
Proline Dehydrogenase/Proline Oxidase (PRODH/POX) Is Involved in the Mechanism of Metformin-Induced Apoptosis in C32 Melanoma Cell Line

 , , , and
, , , and 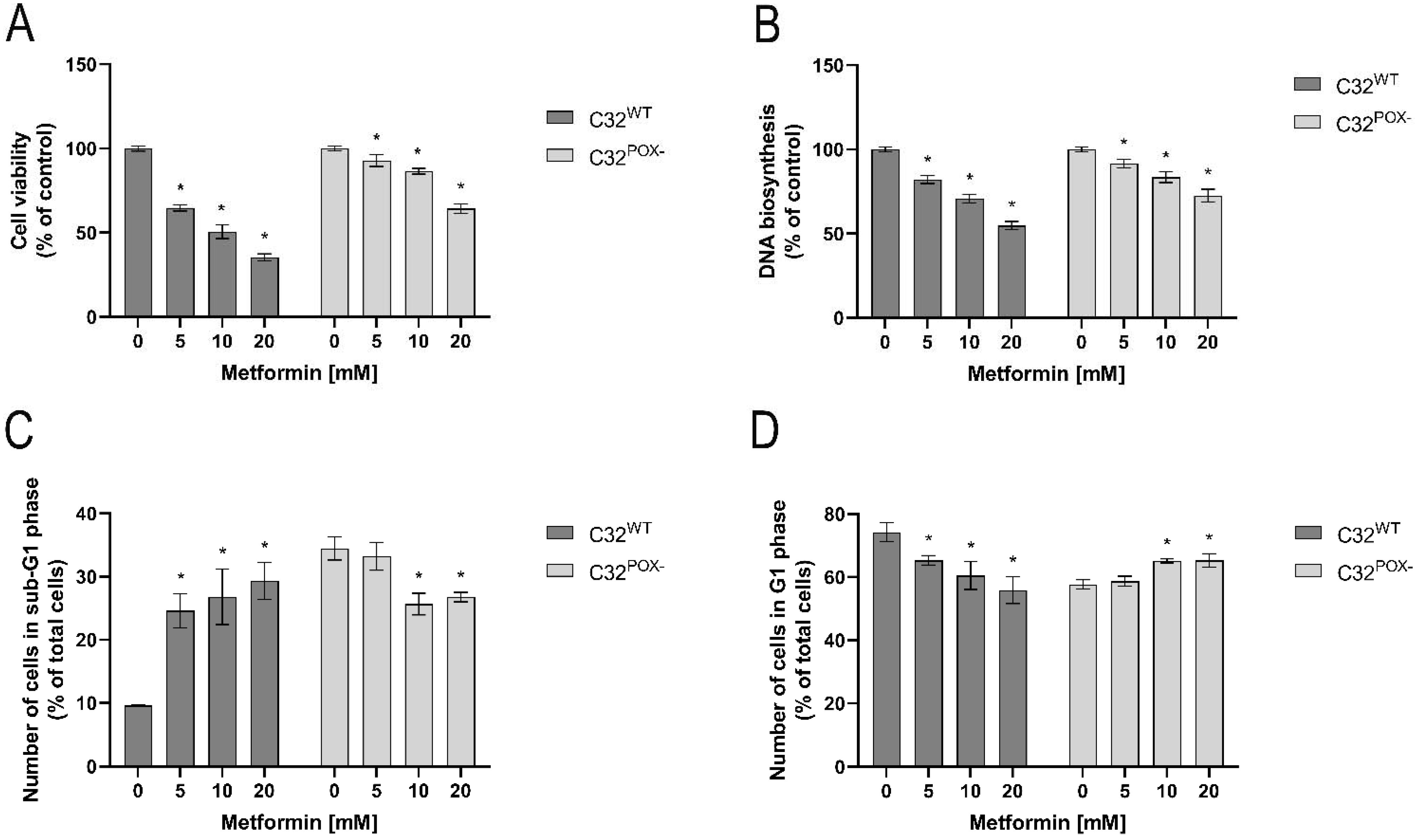{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
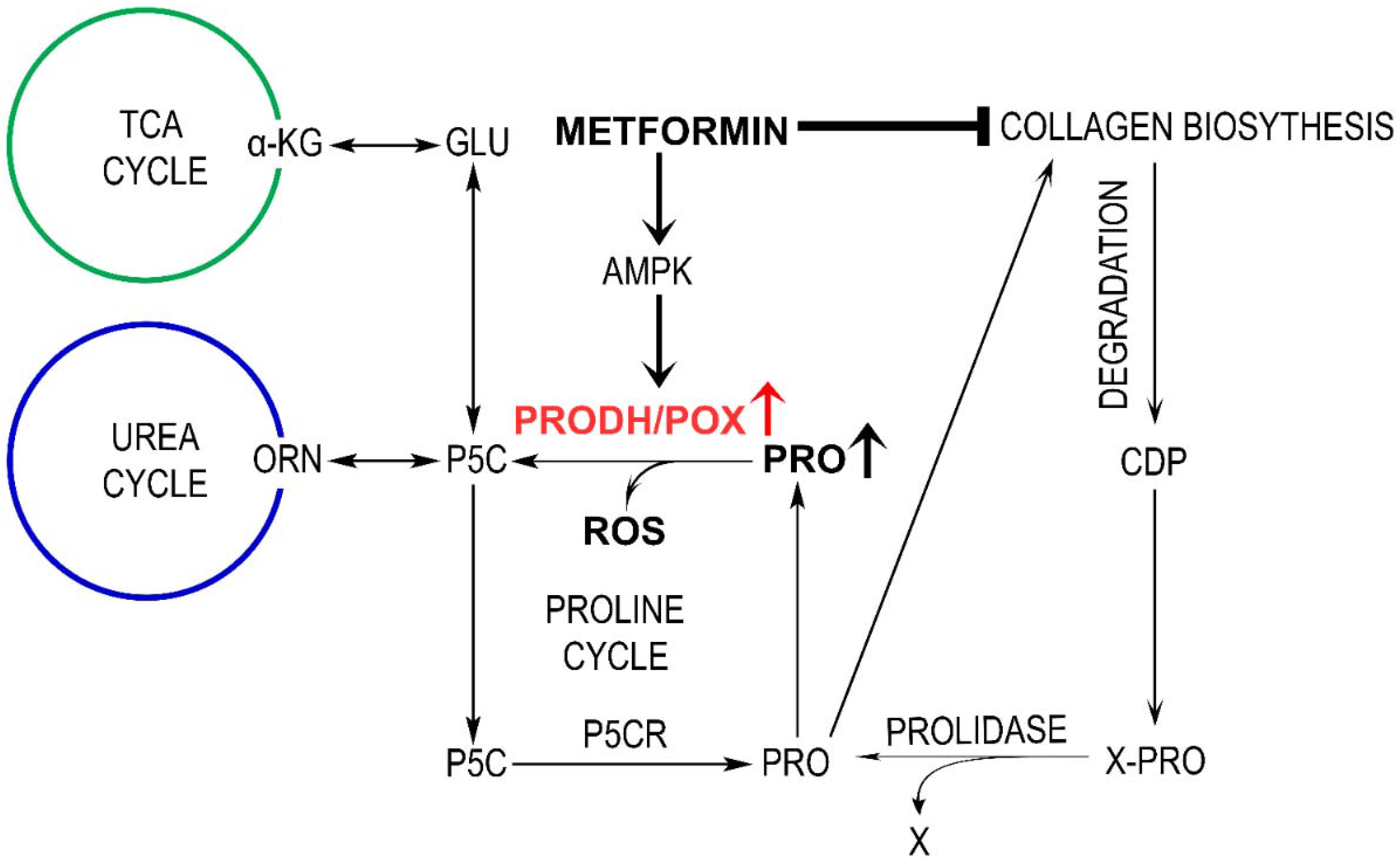
Abstract
:1. Introduction
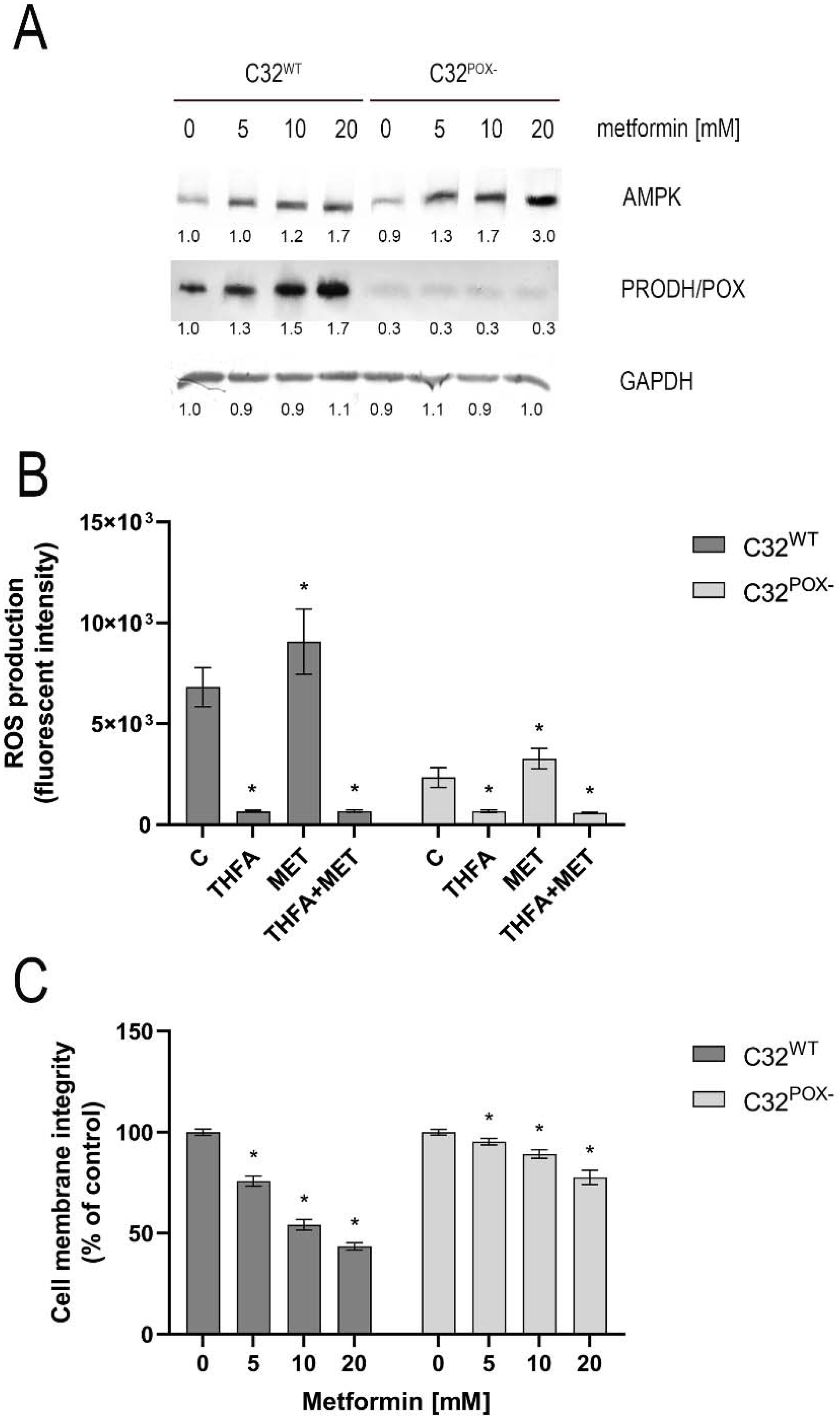
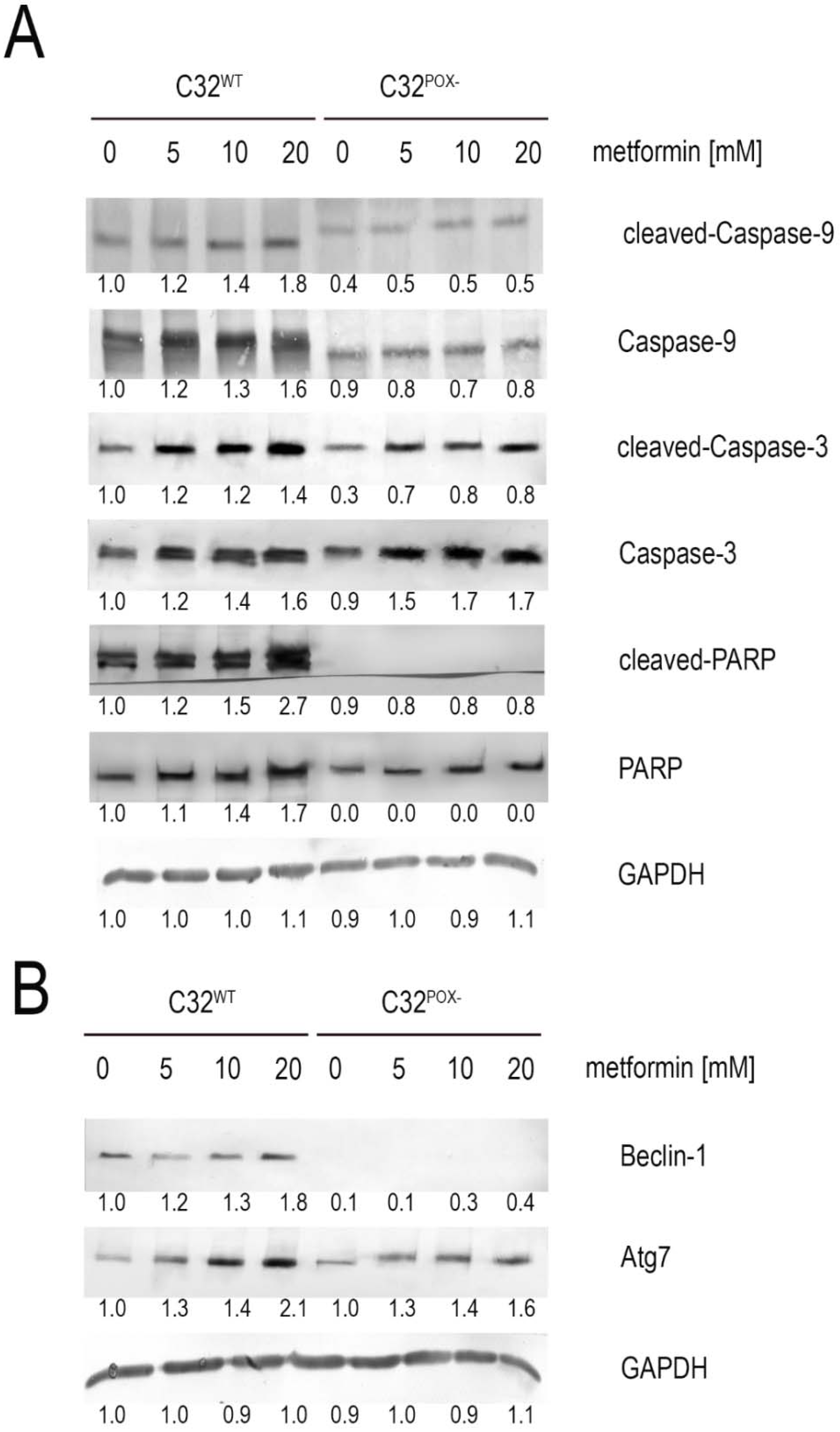
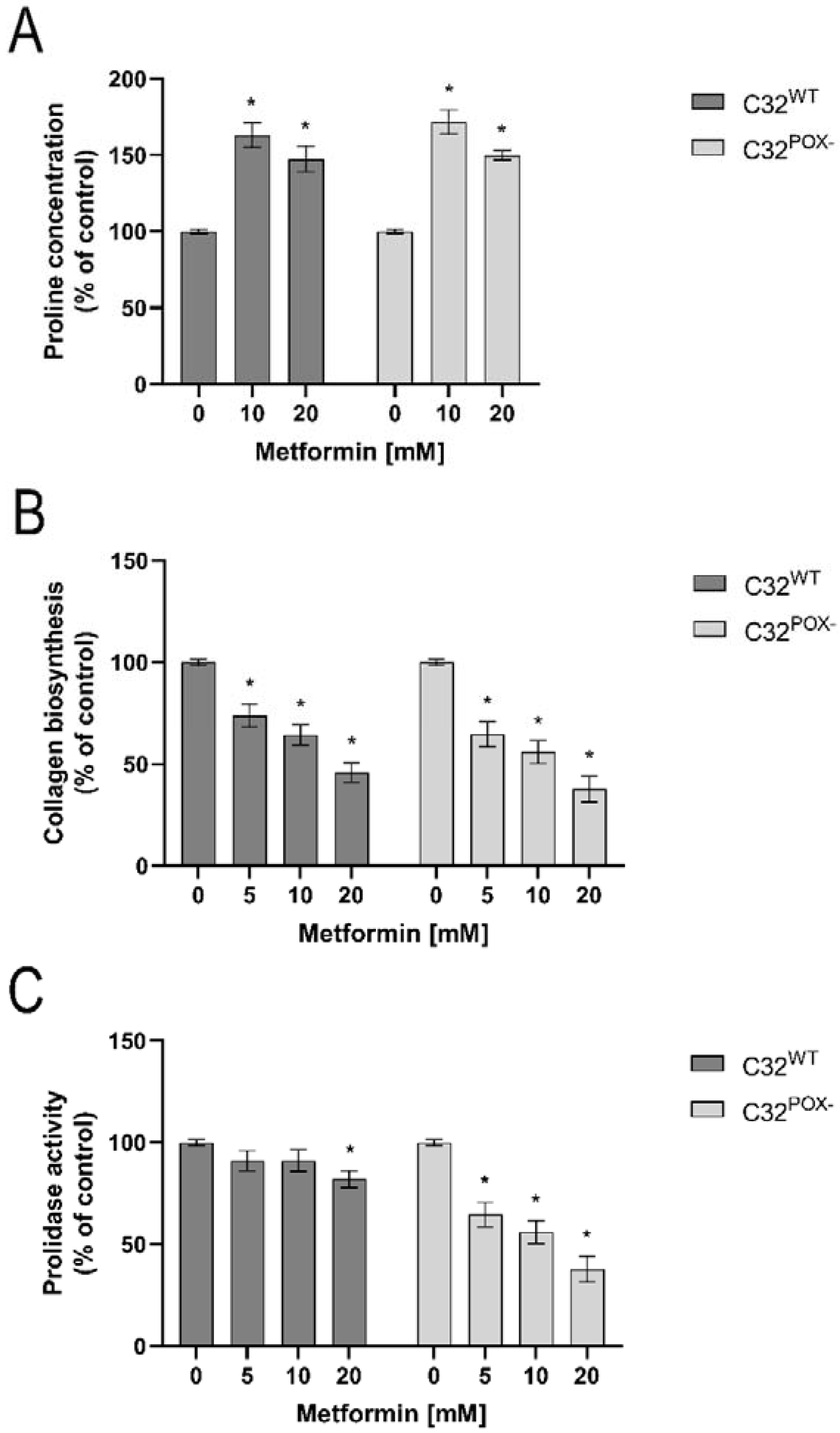
2. Results
3. Discussion
4. Materials and Methods
4.1. PRODH/POX Knock Uut CRISPR-cas9 DNA Plasmid Purification
4.2. Transfection into Melanoma Cell Line
4.3. Cell Culture and Treatment
4.4. Cell Viability
4.5. DNA Biosynthesis Assay
4.6. Cell Cycle Analysis
4.7. ROS Generation Assessment
4.8. Cell Membrane Integrity Assay
4.9. Preparation of Cell Lysates
4.10. Western Immunoblotting
4.11. Determination of Prolidase Activity
4.12. Collagen Biosynthesis Assay
4.13. LC-MS Analysis of Proline Concentration
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AMP | 5′ adenosine monophosphate-activated protein |
| AMPK | AMP kinase |
| ATCC | American Type Culture Collection |
| Atg7 | autophagy-related 7 protein |
| ATP | adenozyno-5′-trifosforan |
| C32 | type of melanoma cells |
| CRISPR | clustered regularly interspaced short palindromic repeats |
| DCFH-DA | 2′-7′dichlorofluorescin diacetate |
| DMEM | Dulbecco’s Modified Eagle Medium |
| DNA | deoxyribonucleic acid |
| FBS | fetal bovine serum |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase |
| HILIC | hydrophilic interaction liquid chromatography |
| HPLC | high-performance liquid chromatography |
| MCF-7 | type of breast cancer cells |
| MET | metformin |
| MMP-2 | metaloproteinase 2 |
| MMP-9 | metaloproteinase 9 |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| NAD+ | nicotinamide adenine dinucleotide |
| NADH | nicotinamide adenine dinucleotide hydrogen |
| NRU | neutral red uptake |
| p53 | transcription factor p53 |
| P5C | ∆1-pyrroline-5-carboxylate |
| P5CR | P5C reductase |
| PARP | poly (ADP-ribose) polymerase |
| PBS | phosphate-buffered saline |
| POX | proline oxidase |
| PRODH | proline dehydrogenase |
| QTOF | quadrupole time-of-flight mass spectroscopy |
| ROS | reactive oxygen species |
| TCA | tricarboxylic acid cycle |
| THFA | tetrahydrofuroic acid |
References
- Wojciechowska, U.; Didkowska, J.; Michałek, I.; Olasek, P.; Ciuba, A. Cancer in Poland in 2018. In Polish National Cancer Registry; Clinical Epidemiology and Global Health: Warsaw, Poland, 2018; pp. 1–92. [Google Scholar]
- Rutkowski, P.; Wysocki, P.; Nowecki, Z.; Rudnicki, L.; Nasierowska-Guttmejer, A.; Fijuth, J.; Kalinka-Warzocha, E.; Świtaj, T.; Jeziorski, A.; Szacht, M.; et al. Cutaneous melanoma—Diagnostic and therapeutic guidelines in 2016. Oncol. Clin. Pract. 2015, 11, 216–231. [Google Scholar]
- Rigel, D.S.; Friedman, R.J.; Kopf, A.W.; Polsky, D. ABCDE—An evolving concept in the early detection of melanoma. Arch. Dermatol. 2005, 141, 1032–1034. [Google Scholar] [CrossRef] [PubMed]
- Clark, W.H., Jr.; Elder, D.E.; Guerry, D.t.; Epstein, M.N.; Greene, M.H.; Van Horn, M. A study of tumor progression: The precursor lesions of superficial spreading and nodular melanoma. Hum. Pathol. 1984, 15, 1147–1165. [Google Scholar] [CrossRef]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaube, B.; Malvi, P.; Singh, S.V.; Mohammad, N.; Meena, A.S.; Bhat, M.K. Targeting metabolic flexibility by simultaneously inhibiting respiratory complex I and lactate generation retards melanoma progression. Oncotarget 2015, 6, 37281–37299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janjetovic, K.; Harhaji-Trajkovic, L.; Misirkic-Marjanovic, M.; Vucicevic, L.; Stevanovic, D.; Zogovic, N.; Sumarac-Dumanovic, M.; Micic, D.; Trajkovic, V. In vitro and in vivo anti-melanoma action of metformin. Eur. J. Pharmacol. 2011, 668, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Peppicelli, S.; Toti, A.; Giannoni, E.; Bianchini, F.; Margheri, F.; Del Rosso, M.; Calorini, L. Metformin is also effective on lactic acidosis-exposed melanoma cells switched to oxidative phosphorylation. Cell Cycle 2016, 15, 1908–1918. [Google Scholar] [CrossRef]
- Jaune, E.; Rocchi, S. Metformin: Focus on Melanoma. Front. Endocrinol. 2018, 9, 472. [Google Scholar] [CrossRef] [Green Version]
- De Flora, S.; Ganchev, G.; Iltcheva, M.; La Maestra, S.; Micale, R.T.; Steele, V.E.; Balansky, R. Pharmacological Modulation of Lung Carcinogenesis in Smokers: Preclinical and Clinical Evidence. Trends Pharmacol. Sci. 2016, 37, 120–142. [Google Scholar] [CrossRef] [Green Version]
- Chae, Y.K.; Arya, A.; Malecek, M.K.; Shin, D.S.; Carneiro, B.; Chandra, S.; Kaplan, J.; Kalyan, A.; Altman, J.K.; Platanias, L.; et al. Repurposing metformin for cancer treatment: Current clinical studies. Oncotarget 2016, 7, 40767–40780. [Google Scholar] [CrossRef] [Green Version]
- Han, D.; Li, S.J.; Zhu, Y.T.; Liu, L.; Li, M.X. LKB1/AMPK/mTOR signaling pathway in non-small-cell lung cancer. Asian Pac. J. Cancer Prev. 2013, 14, 4033–4039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salani, B.; Maffioli, S.; Hamoudane, M.; Parodi, A.; Ravera, S.; Passalacqua, M.; Alama, A.; Nhiri, M.; Cordera, R.; Maggi, D. Caveolin-1 is essential for metformin inhibitory effect on IGF1 action in non-small-cell lung cancer cells. FASEB J. 2012, 26, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Liu, Z.; Jiang, L.; Liu, M.; Ma, J.; Yang, C.; Han, L.; Nan, K.; Liang, X. Metformin inhibits growth of human non-small cell lung cancer cells via liver kinase B-1-independent activation of adenosine monophosphate-activated protein kinase. Mol. Med. Rep. 2016, 13, 2590–2596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Gao, Q.; Wang, D.; Wang, Z.; Hu, C. Metformin inhibits growth of lung adenocarcinoma cells by inducing apoptosis via the mitochondria-mediated pathway. Oncol. Lett. 2015, 10, 1343–1349. [Google Scholar] [CrossRef] [Green Version]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G. Minireview: The AMP-activated protein kinase cascade: The key sensor of cellular energy status. Endocrinology 2003, 144, 5179–5183. [Google Scholar] [CrossRef]
- Liu, W.; Phang, J.M. Proline dehydrogenase (oxidase), a mitochondrial tumor suppressor, and autophagy under the hypoxia microenvironment. Autophagy 2012, 8, 1407–1409. [Google Scholar] [CrossRef] [Green Version]
- Phang, J.M.; Liu, W.; Hancock, C.; Christian, K.J. The proline regulatory axis and cancer. Front. Oncol. 2012, 2, 60. [Google Scholar] [CrossRef] [Green Version]
- Pandhare, J.; Donald, S.P.; Cooper, S.K.; Phang, J.M. Regulation and function of proline oxidase under nutrient stress. J. Cell Biochem. 2009, 107, 759–768. [Google Scholar] [CrossRef] [Green Version]
- Phang, J.M.; Donald, S.P.; Pandhare, J.; Liu, Y. The metabolism of proline, a stress substrate, modulates carcinogenic pathways. Amino Acids 2008, 35, 681–690. [Google Scholar] [CrossRef] [Green Version]
- Donald, S.P.; Sun, X.Y.; Hu, C.A.; Yu, J.; Mei, J.M.; Valle, D.; Phang, J.M. Proline oxidase, encoded by p53-induced gene-6, catalyzes the generation of proline-dependent reactive oxygen species. Cancer Res. 2001, 61, 1810–1815. [Google Scholar] [PubMed]
- Hu, C.A.; Donald, S.P.; Yu, J.; Lin, W.W.; Liu, Z.; Steel, G.; Obie, C.; Valle, D.; Phang, J.M. Overexpression of proline oxidase induces proline-dependent and mitochondria-mediated apoptosis. Mol. Cell. Biochem. 2007, 295, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Borchert, G.L.; Surazynski, A.; Hu, C.A.; Phang, J.M. Proline oxidase activates both intrinsic and extrinsic pathways for apoptosis: The role of ROS/superoxides, NFAT and MEK/ERK signaling. Oncogene 2006, 25, 5640–5647. [Google Scholar] [CrossRef] [Green Version]
- Martindale, J.L.; Holbrook, N.J. Cellular response to oxidative stress: Signaling for suicide and survival. J. Cell. Physiol. 2002, 192, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Raha, S.; Robinson, B.H. Mitochondria, oxygen free radicals, and apoptosis. Am. J. Med. Genet. 2001, 106, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Borchert, G.L.; Surazynski, A.; Phang, J.M. Proline oxidase, a p53-induced gene, targets COX-2/PGE2 signaling to induce apoptosis and inhibit tumor growth in colorectal cancers. Oncogene 2008, 27, 6729–6737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Tang, F.; Deng, Y.; Li, X.; Lan, T.; Zhang, X.; Huang, H.; Liu, P. Berberine reduces fibronectin and collagen accumulation in rat glomerular mesangial cells cultured under high glucose condition. Mol. Cell. Biochem. 2009, 325, 99–105. [Google Scholar] [CrossRef]
- Phang, J.M.; Pandhare, J.; Liu, Y. The metabolism of proline as microenvironmental stress substrate. J. Nutr. 2008, 138, 2008S–2015S. [Google Scholar] [CrossRef]
- Polyak, K.; Xia, Y.; Zweier, J.L.; Kinzler, K.W.; Vogelstein, B. A model for p53-induced apoptosis. Nature 1997, 389, 300–305. [Google Scholar] [CrossRef]
- Maxwell, S.A.; Kochevar, G.J. Identification of a p53-response element in the promoter of the proline oxidase gene. Biochem. Biophys. Res. Commun. 2008, 369, 308–313. [Google Scholar] [CrossRef]
- Myara, I.; Myara, A.; Mangeot, M.; Fabre, M.; Charpentier, C.; Lemonnier, A. Plasma prolidase activity: A possible index of collagen catabolism in chronic liver disease. Clin. Chem. 1984, 30, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Mock, W.L.; Green, P.C.; Boyer, K.D. Specificity and pH dependence for acylproline cleavage by prolidase. J. Biol. Chem. 1990, 265, 19600–19605. [Google Scholar] [CrossRef]
- Wang, S.H.; Zhi, Q.W.; Sun, M.J. Dual activities of human prolidase. Toxicol. In Vitro 2006, 20, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Adibi, S.A.; Mercer, D.W. Protein digestion in human intestine as reflected in luminal, mucosal, and plasma amino acid concentrations after meals. J. Clin. Investig. 1973, 52, 1586–1594. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.H.; Dennis, A.W.; Greenberg, M. Iminodipeptiduria: A genetic defect in recycling collagen; a method for determining prolidase in erythrocytes. Can. Med. Assoc. J. 1975, 113, 759, 762–763. [Google Scholar]
- Priest, R.E.; Davies, L.M. Cellular proliferation and synthesis of collagen. Lab. Investig. 1969, 21, 138–142. [Google Scholar]
- Zhang, M.; White, T.A.; Schuermann, J.P.; Baban, B.A.; Becker, D.F.; Tanner, J.J. Structures of the Escherichia coli PutA proline dehydrogenase domain in complex with competitive inhibitors. Biochemistry 2004, 43, 12539–12548. [Google Scholar] [CrossRef] [Green Version]
- Summitt, C.B.; Johnson, L.C.; Jonsson, T.J.; Parsonage, D.; Holmes, R.P.; Lowther, W.T. Proline dehydrogenase 2 (PRODH2) is a hydroxyproline dehydrogenase (HYPDH) and molecular target for treating primary hyperoxaluria. Biochem. J. 2015, 466, 273–281. [Google Scholar] [CrossRef] [Green Version]
- Hersey, P.; Watts, R.N.; Zhang, X.D.; Hackett, J. Metabolic approaches to treatment of melanoma. Clin. Cancer Res. 2009, 15, 6490–6494. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef]
- Ahn, C.S.; Metallo, C.M. Mitochondria as biosynthetic factories for cancer proliferation. Cancer Metab. 2015, 3, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hay, N. Reprogramming glucose metabolism in cancer: Can it be exploited for cancer therapy? Nat. Rev. Cancer 2016, 16, 635–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frattaruolo, L.; Brindisi, M.; Curcio, R.; Marra, F.; Dolce, V.; Cappello, A.R. Targeting the Mitochondrial Metabolic Network: A Promising Strategy in Cancer Treatment. Int. J. Mol. Sci. 2020, 21, 6014. [Google Scholar] [CrossRef] [PubMed]
- Filipp, F.V.; Ratnikov, B.; De Ingeniis, J.; Smith, J.W.; Osterman, A.L.; Scott, D.A. Glutamine-fueled mitochondrial metabolism is decoupled from glycolysis in melanoma. Pigment. Cell Melanoma Res. 2012, 25, 732–739. [Google Scholar] [CrossRef] [Green Version]
- Kowaloff, E.M.; Phang, J.M.; Granger, A.S.; Downing, S.J. Regulation of proline oxidase activity by lactate. Proc. Natl. Acad. Sci. USA 1977, 74, 5368–5371. [Google Scholar] [CrossRef] [Green Version]
- Phang, J.M.; Liu, W. Proline metabolism and cancer. Front. Biosci. 2012, 17, 1835–1845. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Glunde, K.; Bhujwalla, Z.M.; Raman, V.; Sharma, A.; Phang, J.M. Proline oxidase promotes tumor cell survival in hypoxic tumor microenvironments. Cancer Res. 2012, 72, 3677–3686. [Google Scholar] [CrossRef] [Green Version]
- Catchpole, G.; Platzer, A.; Weikert, C.; Kempkensteffen, C.; Johannsen, M.; Krause, H.; Jung, K.; Miller, K.; Willmitzer, L.; Selbig, J.; et al. Metabolic profiling reveals key metabolic features of renal cell carcinoma. J. Cell. Mol. Med. 2011, 15, 109–118. [Google Scholar] [CrossRef] [Green Version]
- Hirayama, A.; Kami, K.; Sugimoto, M.; Sugawara, M.; Toki, N.; Onozuka, H.; Kinoshita, T.; Saito, N.; Ochiai, A.; Tomita, M.; et al. Quantitative metabolome profiling of colon and stomach cancer microenvironment by capillary electrophoresis time-of-flight mass spectrometry. Cancer Res. 2009, 69, 4918–4925. [Google Scholar] [CrossRef] [Green Version]
- Ii, M.; Yamamoto, H.; Adachi, Y.; Maruyama, Y.; Shinomura, Y. Role of matrix metalloproteinase-7 (matrylisin) in human cancer invasion, apoptosis, growth, and angiogenesis. Exp. Biol. Med. 2006, 231, 20–27. [Google Scholar] [CrossRef]
- Laderoute, K.R.; Amin, K.; Calaoagan, J.M.; Knapp, M.; Le, T.; Orduna, J.; Foretz, M.; Viollet, B. 5’-AMP-activated protein kinase (AMPK) is induced by low-oxygen and glucose deprivation conditions found in solid-tumor microenvironments. Mol. Cell. Biol. 2006, 26, 5336–5347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Guan, K.L. AMP-activated protein kinase and cancer. Acta Physiol. 2009, 196, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. AMPK and Raptor: Matching cell growth to energy supply. Mol. Cell 2008, 30, 263–265. [Google Scholar] [CrossRef]
- Kato, K.; Ogura, T.; Kishimoto, A.; Minegishi, Y.; Nakajima, N.; Miyazaki, M.; Esumi, H. Critical roles of AMP-activated protein kinase in constitutive tolerance of cancer cells to nutrient deprivation and tumor formation. Oncogene 2002, 21, 6082–6090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Aniello, C.; Patriarca, E.J.; Phang, J.M.; Minchiotti, G. Proline Metabolism in Tumor Growth and Metastatic Progression. Front. Oncol. 2020, 10, 776. [Google Scholar] [CrossRef]
- Zareba, I.; Huynh, T.Y.L.; Kazberuk, A.; Teul, J.; Klupczynska, A.; Matysiak, J.; Surazynski, A.; Palka, J. Overexpression of Prolidase Induces Autophagic Death in MCF-7 Breast Cancer Cells. Cell Physiol. Biochem. 2020, 54, 875–887. [Google Scholar] [CrossRef]
- Zareba, I.; Surazynski, A.; Chrusciel, M.; Miltyk, W.; Doroszko, M.; Rahman, N.; Palka, J. Functional Consequences of Intracellular Proline Levels Manipulation Affecting PRODH/POX-Dependent Pro-Apoptotic Pathways in a Novel in Vitro Cell Culture Model. Cell Physiol. Biochem. 2017, 43, 670–684. [Google Scholar] [CrossRef]
- Zareba, I.; Celinska-Janowicz, K.; Surazynski, A.; Miltyk, W.; Palka, J. Proline oxidase silencing induces proline-dependent pro-survival pathways in MCF-7 cells. Oncotarget 2018, 9, 13748–13757. [Google Scholar] [CrossRef] [Green Version]
- Huynh, T.Y.L.; Oscilowska, I.; Saiz, J.; Niziol, M.; Baszanowska, W.; Barbas, C.; Palka, J. Metformin Treatment or PRODH/POX-Knock out Similarly Induces Apoptosis by Reprograming of Amino Acid Metabolism, TCA, Urea Cycle and Pentose Phosphate Pathway in MCF-7 Breast Cancer Cells. Biomolecules 2021, 11, 1888. [Google Scholar] [CrossRef]
- Fearns, C.; Dowdle, E.B. The desmoplastic response: Induction of collagen synthesis by melanoma cells in vitro. Int. J. Cancer 1992, 50, 621–627. [Google Scholar] [CrossRef]
- Gelse, K.; Pfander, D.; Obier, S.; Knaup, K.X.; Wiesener, M.; Hennig, F.F.; Swoboda, B. Role of hypoxia-inducible factor 1 alpha in the integrity of articular cartilage in murine knee joints. Arthritis Res. Ther. 2008, 10, R111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theron, A.E.; Nolte, E.M.; Lafanechère, L.; Joubert, A.M. Molecular crosstalk between apoptosis and autophagy induced by a novel 2-methoxyestradiol analogue in cervical adenocarcinoma cells. Cancer Cell Int. 2013, 13, 87. [Google Scholar] [CrossRef] [PubMed]
- Cechowska-Pasko, M.; Palka, J.; Wojtukiewicz, M.Z. Enhanced prolidase activity and decreased collagen content in breast cancer tissue. Int. J. Exp. Pathol. 2006, 87, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.J.; Xu, J.; Ye, H.Y.; Wang, X.B. Metformin prevents PFKFB3-related aerobic glycolysis from enhancing collagen synthesis in lung fibroblasts by regulating AMPK/mTOR pathway. Exp. Ther. Med. 2021, 21, 581. [Google Scholar] [CrossRef]
- Xu, S.; Yang, Z.; Jin, P.; Yang, X.; Li, X.; Wei, X.; Wang, Y.; Long, S.; Zhang, T.; Chen, G.; et al. Metformin Suppresses Tumor Progression by Inactivating Stromal Fibroblasts in Ovarian Cancer. Mol. Cancer Ther. 2018, 17, 1291–1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karaszewski, J.; Zareba, I.; Guszczyn, T.; Darewicz, B.; Palka, J. Verapamil and collagenase differentially affect collagen metabolism in experimental model of Peyronie’s disease. Mol. Cell. Probes 2020, 49, 101488. [Google Scholar] [CrossRef]
- Baszanowska, W.; Misiura, M.; Oscilowska, I.; Palka, J.; Miltyk, W. Extracellular Prolidase (PEPD) Induces Anabolic Processes through EGFR, beta1-integrin, and IGF-1R Signaling Pathways in an Experimental Model of Wounded Fibroblasts. Int. J. Mol. Sci. 2021, 22, 942. [Google Scholar] [CrossRef]
- Misiura, M.; Guszczyn, T.; Oscilowska, I.; Baszanowska, W.; Palka, J.; Miltyk, W. Platelet-Rich Plasma Promotes the Proliferation of Human Keratinocytes via a Progression of the Cell Cycle. A Role of Prolidase. Int. J. Mol. Sci. 2021, 22, 936. [Google Scholar] [CrossRef]
- Natarajan, S.K.; Zhu, W.; Liang, X.; Zhang, L.; Demers, A.J.; Zimmerman, M.C.; Simpson, M.A.; Becker, D.F. Proline dehydrogenase is essential for proline protection against hydrogen peroxide-induced cell death. Free. Radic. Biol. Med. 2012, 53, 1181–1191. [Google Scholar] [CrossRef] [Green Version]
- Tallarita, E.; Pollegioni, L.; Servi, S.; Molla, G. Expression in Escherichia coli of the catalytic domain of human proline oxidase. Protein Expr. Purif. 2012, 82, 345–351. [Google Scholar] [CrossRef]
- Misiura, M.; Baszanowska, W.; Ościłowska, I.; Pałka, J.; Miltyk, W. Prolidase Stimulates Proliferation and Migration through Activation of the PI3K/Akt/mTOR Signaling Pathway in Human Keratinocytes. Int. J. Mol. Sci. 2020, 21, 9243. [Google Scholar] [CrossRef] [PubMed]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Klupczynska, A.; Misiura, M.; Miltyk, W.; Oscilowska, I.; Palka, J.; Kokot, Z.J.; Matysiak, J. Development of an LC-MS Targeted Metabolomics Methodology to Study Proline Metabolism in Mammalian Cell Cultures. Molecules 2020, 25, 4639. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oscilowska, I.; Rolkowski, K.; Baszanowska, W.; Huynh, T.Y.L.; Lewoniewska, S.; Nizioł, M.; Sawicka, M.; Bielawska, K.; Szoka, P.; Miltyk, W.; et al. Proline Dehydrogenase/Proline Oxidase (PRODH/POX) Is Involved in the Mechanism of Metformin-Induced Apoptosis in C32 Melanoma Cell Line. Int. J. Mol. Sci. 2022, 23, 2354. https://doi.org/10.3390/ijms23042354
Oscilowska I, Rolkowski K, Baszanowska W, Huynh TYL, Lewoniewska S, Nizioł M, Sawicka M, Bielawska K, Szoka P, Miltyk W, et al. Proline Dehydrogenase/Proline Oxidase (PRODH/POX) Is Involved in the Mechanism of Metformin-Induced Apoptosis in C32 Melanoma Cell Line. International Journal of Molecular Sciences. 2022; 23(4):2354. https://doi.org/10.3390/ijms23042354
Chicago/Turabian StyleOscilowska, Ilona, Karol Rolkowski, Weronika Baszanowska, Thi Yen Ly Huynh, Sylwia Lewoniewska, Magdalena Nizioł, Magdalena Sawicka, Katarzyna Bielawska, Paweł Szoka, Wojciech Miltyk, and et al. 2022. "Proline Dehydrogenase/Proline Oxidase (PRODH/POX) Is Involved in the Mechanism of Metformin-Induced Apoptosis in C32 Melanoma Cell Line" International Journal of Molecular Sciences 23, no. 4: 2354. https://doi.org/10.3390/ijms23042354