Zand, L.; Vrana, J.A.; Nasr, S.H.; Theis, J.D.; Dogan, A.; Smith, R.J. C3 glomerulonephritis: Clinicopathological findings, complement abnormalities, glomerular proteomic profile, treatment, and follow-up. Kidney Int. 2012, 82, 465–473. [Google Scholar] [CrossRef] [Green Version]






















| Time from Onset to Biopsy | n | NAPlr | Plasminogen | Fibrinogen | C3 | IgG | IgA | IgM |
|---|---|---|---|---|---|---|---|---|
| 1–14 d | 25 | 25/25 (100%) | 10/25 (40) | 15/25 (60) | 25/25 (100) | 16/25 (64) | 11/25 (44) | 10/25 (40) |
| 15–30 d | 18 | 11/18 (61) | 5/18 (28) | 11/18 (61) | 18/18 (100) | 11/18 (61) | 8/18 (44) | 9/18 (50) |
| 31–90 d | 7 | 0/7 (0) | 0/7 (0) | 4/7 (57) | 6/7 (86) | 3/7 (43) | 3/7 (43) | 3/7 (43) |
| Total | 50 | 36/50 (72) | 15/50 (30) | 30/50 (60) | 49/50 (98) | 30/50 (60) | 22/50 (44) | 22/50 (44) |
| Time from Onset to Biopsy | n | P | C1q | C4 | C5 | C9 | S | MAC |
| 1–14 d | 25 | 23/25 (92%) | 7/25 (28) | 8/25 (32) | 25/25 (100) | 24/25 (96) | 24/25 (96) | 25/25 (100) |
| 15–30 d | 18 | 16/18 (89) | 6/18 (33) | 3/18 (17) | 18/18 (100) | 17/18 (94) | 17/18 (94) | 18/18 (100) |
| 31–90 d | 7 | 5/7 (71) | 2/7 (29) | 1/7 (14) | 6/7 (86) | 5/7 (71) | 5/7 (71) | 6/7 (86) |
| Total | 50 | 44/50 (88) | 15/50 (30) | 12/50 (24) | 49/50 (98) | 46/50 (92) | 46/50 (92) | 49/50 (98) |
| Biopsy Specimens | Onset to Biopsy | Glomerular NAPlr (+) | |
|---|---|---|---|
| PSAGN | 1–14 days | 25/25 | (100%) |
| 15–30 days | 11/18 | (61%) | |
| 31–90 days | 0/7 | (0%) | |
| Total | 36/50 | (72%) | |
| Non-PSAGN | 4/100 | (4%) | |
| Normal kidneys | 0/10 | (0%) | |
| Age in Years Range | Anti-NAPlr Antibody (+) | Anti-NAPlr Antibody Titers | |
|---|---|---|---|
| PSAGN | (5–72 y, mean 29.3) | 46/50 (92%) | 566 ± 106.1 |
| Streptococcal infection | (8–64 y, mean 29.0) | 30/50 (60%) | 227.1 ± 51.2 |
| Pediatric I | (0.2–10 y, mean 7.2) | 13/50 (26%) | 138.9 ± 23.4 |
| Pediatric II | (11–20 y, mean 14.1) | 18/50 (36%) | 166.0 ± 25.7 |
| Normal adults I | (25–35 y, mean 30.0) | 24/50 (48%) | 100.1 ± 18.0 |
| Normal adults II | (52–59 y, mean 53.0) | 36/50 (72%) | 186.0 ± 17.3 |
| Case | Age/ Gender | Diagnosis | Onset | UP | U-RBC (/hpf) | SCr (mg/dL) | CH50 (U/mL) | C3 (mg/dL) | C4 (mg/dL) | CH50 (U/mL) | EP | IF | NAPlr | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 14/F | DDD | AGN | +++ | +++ | 0.48 | <10 | 8 | - | 1090 | +++ | C3 cap | +++ | [71] |
| 2 | 12/M | DDD | AKI on CGN | +++ | 30–49 | 0.44 | 31.1 | 48 | 27 | 333 | +++ | C3 cap | +++ | [70] |
| 3 | 6/F | DDD | Asymptomatic | ± | 20–29 | 0.36 | 23.5 | 36 | 19 | 290 | ++ | C3 cap | + | our case |
| 4 | 62/F | MPGN I | AKI on CGN | +++ | 20–29 | 3.6 | 21.7 | - | - | 170 | ++ | IgG, IgM, C3 cap | ++ | [73] |
| 5 | 6/F | HSPN | Abd pain, purpura | +++ | many | 0.35 | 67.1 | 89 | 32 | 154 | ++ | IgA, C3 mes-cap | ++ | our case |
| 6 | 25/M | HSPN | Abd pain, purpura | + | 30–49 | 1.3 | 28 | 50 | 22 | 743 | +++ | IgA, C3 mes-cap | +++ | [76] |
| 7 | 71/M | SVV | Pain and swelling of the rt. ankle | +++ | 50–99 | 3.28 | - | 42 | 27 | 512 | +++ | C3 mes-cap | +++ | [75] |
| 8 | 17/M | GPA | AGN | ++ | 100< | 7.53 | - | 10 | 26 | 301 | − | C3 mes | + | [74] |
| Case | Age/ Gender | Infection Focus | Pathogen | Complement C3/C4/CH50 | NAPlr /PA | Light Microscopy | IF | EM (Dense Deposits) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 12/F | respiratory infection | Streptococcus pneumoniae | 10/33/- | +/+ | cellular crescent formation, endocapillary proliferative GN | C3 | GBM hump (−) | [77] |
| 2 | 64/M | endocarditis | Aggregatibacter actinomycetemcomitans | 86/16/- | +/− | proliferative GN with inflammatory cell infiltration | IgG IgM C3 C1q IgA | Subend Mes hump (−) | [78] |
| 3 | 7/F | respiratory infection | Mycoplasma pneumoniae | WNL | +/+ | endocapillary proliferation and cellular crescents | IgA IgM C3 | Mes Parames Subendo hump (−) | [79] |
| 4 | 70/M | cellulitis | Staphylococcus aureus (MSSA) | 118/43.9/75.3 | +/+ | endocapillary proliferative GN | IgG IgA IgM C3 C1q | GBM Parames hump (−) | Our case |
| Pathogen | Amino Acid Sequence of GAPDH | |||
|---|---|---|---|---|
| Total Amino Acid | Identity Similarity (%) | C Terminal Amino Acid | ||
| Streptococcal pyogenes | 336 | - | - | T L E Y F A K I A K |
| Streptococcus pneumoniae | 359 | 92 | 99 | T L E Y F A K I A K |
| Aggregatibacter actinomycetemcomitans | 334 | 50 | 85 | L V A H V Y N Y K D |
| Mycoplasma pneumoniae | 337 | 54 | 87 | V R V V N Y C A K L |
| Staphylococcus aureus (MSSA) | 336 | 67 | 92 | T L A Y L A E L S K |
| Case | Age/ Gender | Infection Focus | Underlying Disease | Pathogen | Complement C3/C4/CH50 | NAPlr/ PA | Light Microscopy | IF | EM (Dense Deposits) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| 5 | 68/M | sinusitis | - | unknown | 25.1/20.8/<12 | +/+ | proliferative and exudative GN, MPGN type I | IgG C3 | Parames hump (−) | [89] |
| 6 | 17/F | unknown | C3G | unknown | 6/15/<12 | +/+ | lobulation of the glomerular capillary tufts, proliferation of glomerular mesangial cells, increased mesangial matrix, MPGN type III | C3 | Subendo Subepi | [85] |
| 7 | 55/M | unknown | CFH mutation | unknown | 108/43/61.6 | +/+ | cellular crescents and diffuse endocapillary and mesangial proliferation with double contour formation along the capillary walls | C3 IgG | Subepi Subendo hump (−) | [83] |
| 8 | 70/M | respiratory infection | - | unknown | 80/11.8/15.8 | +/+ | endocapillary proliferative GN | IgG C3 | Subepi hump (+) | [84] |
| 9 | 75/M | unknown | ANCA negative SVV | unknown | 105/26/52.7 | +/+ | cellular crescents/segmental endocapillary proliferation with neutrophils and tuft necrosis | C3 | Mes, GBM, Subepi, hump (+) | [86] |
| 10 | 82/M | unknown | - | unknown | 64/25/- | +/+ | subendothelial deposits including wire loop lesions, endocapillary proliferation, cellular crescents | IgA C3 IgG/IgM C1q/C4 | Subendo, Mes hump (−) | [88] |
| 11 | 27/M | unknown | IgA nephropathy | unknown | 18/23/- | +/+ | mesangial cell proliferation, increased mesangial matrix increasing, endocapillary proliferation | IgA C3 | Mes hump (−) | [87] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoshizawa, N.; Yamada, M.; Fujino, M.; Oda, T. Nephritis-Associated Plasmin Receptor (NAPlr): An Essential Inducer of C3-Dominant Glomerular Injury and a Potential Key Diagnostic Biomarker of Infection-Related Glomerulonephritis (IRGN). Int. J. Mol. Sci. 2022, 23, 9974. https://doi.org/10.3390/ijms23179974
Yoshizawa N, Yamada M, Fujino M, Oda T. Nephritis-Associated Plasmin Receptor (NAPlr): An Essential Inducer of C3-Dominant Glomerular Injury and a Potential Key Diagnostic Biomarker of Infection-Related Glomerulonephritis (IRGN). International Journal of Molecular Sciences. 2022; 23(17):9974. https://doi.org/10.3390/ijms23179974
Chicago/Turabian StyleYoshizawa, Nobuyuki, Muneharu Yamada, Masayuki Fujino, and Takashi Oda. 2022. "Nephritis-Associated Plasmin Receptor (NAPlr): An Essential Inducer of C3-Dominant Glomerular Injury and a Potential Key Diagnostic Biomarker of Infection-Related Glomerulonephritis (IRGN)" International Journal of Molecular Sciences 23, no. 17: 9974. https://doi.org/10.3390/ijms23179974