
Molecular Pathogenesis of Myeloproliferative Neoplasms: From Molecular Landscape to Therapeutic Implications


Abstract
:1. Introduction
2. Driver Mutations in MPN
3. Additional Mutations in MPN
3.1. Epigenetic Regulation
3.2. Messenger RNA Splicing
3.3. Transcriptional Regulation
3.4. Signaling
3.5. DNA Repair
4. Germline Mutations in MPN
5. Role of Mutations in Leukemic Transformation
6. Clinical and Molecular-Integrated Prognostic Scores in MPN
7. Therapeutic Implications
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Campbell, P.J.; Green, A.R. The myeloproliferative disorders. N. Engl. J. Med. 2006, 355, 2452–2466. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M. The 2016 revision to the World Heath Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Barbui, T.; Thiele, J.; Gisslinger, H.; Kvasnicka, H.M.; Vannucchi, A.M.; Guglielmelli, P.; Orazi, A.; Tefferi, A. The 2016 WHO classification and diagnostic criteria for myeloproliferative neoplasms: Document summary and in-depth discussion. Blood Cancer J. 2018, 8, 15. [Google Scholar] [CrossRef]
- Barosi, G.; Mesa, R.A.; Thiele, J.; Cervantes, F.; Campbell, P.J.; Verstovsek, S.; Dupriez, B.; Levine, R.L.; Passamonti, F.; Gotlib, J.; et al. Proposed criteria for the diagnosis of post-polycythemia vera and post-essential thrombocythemia myelofibrosis: A consensus statement from the International Working Group for Myelofibrosis Research and Treatment. Leukemia 2008, 22, 437–438. [Google Scholar] [CrossRef] [Green Version]
- Moulard, O.; Mehta, J.; Fryzek, J.; Olivares, R.; Iqbal, U.; Mesa, R.A. Epidemiology of myelofibrosis, essential thrombocythemia, and polycythemia vera in the European Union. Eur. J. Haematol. 2014, 92, 289–297. [Google Scholar] [CrossRef]
- Nangalia, J.; Green, A.R. Myeloproliferative neoplasms: From origins to outcomes. Blood 2017, 130, 2475–2483. [Google Scholar] [CrossRef]
- Wingelhofer, B.; Neubauer, H.A.; Valent, P.; Han, X.; Constantinescu, S.N.; Gunning, P.T.; Müller, M.; Moriggl, R. Implications of STAT3 and STAT5 signaling on gene regulation and chromatin remodeling in hematopoietic cancer. Leukemia 2018, 32, 1713–1726. [Google Scholar] [CrossRef] [Green Version]
- Kleppe, M.; Kwak, M.; Koppikar, P.; Riester, M.; Keller, M.; Bastian, L.; Hricik, T.; Bhagwat, N.; McKenney, A.S.; Papalexi, E.; et al. JAK-STAT pathway activation in malignant and nonmalignant cells contributes to MPN pathogenesis and therapeutic response. Cancer Discov. 2015, 5, 316–331. [Google Scholar] [CrossRef] [Green Version]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, B.; Huntly, B.; Boggon, T.; Wlodarska, I.; Clark, J.; Moore, S.; et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Grand, F.H.; Hidalgo-Curtis, C.E.; Ernst, T.; Zoi, K.; Zoi, C.; McGuire, C.; Kreil, S.; Jones, A.; Score, J.; Metzgeroth, G.; et al. Frequent CBL mutations associated with 11q acquired uniparental disomy in myeloproliferative neoplasms. Blood 2009, 113, 6182–6192. [Google Scholar] [CrossRef] [Green Version]
- Elliott, J.; Suessmuth, Y.; Scott, L.M.; Nahlik, K.; McMullin, M.F.; Constantinescu, S.N.; Green, A.R.; Johnston, J.A. SOCS3 tyrosine phosphorylation as a potential bio-marker for myeloproliferative neoplasms associated with mutant JAK2 kinases. Haematologica 2009, 94, 576–580. [Google Scholar] [CrossRef] [Green Version]
- Pardanani, A.; Lasho, T.; Finke, C.; Oh, S.T.; Gotlib, J.; Tefferi, A. LNK mutation studies in blast-phase myeloproliferative neoplasms, and in chronic-phase disease with TET2, IDH, JAK2 or MPL mutations. Leukemia 2010, 24, 1713–1718. [Google Scholar] [CrossRef]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- James, C.; Ugo, V.; Le Couedic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garçon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signaling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef]
- Dusa, A.; Mouton, C.; Pecquet, C.; Herman, M.; Constantinescu, S.N. JAK2 V617F constitutive activation requires JH2 residue F595: A pseudokinase domain target for specific inhibitors. PLoS ONE 2010, 5, e11157. [Google Scholar] [CrossRef]
- Li, J.; Kent, D.G.; Chen, E.; Green, A.R. Mouse models of myeloproliferative neoplasms: JAK of all grades. Dis. Model Mech. 2011, 4, 311–317. [Google Scholar] [CrossRef] [Green Version]
- Vannucchi, A.M.; Antonioli, E.; Guglielmelli, P.; Longo, G.; Pancrazzi, A.; Ponziani, V.; Bogani, C.; Ferrini, P.R.; Rambaldi, A.; Guerini, V.; et al. Prospective identification of high-risk polycythemia vera patients based on JAK2(V617F) allele burden. Leukemia 2007, 21, 1952–1959. [Google Scholar] [CrossRef] [Green Version]
- Saliba, J.; Hamidi, S.; Lenglet, G.; Langlois, T.; Yin, J.; Cabagnols, X.; Secardin, L.; Legrand, C.; Galy, A.; Opolon, P.; et al. Heterozygous and homozygous JAK2(V617F) states modeled by induced pluripotent stem cells from myeloproliferative neoplasm patients. PLoS ONE 2013, 8, e74257. [Google Scholar] [CrossRef] [Green Version]
- Scott, L.M.; Tong, W.; Levine, R.L.; Scott, M.A.; Beer, P.A.; Stratton, M.R.; Futreal, P.A.; Erber, W.N.; McMullin, M.F.; Harrison, C.N.; et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N. Engl. J. Med. 2007, 356, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Grisouard, J.; Li, S.; Kubovcakova, L.; Rao, T.N.; Meyer, S.C.; Lundberg, P.; Hao-Shen, H.; Romanet, V.; Murakami, M.; Radimerski, T.; et al. JAK2 exon 12 mutant mice display isolated erythrocytosis and changes in iron metabolism favoring increased erythropoiesis. Blood 2016, 128, 839–851. [Google Scholar] [CrossRef] [Green Version]
- Pardanani, A.D.; Levine, R.L.; Lasho, T.; Pikman, Y.; Mesa, R.A.; Wadleigh, M.; Steensma, D.P.; Elliott, M.A.; Wolanskyj, A.P.; Hogan, W.J.; et al. MPL515 mutations in myeloproliferative and other myeloid disorders: A study of 1182 patients. Blood 2006, 108, 3472–3476. [Google Scholar] [CrossRef] [Green Version]
- de Laval, B.; Pawlikowska, P.; Petit-Cocault, L.; Bilhou-Nabera, C.; Aubin-Houzelstein, G.; Souyri, M.; Pouzoulet, F.; Gaudry, M.; Porteu, F. Thrombopoietin-increased DNA-PK-dependent DNA repair limits hematopoietic stem and progenitor cell mutagenesis in response to DNA damage. Cell Stem Cell 2013, 12, 37–48. [Google Scholar] [CrossRef] [Green Version]
- Tiedt, R.; Coers, J.; Ziegler, S.; Wiestner, A.; Hao-Shen, H.; Bornmann, C.; Schenkel, J.; Karakhanova, S.; de Sauvage, F.J.; Jackson, C.W.; et al. Pronounced thrombocytosis in transgenic mice expressing reduced levels of Mpl in platelets and terminally differentiated megakaryocytes. Blood 2009, 113, 1768–1777. [Google Scholar] [CrossRef]
- Lannutti, B.J.; Epp, A.; Roy, J.; Chen, J.; Josephson, N.C. Incomplete restoration of Mpl expression in the mpl-/- mouse produces partial correction of the stem cell-repopulating defect and paradoxical thrombocytosis. Blood 2009, 113, 1778–1785. [Google Scholar] [CrossRef] [Green Version]
- Pikman, Y.; Lee, B.H.; Mercher, T.; McDowell, E.; Ebert, B.L.; Gozo, M.; Cuker, A.; Wernig, G.; Moore, S.; Galinsky, I.; et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006, 3, e270. [Google Scholar] [CrossRef] [Green Version]
- Rumi, E.; Pietra, D.; Guglielmelli, P.; Bordoni, R.; Casetti, I.; Milanesi, C.; Sant’Antonio, E.; Ferretti, V.; Pancrazzi, A.; Rotunno, G.; et al. Acquired copy-neutral loss of heterozygosity of chromosome 1p as a molecular event associated with marrow fibrosis in MPL-mutated myeloproliferative neoplasms. Blood 2013, 121, 4388–4395. [Google Scholar] [CrossRef] [Green Version]
- Nangalia, J.; Massie, C.E.; Baxter, E.J.; Nice, F.L.; Gundem, G.; Wedge, D.C.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.G.; et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N. Engl. J. Med. 2013, 369, 2391–2405. [Google Scholar] [CrossRef] [Green Version]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef] [Green Version]
- Cabagnols, X.; Defour, J.P.; Ugo, V.; Ianotto, J.C.; Mossuz, P.; Mondet, J.; Girodon, F.; Alexandre, J.H.; Mansier, O.; Viallard, J.F.; et al. Differential association of calreticulin type 1 and type 2 mutations with myelofibrosis and essential thrombocytemia: Relevance for disease evolution. Leukemia 2015, 29, 249–252. [Google Scholar] [CrossRef]
- Elf, S.; Abdelfattah, N.S.; Chen, E.; Perales-Patón, J.; Rosen, E.A.; Ko, A.; Peisker, F.; Florescu, N.; Giannini, S.; Wolach, O.; et al. Mutant Calreticulin Requires Both Its Mutant C-terminus and the Thrombopoietin Receptor for Oncogenic Transformation. Cancer Discov. 2016, 6, 368–381. [Google Scholar] [CrossRef] [Green Version]
- Chachoua, I.; Pecquet, C.; El-Khoury, M.; Nivarthi, H.; Albu, R.-I.; Marty, C.; Gryshkova, V.; Defour, J.P.; Vertenoeil, G.; Ngo, A.; et al. Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood 2016, 127, 1325–1335. [Google Scholar] [CrossRef]
- Cazzola, M. Mutant calreticulin: When a chaperone becomes intrusive. Blood 2016, 127, 1219–1221. [Google Scholar] [CrossRef] [Green Version]
- Balligand, T.; Achouri, Y.; Pecquet, C.; Chachoua, I.; Nivarthi, H.; Marty, C.; Plo, I.; Kralovics, R.; Constantinescu, S.N. Pathologic activation of thrombopoietin receptor and JAK2-STAT5 pathway by frameshift mutants of mouse calreticulin. Leukemia 2016, 30, 1775–1778. [Google Scholar] [CrossRef] [Green Version]
- Masubuchi, N.; Araki, M.; Yang, Y.; Hayashi, E.; Imai, M.; Edahiro, Y.; Hironaka, Y.; Mizukami, Y.; Kihara, Y.; Takei, H.; et al. Mutant calreticulin interacts with MPL in the secretion pathway for activation on the cell surface. Leukemia 2020, 34, 499–509. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Finke, C.; Belachew, A.A.; Wassie, E.A.; Ketterling, R.P.; Hanson, C.A.; Pardanani, A. Type 1 vs. type 2 calreticulin mutations in primary myelofibrosis: Differences in phenotype and prognostic impact. Leukemia 2014, 28, 1568–1570. [Google Scholar] [CrossRef]
- Tefferi, A.; Wassie, E.A.; Guglielmelli, P.; Gangat, N.; Belachew, A.A.; Lasho, T.L.; Finke, C.; Ketterling, R.P.; Hanson, C.A.; Pardanani, A.; et al. Type 1 versus Type 2 calreticulin mutations in essential thrombocythemia: A collaborative study of 1027 patients. Am. J. Hematol. 2014, 89, 121–124. [Google Scholar] [CrossRef]
- Tefferi, A.; Thiele, J.; Vannucchi, A.M.; Barbui, T. An overview on CALR and CSF3R mutations and a proposal for revision of WHO diagnostic criteria for myeloproliferative neoplasms. Leukemia 2014, 28, 1407–1413. [Google Scholar] [CrossRef] [Green Version]
- Marneth, A.E.; Jutzi, J.S.; Guerra-Moreno, A.; Ciboddo, M.; Santos, M.J.J.; Kosmidou, A.; Hamel, R.; Lozano, P.; Rumi, E.; Doench, J.G.; et al. Whole-Genome CRISPR Screening Identifies N -Glycosylation As an Essential Pathway and a Potential Novel Therapeutic Target in CALR -Mutant MPN. Blood 2021, 138 (Suppl. S1), 58. [Google Scholar] [CrossRef]
- Greenbaum, H.S.; Evers, M.; Rosencrance, A.; Maxwell, L.; Kurylowicz, K.; Arellano, N.S.; Ciboddo, M.; Gao, J.; Ibarra, J.; Elf, S. Type I Calreticulin Mutations Result in Hyperactivation of Its Acetyltransferase Function and Iron Metabolism, Inducing a Susceptibility to Ferroptosis. Blood 2021, 138 (Suppl. S1), 3593. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Finke, C.M.; Knudson, R.A.; Ketterling, R.; Hanson, C.H.; Maffioli, M.; Caramazza, D.; Passamonti, F.; Pardanani, A. CALR vs JAK2 vs MPL-mutated or triple negative myelofibrosis: Clinical, cytogenetic and molecular comparisons. Leukemia 2014, 28, 1472–1477. [Google Scholar] [CrossRef]
- Lee, J.; Godfrey, A.L.; Nangalia, J. Genomic heterogeneity in myeloproliferative neoplasms and applications to clinical practice. Blood Rev. 2020, 42, 100708. [Google Scholar] [CrossRef]
- Stuckey, R.; Gómez-Casares, M.T. Recent Advances in the Use of Molecular Analyses to Inform the Diagnosis and Prognosis of Patients with Polycythaemia Vera. Int. J. Mol. Sci. 2021, 22, 5042. [Google Scholar] [CrossRef]
- Jia, R.; Kralovics, R. Progress in elucidation of molecular pathophysiology of myeloproliferative neoplasms and its application to therapeutic decisions. Int. J. Hematol. 2020, 111, 182–191. [Google Scholar] [CrossRef] [Green Version]
- Takenaka, K. Progress in elucidation of molecular pathophysiology and its application in therapeutic decision-making for myeloproliferative neoplasms. Int. J. Hematol. 2020, 111, 180–181. [Google Scholar] [CrossRef] [Green Version]
- Lundberg, P.; Karow, A.; Nienhold, R.; Looser, R.; Hao-Shen, H.; Nissen, I.; Girsberger, S.; Lehmann, T.; Passweg, J.; Stern, M.; et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood 2014, 123, 2220–2228. [Google Scholar] [CrossRef] [Green Version]
- Szybinski, J.; Meyer, S.C. Genetics of Myeloproliferative Neoplasms. Hematol. Oncol. Clin. N. Am. 2021, 35, 217–236. [Google Scholar] [CrossRef]
- Marneth, A.E.; Mullally, A. The Molecular Genetics of Myeloproliferative Neoplasms. Cold Spring Harb. Perspect. Med. 2020, 10, a034876. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Wahab, O.; Pardanani, A.; Rampal, R.; Lasho, T.L.; Levine, R.L.; Tefferi, A. DNMT3A mutational analysis in primary myelofibrosis, chronic myelomonocytic leukemia and advanced phases of myeloproliferative neoplasms. Leukemia 2011, 25, 1219–1220. [Google Scholar] [CrossRef] [Green Version]
- Stegelmann, F.; Bullinger, L.; Schlenk, R.F.; Paschka, P.; Griesshammer, M.; Blersch, C.; Kuhn, S.; Schauer, S.; Döhner, H.; Döhne, K. DNMT3A mutations in myeloproliferative neoplasms. Leukemia 2011, 25, 1217–1219. [Google Scholar] [CrossRef] [Green Version]
- Challen, G.A.; Sun, D.; Jeong, M.; Luo, M.; Jelinek, J.; Berg, J.S.; Bock, C.; Vasanthakumar, A.; Gu, H.; Xi, Y.; et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat. Genet. 2011, 44, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Jacquelin, S.; Straube, J.; Cooper, L.; Vu, T.; Song, A.; Bywater, M.; Baxter, E.; Heidecker, M.; Wackrow, B.; Porter, A.; et al. Jak2V617F and Dnmt3a loss cooperate to induce myelofibrosis through activated enhancer-driven inflammation. Blood 2018, 132, 2707–2721. [Google Scholar] [CrossRef] [Green Version]
- Nangalia, J.; Nice, F.L.; Wedge, D.C.; Godfrey, A.L.; Grinfeld, J.; Thakker, C.; Massie, C.E.; Baxter, J.; Sewell, D.; Silber, Y.; et al. DNMT3A mutations occur early or late in patients with myeloproliferative neoplasms and mutation order influences phenotype. Haematologica 2015, 100, e438–e442. [Google Scholar] [CrossRef] [Green Version]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Wahab, O.; Manshouri, T.; Patel, J.; Harris, K.; Yao, J.; Hedvat, C.; Hedvat, C.; Heguy, A.; Bueso-Ramos, C.; Kantarjian, H.; et al. Genetic analysis of transforming events that convert chronic myeloproliferative neoplasms to leukemias. Cancer Res. 2010, 70, 447–452. [Google Scholar] [CrossRef] [Green Version]
- Chen, E.; Schneider, R.K.; Breyfogle, L.J.; Rosen, E.A.; Poveromo, L.; Elf, S.; Ko, A.; Brumme, K.; Levine, R.; Ebert, B.J.; et al. Distinct effects of concomitant Jak2V617F expression and Tet2 loss in mice promote disease progression in myeloproliferative neoplasms. Blood 2015, 125, 327–335. [Google Scholar] [CrossRef] [Green Version]
- Ortmann, C.A.; Kent, D.G.; Nangalia, J.; Silber, Y.; Wedge, D.C.; Grinfeld, J.; Baxter, E.J.; Massie, C.E.; Papaemmanuil, E.; Menon, S.; et al. Effect of mutation order on myeloproliferative neoplasms. N. Engl. J. Med. 2015, 372, 601–612. [Google Scholar] [CrossRef] [Green Version]
- Shih, A.H.; Abdel-Wahab, O.; Patel, J.P.; Levine, R.L. The role of mutations in epigenetic regulators in myeloid malignancies. Nat. Rev. Cancer 2012, 12, 599–612. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Abdel-Wahab, O.; Guglielmelli, P.; Patel, J.; Caramazza, D.; Pieri, L.; Finke, C.M.; Kilpivaara, O.; Wadleigh, M.; et al. IDH1 and IDH2 mutation studies in 1473 patients with chronic-, fibrotic- or blast-phase essential thrombocythemia, polycythemia vera or myelofibrosis. Leukemia 2010, 24, 1302–1309. [Google Scholar] [CrossRef]
- McKenney, A.S.; Lau, A.N.; Somasundara, A.V.H.; Spitzer, B.; Intlekofer, A.M.; Ahn, J.; Shank, K.; Rapaport, F.T.; Patel, M.A.; Papalexi, E.; et al. JAK2/IDH-mutant-driven myeloproliferative neoplasm is sensitive to combined targeted inhibition. J. Clin. Investig. 2018, 128, 789–804. [Google Scholar] [CrossRef]
- Yoshimi, A.; Lin, K.-T.; Wiseman, D.H.; Rahman, M.A.; Pastore, A.; Wang, B.; Chun-Wei Lee, S.; Micol, J.-B.; Zhang, X.J.; de Botton, S.; et al. Coordinated alterations in RNA splicing and epigenetic regulation drive leukaemogenesis. Nature 2019, 574, 273–277. [Google Scholar] [CrossRef]
- Cho, Y.-S.; Kim, E.-J.; Park, U.-H.; Sin, H.-S.; Um, S.-J. Additional sex comb-like 1 (ASXL1), in cooperation with SRC-1, acts as a ligand-dependent coactivator for retinoic acid receptor. J. Biol. Chem. 2006, 281, 17588–17598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Wahab, O.; Adli, M.; LaFave, L.M.; Gao, J.; Hricik, T.; Shih, A.H.; Pandey, S.; Patel, J.P.; Chung, Y.R.; Koche, R.; et al. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell 2012, 22, 180–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Wahab, O.; Tefferi, A.; Levine, R.L. Role of TET2 and ASXL1 mutations in the pathogenesis of myeloproliferative neoplasms. Hematol. Oncol. Clin. N. Am. 2012, 26, 1053–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, C.L.; Pineault, N.; Brookes, C.; Helgason, C.D.; Ohta, H.; Bodner, C.; Hess, J.L.; Humphries, R.K.; Brock, H.W. Loss-of-function Additional sex combs like 1 mutations disrupt hematopoiesis but do not cause severe myelodysplasia or leukemia. Blood 2010, 115, 38–46. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Yamamoto, S.; Sheng, M.; Bai, J.; Zhang, P.; Chen, R.; Chen, S.; Shi, L.; Abdel-Wahab, O.; Xu, M.; et al. ASXL1 plays an important role in erythropoiesis. Sci. Rep. 2016, 6, 28789. [Google Scholar] [CrossRef]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Finke, C.; Mannarelli, C.; Belachew, A.A.; Pancrazzi, A.; Wassie, E.A.; Ketterling, R.P.; et al. CALR and ASXL1 mutations-based molecular prognostication in primary myelofibrosis: An international study of 570 patients. Leukemia 2014, 28, 1494–1500. [Google Scholar] [CrossRef]
- Paz, D.L.; Riou, J.; Verger, E.; Cassinat, B.; Chauveau, A.; Ianotto, J.-C.; Dupriez, B.; Boyer, F.; Renard, M.; Mansier, O.; et al. Genomic analysis of primary and secondary myelofibrosis redefines the prognostic impact of ASXL1 mutations: A FIM study. Blood Adv. 2021, 5, 1442–1451. [Google Scholar]
- Elshoury, A.; Yu, H.; Ji, W.; Thompson, J.E.; Griffiths, E.A.; Przespolewski, A.; Green, S.D.; Sung, P.J.; Aqeel, S.; Awada, H.; et al. ASXL1 Mutation Is a Novel Risk Factor for Bleeding in Patients with Philadelphia-Negative Myeloproliferative Neoplasms (MPN). Blood 2021, 138 (Suppl. S1), 3637. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Biamonte, F.; Score, J.; Hidalgo-Curtis, C.; Cervantes, F.; Maffioli, M.; Fanelli, T.; Ernst, T.; Winkelman, N.; Jones, A.V.; et al. EZH2 mutational status predicts poor survival in myelofibrosis. Blood 2011, 118, 5227–5234. [Google Scholar] [CrossRef]
- Shimizu, T.; Kubovcakova, L.; Nienhold, R.; Zmajkovic, J.; Meyer, S.C.; Hao-Shen, H.; Geier, F.; Dirnhofer, S.; Guglielmelli, P.; Vannucchi, A.M.; et al. Loss of Ezh2 synergizes with JAK2-V617F in initiating myeloproliferative neoplasms and promoting myelofibrosis. J. Exp. Med. 2016, 213, 1479–1496. [Google Scholar] [CrossRef]
- Pastore, F.; Bhagwat, N.; Pastore, A.; Radzisheuskaya, A.; Karzai, A.; Krishnan, A.; Li, B.; Bowman, R.H.; Xiao, W.; Viny, A.D.; et al. PRMT5 Inhibition Modulates E2F1 Methylation and Gene-Regulatory Networks Leading to Therapeutic Efficacy in JAK2(V617F)-Mutant MPN. Cancer Discov. 2020, 10, 1742–1757. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Kim, J.-H.; Lu, W.; Williams, D.M.; Kim, J.; Cope, L.; Rampal, R.; Koche, R.; Xian, L.; Luo, L.Z.; et al. HMGA1 Chromatin Regulators Induce Transcriptional Networks Involved in GATA2 and Proliferation During MPN Progression. Blood 2022. [Google Scholar] [CrossRef] [PubMed]
- Hautin, M.; Mornet, C.; Chauveau, A.; Bernard, D.G.; Corcos, L.; Lippert, E. Splicing Anomalies in Myeloproliferative Neoplasms: Paving the Way for New Therapeutic Venues. Cancers 2020, 12, 2216. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, J.-Y.; Huang, Y.-J.; Gu, Y.; Qiu, J.; Qian, H.; Shao, C.; Zhang, X.; Hu, J.; Li, H.; et al. The Augmented R-Loop Is a Unifying Mechanism for Myelodysplastic Syndromes Induced by High-Risk Splicing Factor Mutations. Mol. Cell 2018, 69, 412–425.e6. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, H.; Harada, Y.; Ogata, Y.; Kagiyama, Y.; Shingai, N.; Doki, N.; Ohashi, K.; Kitamura, T.; Komatsu, N.; Harada, H. Overexpression of RUNX1 short isoform has an important role in the development of myelodysplastic/myeloproliferative neoplasms. Blood Adv. 2017, 1, 1382–1386. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhang, Q.; Zhang, D.-E.; Zhou, C.; Xing, H.; Tian, Z.; Rao, Q.; Wang, M.; Wang, J. Overexpression of an isoform of AML1 in acute leukemia and its potential role in leukemogenesis. Leukemia 2009, 23, 739–745. [Google Scholar] [CrossRef]
- Lasho, T.L.; Jimma, T.; Finke, C.M.; Patnaik, M.; Hanson, C.A.; Ketterling, R.P.; Pardanani, A.; Tefferi, A. SRSF2 mutations in primary myelofibrosis: Significant clustering with IDH mutations and independent association with inferior overall and leukemia-free survival. Blood 2012, 120, 4168–4171. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Pardanani, A. Screening for ASXL1 and SRSF2 mutations is imperative for treatment decision-making in otherwise low or intermediate-1 risk patients with myelofibrosis. Br. J. Haematology 2018, 183, 678–681. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Nicolosi, M.; Mudireddy, M.; Szuber, N.; Finke, C.M.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; Pardanani, A.; Gangat, N.; et al. Driver mutations and prognosis in primary myelofibrosis: Mayo-Careggi MPN alliance study of 1095 patients. Am. J. Hematol. 2018, 93, 348–355. [Google Scholar] [CrossRef] [Green Version]
- Willekens, C.; Laplane, L.; Dagher, T.; Benlabiod, C.; Lacout, C.; Rameau, P.; Catelain, C.; Alfaro, A.; Edmond, V.; Signolle, N.; et al. SRSF2-P95Hdelays Myelofibrosis Development through Altered JAK/STAT Signaling in JAK2-V617F Megakaryocytes. Blood 2021, 138 (Suppl. S1), 2544. [Google Scholar] [CrossRef]
- Tefferi, A.; Finke, C.M.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Pardanai, A. U2AF1 mutation types in primary myelofibrosis: Phenotypic and prognostic distinctions. Leukemia 2018, 32, 2274–2278. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Lasho, T.L.; Finke, C.M.; Elala, Y.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Pardanai, A. Targeted deep sequencing in primary myelofibrosis. Blood Adv. 2016, 1, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palangat, M.; Anastasakis, D.G.; Fei, D.L.; Lindblad, K.E.; Bradley, R.; Hourigan, C.S.; Hagner, M.; Larson, D.R. The splicing factor U2AF1 contributes to cancer progression through a noncanonical role in translation regulation. Genes Dev. 2019, 33, 482–497. [Google Scholar] [CrossRef] [PubMed]
- Broséus, J.; Alpermann, T.; Wulfert, M.; Florensa Brichs, L.; Jeromin, S.; Lippert, E.; Rozman, M.; Lifermann, F.; Grossmann, V.; Haferlach, T.; et al. Age, JAK2(V617F) and SF3B1 mutations are the main predicting factors for survival in refractory anaemia with ring sideroblasts and marked thrombocytosis. Leukemia 2013, 27, 1826–1831. [Google Scholar] [CrossRef] [Green Version]
- Jayavelu, A.K.; Schnöder, T.M.; Perner, F.; Herzog, C.; Meiler, A.; Krishnamoorthy, G.; Huber, N.; Mohr, J.; Edelmann-Stephan, B.; Austin, R.; et al. Splicing factor YBX1 mediates persistence of JAK2-mutated neoplasms. Nature 2020, 588, 157–163. [Google Scholar] [CrossRef]
- Schischlik, F.; Jäger, R.; Rosebrock, F.; Hug, E.; Schuster, M.; Holly, R.; Fuchs, E.; Milosevic, J.D.; Bogner, E.; Gisslinger, B.; et al. Mutational landscape of the transcriptome offers putative targets for immunotherapy of myeloproliferative neoplasms. Blood 2019, 134, 199–210. [Google Scholar] [CrossRef] [Green Version]
- Peeken, J.C.; Jutzi, J.S.; Wehrle, J.; Koellerer, C.; Staehle, H.F.; Becker, H.; Schoenwandt, E.; Seeger, T.S.; Schanne, D.H.; Gothwal, M.; et al. Epigenetic regulation of NFE2 overexpression in myeloproliferative neoplasms. Blood 2018, 131, 2065–2073. [Google Scholar] [CrossRef] [Green Version]
- Lasho, T.L.; Mudireddy, M.; Finke, C.M.; Hanson, C.A.; Ketterling, R.P.; Szuber, N.; Begna, K.H.; Patnaik, M.M.; Gangat, N.; Pardanani, A.; et al. Targeted next-generation sequencing in blast phase myeloproliferative neoplasms. Blood Adv. 2018, 2, 370–380. [Google Scholar] [CrossRef] [Green Version]
- Milosevic, J.D.; Puda, A.; Malcovati, L.; Berg, T.; Hofbauer, M.; Stukalov, A.; Klampfl, T.; Harutyunyan, A.S.; Gisslinger, H.; Gisslinger, B.; et al. Clinical significance of genetic aberrations in secondary acute myeloid leukemia. Am. J. Hematol. 2012, 87, 1010–1016. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Harada, Y.; Imagawa, J.; Kimura, A.; Harada, H. AML1/RUNX1 point mutation possibly promotes leukemic transformation in myeloproliferative neoplasms. Blood 2009, 114, 5201–5205. [Google Scholar] [CrossRef] [Green Version]
- Ward, A.F.; Braun, B.S.; Shannon, K.M. Targeting oncogenic Ras signaling in hematologic malignancies. Blood 2012, 120, 3397–3406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, I.T.; Kutok, J.L.; Williams, I.R.; Cohen, S.; Kelly, L.; Shigematsu, H.; Johnson, L.; Akashi, K.; Tuveson, D.A.; Jacks, T.; et al. Conditional expression of oncogenic K-ras from its endogenous promoter induces a myeloproliferative disease. J. Clin. Investig. 2004, 113, 528–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, X.; Kong, G.; Ranheim, E.A.; Yang, D.; Zhou, Y.; Zhang, J. Unique dependence on Sos1 in Kras (G12D)-induced leukemogenesis. Blood 2018, 132, 2575–2579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cargo, C.; Cullen, M.; Taylor, J.; Short, M.; Glover, P.; Van Hoppe, S.; Smith, A.; Evans, P.; Crouch, S. The use of targeted sequencing and flow cytometry to identify patients with a clinically significant monocytosis. Blood 2019, 133, 1325–1334. [Google Scholar] [CrossRef] [Green Version]
- Belizaire, R.; Koochaki, S.H.J.; Udeshi, N.D.; Vedder, A.; Sun, L.; Svinkina, T.; Hartigan, C.; McConkey, M.; Kovalcik, V.; Bizuayehu, A.; et al. CBL mutations drive PI3K/AKT signaling via increased interaction with LYN and PIK3R1. Blood 2021, 137, 2209–2220. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Guglielmelli, P.; Finke, C.M.; Rotunno, G.; Elala, Y.; Pacilli, A.; Hanson, C.A.; Pancrazzi, A.; Ketterling, R.P.; et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016, 1, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Gery, S.; Cao, Q.; Gueller, S.; Xing, H.; Tefferi, A.; Koeffler, H.P. Lnk inhibits myeloproliferative disorder-associated JAK2 mutant, JAK2V617F. J. Leukoc. Biol. 2009, 85, 957–965. [Google Scholar] [CrossRef] [Green Version]
- Sadler, B.; Chorzalska, A.D.; Bonal, D.M.; Haller, G.; Oakes, A.; Petersen, M.; Liu, Y.; Olszewski, A.; Reagan, J.L.; Egan, P.C.; et al. Whole Genome Sequencing Identifies a Recurrent Mutation in Complement Factor I (CFI) in Primary Myelofibrosis (PMF). Blood 2021, 138 (Suppl. S1), 1472. [Google Scholar] [CrossRef]
- Nakatake, M.; Monte-Mor, B.; Debili, N.; Casadevall, N.; Ribrag, V.; Solary, E.; Vainchenker, W.; Plo, I.J. AK2 V617F negatively regulates p53 stabilization by enhancing MDM2 via la expression in myeloproliferative neoplasms. Oncogene 2012, 31, 1323–1333. [Google Scholar] [CrossRef] [Green Version]
- Rampal, R.; Ahn, J.; Abdel-Wahab, O.; Nahas, M.; Wang, K.; Lipson, D.; Otto, G.A.; Yelensky, R.; Hricik, T.; McKenney, A.S.; et al. Genomic and functional analysis of leukemic transformation of myeloproliferative neoplasms. Proc. Natl. Acad. Sci. USA 2014, 111, e5401–e5410. [Google Scholar] [CrossRef] [Green Version]
- Harutyunyan, A.; Klampfl, T.; Cazzola, M.; Kralovics, R. p53 Lesions in Leukemic Transformation. N. Engl. J. Med. 2011, 364, 488–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahn, J.D.; Miller, P.G.; Silver, A.J.; Sellar, R.S.; Bhatt, S.; Gibson, C.; McConkey, M.; Adams, D.; Mar, B.; Mertins, P.; et al. PPM1D-truncating mutations confer resistance to chemotherapy and sensitivity to PPM1D inhibition in hematopoietic cells. Blood 2018, 132, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Grinfeld, J.; Nangalia, J.; Baxter, E.J.; Wedge, D.C.; Angelopoulos, N.; Cantrill, R.; Godfrey, A.L.; Papaemmanuil, E.; Gundem, G.; MacLean, C.; et al. Classification and Personalized Prognosis in Myeloproliferative Neoplasms. N. Engl. J. Med. 2018, 379, 1416–1430. [Google Scholar] [CrossRef] [PubMed]
- Milosevic Feenstra, J.D.; Nivarthi, H.; Gisslinger, H.; Leroy, E.; Rumi, E.; Chachoua, I.; Bagienski, K.; Kubesova, B.; Pietra, D.; Gisslinger, B.; et al. Whole-exome sequencing identifies novel MPL and JAK2 mutations in triple-negative myeloproliferative neoplasms. Blood 2016, 127, 325–332. [Google Scholar] [CrossRef]
- Rodriguez-Meira, A.; Buck, G.; Clark, S.-A.; Povinelli, B.J.; Alcolea, V.; Louka, E.; McGowan, S.; Hamblin, A.; Sousos, N.; Barkas, N.; et al. Unravelling Intratumoral Heterogeneity through High-Sensitivity Single-Cell Mutational Analysis and Parallel RNA Sequencing. Mol. Cell 2019, 73, 1292–1305.e8. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bick, A.G.; Weinstock, J.S.; Nandakumar, S.K.; Fulco, C.P.; Bao, E.L.; Zekavat, S.M.; Szeto, M.D.; Liao, X.; Leventhal, M.J.; Nasser, J.; et al. Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature 2020, 586, 763–768. [Google Scholar] [CrossRef]
- Zhao, L.-P.; Cazaux, M.; Maslah, N.; Daltro De Oliveira, R.; Verger, E.; Soret-Dulphy, J.; Marcault, C.; Parquet, N.; Dosquet, C.; Vainchenker, W.; et al. Myeloproliferative Neoplasms (MPN) Clonal Evolution Landscape and Its Impact on Patients’ Prognosis. Blood 2021, 138 (Suppl. S1), 317. [Google Scholar] [CrossRef]
- Rumi, E.; Passamonti, F.; Della Porta, M.G.; Elena, C.; Arcaini, L.; Vanelli, L.; Del Curto, C.; Pietra, D.; Boveri, E.; Pascutto, C.; et al. Familial chronic myeloproliferative disorders: Clinical phenotype and evidence of disease anticipation. J. Clin. Oncol. 2007, 25, 5630–5635. [Google Scholar] [CrossRef]
- Rumi, E.; Harutyunyan, A.S.; Pietra, D.; Feenstra, J.D.M.; Cavalloni, C.; Roncoroni, E.; Casetti, I.; Bellini, M.; Milanesi, C.; Renna, M.C.; et al. LNK mutations in familial myeloproliferative neoplasms. Blood 2016, 128, 144–145. [Google Scholar] [CrossRef] [Green Version]
- Hinds, D.A.; Barnholt, K.E.; Mesa, R.A.; Kiefer, A.K.; Do, C.B.; Eriksson, N.; Mountain, L.J.; Francke, U.; Tung, J.Y.; Nguyen, H.; et al. Germ line variants predispose to both JAK2 V617F clonal hematopoiesis and myeloproliferative neoplasms. Blood 2016, 128, 1121–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunbar, A.J.; Rampal, R.K.; Levine, R. Leukemia secondary to myeloproliferative neoplasms. Blood 2020, 136, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Vannucchi, A.M.; Lasho, T.L.; Guglielmelli, P.; Biamonte, F.; Pardanani, A.; Pereira, A.; Finke, C.; Score, J.; Gangat, N.; Mannarelli, C.; et al. Mutations and prognosis in primary myelofibrosis. Leukemia 2013, 27, 1861–1869. [Google Scholar] [CrossRef] [PubMed]
- Paz, D.L.; Jouanneau-Courville, R.; Riou, J.; Ianotto, J.-C.; Boyer, F.; Chauveau, A.; Renard, M.; Chomel, J.-C.; Cayssials, E.; Gallego-Hernanz, M.-P.; et al. Leukemic evolution of polycythemia vera and essential thrombocythemia: Genomic profiles predict time to transformation. Blood Adv. 2020, 4, 4887–4897. [Google Scholar]
- Theocharides, A.; Boissinot, M.; Girodon, F.; Garand, R.; Teo, S.-S.; Lippert, E.; Talmant, P.; Tichelli, A.; Hermouet, S.; Skoda, R.C. Leukemic blasts in transformed JAK2-V617F-positive myeloproliferative disorders are frequently negative for the JAK2-V617F mutation. Blood 2007, 110, 375–379. [Google Scholar] [CrossRef] [Green Version]
- Kong, T.; Laranjeira, A.B.; Yang, K.; Fisher, D.A.; Yu, L.; Wang, A.Z.; Ruzinova, M.B.; Fowles, J.S.; Allen, M.J.; Celik, H.; et al. DUSP6 Mediates Resistance to JAK2 Inhibition and Drives Myeloproliferative Neoplasm Disease Progression. Blood 2021, 138 (Suppl. S1), 55. [Google Scholar] [CrossRef]
- Patel, K.P.; Newberry, K.J.; Luthra, R.; Jabbour, E.; Pierce, S.; Cortes, J.; Singh, R.; Mehrotra, M.; Routbort, M.J.; Luthra, M.; et al. Correlation of mutation profile and response in patients with myelofibrosis treated with ruxolitinib. Blood 2015, 126, 790–797. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Guglielmelli, P.; Larson, D.R.; Finke, C.; Wassie, E.A.; Pieri, L.; Gangat, N.; Fjerza, R.; Belachew, A.A.; Lasho, T.; et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood 2014, 124, 2507–2513. [Google Scholar] [CrossRef]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Coltro, G.; Finke, C.M.; Loscocco, G.G.; Sordi, B.; Szuber, N.; Rotunno, G.; Pacilli, A.; et al. Mutation-enhanced international prognostic systems for essential thrombocythaemia and polycythaemia vera. Br. J. Haematol. 2020, 189, 291–302. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Gangat, N.; Coltro, G.; Lasho, T.L.; Loscocco, G.G.; Finke, C.M.; Morsia, E.; Sordi, B.; Szuber, N.; Hanson, C.A.; et al. Mutations and thrombosis in essential thrombocythemia. Blood Cancer J. 2021, 11, 77. [Google Scholar] [CrossRef]
- Alvarez-Larrán, A.; Bellosillo, B.; Pereira, A.; Kerguelen, A.; Hernández-Boluda, J.C.; Martínez-Avilés, L.; Fernández-Rodríguez, C.; Gómez, M.; Lombardía, L.; Angona, A.; et al. JAK2V617F monitoring in polycythemia vera and essential thrombocythemia: Clinical usefulness for predicting myelofibrotic transformation and thrombotic events. Am. J. Hematol. 2014, 89, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Guglielmelli, P.; Nicolosi, M.; Mannelli, F.; Mudireddy, M.; Bartalucci, N.; Finke, C.M.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; et al. GIPSS: Genetically inspired prognostic scoring system for primary myelofibrosis. Leukemia 2018, 32, 1631–1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuykendall, A.T.; Talati, C.; Padron, E.; Sweet, K.; Sallman, D.; List, A.F.; Lancet, J.E.; Komrokji, R.S. Genetically inspired prognostic scoring system (GIPSS) outperforms dynamic international prognostic scoring system (DIPSS) in myelofibrosis patients. Am. J. Hematol. 2019, 94, 87–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gagelmann, N.; Ditschkowski, M.; Bogdanov, R.; Bredin, S.; Robin, M.; Cassinat, B.; Shahswar, R.; Thol, F.; Heuser, M.; Socié, G.; et al. Comprehensive clinical-molecular transplant scoring system for myelofibrosis undergoing stem cell transplantation. Blood 2019, 133, 2233–2242. [Google Scholar] [CrossRef]
- Coltro, G.; Guglielmelli, P.; Rotunno, G.; Mannarelli, C.; Maccari, C.; Salvadori, C.; Vanderwert, F.I.; Mannelli, F.; Salvati, C.; Vannucchi, A. Mutation Landscape and Prognostic Correlates of ASXL1 Variants in Primary and Secondary Myelofibrosis. Blood 2021, 138 (Suppl. S1), 2578. [Google Scholar] [CrossRef]
- Mascarenhas, J.; Mehra, M.; He, J.; Potluri, R.; Loefgren, C. Patient characteristics and outcomes after ruxolitinib discontinuation in patients with myelofibrosis. J. Med. Econ. 2020, 23, 721–727. [Google Scholar] [CrossRef]
- Newberry, K.J.; Patel, K.; Masarova, L.; Luthra, R.; Manshouri, T.; Jabbour, E.; Newberry, K.J.; Patel, K.; Masarova, L.; Luthra, R.; et al. Clonal evolution and outcomes in myelofibrosis after ruxolitinib discontinuation. Blood 2017, 130, 1125–1131. [Google Scholar] [CrossRef] [Green Version]
- Mullally, A.; Hood, J.; Harrison, C.; Mesa, R. Fedratinib in myelofibrosis. Blood Adv. 2020, 4, 1792–1800. [Google Scholar] [CrossRef]
- Tefferi, A.; Barraco, D.; Lasho, T.L.; Shah, S.; Begna, K.H.; Al-Kali, A.; Hogan, W.J.; Litzow, M.R.; Hanson, C.A.; Ketterling, R.P.; et al. Momelotinib therapy for myelofibrosis: A 7-year follow-up. Blood Cancer J. 2018, 8, 4–8. [Google Scholar] [CrossRef]
- Mascarenhas, J.; Hoffman, R.; Talpaz, M.; Gerds, A.T.; Stein, B.; Gupta, V.; Szoke, A.; Drummond, M.; Pristupa, A.; Granston, T.; et al. Pacritinib vs best available therapy, including ruxolitinib, in patients with myelofibrosis: A randomized clinical trial. JAMA Oncol. 2018, 4, 652–659. [Google Scholar] [CrossRef]
- Stivala, S.; Codilupi, T.; Brkic, S.; Baerenwaldt, A.; Ghosh, N.; Hao-Shen, H.; Dirnhofer, S.; Dettmer, M.S.; Simillion, C.; Kaufmann, B.A.; et al. Targeting compensatory MEK/ERK activation increases JAK inhibitor efficacy in myeloproliferative neoplasms. J. Clin. Investig. 2019, 129, 1596–1611. [Google Scholar] [CrossRef] [PubMed]
- Truong, B.; Zhang, Y.; Fahl, S.; Cai, K.Q.; Martinez, E.; Al-Saleem, E.D.; Gong, Y.; Liebermann, D.; Soboloff, J.; Dunbrack, R.; et al. ERK2 Substrate Binding Domains Perform Opposing Roles in Pathogenesis of a JAK2V617F-Driven Myeloproliferative Neoplasm. Blood 2021, 138 (Suppl. S1), 2547. [Google Scholar] [CrossRef]
- Gurska, L.M.; Okabe, R.; Tong, M.M.; Choi, D.; Ames, K.; Glushakow-Smith, S.; Montoya, A.; Tein, E.; Cheng, H.; Goel, S.; et al. RON Kinase Is a Novel Therapeutic Target for Philadelphia-Negative Myeloproliferative Neoplasms. Blood 2021, 138 (Suppl. S1), 1462. [Google Scholar] [CrossRef]
- Mylonas, E.; Yoshida, K.; Frick, M.; Hoyer, K.; Christen, F.; Kaeda, J.; Obenaus, M.; Noerenberg, D.; Hennch, C.; Chan, W.; et al. Single-cell analysis based dissection of clonality in myelofibrosis. Nat. Commun. 2020, 11, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tremblay, D.; Mascarenhas, J. Next Generation Therapeutics for the Treatment of Myelofibrosis. Cells 2021, 10, 1034. [Google Scholar] [CrossRef]
- Bose, P.; Verstovsek, S. Management of myelofibrosis after ruxolitinib failure. Ann. Hematol. 2020, 99, 1177–1191. [Google Scholar] [CrossRef]
- Morsia, E.; Gangat, N. Myelofibrosis: Challenges for preclinical models and emerging therapeutic targets. Expert Opin. Targets 2021, 25, 211–222. [Google Scholar] [CrossRef]
- Miyauchi, M.; Sasaki, K.; Kagoya, Y.; Taoka, K.; Masamoto, Y.; Yamazaki, S.; Arai, S.; Mizuno, H.; Kurokawa, M. CAMK2G is identified as a novel therapeutic target for myelofibrosis. Blood Adv. 2022, 6, 1585–1597. [Google Scholar] [CrossRef]
- Pronier, E.; Cifani, P.; Merlinsky, T.R.; Berman, K.B.; Somasundara, A.V.H.; Rampal, R.K.; LaCava, J.; Wei, K.E.; Pastore, F.; Maag, J.L.V.; et al. Targeting the CALR interactome in myeloproliferative neoplasms. JCI Insight 2018, 3, e122703. [Google Scholar] [CrossRef]
- Kihara, Y.; Araki, M.; Imai, M.; Mori, Y.; Horino, M.; Ogata, S.; Yoshikawa, S.; Taguchi, T.; Masubuchi, N.; Mabuchi, Y.; et al. Therapeutic Potential of an Antibody Targeting the Cleaved Form of Mutant Calreticulin in Myeloproliferative Neoplasms. Blood 2020, 136 (Suppl. S1), 9–10. [Google Scholar] [CrossRef]


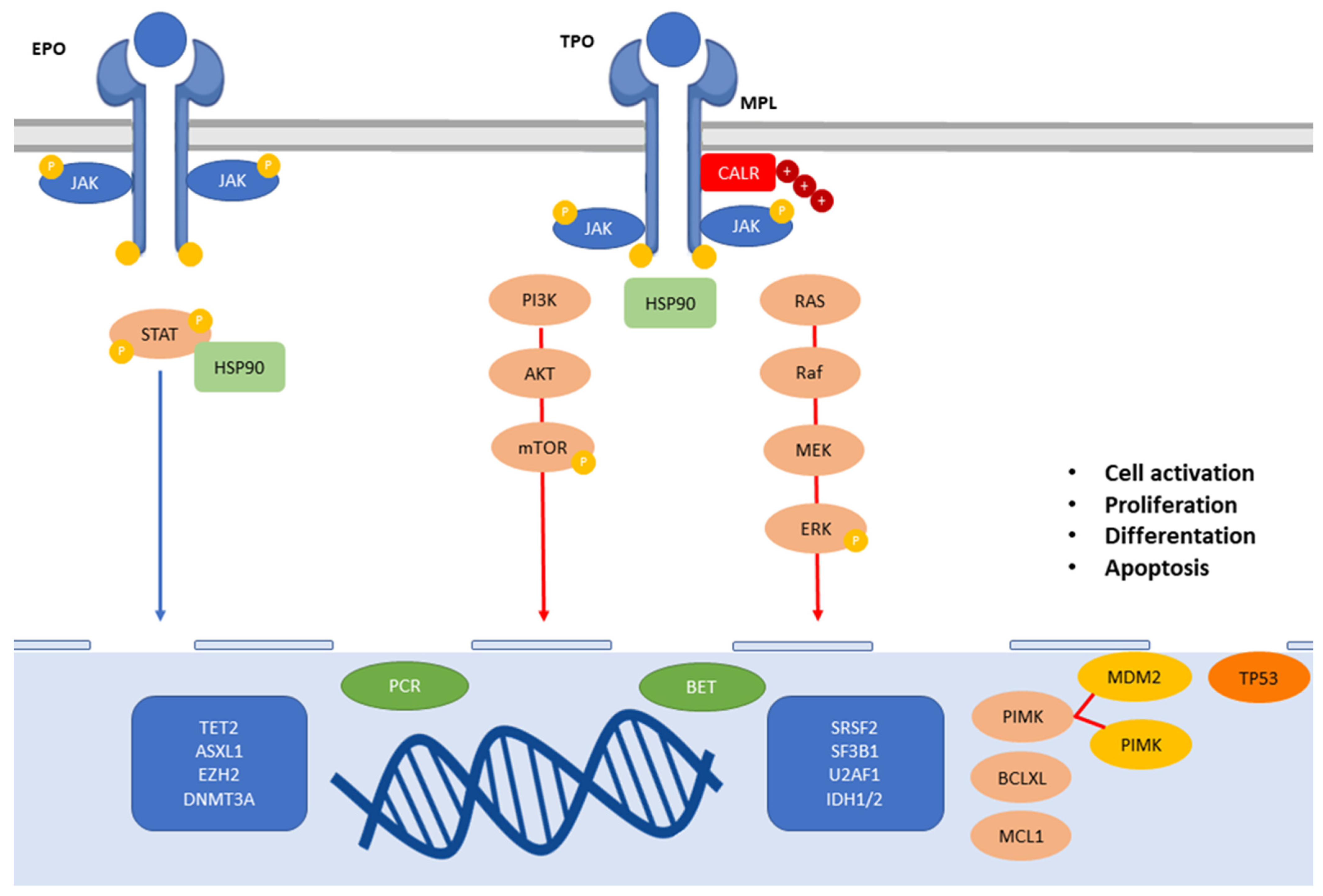{kind=link}
| Class | Mutated Genes | Frequency (%) | |||
|---|---|---|---|---|---|
| ET | PV | PMF | Blast Phase | ||
| Epigenetic regulation | DNMT3A | 0–9 | 0–7 | 3–15 | 2–14 |
| TET2 | 7–16 | 19–22 | 10–18 | 19–28 | |
| IDH1/IDH2 | 1 | 2 | 0–6 | 19–31 | |
| ASXL1 | 1–11 | 3–12 | 18–37 | 17–47 | |
| EZH2 | 1–3 | 0–3 | 0–9 | 13–15 | |
| Messenger RNA splicing | SRSF2 | 2 | 3 | 8–18 | 13–22 |
| U2AF1 | 1 | <1 | 16 | 5–6 | |
| SF3B1 | 5 | 3 | 9–10 | 4–7 | |
| ZRSR2 | 3 | 5 | 10 | 2 | |
| Transcriptional regulation | NFE2 | <1 | 2–3 | 0–3 | <1 |
| RUNX1 | 0–2 | 0–2 | 3–4 | 4–13 | |
| Signaling | NRAS/KRAS | <1 | 0–1 | 3–4 | 7–15 |
| PTPN11 | 0–2 | <1 | 0–2 | 6–8 | |
| CBL | 0–1 | 0–2 | 0–6 | 4 | |
| LNK (SH2B3) | 1–3 | 0–9 | 0–6 | 11 | |
| DNA repair | TP53 | 2–6 | 1 | 1–3 | 11–36 |
| PPM1D | 2 | 1 | 1 | NA | |
| Prognostic Score | Variables (Points) | Risk Categories (Median OS, Years) |
|---|---|---|
| MIPSS70 | Hemoglobin < 10 g/dL (1) Blasts >2% (1) Constitutional symptoms (1) Leukocytes > 25 × 10*9/L (2) Platelet < 100 × 10*9/L (2) Bone marrow fibrosis ≥ 2 (1) Non type-1 CALR (1) HMR = 1 (1) HMR ≥ 2 (2) | 0–1: Low (27.7) 2–4: Intermediate (7.1) 5–12: High (2.3) |
| MIPSS70+ | Hemoglobin < 10 g/dL (1) Blasts >2% (1) Constitutional symptoms (1) Non type-1 CALR (2) HMR = 1 (1) HMR ≥ 2 (2) Unfavourable karyotype (3) | 0–2: Low (20.0) 3: Intermediate (6.3) 4–6: High (3.9) 7–11: Very high (1.7) |
| MIPSS70+ v2.0 | Hemoglobin <8–10 g/dL (1) Hemoglobin < 8 g/dL (2) Blasts >2% (1) Constitutional symptoms (2) Non type-1 CALR (2) HMR+ U2AF1Q157 = 1 (2) HMR+ U2AF1Q157 ≥ 2 (3) HR karyotype (3) VHR karyotype (4) | 0: Very low (Not reached) 1–2: Low (10.3) 3–4: Intermediate (7) 5–8: High (3.5) 9–14: Very high (1.8) |
| GIPSS | Non type-1 CALR (1) ASXL1 mutated (1) SRSF2 mutated (1) U2AF1Q157 (1) HR karyotype (1) VHR karyotype (2) | 0: Low (26.4) 1: Intermediate-1 (8) 2: Intermediate-2 (4.2) 3–6: High (2) |
| MYSEC-PM | Hemoglobin < 11 g/dL (1) Blasts >3% (1) Constitutional symptoms (2) Platelet < 150 × 10*9/L (1) Age at secondary MF (0.15 point/year) CALR unmutated (2) | <11: Low (Not reached) 11-<14: Intermediate-1 (9.3) 14-<16: Intermediate-2 (4.4) ≥ 16: High (2) |
| MTSS | Leukocytes > 25 × 10*9/L (1) Platelet < 150 × 10*9/L (1) Karnofsky performance status <90% (1) Age ≥ 57 years (1) HLA-mismatched unrelated donor (2) Non CALR/MPL mutations (2) ASXL1 mutated (1) | 0–2: Low (5-years overall survival 83%) 3–4: Intermediate (5-years overall survival 64%) 5: High (5-years overall survival 37%) 6–9: Very high (5-years overall survival 22%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morsia, E.; Torre, E.; Poloni, A.; Olivieri, A.; Rupoli, S. Molecular Pathogenesis of Myeloproliferative Neoplasms: From Molecular Landscape to Therapeutic Implications. Int. J. Mol. Sci. 2022, 23, 4573. https://doi.org/10.3390/ijms23094573
Morsia E, Torre E, Poloni A, Olivieri A, Rupoli S. Molecular Pathogenesis of Myeloproliferative Neoplasms: From Molecular Landscape to Therapeutic Implications. International Journal of Molecular Sciences. 2022; 23(9):4573. https://doi.org/10.3390/ijms23094573
Chicago/Turabian StyleMorsia, Erika, Elena Torre, Antonella Poloni, Attilio Olivieri, and Serena Rupoli. 2022. "Molecular Pathogenesis of Myeloproliferative Neoplasms: From Molecular Landscape to Therapeutic Implications" International Journal of Molecular Sciences 23, no. 9: 4573. https://doi.org/10.3390/ijms23094573