
An Update on Familial Mediterranean Fever
 , , ,
, , ,
Abstract
:1. The Historical Background
2. Epidemiology
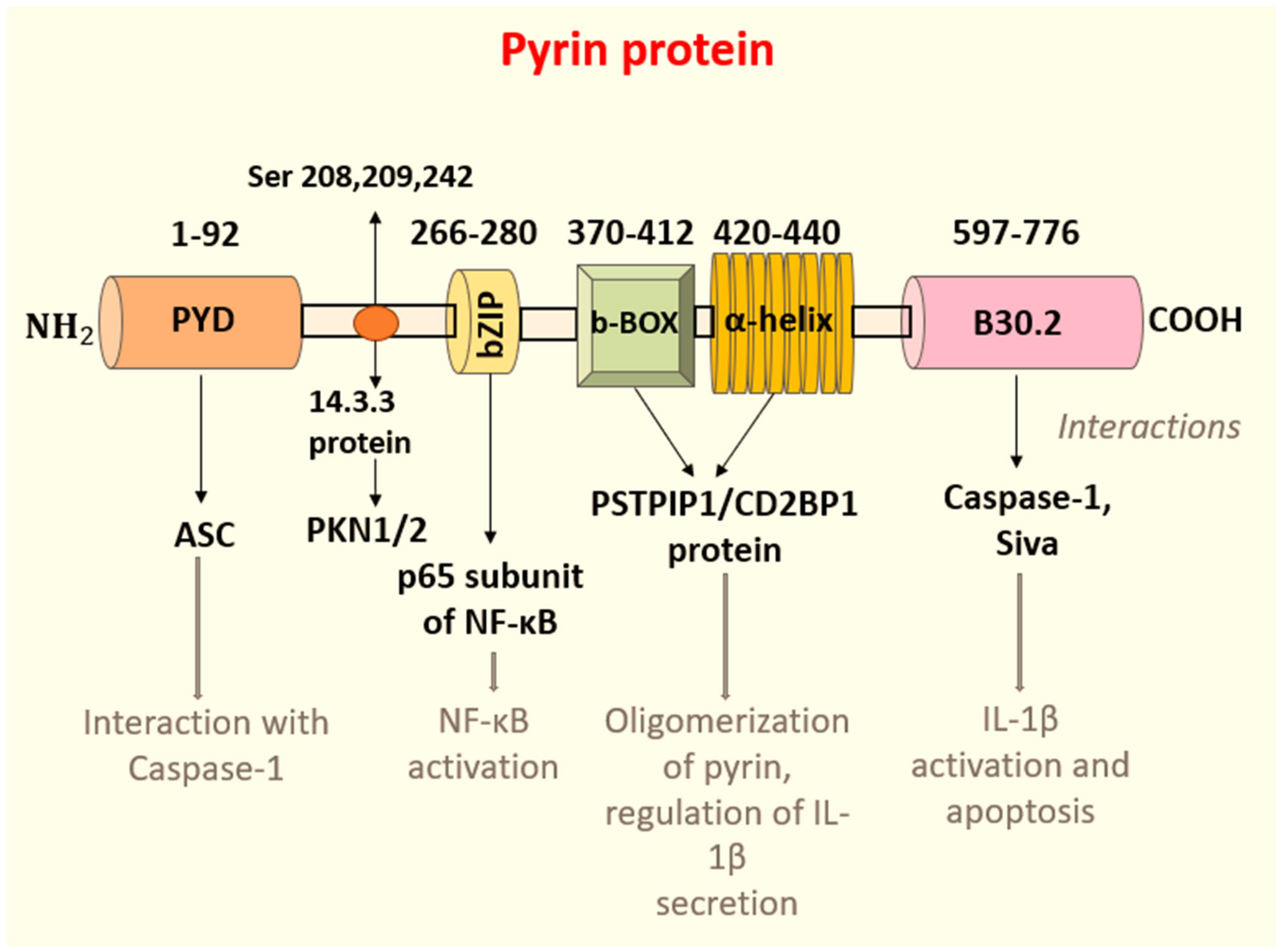
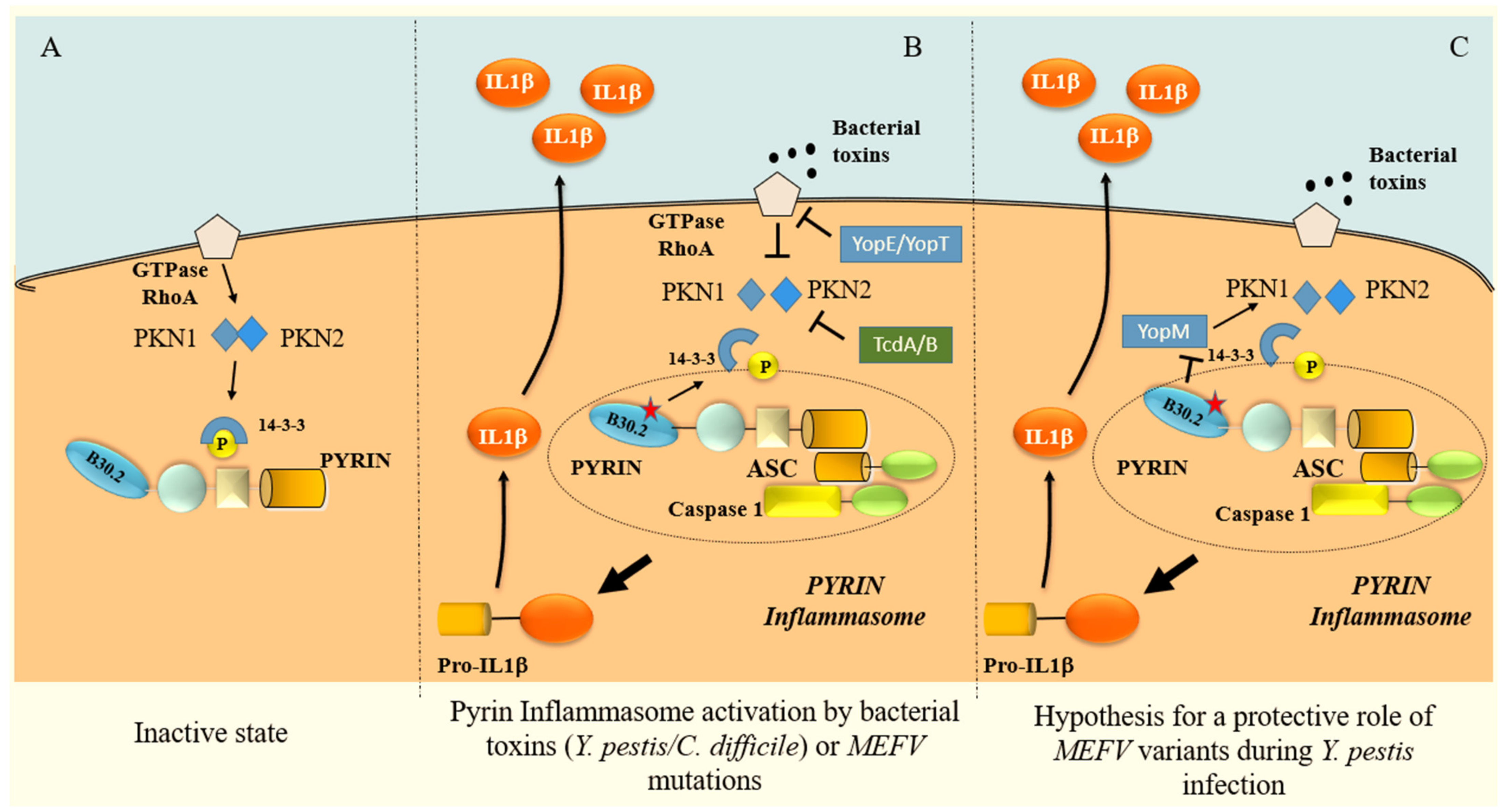
3. Pathogenesis
IL-1β
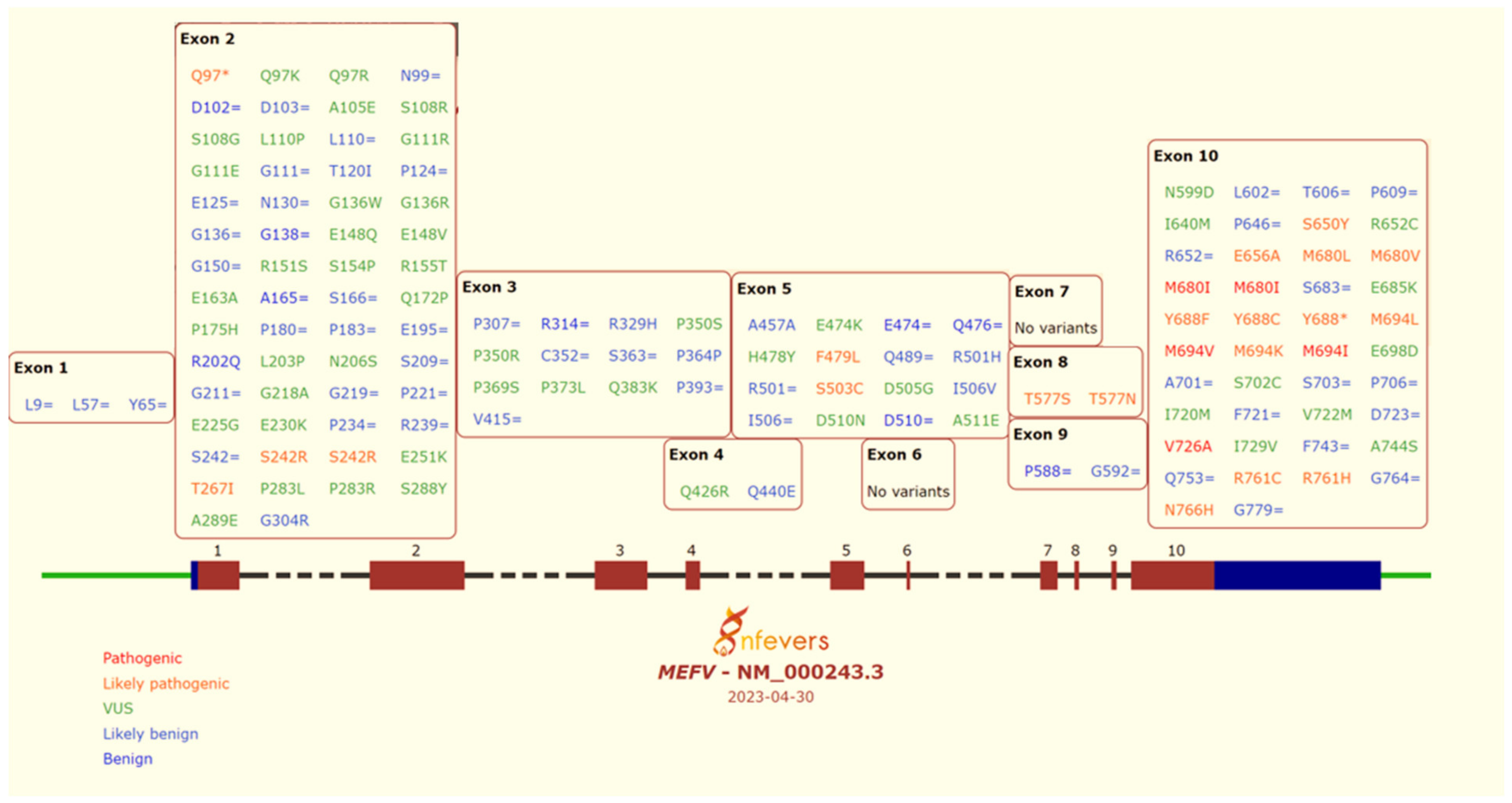
4. Genetics and Genotype–Phenotype Correlations
5. Clinical Features
5.1. Fever
5.2. Abdominal Manifestations
5.3. Pleurisy and Pericarditis
5.4. Musculoskeletal Symptoms
5.5. Other Manifestations
6. Associated Diseases
7. Diagnostic and Classification Criteria
8. Interpretation of MEFV Gene Variants
9. Treatment
9.1. Colchicine: Mechanism of Action
9.2. Colchicine: Metabolism and Toxicity
9.3. Colchicine: Management in Familial Mediterranean Fever
10. Colchicine Resistance
11. Interlukin-1 Inhibitors
11.1. Anakinra
11.2. Rilonacept
11.3. Canakinumab
12. Anti-IL-6 Drugs
Conclusive Remarks and Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Alghamdi, M. Familial Mediterranean Fever, Review of the Literature. Clin. Rheumatol. 2017, 36, 1707–1713. [Google Scholar] [CrossRef]
- French FMF Consortium A Candidate Gene for Familial Mediterranean Fever. Nat. Genet. 1997, 17, 25–31. [CrossRef] [PubMed]
- Siegal, S. Benign Paroxysmal Peritonitis. Gastroenterology 1949, 12, 234–247. [Google Scholar] [CrossRef] [PubMed]
- Goldfinger, S.E. Colchicine for Familial Mediterranean Fever. N. Engl. J. Med. 1972, 287, 1302. [Google Scholar] [CrossRef]
- Zemer, D.; Revach, M.; Pras, M.; Modan, B.; Schor, S.; Sohar, E.; Gafni, J. A Controlled Trial of Colchicine in Preventing Attacks of Familial Mediterranean Fever. N. Engl. J. Med. 1974, 191, 932–934. [Google Scholar] [CrossRef]
- Tunca, M.; Ozdogan, H.; Kasapcopur, O.; Yalcinkaya, F.; Tutar, E.; Topaloglu, R.; Yilmaz, E.; Arici, M.; Bakkaloglu, A.; Besbas, N.; et al. Familial Mediterranean Fever (FMF) in Turkey: Results of a Nationwide Multicenter Study. Medicine 2005, 84, 1–11. [Google Scholar] [CrossRef]
- Cakır, N.; Pamuk, Ö.N.; Derviş, E.; Imeryüz, N.; Uslu, H.; Benian, Ö.; Elelçi, E.; Erdem, G.; Sarvan, F.O.; Senocak, M. The Prevalences of Some Rheumatic Diseases in Western Turkey: Havsa Study. Rheumatol. Int. 2012, 32, 895–908. [Google Scholar] [CrossRef]
- Piazza, A.; Cappello, N.; Olivetti, E.; Rendine, S. A Genetic History of Italy. Ann. Hum. Genet. 1988, 52, 203–213. [Google Scholar] [CrossRef]
- Papadopoulos, V.P.; Giaglis, S.; Mitroulis, I.; Ritis, K. The Population Genetics of Familial Mediterranean Fever: A Meta-Analysis Study. Ann. Hum. Genet. 2008, 72, 752–761. [Google Scholar] [CrossRef] [PubMed]
- Sarkisian, T.; Ajrapetian, H.; Beglarian, A.; Shahsuvarian, G.; Egiazarian, A. Familial Mediterranean Fever in Armenian Population. Georgian Med. News 2008, 156, 105–111. [Google Scholar]
- Rigante, D.; Frediani, B.; Galeazzi, M.; Cantarini, L. From the Mediterranean to the Sea of Japan: The Transcontinental Odyssey of Autoinflammatory Diseases. Biomed. Res. Int. 2013, 2013, 485103. [Google Scholar] [CrossRef] [PubMed]
- Cattan, D. Familial Mediterranean Fever: Is Low Mortality from Tuberculosis a Specific Advantage for MEFV Mutations Carriers? Mortality from Tuberculosis among Muslims, Jewish, French, Italian and Maltese Patients in Tunis (Tunisia) in the First Half of the 20th Century. Clin. Exp. Rheumatol. 2003, 21, S53–S54. [Google Scholar]
- Ross, J.J. Goats, Germs, and Fever: Are the Pyrin Mutations Responsible for Familial Mediterranean Fever Protective against Brucellosis? Med. Hypotheses 2007, 68, 499–501. [Google Scholar] [CrossRef]
- McDermott, M.F.; Aksentijevich, I.; Galon, J.; McDermott, E.M.; Ogunkolade, B.W.; Centola, M.; Mansfield, E.; Gadina, M.; Karenko, L.; Pettersson, T.; et al. Germline Mutations in the Extracellular Domains of the 55 KDa TNF Receptor, TNFR1, Define a Family of Dominantly Inherited Autoinflammatory Syndromes. Cell 1999, 97, 133–144. [Google Scholar] [CrossRef]
- Touitou, I.; Sarkisian, T.; Medlej-Hashim, M.; Tunca, M.; Livneh, A.; Cattan, D.; Yalçinkaya, F.; Ozen, S.; Majeed, H.; Ozdogan, H.; et al. Country as the Primary Risk Factor for Renal Amyloidosis in Familial Mediterranean Fever. Arthritis Rheum. 2007, 56, 1706–1712. [Google Scholar] [CrossRef]
- Ozen, S.; Demirkaya, E.; Amaryan, G.; Koné-Paut, I.; Polat, A.; Woo, P.; Uziel, Y.; Modesto, C.; Finetti, M.; Quartier, P.; et al. Results from a Multicentre International Registry of Familial Mediterranean Fever: Impact of Environment on the Expression of a Monogenic Disease in Children. Ann. Rheum. Dis. 2014, 73, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya-Suzuki, A.; Yazaki, M.; Nakamura, A.; Yamazaki, K.; Agematsu, K.; Matsuda, M.; Ikeda, S.-I. Clinical and Genetic Features of Familial Mediterranean Fever in Japan. J. Rheumatol. 2009, 36, 1671–1676. [Google Scholar] [CrossRef]
- Diaz, A.; Hu, C.; Kastner, D.L.; Schaner, P.; Reginato, A.M.; Richards, N.; Gumucio, D.L. Lipopolysaccharide-Induced Expression of Multiple Alternatively Spliced MEFV Transcripts in Human Synovial Fibroblasts: A Prominent Splice Isoform Lacks the C-Terminal Domain That Is Highly Mutated in Familial Mediterranean Fever. Arthritis Rheum. 2004, 50, 3679–3689. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.H.; Wood, G.; Kastner, D.L.; Chae, J.J. Pyrin Inflammasome Activation and RhoA Signaling in the Autoinflammatory Diseases FMF and HIDS. Nat. Immunol. 2016, 17, 914–921. [Google Scholar] [CrossRef]
- Schnappauf, O.; Chae, J.J.; Kastner, D.L.; Aksentijevich, I. The Pyrin Inflammasome in Health and disease. Front. Immunol. 2019, 10, 1745. [Google Scholar] [CrossRef]
- Richards, N.; Schaner, P.; Diaz, A.; Stuckey, J.; Shelden, E.; Wadhwa, A.; Gumucio, D.L. Interaction between Pyrin and the Apoptotic Speck Protein (ASC) Modulates ASC-Induced Apoptosis. J. Biol. Chem. 2001, 276, 39320–39329. [Google Scholar] [CrossRef] [PubMed]
- Chae, J.J.; Wood, G.; Masters, S.L.; Richard, K.; Park, G.; Smith, B.J.; Kastner, D.L. The B30.2 Domain of Pyrin, the Familial Mediterranean Fever Protein, Interacts Directly with Caspase-1 to Modulate IL-1beta Production. Proc. Natl. Acad. Sci. USA 2006, 103, 9982–9987. [Google Scholar] [CrossRef]
- Papin, S.; Cuenin, S.; Agostini, L.; Martinon, F.; Werner, S.; Beer, H.-D.; Grütter, C.; Grütter, M.; Tschopp, J. The SPRY Domain of Pyrin, Mutated in Familial Mediterranean Fever Patients, Interacts with Inflammasome Components and Inhibits ProIL-1beta Processing. Cell Death Differ. 2007, 14, 1457–1466. [Google Scholar] [CrossRef]
- Seshadri, S.; Duncan, M.D.; Hart, J.M.; Gavrilin, M.A.; Wewers, M.D. Pyrin Levels in Human Monocytes and Monocyte-Derived Macrophages Regulate IL-1beta Processing and Release. J. Immunol. 2007, 179, 1274–1281. [Google Scholar] [CrossRef]
- Yu, J.-W.; Fernandes-Alnemri, T.; Datta, P.; Wu, J.; Juliana, C.; Solorzano, L.; McCormick, M.; Zhang, Z.; Alnemri, E.S. Pyrin Activates the ASC Pyroptosome in Response to Engagement by Autoinflammatory PSTPIP1 Mutants. Mol. Cell 2007, 28, 214–227. [Google Scholar] [CrossRef] [PubMed]
- Booty, M.G.; Chae, J.J.; Masters, S.L.; Remmers, E.F.; Barham, B.; Le, J.M.; Barron, K.S.; Holland, S.M.; Kastner, D.L.; Aksentijevich, I. Familial Mediterranean Fever with a Single MEFV Mutation: Where Is the Second Hit? Arthritis Rheum. 2009, 60, 1851–1861. [Google Scholar] [CrossRef]
- Soriano, A.; Manna, R. Familial Mediterranean Fever: New Phenotypes. Autoimmun. Rev. 2012, 12, 31–37. [Google Scholar] [CrossRef]
- Lachmann, H.J.; Sengül, B.; Yavuzşen, T.U.; Booth, D.R.; Booth, S.E.; Bybee, A.; Gallimore, J.R.; Soytürk, M.; Akar, S.; Tunca, M.; et al. Clinical and Subclinical Inflammation in Patients with Familial Mediterranean Fever and in Heterozygous Carriers of MEFV Mutations. Rheumatology 2006, 45, 746–750. [Google Scholar] [CrossRef]
- Chae, J.J.; Cho, Y.-H.; Lee, G.-S.; Cheng, J.; Liu, P.P.; Feigenbaum, L.; Katz, S.I.; Kastner, D.L. Gain-of-Function Pyrin Mutations Induce NLRP3 Protein-Independent Interleukin-1β Activation and Severe Autoinflammation in Mice. Immunity 2011, 34, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Federici, S.; Calcagno, G.; Finetti, M.; Gallizzi, R.; Meini, A.; Vitale, A.; Caroli, F.; Cattalini, M.; Caorsi, R.; Zulian, F.; et al. Clinical Impact of MEFV Mutations in Children with Periodic Fever in a Prevalent Western European Caucasian Population. Ann. Rheum. Dis. 2012, 71, 1961–1965. [Google Scholar] [CrossRef] [PubMed]
- Omenetti, A.; Carta, S.; Delfino, L.; Martini, A.; Gattorno, M.; Rubartelli, A. Increased NLRP3-Dependent Interleukin 1β Secretion in Patients with Familial Mediterranean Fever: Correlation with MEFV Genotype. Ann. Rheum. Dis. 2014, 73, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Masters, S.L.; Lagou, V.; Jéru, I.; Baker, P.J.; Van Eyck, L.; Parry, D.A.; Lawless, D.; De Nardo, D.; Garcia-Perez, J.E.; Dagley, L.F.; et al. Familial Autoinflammation with Neutrophilic Dermatosis Reveals a Regulatory Mechanism of Pyrin Activation. Sci. Transl. Med. 2016, 8, 332ra45. [Google Scholar] [CrossRef]
- Loeven, N.A.; Medici, N.P.; Bliska, J.B. The Pyrin Inflammasome in Host-Microbe Interactions. Curr. Opin. Microbiol. 2020, 54, 77–86. [Google Scholar] [CrossRef]
- Park, Y.H.; Remmers, E.F.; Lee, W.; Ombrello, A.K.; Chung, L.K.; Shilei, Z.; Stone, D.L.; Ivanov, M.I.; Loeven, N.A.; Barron, K.S.; et al. Ancient Familial Mediterranean Fever Mutations in Human Pyrin and Resistance to Yersinia Pestis. Nat. Immunol. 2020, 21, 857–867. [Google Scholar] [CrossRef]
- Dinarello, C.A.; Cannon, J.G.; Mier, J.W.; Bernheim, H.A.; LoPreste, G.; Lynn, D.L.; Love, R.N.; Webb, A.C.; Auron, P.E.; Reuben, R.C. Multiple Biological Activities of Human Recombinant Interleukin 1. J. Clin. Investig. 1986, 77, 1734–1739. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Okamura, H.; Wada, M.; Nagata, K.; Tamura, T. Endotoxin-Induced Serum Factor That Stimulates Gamma Interferon Production. Infect. Immun. 1989, 57, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Rigante, D.; Vitale, A.; Lucherini, O.M.; Cantarini, L. The Hereditary Autoinflammatory Disorders Uncovered. Autoimmun. Rev. 2014, 13, 892–900. [Google Scholar] [CrossRef]
- Rigante, D. The Fresco of Autoinflammatory Diseases from the Pediatric Perspective. Autoimmun. Rev. 2012, 11, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Magal, N.; Lotan, R.; Allon-Shalev, S.; Khamaysi, N.; Shohat, M. A New Hot Spot in the Gene Causing Familial Mediterranean Fever. Am. J. Hum. Genet. 1998, 63, A372. [Google Scholar]
- Bernot, A.; da Silva, C.; Petit, J.L.; Cruaud, C.; Caloustian, C.; Castet, V.; Ahmed-Arab, M.; Dross, C.; Dupont, M.; Cattan, D.; et al. Non-Founder Mutations in the MEFV Gene Establish This Gene as the Cause of Familial Mediterranean Fever (FMF). Hum. Mol. Genet. 1998, 7, 1317–1325. [Google Scholar] [CrossRef]
- Dundar, M.; Kiraz, A.; Emirogullari, E.F.; Saatci, C.E.; Taheri, S.; Baskol, M.; Polat, S.; Ozkul, Y. A Molecular Analysis of Familial Mediterranean Fever Disease in a Cohort of Turkish Patients. Ann. Saudi Med. 2012, 32, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Sharkia, R.; Mahajnah, M.; Zalan, A.; Athamna, M.; Azem, A.; Badarneh, K.; Faris, F. Comparative Screening of FMF Mutations in Various Communities of the Israeli Society. Eur. J. Med. Genet. 2013, 56, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Lidar, M.; Kedem, R.; Berkun, Y.; Langevitz, P.; Livneh, A. Familial Mediterranean Fever in Ashkenazi Jews: The Mild End of the Clinical Spectrum. J. Rheumatol. 2010, 37, 422–425. [Google Scholar] [CrossRef]
- Aksentijevich, I.; Torosyan, Y.; Samuels, J.; Centola, M.; Pras, E.; Chae, J.J.; Oddoux, C.; Wood, G.; Azzaro, M.P.; Palumbo, G.; et al. Mutation and Haplotype Studies of Familial Mediterranean Fever Reveal New Ancestral Relationships and Evidence for a High Carrier Frequency with Reduced Penetrance in the Ashkenazi Jewish Population. Am. J. Hum. Genet. 1999, 64, 949–962. [Google Scholar] [CrossRef]
- Aldea, A.; Calafell, F.; Aróstegui, J.I.; Lao, O.; Rius, J.; Plaza, S.; Masó, M.; Vives, J.; Buades, J.; Yagüe, J. The West Side Story: MEFV Haplotype in Spanish FMF Patients and Controls, and Evidence of High LD and a Recombination “Hot-Spot” at the MEFV Locus. Hum. Mutat. 2004, 23, 399. [Google Scholar] [CrossRef]
- Booth, D.R.; Gillmore, J.D.; Lachmann, H.J.; Booth, S.E.; Bybee, A.; Soytürk, M.; Akar, S.; Pepys, M.B.; Tunca, M.; Hawkins, P.N. The Genetic Basis of Autosomal Dominant Familial Mediterranean Fever. QJM 2000, 93, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Brik, R.; Shinawi, M.; Kasinetz, L.; Gershoni-Baruch, R. The Musculoskeletal Manifestations of Familial Mediterranean Fever in Children Genetically Diagnosed with the Disease. Arthritis Rheum. 2001, 44, 1416–1419. [Google Scholar] [CrossRef] [PubMed]
- Cazeneuve, C.; Sarkisian, T.; Pêcheux, C.; Dervichian, M.; Nédelec, B.; Reinert, P.; Ayvazyan, A.; Kouyoumdjian, J.C.; Ajrapetyan, H.; Delpech, M.; et al. MEFV-Gene Analysis in Armenian Patients with Familial Mediterranean Fever: Diagnostic Value and Unfavorable Renal Prognosis of the M694V Homozygous Genotype-Genetic and Therapeutic Implications. Am. J. Hum. Genet. 1999, 65, 88–97. [Google Scholar] [CrossRef]
- Lidar, M.; Yonath, H.; Shechter, N.; Sikron, F.; Sadetzki, S.; Langevitz, P.; Livneh, A.; Pras, E. Incomplete Response to Colchicine in M694V Homozygote FMF Patients. Autoimmun. Rev. 2012, 12, 72–76. [Google Scholar] [CrossRef]
- Livneh, A.; Langevitz, P.; Shinar, Y.; Zaks, N.; Kastner, D.L.; Pras, M.; Pras, E. MEFV Mutation Analysis in Patients Suffering from Amyloidosis of Familial Mediterranean Fever. Amyloid 1999, 6, 1–6. [Google Scholar] [CrossRef]
- Shohat, M.; Livneh, A.; Zemer, D.; Pras, M.; Sohar, E. Twin Studies in Familial Mediterranean Fever. Am. J. Med. Genet. 1992, 44, 179–182. [Google Scholar] [CrossRef] [PubMed]
- Touitou, I.; Picot, M.C.; Domingo, C.; Notarnicola, C.; Cattan, D.; Demaille, J.; Koné-Paut, I. The MICA Region Determines the First Modifier Locus in Familial Mediterranean Fever. Arthritis Rheum. 2001, 44, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Cazeneuve, C.; Ajrapetyan, H.; Papin, S.; Roudot-Thoraval, F.; Geneviève, D.; Mndjoyan, E.; Papazian, M.; Sarkisian, A.; Babloyan, A.; Boissier, B.; et al. Identification of MEFV-Independent Modifying Genetic Factors for Familial Mediterranean Fever. Am. J. Hum. Genet. 2000, 67, 1136–1143. [Google Scholar] [CrossRef]
- Rigante, D.; La Torraca, I.; Ansuini, V.; Compagnone, A.; Sallì, A.; Stabile, A. The Multi-Face Expression of Familial Mediterranean Fever in the Child. Eur. Rev. Med. Pharmacol. Sci. 2006, 10, 163–171. [Google Scholar] [PubMed]
- Livneh, A.; Langevitz, P.; Zemer, D.; Padeh, S.; Migdal, A.; Sohar, E.; Pras, M. The Changing Face of Familial Mediterranean Fever. Semin. Arthritis Rheum. 1996, 26, 612–627. [Google Scholar] [CrossRef]
- Yenokyan, G.; Armenian, H.K. Triggers for Attacks in Familial Mediterranean Fever: Application of the Case-Crossover Design. Am. J. Epidemiol. 2012, 175, 1054–1061. [Google Scholar] [CrossRef]
- Karadag, O.; Tufan, A.; Yazisiz, V.; Ureten, K.; Yilmaz, S.; Cinar, M.; Akdogan, A.; Erdem, H.; Ozturk, M.A.; Pay, S.; et al. The Factors Considered as Trigger for the Attacks in Patients with Familial Mediterranean Fever. Rheumatol. Int. 2013, 33, 893–897. [Google Scholar] [CrossRef]
- Ben-Chetrit, E.; Ben-Chetrit, A. Familial Mediterranean Fever and Menstruation. BJOG 2001, 108, 403–407. [Google Scholar] [CrossRef]
- Lidar, M.; Yaqubov, M.; Zaks, N.; Ben-Horin, S.; Langevitz, P.; Livneh, A. The Prodrome: A Prominent yet Overlooked Pre-Attack Manifestation of Familial Mediterranean Fever. J. Rheumatol. 2006, 33, 1089–1092. [Google Scholar]
- Sohar, E.; Gafni, J.; Pras, M.; Heller, H. Familial Mediterranean Fever. A Survey of 470 Cases and Review of the Literature. Am. J. Med. 1967, 43, 227–253. [Google Scholar] [CrossRef]
- Padeh, S.; Livneh, A.; Pras, E.; Shinar, Y.; Lidar, M.; Feld, O.; Berkun, Y. Familial Mediterranean Fever in the First Two Years of Life: A Unique Phenotype of Disease in Evolution. J. Pediatr. 2010, 156, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Tanatar, A.; Karadağ, Ş.G.; Çakan, M.; Sönmez, H.E.; Ayaz, N.A. Age of Onset as an Influencing Factor for Disease Severity in Children with Familial Mediterranean Fever. Mod. Rheumatol. 2021, 31, 219–222. [Google Scholar] [CrossRef]
- Hernández-Rodríguez, J.; Ruíz-Ortiz, E.; Tomé, A.; Espinosa, G.; González-Roca, E.; Mensa-Vilaró, A.; Prieto-González, S.; Espígol-Frigolé, G.; Mensa, J.; Cardellach, F.; et al. Clinical and Genetic Characterization of the Autoinflammatory Diseases Diagnosed in an Adult Reference Center. Autoimmun. Rev. 2016, 15, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Muscari, I.; Iacoponi, F.; Cantarini, L.; Lucherini, O.M.; Simonini, G.; Brizi, M.G.; Vitale, A.; Frediani, B.; Cimaz, R.; Galeazzi, M. The Diagnostic Evaluation of Patients with Potential Adult-Onset Autoinflammatory Disorders: Our Experience and Review of the Literature. Autoimmun. Rev. 2012, 12, 10–13. [Google Scholar] [CrossRef]
- Varan, O.; Kucuk, H.; Babaoglu, H.; Tecer, D.; Atas, N.; Bilici Salman, R.; Satıs, H.; Ozturk, M.A.; Haznedaroglu, S.; Goker, B.; et al. Chronic Inflammation in Adult Familial Mediterranean Fever Patients: Underlying Causes and Association with Amyloidosis. Scand. J. Rheumatol. 2019, 48, 315–319. [Google Scholar] [CrossRef]
- Yilmaz, H.; Inan, O.; Darcin, T.; Bilgic, M.A.; Akcay, A. Serum Galectin-3 Levels Were Associated with Proteinuria in Patients with Familial Mediterranean Fever. Clin. Exp. Nephrol. 2015, 19, 436–442. [Google Scholar] [CrossRef]
- Saccon, F.; Gatto, M.; Ghirardello, A.; Iaccarino, L.; Punzi, L.; Doria, A. Role of Galectin-3 in Autoimmune and Non-Autoimmune Nephropathies. Autoimmun. Rev. 2017, 16, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Ben-Chetrit, E.; Levy, M. Familial Mediterranean Fever. Lancet 1998, 351, 659–664. [Google Scholar] [CrossRef]
- Lidar, M.; Doron, A.; Kedem, R.; Yosepovich, A.; Langevitz, P.; Livneh, A. Appendectomy in Familial Mediterranean Fever: Clinical, Genetic and Pathological Findings. Clin. Exp. Rheumatol. 2008, 26, 568–573. [Google Scholar]
- Yanmaz, M.N.; Özcan, A.J.; Savan, K. The Impact of Familial Mediterranean Fever on Reproductive System. Clin. Rheumatol. 2014, 33, 1385–1388. [Google Scholar] [CrossRef]
- Aharoni, D.; Hiller, N.; Hadas-Halpern, I. Familial Mediterranean Fever: Abdominal Imaging Findings in 139 Patients and Review of the Literature. Abdom. Imaging 2000, 25, 197–300. [Google Scholar] [CrossRef]
- Zadeh, N.; Getzug, T.; Grody, W.W. Diagnosis and Management of Familial Mediterranean Fever: Integrating Medical Genetics in a Dedicated Interdisciplinary Clinic. Genet. Med. 2011, 13, 263–269. [Google Scholar] [CrossRef]
- Kees, S.; Langevitz, P.; Zemer, D.; Padeh, S.; Pras, M.; Livneh, A. Attacks of Pericarditis as a Manifestation of Familial Mediterranean Fever (FMF). QJM 1997, 90, 643–647. [Google Scholar] [CrossRef] [PubMed]
- Cantarini, L.; Lopalco, G.; Selmi, C.; Napodano, S.; De Rosa, G.; Caso, F.; Costa, L.; Iannone, F.; Rigante, D. Autoimmunity and Autoinflammation as the Yin and Yang of Idiopathic Recurrent Acute Pericarditis. Autoimmun. Rev. 2015, 14, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Maestroni, S.; Di Corato, P.R.; Cumetti, D.; Chiara, D.B.L.C.; Ghidoni, S.; Prisacaru, L.; Cantarini, L.; Imazio, M.; Penco, S.; Pedrotti, P.; et al. Recurrent Pericarditis: Autoimmune or Autoinflammatory? Autoimmun. Rev. 2012, 12, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Majeed, H.A.; Rawashdeh, M. The Clinical Patterns of Arthritis in Children with Familial Mediterranean Fever. QJM 1997, 90, 37–43. [Google Scholar] [CrossRef]
- Lidar, M.; Kedem, R.; Mor, A.; Levartovsky, D.; Langevitz, P.; Livneh, A. Arthritis as the Sole Episodic Manifestation of Familial Mediterranean Fever. J. Rheumatol. 2005, 32, 859–862. [Google Scholar]
- Kushnir, T.; Eshed, I.; Heled, Y.; Livneh, A.; Langevitz, P.; Ben Zvi, I.; Konen, E.; Lidar, M. Exertional Muscle Pain in Familial Mediterranean Fever Patients Evaluated by MRI and 31P Magnetic Resonance Spectroscopy. Clin. Radiol. 2013, 68, 371–375. [Google Scholar] [CrossRef]
- Kotevoglu, N.; Sahin, F.; Ozkiris, S.O.; Bankaoglu, M.; Sakiz, D.; Kuran, B. Protracted Febrile Myalgia of Familial Mediterranean Fever. Clin. Exp. Rheumatol. 2004, 22, S69–S70. [Google Scholar]
- Yıldırım, D.G.; Bakkaloglu, S.A.; Buyan, N. Protracted Febrile Myalgia as a Challenging Manifestation of Familial Mediterranean Fever: Case-Based Review. Rheumatol. Int. 2019, 39, 147–152. [Google Scholar] [CrossRef]
- Majeed, H.A.; Ghandour, K.; Shahin, H.M. The Acute Scrotum in Arab Children with Familial Mediterranean Fever. Pediatr. Surg. Int. 2000, 16, 72–74. [Google Scholar] [CrossRef]
- Gezgin Yildirim, D.; Seven, M.B.; Gönen, S.; Söylemezoğlu, O. Erysipelas-like Erythema in Children with Familial Mediterranean Fever. Clin. Exp. Rheumatol. 2020, 38 (Suppl. 127), 101–104. [Google Scholar] [PubMed]
- Koné Paut, I.; Dubuc, M.; Sportouch, J.; Minodier, P.; Garnier, J.M.; Touitou, I. Phenotype-Genotype Correlation in 91 Patients with Familial Mediterranean Fever Reveals a High Frequency of Cutaneomucous Features. Rheumatology 2000, 39, 1275–1279. [Google Scholar] [CrossRef] [PubMed]
- Abbara, S.; Grateau, G.; Ducharme-Bénard, S.; Saadoun, D.; Georgin-Lavialle, S. Association of Vasculitis and Familial Mediterranean Fever. Front. Immunol. 2019, 10, 763. [Google Scholar] [CrossRef]
- Yahalom, G.; Kivity, S.; Lidar, M.; Vaknin-Dembinsky, A.; Karussis, D.; Flechter, S.; Ben-Chetrit, E.; Livneh, A. Familial Mediterranean Fever (FMF) and Multiple Sclerosis: An Association Study in One of the World’s Largest FMF Cohorts. Eur. J. Neurol. 2011, 18, 1146–1150. [Google Scholar] [CrossRef] [PubMed]
- Akman-Demir, G.; Gul, A.; Gurol, E.; Ozdogan, H.; Bahar, S.; Oge, A.E.; Gurvit, H.; Saruhan-Direskeneli, G.; Yazici, H.; Eraksoy, M. Inflammatory/Demyelinating Central Nervous System Involvement in Familial Mediterranean Fever (FMF): Coincidence or Association? J. Neurol. 2006, 253, 928–934. [Google Scholar] [CrossRef] [PubMed]
- Balcı-Peynircioğlu, B.; Kaya-Akça, Ü.; Arıcı, Z.S.; Avcı, E.; Akkaya-Ulum, Z.Y.; Karadağ, Ö.; Kalyoncu, U.; Bilginer, Y.; Yılmaz, E.; Özen, S. Comorbidities in Familial Mediterranean Fever: Analysis of 2000 Genetically Confirmed Patients. Rheumatology 2020, 59, 1372–1380. [Google Scholar] [CrossRef]
- Hodak, E.; Atzmony, L.; Pavlovsky, L.; Comaneshter, D.; Cohen, A.D. Hidradenitis Suppurativa Is Associated with Familial Mediterranean Fever-A Population-Based Study. J. Investig. Dermatol. 2017, 137, 2019–2021. [Google Scholar] [CrossRef]
- Livneh, A.; Langevitz, P.; Zemer, D.; Zaks, N.; Kees, S.; Lidar, T.; Migdal, A.; Padeh, S.; Pras, M. Criteria for the Diagnosis of Familial Mediterranean Fever. Arthritis Rheum. 1997, 40, 1879–1885. [Google Scholar] [CrossRef]
- Yalçinkaya, F.; Ozen, S.; Ozçakar, Z.B.; Aktay, N.; Cakar, N.; Düzova, A.; Kasapçopur, O.; Elhan, A.H.; Doganay, B.; Ekim, M.; et al. A New Set of Criteria for the Diagnosis of Familial Mediterranean Fever in Childhood. Rheumatology 2009, 48, 395–398. [Google Scholar] [CrossRef]
- Demirkaya, E.; Saglam, C.; Turker, T.; Koné-Paut, I.; Woo, P.; Doglio, M.; Amaryan, G.; Frenkel, J.; Uziel, Y.; Insalaco, A.; et al. Performance of Different Diagnostic Criteria for Familial Mediterranean Fever in Children with Periodic Fevers: Results from a Multicenter International Registry. J. Rheumatol. 2016, 43, 154–160. [Google Scholar] [CrossRef]
- Berkun, Y.; Eisenstein, E.M. Diagnostic Criteria of Familial Mediterranean Fever. Autoimmun. Rev. 2014, 13, 388–390. [Google Scholar] [CrossRef] [PubMed]
- Giancane, G.; Ter Haar, N.M.; Wulffraat, N.; Vastert, S.J.; Barron, K.; Hentgen, V.; Kallinich, T.; Ozdogan, H.; Anton, J.; Brogan, P.; et al. Evidence-Based Recommendations for Genetic Diagnosis of Familial Mediterranean Fever. Ann. Rheum. Dis. 2015, 74, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Gattorno, M.; Hofer, M.; Federici, S.; Vanoni, F.; Bovis, F.; Aksentijevich, I.; Anton, J.; Arostegui, J.I.; Barron, K.; Ben-Cherit, E.; et al. Classification Criteria for Autoinflammatory Recurrent Fevers. Ann. Rheum. Dis. 2019, 78, 1025–1032. [Google Scholar] [CrossRef] [PubMed]
- Shinar, Y.; Obici, L.; Aksentijevich, I.; Bennetts, B.; Austrup, F.; Ceccherini, I.; Costa, J.M.; De Leener, A.; Gattorno, M.; Kania, U.; et al. Guidelines for the Genetic Diagnosis of Hereditary Recurrent Fevers. Ann. Rheum. Dis. 2012, 71, 1599–1605. [Google Scholar] [CrossRef]
- Federici, L.; Rittore-Domingo, C.; Koné-Paut, I.; Jorgensen, C.; Rodière, M.; Le Quellec, A.; Touitou, I. A Decision Tree for Genetic Diagnosis of Hereditary Periodic Fever in Unselected Patients. Ann. Rheum. Dis. 2006, 65, 1427–1432. [Google Scholar] [CrossRef]
- Touitou, I.; Lesage, S.; McDermott, M.; Cuisset, L.; Hoffman, H.; Dode, C.; Shoham, N.; Aganna, E.; Hugot, J.-P.; Wise, C.; et al. Infevers: An Evolving Mutation Database for Auto-Inflammatory Syndromes. Hum. Mutat. 2004, 24, 194–198. [Google Scholar] [CrossRef]
- Touitou, I. New Genetic Interpretation of Old Diseases. Autoimmun. Rev. 2012, 12, 5–9. [Google Scholar] [CrossRef]
- Liantinioti, G.; Argyris, A.A.; Protogerou, A.D.; Vlachoyiannopoulos, P. The Role of Colchicine in the Treatment of Autoinflammatory Diseases. Curr. Pharm. Des. 2018, 24, 690–694. [Google Scholar] [CrossRef]
- Taylor, E.W. The mechanism of colchicine inhibition of mitosis. I. kinetics of inhibition and the binding of h3-colchicine. J. Cell Biol. 1965, 25, 145–160. [Google Scholar] [CrossRef]
- Dalbeth, N.; Lauterio, T.J.; Wolfe, H.R. Mechanism of Action of Colchicine in the Treatment of Gout. Clin. Ther. 2014, 36, 1465–1479. [Google Scholar] [CrossRef]
- Dasgeb, B.; Kornreich, D.; McGuinn, K.; Okon, L.; Brownell, I.; Sackett, D.L. Colchicine: An Ancient Drug with Novel Applications. Br. J. Dermatol. 2018, 178, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Cronstein, B.N.; Molad, Y.; Reibman, J.; Balakhane, E.; Levin, R.I.; Weissmann, G. Colchicine Alters the Quantitative and Qualitative Display of Selectins on Endothelial Cells and Neutrophils. J. Clin. Investig. 1995, 96, 994–1002. [Google Scholar] [CrossRef] [PubMed]
- Chia, E.W.; Grainger, R.; Harper, J.L. Colchicine Suppresses Neutrophil Superoxide Production in a Murine Model of Gouty Arthritis: A Rationale for Use of Low-Dose Colchicine. Br. J. Pharmacol. 2008, 153, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Oka, T.; Hori, M.; Ozaki, H. Microtubule Disruption Suppresses Allergic Response through the Inhibition of Calcium Influx in the Mast Cell Degranulation Pathway. J. Immunol. 2005, 174, 4584–4589. [Google Scholar] [CrossRef]
- Ding, A.H.; Porteu, F.; Sanchez, E.; Nathan, C.F. Downregulation of Tumor Necrosis Factor Receptors on Macrophages and Endothelial Cells by Microtubule Depolymerizing Agents. J. Exp. Med. 1990, 171, 715–727. [Google Scholar] [CrossRef]
- Jackman, R.W.; Rhoads, M.G.; Cornwell, E.; Kandarian, S.C. Microtubule-Mediated NF-KappaB Activation in the TNF-Alpha Signaling Pathway. Exp. Cell Res. 2009, 315, 3242–3249. [Google Scholar] [CrossRef]
- Martinon, F.; Pétrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-Associated Uric Acid Crystals Activate the NALP3 Inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef]
- Slobodnick, A.; Shah, B.; Krasnokutsky, S.; Pillinger, M.H. Update on Colchicine, 2017. Rheumatology 2018, 57, i4–i11. [Google Scholar] [CrossRef]
- Niel, E.; Scherrmann, J.-M. Colchicine Today. Joint Bone Spine 2006, 73, 672–678. [Google Scholar] [CrossRef]
- Slobodnick, A.; Shah, B.; Pillinger, M.H.; Krasnokutsky, S. Colchicine: Old and New. Am. J. Med. 2015, 128, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Kuncl, R.W.; Duncan, G. Chronic Human Colchicine Myopathy and Neuropathy. Arch. Neurol. 1988, 45, 245–246. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, Y.; Aks, S.E.; Hutson, J.R.; Juurlink, D.N.; Nguyen, P.; Dubnov-Raz, G.; Pollak, U.; Koren, G.; Bentur, Y. Colchicine Poisoning: The Dark Side of an Ancient Drug. Clin. Toxicol. 2010, 48, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Putterman, C.; Ben-Chetrit, E.; Caraco, Y.; Levy, M. Colchicine Intoxication: Clinical Pharmacology, Risk Factors, Features, and Management. Semin. Arthritis Rheum. 1991, 21, 143–155. [Google Scholar] [CrossRef]
- Fradkin, A.; Yahav, J.; Zemer, D.; Jonas, A. Colchicine-Induced Lactose Malabsorption in Patients with Familial Mediterranean Fever. Isr. J. Med. Sci. 1995, 31, 616–620. [Google Scholar]
- Sarica, K.; Süzer, O.; Gürler, A.; Baltaci, S.; Ozdiler, E.; Dinçel, C. Urological Evaluation of Behçet Patients and the Effect of Colchicine on Fertility. Eur. Urol. 1995, 27, 39–42. [Google Scholar] [CrossRef]
- Ben-Chetrit, E.; Backenroth, R.; Haimov-Kochman, R.; Pizov, G. Azoospermia in Familial Mediterranean Fever Patients:The Role of Colchicine and Amyloidosis. Ann. Rheum. Dis. 1998, 57, 259–260. [Google Scholar] [CrossRef]
- Bremner, W.J.; Paulsen, C.A. Colchicine and Testicular Function in Man. N. Engl. J. Med. 1976, 194, 1384–1385. [Google Scholar] [CrossRef]
- Ben-Chetrit, A.; Ben-Chetrit, E.; Nitzan, R.; Ron, M. Colchicine Inhibits Spermatozoal Motility in Vitro. Int. J. Fertil. Menopausal. Stud. 1993, 38, 301–304. [Google Scholar]
- Diav-Citrin, O.; Shechtman, S.; Schwartz, V.; Avgil-Tsadok, M.; Finkel-Pekarsky, V.; Wajnberg, R.; Arnon, J.; Berkovitch, M.; Ornoy, A. Pregnancy Outcome after in Utero Exposure to Colchicine. Am. J. Obstet. Gynecol. 2010, 203, 144.e1–144.e6. [Google Scholar] [CrossRef]
- Ben-Chetrit, E.; Ben-Chetrit, A.; Berkun, Y.; Ben-Chetrit, E. Pregnancy Outcomes in Women with Familial Mediterranean Fever Receiving Colchicine: Is Amniocentesis Justified? Arthritis Care Res. 2010, 62, 143–148. [Google Scholar] [CrossRef]
- Dinarello, C.A.; Wolff, S.M.; Goldfinger, S.E.; Dale, D.C.; Alling, D.W. Colchicine Therapy for Familial Mediterranean Fever. A Double-Blind Trial. N. Engl. J. Med. 1974, 291, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Ozen, S.; Demirkaya, E.; Erer, B.; Livneh, A.; Ben-Chetrit, E.; Giancane, G.; Ozdogan, H.; Abu, I.; Gattorno, M.; Hawkins, P.N.; et al. EULAR Recommendations for the Management of Familial Mediterranean Fever. Ann. Rheum. Dis. 2016, 75, 644–651. [Google Scholar] [CrossRef] [PubMed]
- Ben-Chetrit, E.; Ozdogan, H. Non-Response to Colchicine in FMF--Definition, Causes and Suggested Solutions. Clin. Exp. Rheumatol. 2008, 26, S49–S51. [Google Scholar] [PubMed]
- Ozkaya, N.; Yalçinkaya, F. Colchicine Treatment in Children with Familial Mediterranean Fever. Clin. Rheumatol. 2003, 22, 314–317. [Google Scholar] [CrossRef]
- Knieper, A.-M.; Klotsche, J.; Lainka, E.; Berger, T.; Dressler, F.; Jansson, A.F.; Rietschel, C.; Oommen, P.T.; Berendes, R.; Niehues, T.; et al. Familial Mediterranean Fever in Children and Adolescents: Factors for Colchicine Dosage and Predicting Parameters for Dose Increase. Rheumatology 2017, 56, 1597–1606. [Google Scholar] [CrossRef]
- Özen, S.; Sag, E.; Ben-Chetrit, E.; Gattorno, M.; Gül, A.; Hashkes, P.J.; Kone-Paut, I.; Lachmann, H.J.; Tsitsami, E.; Twilt, M.; et al. Defining Colchicine Resistance/Intolerance in Patients with Familial Mediterranean Fever: A Modified-Delphi Consensus Approach. Rheumatology 2021, 60, 3799–3808. [Google Scholar] [CrossRef]
- Bustaffa, M.; Mazza, F.; Sutera, D.; Carrabba, M.D.; Alessio, M.; Cantarini, L.; Obici, L.; Rigante, D.; Maggio, M.C.; Insalaco, A.; et al. Persistence of Disease Flares Is Associated with an Inadequate Colchicine Dose in Familial Mediterranean Fever: A National Multicenter Longitudinal Study. J. Allergy Clin. Immunol. Pract. 2021, 9, 3218–3220. [Google Scholar] [CrossRef]
- Alpay, N.; Sumnu, A.; Calışkan, Y.; Yazıcı, H.; Türkmen, A.; Gül, A. Efficacy of Anakinra Treatment in a Patient with Colchicine-Resistant Familial Mediterranean Fever. Rheumatol. Int. 2012, 32, 3277–3279. [Google Scholar] [CrossRef]
- Dhimolea, E. Canakinumab. MAbs 2010, 2, 3–13. [Google Scholar] [CrossRef]
- van der Hilst, J.C.; Moutschen, M.; Messiaen, P.E.; Lauwerys, B.R.; Vanderschueren, S. Efficacy of Anti-IL-1 Treatment in Familial Mediterranean Fever: A Systematic Review of the Literature. Biologics 2016, 10, 75–80. [Google Scholar] [CrossRef] [PubMed]
- ter Haar, N.; Lachmann, H.; Özen, S.; Woo, P.; Uziel, Y.; Modesto, C.; Koné-Paut, I.; Cantarini, L.; Insalaco, A.; Neven, B.; et al. Treatment of Autoinflammatory Diseases: Results from the Eurofever Registry and a Literature Review. Ann. Rheum. Dis. 2013, 72, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Ben-Zvi, I.; Kukuy, O.; Giat, E.; Pras, E.; Feld, O.; Kivity, S.; Perski, O.; Bornstein, G.; Grossman, C.; Harari, G.; et al. Anakinra for Colchicine-Resistant Familial Mediterranean Fever: A Randomized, Double-Blind, Placebo-Controlled Trial. Arthritis Rheumatol. 2017, 69, 854–862. [Google Scholar] [CrossRef] [PubMed]
- Sevillano, Á.M.; Hernandez, E.; Gonzalez, E.; Mateo, I.; Gutierrez, E.; Morales, E.; Praga, M. Anakinra Induce La Remisión Completa Del Síndrome Nefrótico En Un Paciente Con Fiebre Mediterránea Familiar y Amiloidosis. Nefrología 2016, 36, 63–66. [Google Scholar] [CrossRef] [PubMed]
- İlgen, U.; Küçükşahin, O. Anakinra Use during Pregnancy: Report of a Case with Familial Mediterranean Fever and Infertility. Eur. J. Rheumatol. 2017, 4, 66–67. [Google Scholar] [CrossRef]
- Venhoff, N.; Voll, R.E.; Glaser, C.; Thiel, J. IL-1-Blockade mit Anakinra in der Schwangerschaft. Z. Rheumatol. 2018, 77, 127–134. [Google Scholar] [CrossRef]
- Ugurlu, S.; Ergezen, B.; Egeli, B.H.; Selvi, O.; Ozdogan, H. Anakinra Treatment in Patients with Familial Mediterranean Fever: A Single-Centre Experience. Rheumatology 2021, 60, 2327–2332. [Google Scholar] [CrossRef]
- Ozdogan, H.; Ugurlu, S. Familial Mediterranean Fever. Presse Med. 2019, 48, e61–e76. [Google Scholar] [CrossRef]
- Cavalli, G.; Dinarello, C.A. Anakinra Therapy for Non-Cancer Inflammatory Diseases. Front. Pharmacol. 2018, 9, 1157. [Google Scholar] [CrossRef]
- Economides, A.N.; Carpenter, L.R.; Rudge, J.S.; Wong, V.; Koehler-Stec, E.M.; Hartnett, C.; Pyles, E.A.; Xu, X.; Daly, T.J.; Young, M.R.; et al. Cytokine Traps: Multi-Component, High-Affinity Blockers of Cytokine Action. Nat. Med. 2003, 9, 47–52. [Google Scholar] [CrossRef]
- Hashkes, P.J.; Spalding, S.J.; Giannini, E.H.; Huang, B.; Johnson, A.; Park, G.; Barron, K.S.; Weisman, M.H.; Pashinian, N.; Reiff, A.O.; et al. Rilonacept for Colchicine-Resistant or -Intolerant Familial Mediterranean Fever: A Randomized Trial. Ann. Intern. Med. 2012, 157, 533–541. [Google Scholar] [CrossRef]
- El Hasbani, G.; Jawad, A.; Uthman, I. Update on the Management of Colchicine Resistant Familial Mediterranean Fever (FMF). Orphanet J. Rare Dis. 2019, 14, 224. [Google Scholar] [CrossRef] [PubMed]
- Mitroulis, I.; Skendros, P.; Oikonomou, A.; Tzioufas, A.G.; Ritis, K. The Efficacy of Canakinumab in the Treatment of a Patient with Familial Mediterranean Fever and Longstanding Destructive Arthritis. Ann. Rheum. Dis. 2011, 70, 1347–1348. [Google Scholar] [CrossRef] [PubMed]
- Cetin, P.; Sari, I.; Sozeri, B.; Cam, O.; Birlik, M.; Akkoc, N.; Onen, F.; Akar, S. Efficacy of Interleukin-1 Targeting Treatments in Patients with Familial Mediterranean Fever. Inflammation 2015, 38, 27–31. [Google Scholar] [CrossRef]
- Başaran, Ö.; Uncu, N.; Çelikel, B.A.; Taktak, A.; Gür, G.; Cakar, N. Interleukin-1 Targeting Treatment in Familial Mediterranean Fever: An Experience of Pediatric Patients. Mod. Rheumatol. 2015, 25, 621–624. [Google Scholar] [CrossRef] [PubMed]
- Eroglu, F.K.; Beşbaş, N.; Topaloglu, R.; Ozen, S. Treatment of Colchicine-Resistant Familial Mediterranean Fever in Children and Adolescents. Rheumatol. Int. 2015, 35, 1733–1737. [Google Scholar] [CrossRef]
- Köhler, B.M.; Lorenz, H.-M.; Blank, N. IL1-Blocking Therapy in Colchicine-Resistant Familial Mediterranean Fever. Eur. J. Rheumatol. 2018, 5, 230–234. [Google Scholar] [CrossRef]
- De Benedetti, F.; Gattorno, M.; Anton, J.; Ben-Chetrit, E.; Frenkel, J.; Hoffman, H.M.; Koné-Paut, I.; Lachmann, H.J.; Ozen, S.; Simon, A.; et al. Canakinumab for the Treatment of Autoinflammatory Recurrent Fever Syndromes. N. Engl. J. Med. 2018, 378, 1908–1919. [Google Scholar] [CrossRef]
- Ozen, S.; Ben-Cherit, E.; Foeldvari, I.; Amarilyo, G.; Ozdogan, H.; Vanderschueren, S.; Marzan, K.; Kahlenberg, J.M.; Dekker, E.; De Benedetti, F.; et al. Long-Term Efficacy and Safety of Canakinumab in Patients with Colchicine-Resistant Familial Mediterranean Fever: Results from the Randomised Phase III CLUSTER Trial. Ann. Rheum. Dis. 2020, 79, 1362–1369. [Google Scholar] [CrossRef]
- Koga, T.; Migita, K.; Sato, S.; Umeda, M.; Nonaka, F.; Kawashiri, S.-Y.; Iwamoto, N.; Ichinose, K.; Tamai, M.; Nakamura, H.; et al. Multiple Serum Cytokine Profiling to Identify Combinational Diagnostic Biomarkers in Attacks of Familial Mediterranean Fever. Medicine 2016, 95, e3449. [Google Scholar] [CrossRef]
- Ugurlu, S.; Hacioglu, A.; Adibnia, Y.; Hamuryudan, V.; Ozdogan, H. Tocilizumab in the Treatment of Twelve Cases with Aa Amyloidosis Secondary to Familial Mediterranean Fever. Orphanet J. Rare Dis. 2017, 12, 105. [Google Scholar] [CrossRef] [PubMed]
- Koga, T.; Hagimori, N.; Sato, S.; Morimoto, S.; Hosogaya, N.; Fukushima, C.; Yamamoto, H.; Kawakami, A. An Open-Label Continuation Trial of Tocilizumab for Familial Mediterranean Fever with Colchicine Ineffective or Intolerance: Study Protocol for Investigator-Initiated, Multicenter, Open-Label Trial. Medicine 2020, 99, e18328. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tel Hashomer criteria (at least two major criteria or one major criterion plus two minor criteria) |
| Major criteria |
|
| Minor criteria |
|
| Livneh criteria (at least one major criterion, or two minor criteria, or one minor criterion plus at least five supportive criteria, or one minor criterion plus at least four of the “first” five supportive criteria) |
| Major criteria |
|
| Minor criteria |
|
| Supportive criteria |
|
| Yalcinkaya-Ozen (childhood) criteria (at least two out of five criteria) |
|
| Eurofever/PRINTO Classification Criteria for FMF |
|---|
| Presence of confirmatory MEFV genotype * and at least one, or not confirmatory MEFV genotype ** and at least two, of the following: |
|
| Delphi Expert Consensus Statements (Özen et al., 2021 [124]) | Results of a National Multicentre Longitudinal Study (Bustaffa et al., 2021 [125]) | |
|---|---|---|
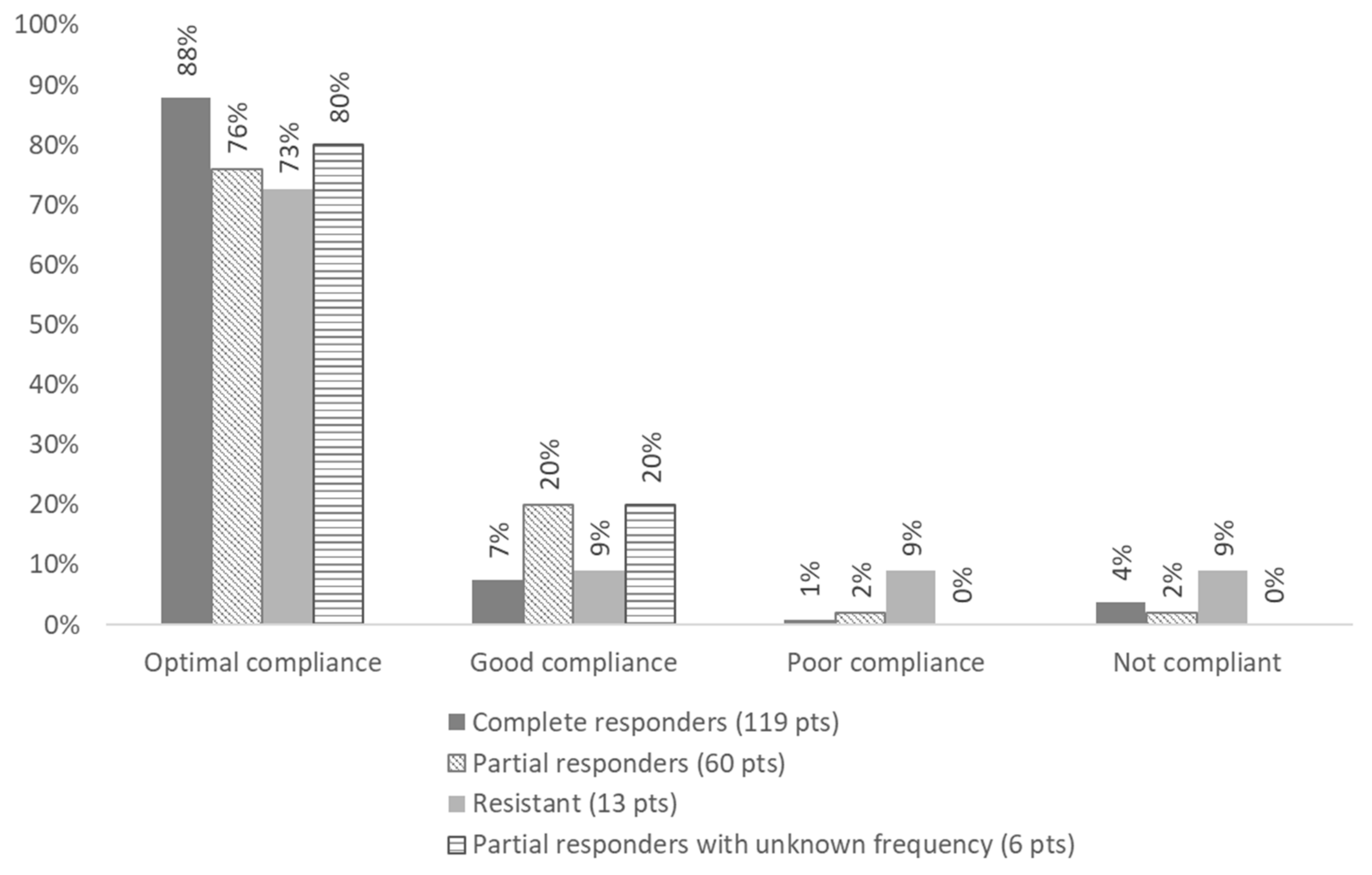
| Adherence | Statement 1: Colchicine is the drug of choice for the treatment of FMF, and adherence is a critical issue. For the following statements, it is assumed that the patient is adherent to their prescribed colchicine treatment. | Overall, 83.8% displayed optimal adherence (>90% of prescription); 10.6% displayed good adherence (50–89% of prescriptions); 2.0% displayed poor adherence (<50% of prescriptions); and 3.5% displayed no adherence. |
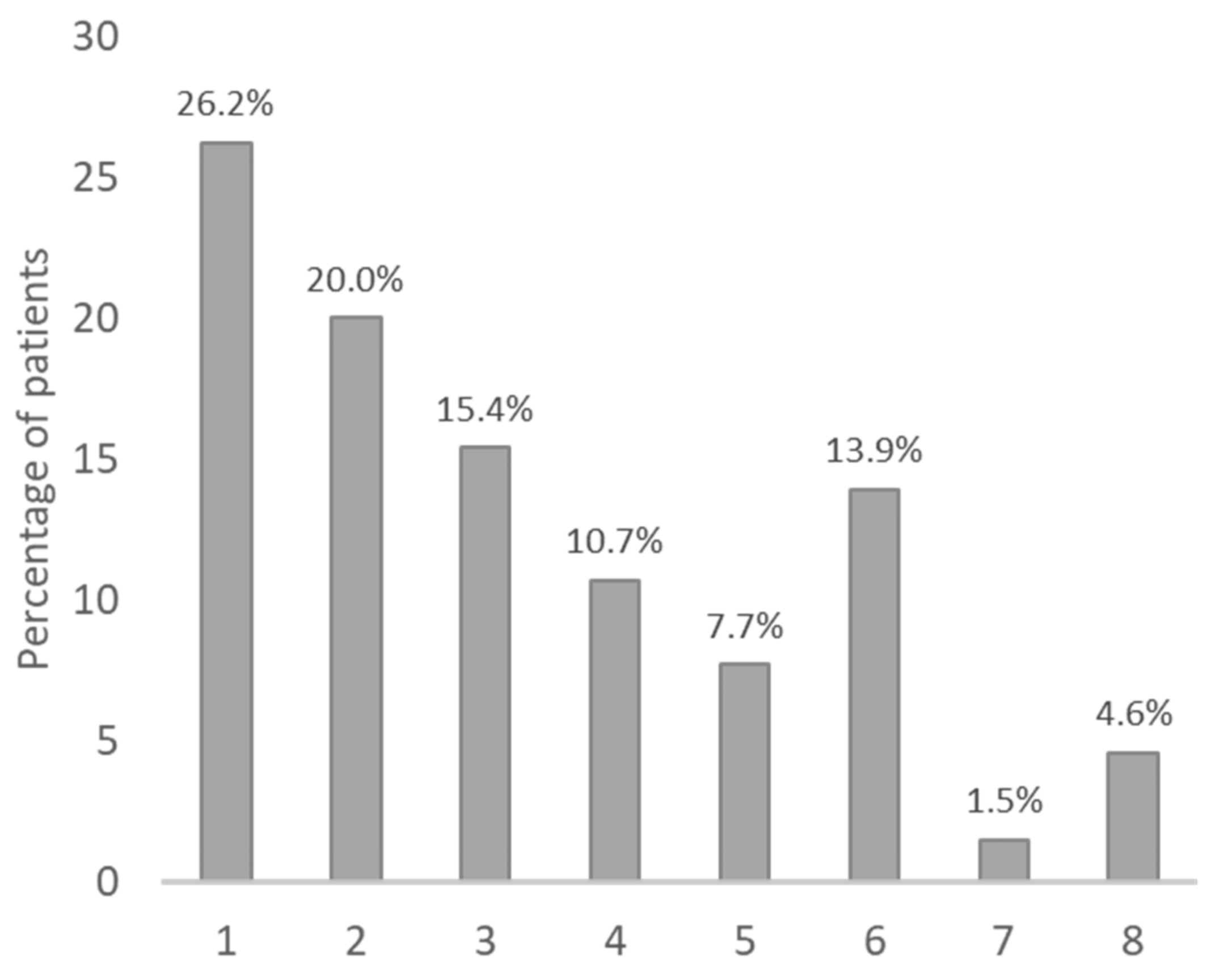
| Dose adjustment criteria | Statement 2: When utilising colchicine to treat FMF, it is recommended to adjust the dose based on disease activity, with the adjustment of maximal dose for children depending on age (and weight). | The percentage of patients taking a colchicine dose without adjustments were:
|
| Recommended maximum colchicine dose | Statement 3: The maximum recommended colchicine dose for the treatment of FMF is 1–3 mg per day, depending on age and weight, limited by signs of toxicity and tolerability (see below). | No patient reached the maximum recommended dose. |
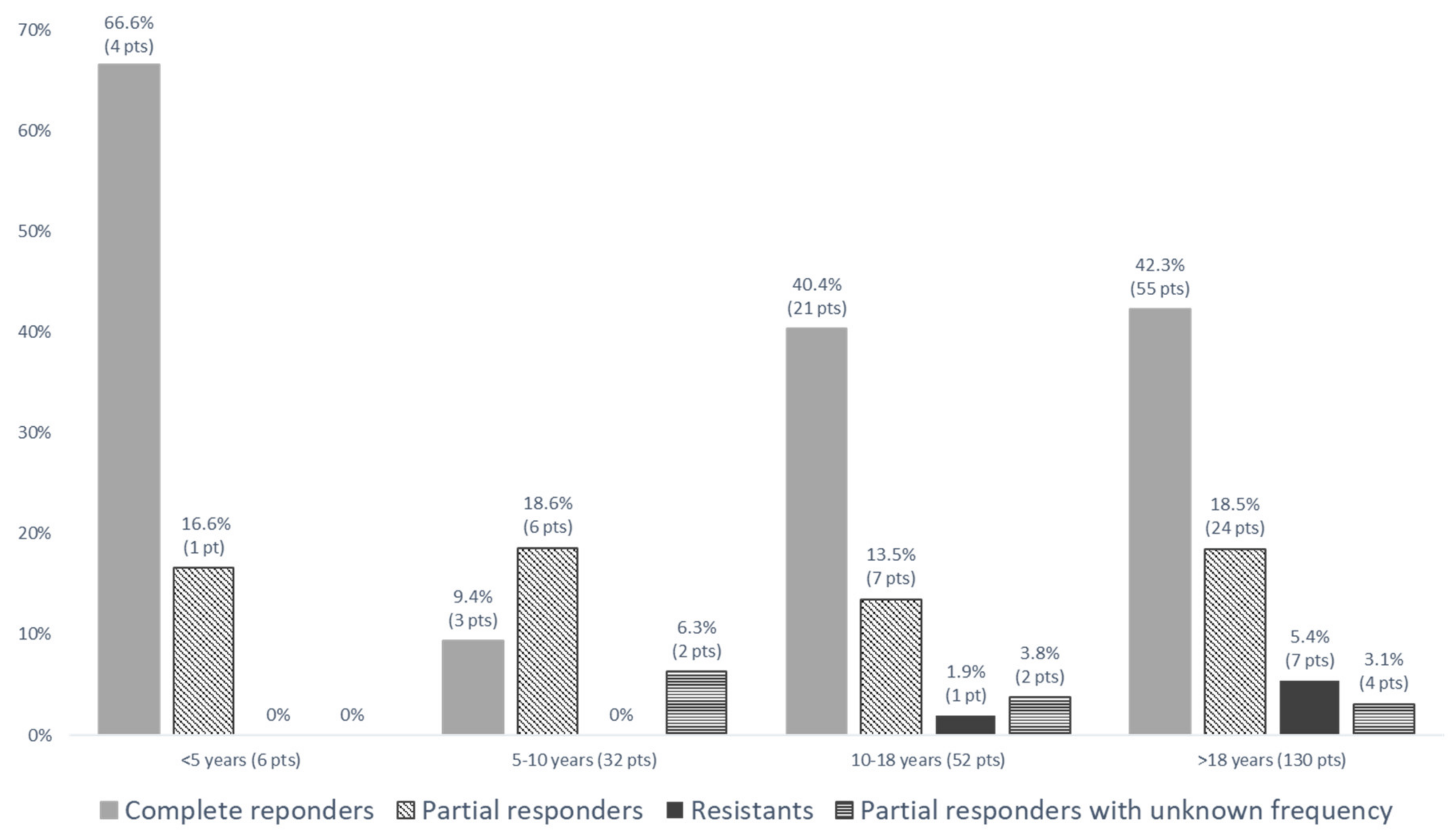
| Resistance to colchicine | Statement 4: For a patient receiving the maximum tolerated dose of colchicine, resistance to colchicine is defined as ongoing disease activity (as reflected by either recurrent clinical attacks (average one or more attacks per month over a 3-month period), or persistently elevated CRP or SAA in between attacks (depending on which is available locally)) in the absence of any other plausible explanation. | Resistance was be defined as the persistence of fever attacks, despite optimal treatment. Overall, 54.2% patients had a complete disease control; 30.1% patients had < one episode/month for 3 months; 8.5% had ≥ one episode/month for 3 month; and 7.2% had persisting disease with an unknown frequency of attacks. |
| Inclusion of secondary amyloidosis in the definition of colchicine resistance | Statement 5: AA amyloidosis develops as a consequence of persistent inflammation, which may be a complication of colchicine resistance. | Five adult patients (2.1%) displayed amyloidosis, two of which were prescribed anti-Il1 treatment. |
| Colchicine intolerance | Statement 6: Colchicine intolerance, which generally manifests as mild gastrointestinal symptoms (such as diarrhoea and nausea), is common but can limit the ability to achieve or maintain the effective dose. Dose-limiting toxicity is rare and may include serious gastrointestinal manifestations, such as persistent diarrhoea, elevated liver enzymes, leukopenia, azoospermia, neuromyopathy, etc. | Eight patients (3.4%) with follow-up had persistent manifestations of intolerance to colchicine. No patient experienced real toxicity. |
| Patient quality of life and self-reported outcome | Statement 7: Active disease and intolerance to colchicine affect quality of life. | Overall, 20.1% of patients experienced fatigue or chronic pain, 26.6% experienced limitations in daily activities, and 19.6% lost school/work days. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lancieri, M.; Bustaffa, M.; Palmeri, S.; Prigione, I.; Penco, F.; Papa, R.; Volpi, S.; Caorsi, R.; Gattorno, M. An Update on Familial Mediterranean Fever. Int. J. Mol. Sci. 2023, 24, 9584. https://doi.org/10.3390/ijms24119584
Lancieri M, Bustaffa M, Palmeri S, Prigione I, Penco F, Papa R, Volpi S, Caorsi R, Gattorno M. An Update on Familial Mediterranean Fever. International Journal of Molecular Sciences. 2023; 24(11):9584. https://doi.org/10.3390/ijms24119584
Chicago/Turabian StyleLancieri, Maddalena, Marta Bustaffa, Serena Palmeri, Ignazia Prigione, Federica Penco, Riccardo Papa, Stefano Volpi, Roberta Caorsi, and Marco Gattorno. 2023. "An Update on Familial Mediterranean Fever" International Journal of Molecular Sciences 24, no. 11: 9584. https://doi.org/10.3390/ijms24119584