
Whole-Exome Sequencing in a Family with an Unexplained Tendency for Venous Thromboembolism: Multicomponent Prediction of Low-Frequency Variant Deleteriousness and of Individual Protein Interaction
 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Results
2.1. WES Analysis
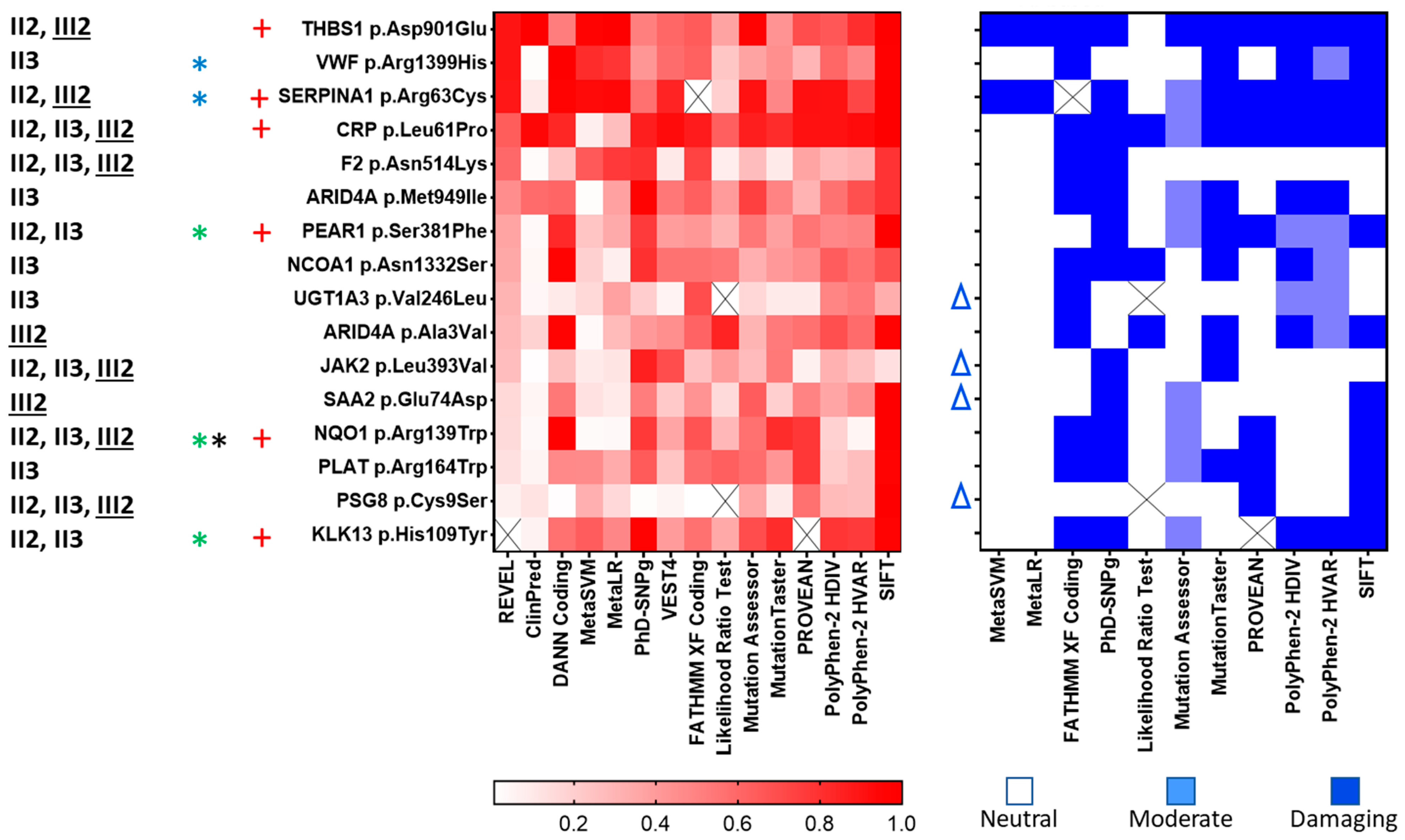
2.2. Observed/Predicted Functional Impact of Variants
2.3. Predicted Impact of Variants on Protein Structure and Function
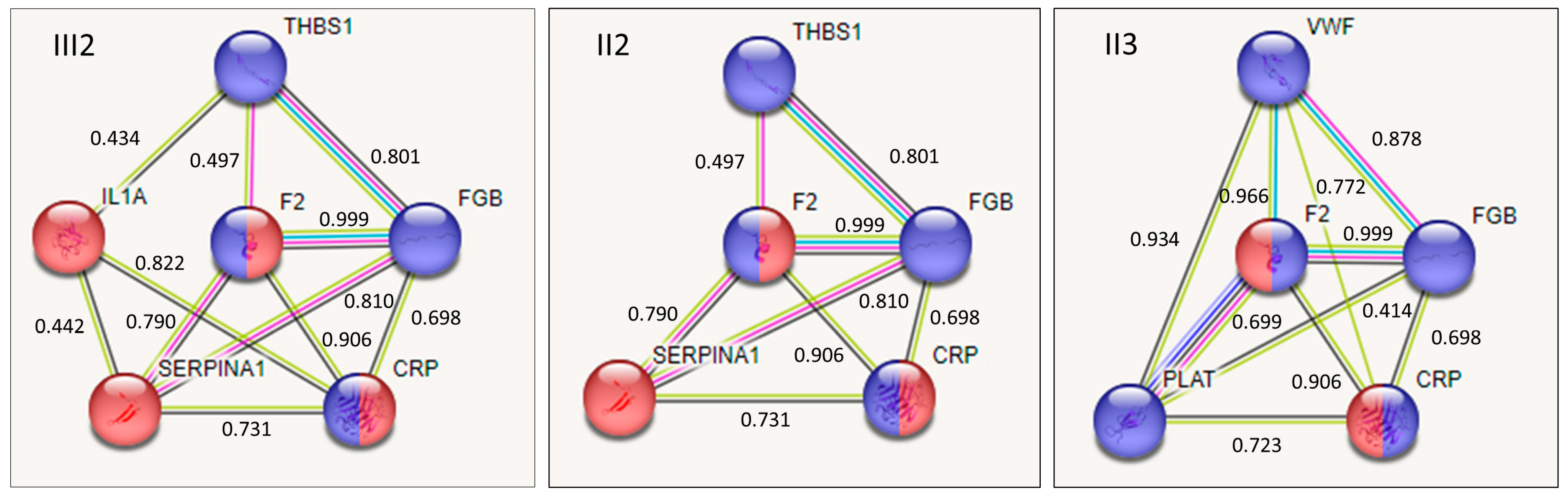
2.4. Interaction among Proteins Containing Variants in the Affected Family Members
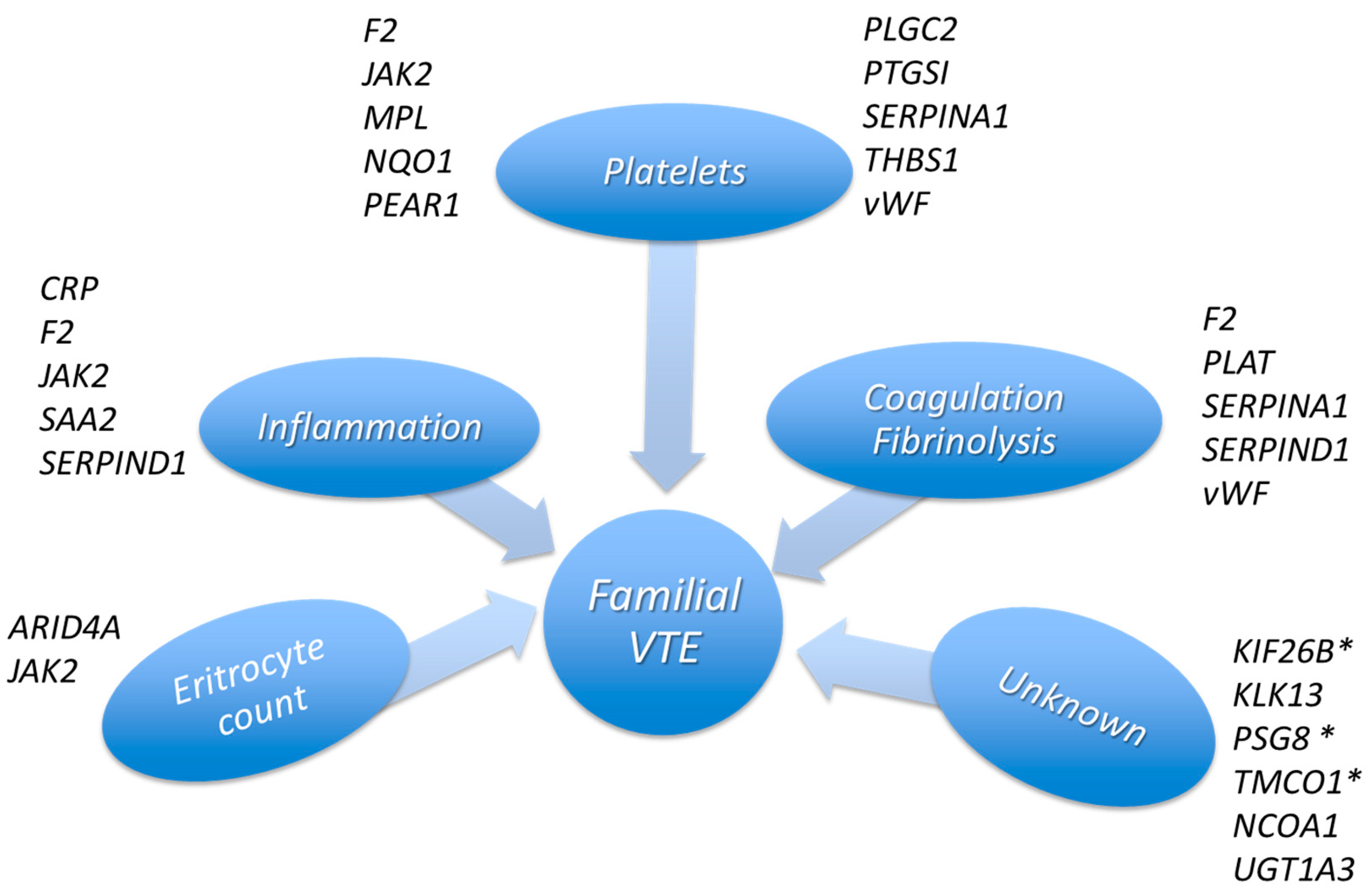
3. Discussion
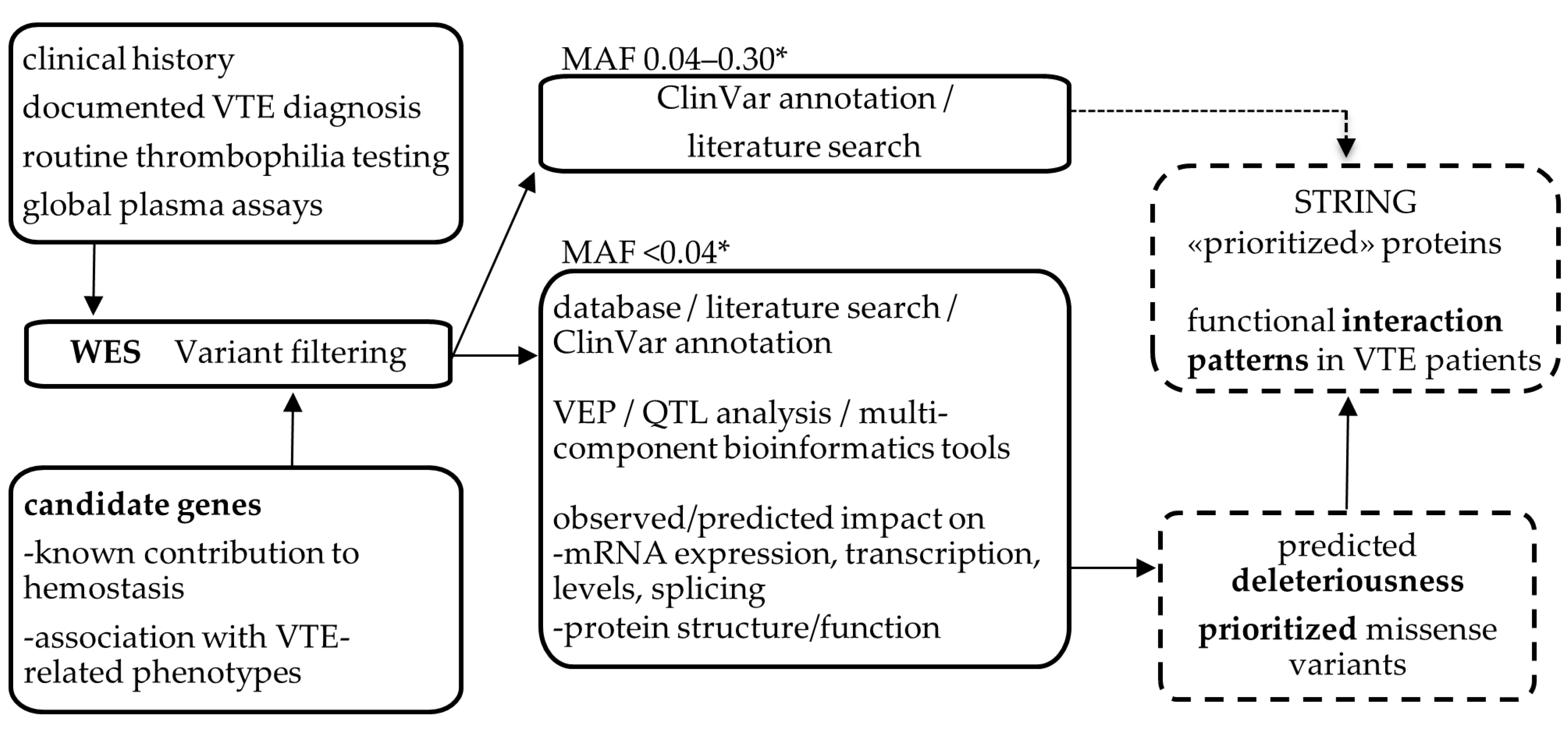
4. Materials and Methods
4.1. Clinical History of the Family and Laboratory Assays
4.2. Global Functional Assays
4.3. Selection of Candidates Genes
4.4. Whole-Exome Sequencing and Analysis
4.5. Analysis of the Functional Impact of Variants
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rosendaal, F.R. Venous Thrombosis: A Multicausal Disease. Lancet 1999, 353, 1167–1173. [Google Scholar] [CrossRef] [PubMed]
- Pastori, D.; Cormaci, V.M.; Marucci, S.; Franchino, G.; Del Sole, F.; Capozza, A.; Fallarino, A.; Corso, C.; Valeriani, E.; Menichelli, D.; et al. A Comprehensive Review of Risk Factors for Venous Thromboembolism: From Epidemiology to Pathophysiology. Int. J. Mol. Sci. 2023, 24, 3169. [Google Scholar] [CrossRef] [PubMed]
- Zöller, B.; García de Frutos, P.; Hillarp, A.; Dahlbäck, B. Thrombophilia as a Multigenic Disease. Haematologica 1999, 84, 59–70. [Google Scholar] [PubMed]
- Souto, J.C.; Almasy, L.; Borrell, M.; Blanco-Vaca, F.; Mateo, J.; Soria, J.M.; Coll, I.; Felices, R.; Stone, W.; Fontcuberta, J.; et al. Genetic Susceptibility to Thrombosis and Its Relationship to Physiological Risk Factors: The GAIT Study. Genetic Analysis of Idiopathic Thrombophilia. Am. J. Hum. Genet. 2000, 67, 1452–1459. [Google Scholar] [CrossRef]
- Seligsohn, U.; Lubetsky, A. Genetic Susceptibility to Venous Thrombosis. N. Engl. J. Med. 2001, 344, 1222–1231. [Google Scholar] [CrossRef]
- Egeberg, O. Inherited Antithrombin Deficiency Causing Thrombophilia. Thromb. Diath. Haemorrh. 1965, 13, 516–530. [Google Scholar] [CrossRef]
- Griffin, J.H.; Evatt, B.; Zimmerman, T.S.; Kleiss, A.J.; Wideman, C. Deficiency of Protein C in Congenital Thrombotic Disease. J. Clin. Invest. 1981, 68, 1370–1373. [Google Scholar] [CrossRef]
- Dahlbäck, B.; Carlsson, M.; Svensson, P.J. Familial Thrombophilia Due to a Previously Unrecognized Mechanism Characterized by Poor Anticoagulant Response to Activated Protein C: Prediction of a Cofactor to Activated Protein C. Proc. Natl. Acad. Sci. USA 1993, 90, 1004–1008. [Google Scholar] [CrossRef]
- Bertina, R.M.; Koeleman, B.P.; Koster, T.; Rosendaal, F.R.; Dirven, R.J.; de Ronde, H.; van der Velden, P.A.; Reitsma, P.H. Mutation in Blood Coagulation Factor V Associated with Resistance to Activated Protein C. Nature 1994, 369, 64–67. [Google Scholar] [CrossRef]
- Poort, S.R.; Rosendaal, F.R.; Reitsma, P.H.; Bertina, R.M. A Common Genetic Variation in the 3′-Untranslated Region of the Prothrombin Gene Is Associated with Elevated Plasma Prothrombin Levels and an Increase in Venous Thrombosis. Blood 1996, 88, 3698–3703. [Google Scholar] [CrossRef]
- Bernardi, F.; Faioni, E.M.; Castoldi, E.; Lunghi, B.; Castaman, G.; Sacchi, E.; Mannucci, P.M. A Factor V Genetic Component Differing from Factor V R506Q Contributes to the Activated Protein C Resistance Phenotype. Blood 1997, 90, 1552–1557. [Google Scholar] [CrossRef] [PubMed]
- Reitsma, P.H.; Rosendaal, F.R. Past and Future of Genetic Research in Thrombosis. J. Thromb. Haemost. 2007, 5 (Suppl. S1), 264–269. [Google Scholar] [CrossRef]
- De Stefano, V.; Martinelli, I.; Mannucci, P.M.; Paciaroni, K.; Chiusolo, P.; Casorelli, I.; Rossi, E.; Leone, G. The Risk of Recurrent Deep Venous Thrombosis among Heterozygous Carriers of Both Factor V Leiden and the G20210A Prothrombin Mutation. N. Engl. J. Med. 1999, 341, 801–806. [Google Scholar] [CrossRef] [PubMed]
- Castoldi, E.; Simioni, P.; Kalafatis, M.; Lunghi, B.; Tormene, D.; Girelli, D.; Girolami, A.; Bernardi, F. Combinations of 4 Mutations (FV R506Q, FV H1299R, FV Y1702C, PT 20210G/A) Affecting the Prothrombinase Complex in a Thrombophilic Family. Blood 2000, 96, 1443–1448. [Google Scholar] [CrossRef] [PubMed]
- Morange, P.-E.; Tregouet, D.-A. Deciphering the Molecular Basis of Venous Thromboembolism: Where Are We and Where Should We Go? Br. J. Haematol. 2010, 148, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Trégouët, D.-A.; Heath, S.; Saut, N.; Biron-Andreani, C.; Schved, J.-F.; Pernod, G.; Galan, P.; Drouet, L.; Zelenika, D.; Juhan-Vague, I.; et al. Common Susceptibility Alleles Are Unlikely to Contribute as Strongly as the FV and ABO Loci to VTE Risk: Results from a GWAS Approach. Blood 2009, 113, 5298–5303. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Basu, S.; Kong, X.; Pankow, J.S.; Aleksic, N.; Tan, A.; Cushman, M.; Boerwinkle, E.; Folsom, A.R. Genome-Wide Association Study Identifies Novel Loci for Plasma Levels of Protein C: The ARIC Study. Blood 2010, 116, 5032–5036. [Google Scholar] [CrossRef]
- Heit, J.A.; Armasu, S.M.; Asmann, Y.W.; Cunningham, J.M.; Matsumoto, M.E.; Petterson, T.M.; De Andrade, M. A Genome-Wide Association Study of Venous Thromboembolism Identifies Risk Variants in Chromosomes 1q24.2 and 9q. J. Thromb. Haemost. 2012, 10, 1521–1531. [Google Scholar] [CrossRef]
- Germain, M.; Chasman, D.I.; de Haan, H.; Tang, W.; Lindström, S.; Weng, L.-C.; de Andrade, M.; de Visser, M.C.H.; Wiggins, K.L.; Suchon, P.; et al. Meta-Analysis of 65,734 Individuals Identifies TSPAN15 and SLC44A2 as Two Susceptibility Loci for Venous Thromboembolism. Am. J. Hum. Genet. 2015, 96, 532–542. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.I.; Abecasis, G.R.; Cardon, L.R.; Goldstein, D.B.; Little, J.; Ioannidis, J.P.A.; Hirschhorn, J.N. Genome-Wide Association Studies for Complex Traits: Consensus, Uncertainty and Challenges. Nat. Rev. Genet. 2008, 9, 356–369. [Google Scholar] [CrossRef]
- Trégouët, D.-A.; Morange, P.-E. What Is Currently Known about the Genetics of Venous Thromboembolism at the Dawn of next Generation Sequencing Technologies. Br. J. Haematol. 2018, 180, 335–345. [Google Scholar] [CrossRef]
- Lotta, L.A.; Wang, M.; Yu, J.; Martinelli, I.; Yu, F.; Passamonti, S.M.; Consonni, D.; Pappalardo, E.; Menegatti, M.; Scherer, S.E.; et al. Identification of Genetic Risk Variants for Deep Vein Thrombosis by Multiplexed Next-Generation Sequencing of 186 Hemostatic/pro-Inflammatory Genes. BMC Med. Genom. 2012, 5, 7. [Google Scholar] [CrossRef]
- Lee, E.-J.; Dykas, D.J.; Leavitt, A.D.; Camire, R.M.; Ebberink, E.; García de Frutos, P.; Gnanasambandan, K.; Gu, S.X.; Huntington, J.A.; Lentz, S.R.; et al. Whole-Exome Sequencing in Evaluation of Patients with Venous Thromboembolism. Blood Adv. 2017, 1, 1224–1237. [Google Scholar] [CrossRef] [PubMed]
- Lindström, S.; Brody, J.A.; Turman, C.; Germain, M.; Bartz, T.M.; Smith, E.N.; Chen, M.-H.; Puurunen, M.; Chasman, D.; Hassler, J.; et al. A Large-Scale Exome Array Analysis of Venous Thromboembolism. Genet. Epidemiol. 2019, 43, 449–457. [Google Scholar] [CrossRef]
- Desch, K.C.; Ozel, A.B.; Halvorsen, M.; Jacobi, P.M.; Golden, K.; Underwood, M.; Germain, M.; Tregouet, D.-A.; Reitsma, P.H.; Kearon, C.; et al. Whole-Exome Sequencing Identifies Rare Variants in STAB2 Associated with Venous Thromboembolic Disease. Blood 2020, 136, 533–541. [Google Scholar] [CrossRef]
- Lindström, S.; Wang, L.; Smith, E.N.; Gordon, W.; van Hylckama Vlieg, A.; de Andrade, M.; Brody, J.A.; Pattee, J.W.; Haessler, J.; Brumpton, B.M.; et al. Genomic and Transcriptomic Association Studies Identify 16 Novel Susceptibility Loci for Venous Thromboembolism. Blood 2019, 134, 1645–1657. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Rivero, M.; Stoll, M.; Hegenbarth, J.-C.; Rühle, F.; Limperger, V.; Junker, R.; Franke, A.; Hoffmann, P.; Shneyder, M.; Stach, M.; et al. Single- and Multimarker Genome-Wide Scans Evidence Novel Genetic Risk Modifiers for Venous Thromboembolism. Thromb. Haemost. 2021, 121, 1169–1180. [Google Scholar] [CrossRef] [PubMed]
- Thibord, F.; Klarin, D.; Brody, J.A.; Chen, M.-H.; Levin, M.G.; Chasman, D.I.; Goode, E.L.; Hveem, K.; Teder-Laving, M.; Martinez-Perez, A.; et al. Cross-Ancestry Investigation of Venous Thromboembolism Genomic Predictors. Circulation 2022, 146, 1225–1242. [Google Scholar] [CrossRef] [PubMed]
- Zöller, B. Genetics of Venous Thromboembolism Revised. Blood 2019, 134, 1568–1570. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, G.; Margaglione, M. Rare Defects: Looking at the Dark Face of the Thrombosis. Int. J. Environ. Res. Public Health 2021, 18, 9146. [Google Scholar] [CrossRef] [PubMed]
- Mulder, R.; Lisman, T.; Meijers, J.C.M.; Huntington, J.A.; Mulder, A.B.; Meijer, K. Linkage Analysis Combined with Whole-Exome Sequencing Identifies a Novel Prothrombin (F2) Gene Mutation in a Dutch Caucasian Family with Unexplained Thrombosis. Haematologica 2020, 105, e370–e372. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.-A.; Sheu, C.-C.; Liu, K.-T.; Shen, J.-H.; Yen, M.-C.; Kuo, P.-L. Identification of Mutations in SLC4A1, GP1BA and HFE in a Family with Venous Thrombosis of Unknown Cause by next-Generation Sequencing. Exp. Ther. Med. 2018, 16, 4172–4180. [Google Scholar] [CrossRef] [PubMed]
- Morange, P.-E.; Peiretti, F.; Gourhant, L.; Proust, C.; Soukarieh, O.; Pulcrano-Nicolas, A.-S.; Saripella, G.-V.; Stefanucci, L.; Lacroix, R.; Ibrahim-Kosta, M.; et al. A Rare Coding Mutation in the MAST2 Gene Causes Venous Thrombosis in a French Family with Unexplained Thrombophilia: The Breizh MAST2 Arg89Gln Variant. PLoS Genet. 2021, 17, e1009284. [Google Scholar] [CrossRef]
- Cunha, M.L.R.; Meijers, J.C.M.; Rosendaal, F.R.; Vlieg, A.V.; Reitsma, P.H.; Middeldorp, S. Whole Exome Sequencing in Thrombophilic Pedigrees to Identify Genetic Risk Factors for Venous Thromboembolism. PLoS ONE 2017, 12, e0187699. [Google Scholar] [CrossRef]
- Ajjan, R.; Lim, B.C.B.; Standeven, K.F.; Harrand, R.; Dolling, S.; Phoenix, F.; Greaves, R.; Abou-Saleh, R.H.; Connell, S.; Smith, D.A.M.; et al. Common Variation in the C-Terminal Region of the Fibrinogen Beta-Chain: Effects on Fibrin Structure, Fibrinolysis and Clot Rigidity. Blood 2008, 111, 643–650. [Google Scholar] [CrossRef]
- Kotzé, R.C.; Nienaber-Rousseau, C.; De Lange, Z.; De Maat, M.P.; Hoekstra, T.; Pieters, M. Genetic Polymorphisms Influencing Total and γ’ Fibrinogen Levels and Fibrin Clot Properties in Africans. Br. J. Haematol. 2015, 168, 102–112. [Google Scholar] [CrossRef]
- Klovaite, J.; Nordestgaard, B.G.; Tybjærg-Hansen, A.; Benn, M. Elevated Fibrinogen Levels Are Associated with Risk of Pulmonary Embolism, but Not with Deep Venous Thrombosis. Am. J. Respir. Crit. Care Med. 2013, 187, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Heit, J.A.; Cunningham, J.M.; Petterson, T.M.; Armasu, S.M.; Rider, D.N.; DE Andrade, M. Genetic Variation within the Anticoagulant, Procoagulant, Fibrinolytic and Innate Immunity Pathways as Risk Factors for Venous Thromboembolism. J. Thromb. Haemost. 2011, 9, 1133–1142. [Google Scholar] [CrossRef]
- Turcot, V.; Lu, Y.; Highland, H.M.; Schurmann, C.; Justice, A.E.; Fine, R.S.; Bradfield, J.P.; Esko, T.; Giri, A.; Graff, M.; et al. Publisher Correction: Protein-Altering Variants Associated with Body Mass Index Implicate Pathways That Control Energy Intake and Expenditure in Obesity. Nat. Genet. 2018, 50, 766–767. [Google Scholar] [CrossRef]
- Rohmann, J.L.; de Haan, H.G.; Algra, A.; Vossen, C.Y.; Rosendaal, F.R.; Siegerink, B. Genetic Determinants of Activity and Antigen Levels of Contact System Factors. J. Thromb. Haemost. 2019, 17, 157–168. [Google Scholar] [CrossRef]
- Sabater-Lleal, M.; Martinez-Perez, A.; Buil, A.; Folkersen, L.; Souto, J.C.; Bruzelius, M.; Borrell, M.; Odeberg, J.; Silveira, A.; Eriksson, P.; et al. A Genome-Wide Association Study Identifies KNG1 as a Genetic Determinant of Plasma Factor XI Level and Activated Partial Thromboplastin Time. Arter. Thromb. Vasc. Biol. 2012, 32, 2008–2016. [Google Scholar] [CrossRef] [PubMed]
- Wiggins, K.A.; Pyrillou, K.; Humphry, M.; Butterworth, A.S.; Clarke, M.C. The Common IL1A Single Nucleotide Polymorphism Rs17561 Is a Hypomorphic Mutation That Significantly Reduces Interleukin-1α Release from Human Blood Cells. Immunology 2023, 168, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Lienhart, W.-D.; Strandback, E.; Gudipati, V.; Koch, K.; Binter, A.; Uhl, M.K.; Rantasa, D.M.; Bourgeois, B.; Madl, T.; Zangger, K.; et al. Catalytic Competence, Structure and Stability of the Cancer-Associated R139W Variant of the Human NAD(P)H:Quinone Oxidoreductase 1 (NQO1). FEBS J. 2017, 284, 1233–1245. [Google Scholar] [CrossRef] [PubMed]
- Ellinghaus, D.; Jostins, L.; Spain, S.L.; Cortes, A.; Bethune, J.; Han, B.; Park, Y.R.; Raychaudhuri, S.; Pouget, J.G.; Hübenthal, M.; et al. Analysis of Five Chronic Inflammatory Diseases Identifies 27 New Associations and Highlights Disease-Specific Patterns at Shared Loci. Nat. Genet. 2016, 48, 510–518. [Google Scholar] [CrossRef]
- van der Harst, P.; Verweij, N. Identification of 64 Novel Genetic Loci Provides an Expanded View on the Genetic Architecture of Coronary Artery Disease. Circ. Res. 2018, 122, 433–443. [Google Scholar] [CrossRef]
- Pagel, K.A.; Kim, R.; Moad, K.; Busby, B.; Zheng, L.; Tokheim, C.; Ryan, M.; Karchin, R. Integrated Informatics Analysis of Cancer-Related Variants. JCO Clin. Cancer Inform. 2020, 4, 310–317. [Google Scholar] [CrossRef]
- Huang, J.; Huffman, J.E.; Huang, Y.; Do Valle, Í.; Assimes, T.L.; Raghavan, S.; Voight, B.F.; Liu, C.; Barabási, A.-L.; Huang, R.D.L.; et al. Genomics and Phenomics of Body Mass Index Reveals a Complex Disease Network. Nat. Commun. 2022, 13, 7973. [Google Scholar] [CrossRef]
- Richardson, B.C.; Smith, R.D.; Ungar, D.; Nakamura, A.; Jeffrey, P.D.; Lupashin, V.V.; Hughson, F.M. Structural Basis for a Human Glycosylation Disorder Caused by Mutation of the COG4 Gene. Proc. Natl. Acad. Sci. USA 2009, 106, 13329–13334. [Google Scholar] [CrossRef]
- Seixas, S.; Marques, P.I. Known Mutations at the Cause of Alpha-1 Antitrypsin Deficiency an Updated Overview of SERPINA1 Variation Spectrum. Appl. Clin. Genet. 2021, 14, 173–194. [Google Scholar] [CrossRef]
- Pagliari, M.T.; Cairo, A.; Boscarino, M.; Mancini, I.; Pappalardo, E.; Bucciarelli, P.; Martinelli, I.; Rosendaal, F.R.; Peyvandi, F. Role of ADAMTS13, VWF and F8 Genes in Deep Vein Thrombosis. PLoS ONE 2021, 16, e0258675. [Google Scholar] [CrossRef]
- Ansari, N.; Najafi, S.; Shahrabi, S.; Saki, N. PEAR1 Polymorphisms as a Prognostic Factor in Hemostasis and Cardiovascular Diseases. J. Thromb. Thrombolysis 2021, 51, 89–95. [Google Scholar] [CrossRef]
- Chen, M.-H.; Raffield, L.M.; Mousas, A.; Sakaue, S.; Huffman, J.E.; Moscati, A.; Trivedi, B.; Jiang, T.; Akbari, P.; Vuckovic, D.; et al. Trans-Ethnic and Ancestry-Specific Blood-Cell Genetics in 746,667 Individuals from 5 Global Populations. Cell 2020, 182, 1198–1213.e14. [Google Scholar] [CrossRef]
- Vuckovic, D.; Bao, E.L.; Akbari, P.; Lareau, C.A.; Mousas, A.; Jiang, T.; Chen, M.-H.; Raffield, L.M.; Tardaguila, M.; Huffman, J.E.; et al. The Polygenic and Monogenic Basis of Blood Traits and Diseases. Cell 2020, 182, 1214–1231. [Google Scholar] [CrossRef] [PubMed]
- Frezzato, M.; Tosetto, A.; Rodeghiero, F. Validated Questionnaire for the Identification of Previous Personal or Familial Venous Thromboembolism. Am. J. Epidemiol. 1996, 143, 1257–1265. [Google Scholar] [CrossRef] [PubMed]
- Consolo, F.; Pozzi, L.; Pieri, M.; Della Valle, P.; Redaelli, A.; D’Angelo, A.; Pappalardo, F. Influence of Different Antithrombotic Regimens on Platelet-Mediated Thrombin Generation in Patients with Left Ventricular Assist Devices. ASAIO J. 2020, 66, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Panigada, M.; Zacchetti, L.; L’Acqua, C.; Cressoni, M.; Anzoletti, M.B.; Bader, R.; Protti, A.; Consonni, D.; D’Angelo, A.; Gattinoni, L. Assessment of Fibrinolysis in Sepsis Patients with Urokinase Modified Thromboelastography. PLoS ONE 2015, 10, e0136463. [Google Scholar] [CrossRef]
- Ziliotto, N.; Marchetti, G.; Scapoli, C.; Bovolenta, M.; Meneghetti, S.; Benazzo, A.; Lunghi, B.; Balestra, D.; Laino, L.A.; Bozzini, N.; et al. C6orf10 Low-Frequency and Rare Variants in Italian Multiple Sclerosis Patients. Front. Genet. 2019, 10, 573. [Google Scholar] [CrossRef] [PubMed]
- Scapoli, C.; Ziliotto, N.; Lunghi, B.; Menegatti, E.; Salvi, F.; Zamboni, P.; Baroni, M.; Mascoli, F.; Bernardi, F.; Marchetti, G. Combination of Genomic and Transcriptomic Approaches Highlights Vascular and Circadian Clock Components in Multiple Sclerosis. Int. J. Mol. Sci. 2021, 23, 310. [Google Scholar] [CrossRef]
- Ittisoponpisan, S.; Islam, S.A.; Khanna, T.; Alhuzimi, E.; David, A.; Sternberg, M.J.E. Can Predicted Protein 3D Structures Provide Reliable Insights into Whether Missense Variants Are Disease Associated? J. Mol. Biol. 2019, 431, 2197–2212. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | rsID dbSNP | cDNA | Protein Change | Frequency | Clinical Significance | Family Carriers |
|---|---|---|---|---|---|---|
| Non synonymous | ||||||
| ARID4A | rs146509016 | c.8C > T | p.Ala3Val | 4.18819 × 10−5 | NR | II1, III1, III2 |
| ARID4A | rs1051029502 | c. 2847G > C | p.Met949Ile | 6.97876 × 10−6 | NR | I1, II3 |
| CRP | rs1376711485 | c.182T > C | Leu61Pro | 6.98295 × 10−6 | NR | I1, II2, II3, III1, III2 |
| F2 | rs199772906 | c.1542C > A | p.Asn514Lys | 0.000244325 | NR | I1, II2, II3, III2 |
| JAK2 | rs2230723 | c.1177C > G | p.Leu393Val | 0.0134624 | B | I1, II2, II3, III2 |
| KLK13 | rs34089525 | c.325C > T | p.His109Tyr | 0.0217616 | NR | II2, II3, III1 |
| NCOA1 | rs150066931 | c.3995A > G | p.Asn1332Ser | 0.0015095 | NR | I1, II3 |
| NQO1 | rs1131341 | c.415C > T | p.Arg139Trp | 0.0255606 | NR | I1, II2, II3, III2 |
| PEAR1 | rs77795865 | c.1142 C > T | p.Ser381Phe | 0.0230726 | NR | II2, II3 |
| PLAT | rs2020921 | c.490 C > T | p.Arg164Trp | 0.012879 | NR | II3 |
| PSG8 | - | c.26G > C | p.Cys9Ser | - | - | I1, II2, II3, III1, III2 |
| SAA2, SAA2-SAA4 | rs138605229 | c.222A > C | p.Glu74Asp | 0.00552043 | NR | II1, III1, III2 |
| SERPINA1 | rs28931570 | c.187C > T | p.Arg63Cys | 0.00151399 | P; LP | II2, III1, III2 |
| THBS1 | - | c.2703T > A | p.Asp901Glu | - | - | I1, II2, III2 |
| UGT1A3 | rs146461519 | c.736G > T | p.Val246Leu | 0.00043999 | NR | II3 |
| VWF | rs1800382 | c.4196G > A | p.Arg1399His | 0.00895051 | B; LB; LP; P | II3 |
| Synonymous | ||||||
| KIF26B | rs201717788 | c.507C > T | p.Val169= | 0.00295131 | NR | II1, III1, III2 |
| MPL | rs544064034 | c.1242G > A | p.Ser414= | 6.98227 × 10−6 | LB | II2, II3, III2 |
| PLCG2 | rs138637229 | c.1146T > C | p.Phe382= | 0.0071625 | B; LB | II2 |
| PTGIS | rs61322884 | c.531C > T | p.Tyr177= | 0.0221135 | NR | II3 |
| SERPIND1 | rs35646566 | c.423G > A | p.Leu141= | 0.0173203 | NR | I1, II3 |
| TMCO1 | rs78363884 | c.486C > T | p.Leu162= | 0.033832 | B | II2 |
| Gene | rsID_dbSNP | Variant Region Features | Splicing Process * | QTL | |
|---|---|---|---|---|---|
| eQTL | sQTL | ||||
| ARID4A | rs146509016 | missense; splice region # | ESE disruption | no data | no data |
| ARID4A | rs1051029502 | missense | ESE disruption | no data | no data |
| F2 | rs199772906 | missense | ESE disruption | no data | no data |
| JAK2 | rs2230723 | missense | new ESS; ESE disruption; new donor | no data | no data |
| KLK13 | rs34089525 | missense | new ESS | IGLON5 | not found |
| MPL | rs544064034 | synonymous; enhancer | no predicted effect | no data | no data |
| NCOA1 | rs150066931 | missense | ESE disruption | no data | no data |
| NQO1 | rs1131341 | missense; splice region # open chromatin | donor site disruption | NOB1; COG4; PDXDC2P | NQO1; NOB1; NPIPB14P |
| PEAR1 | rs77795865 | missense | no predicted effect | LRRC71 | not found |
| PLAT | rs2020921 | missense | new ESE, new donor | PLAT; POLB; AP3M2 | SLC20A2; POLB |
| SAA2-SAA4 | rs138605229 | missense | ESE disruption | ns | not found |
| UGT1A3 | rs146461519 | missense | new donor | no data | no data |
| PLGC2 | rs138637229 | synonymous ; CTCF site | new ESS | ns | not found |
| PTGIS | rs61322884 | synonymous | ESE disruption | SLC9A8 | not found |
| SERPIND1 | rs35646566 | synonymous; enhancer | ESE disruption | AC000089.3 | not found |
| TMCO1 | rs78363884 | synonymous | new ESS | RP11-466F5.10 | not found |
| Reference/Strategy | Population | Gene | rsID_dbSNP | MAF | Variant |
|---|---|---|---|---|---|
| Cunha MLR et al., 2017 [34] - Candidate genes (n = 126) - Variant MAF < 5% | Dutch 2 Families na = 5 + 5 | STX2 | rs137928907 | 0.014 | Phe32Val |
| ITGB3 | rs5918 | 0.121 | Leu59Pro | ||
| APOH | rs4581 | 0.353 | Val266Leu | ||
| KLK8 | rs16988799 | 0.046 | Val154Ile | ||
| KLK11 | rs3745539 | 0.063 | Gly17Glu | ||
| Chang WA et al., 2018 [32] - Variant MAF < 1% - ClinVar annotation | Asian na = 3 | SLC4A1 | rs121912749 | 0.000135 | Gly130Arg |
| GP1BA | rs770089708 | 0.109 | Ser441fs | ||
| Mulder R et al., 2020 [31] - GWLA—Pathogenicity prediction - Rec. proteins assays—Molec. dynamics | Dutch na = 5 | F2 | rs886048338 | 0.000004 | Arg541Trp |
| Morange PE et al., 2021 [33] - MAF < 0.1%—Deleteriousness prediction-RNA seq in siRNAs targeted EC | French na = 4 | MAST2 | rs1387081220 | 0.000004 | Arg89Gln |
| Present study - Candidate genes (n = 192) - MAF < 4%—Deleteriousness prediction—QTL analysis - Protein–protein interaction analysis | Italian na = 3 | THBS1 | -- | -- | Asp901Glu |
| VWF | rs1800382 | 0.008950 | Arg1399His | ||
| SERPINA1 | rs28931570 | 0.001513 | Arg63Cys | ||
| CRP | rs1376711485 | 0.000007 | Leu61Pro | ||
| F2 | rs199772906 | 0.000244 | Asn514Lys | ||
| PLAT | rs2020921 | 0.013 | Arg164Trp |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lunghi, B.; Ziliotto, N.; Balestra, D.; Rossi, L.; Della Valle, P.; Pignatelli, P.; Pinotti, M.; D’Angelo, A.; Marchetti, G.; Bernardi, F. Whole-Exome Sequencing in a Family with an Unexplained Tendency for Venous Thromboembolism: Multicomponent Prediction of Low-Frequency Variant Deleteriousness and of Individual Protein Interaction. Int. J. Mol. Sci. 2023, 24, 13809. https://doi.org/10.3390/ijms241813809
Lunghi B, Ziliotto N, Balestra D, Rossi L, Della Valle P, Pignatelli P, Pinotti M, D’Angelo A, Marchetti G, Bernardi F. Whole-Exome Sequencing in a Family with an Unexplained Tendency for Venous Thromboembolism: Multicomponent Prediction of Low-Frequency Variant Deleteriousness and of Individual Protein Interaction. International Journal of Molecular Sciences. 2023; 24(18):13809. https://doi.org/10.3390/ijms241813809
Chicago/Turabian StyleLunghi, Barbara, Nicole Ziliotto, Dario Balestra, Lucrezia Rossi, Patrizia Della Valle, Pasquale Pignatelli, Mirko Pinotti, Armando D’Angelo, Giovanna Marchetti, and Francesco Bernardi. 2023. "Whole-Exome Sequencing in a Family with an Unexplained Tendency for Venous Thromboembolism: Multicomponent Prediction of Low-Frequency Variant Deleteriousness and of Individual Protein Interaction" International Journal of Molecular Sciences 24, no. 18: 13809. https://doi.org/10.3390/ijms241813809