
Antiphospholipid Syndrome in Pregnancy: New and Old Pathogenetic Mechanisms

 , ,
, ,
Abstract
:1. Introduction
2. Classification Criteria of Antiphospholipid Syndrome
3. Obstetric Clinical Manifestations
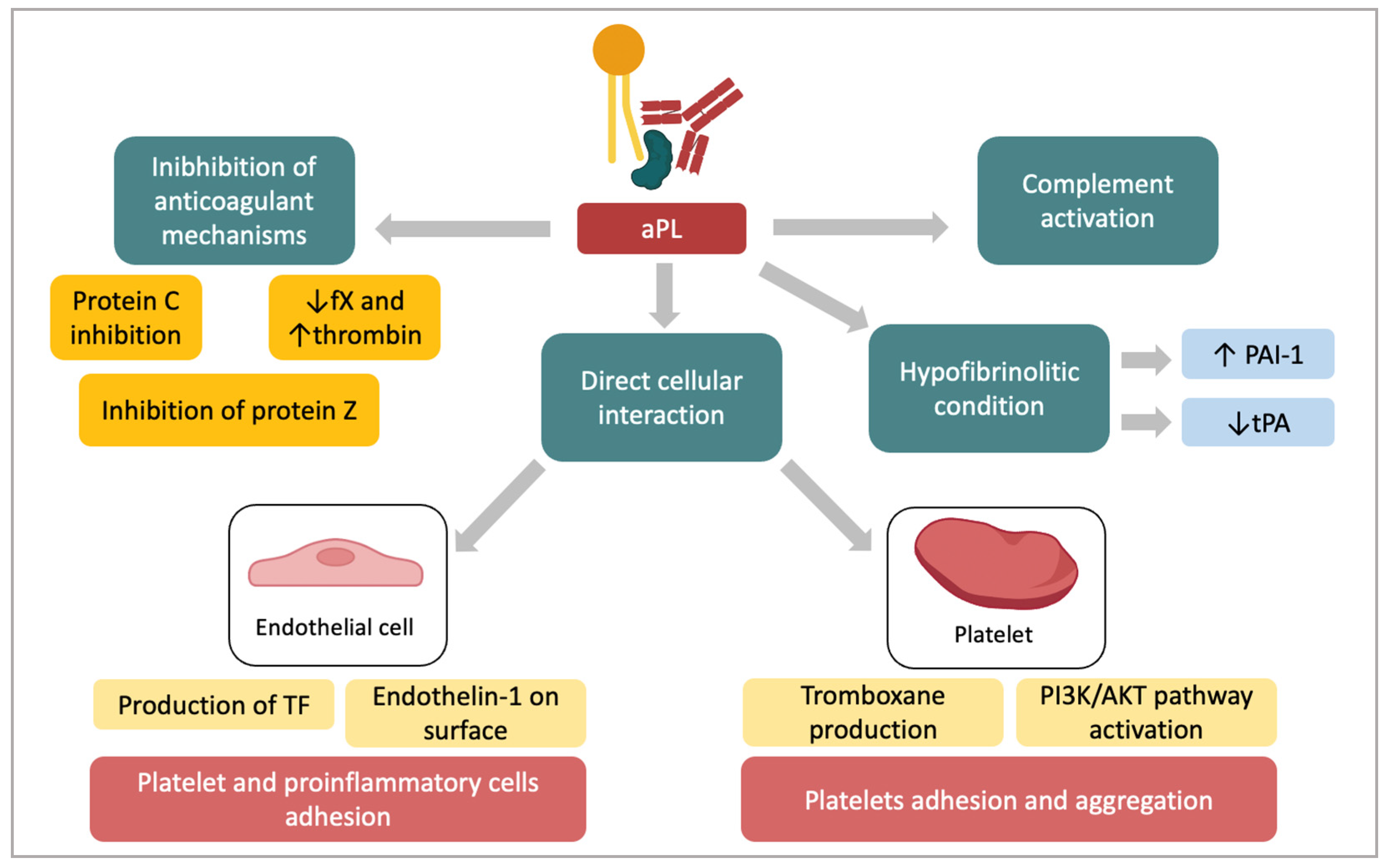
4. Pathogenesis of the Prothrombotic State in APS
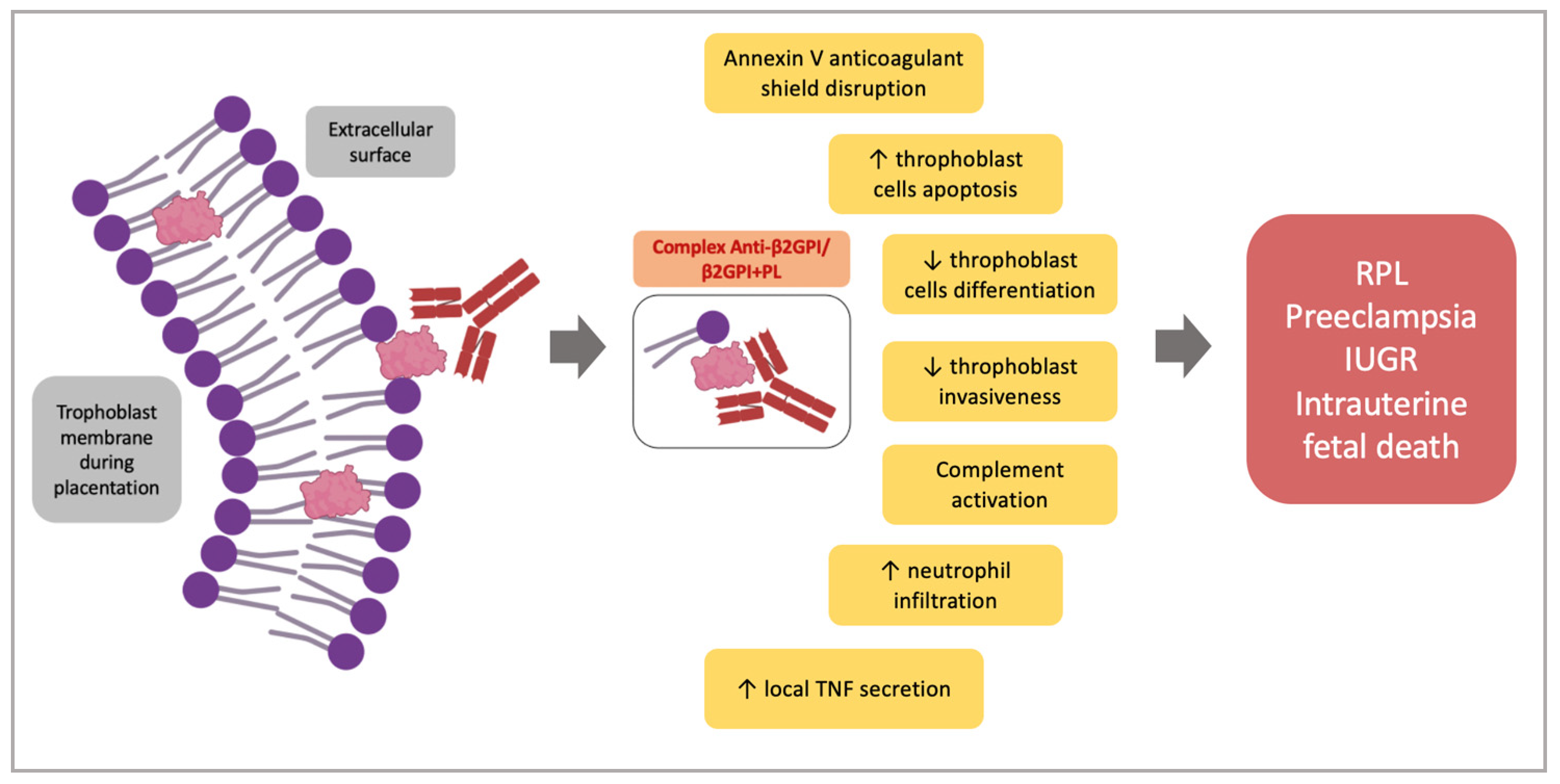
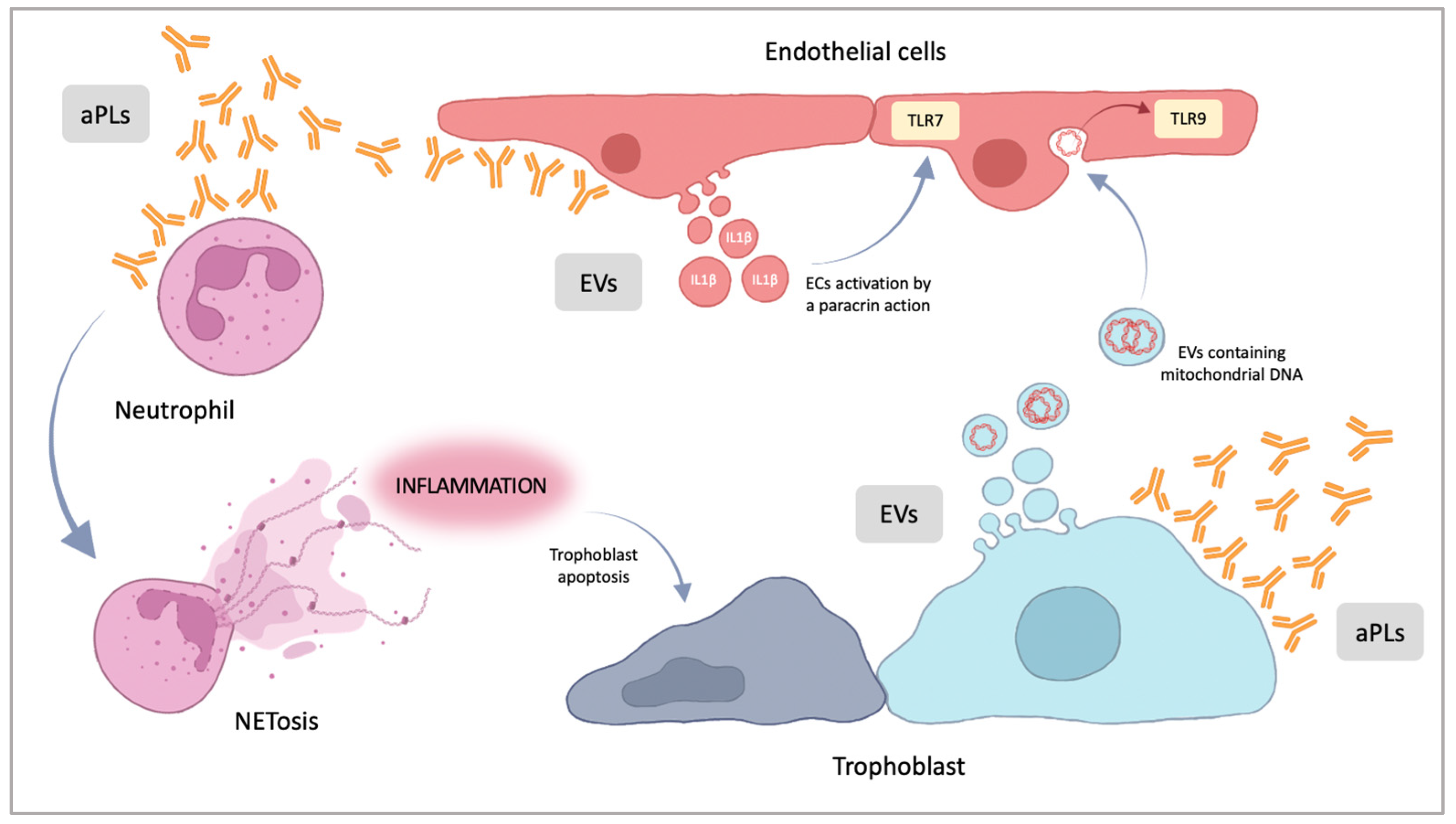
5. Pathogenesis of the Placental Damage in APS
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mehdi, A.; Uthman, I.; Khamashta, M. Antiphospholipid syndrome: Pathogenesis and a window of treatment opportunities in the future. Eur. J. Clin. Investig. 2010, 40, 451–464. [Google Scholar] [CrossRef]
- Arachchillage, D.R.J.; Laffan, M. Pathogenesis and management of antiphospholipid syndrome. Br. J. Haematol. 2017, 178, 181–195. [Google Scholar] [CrossRef]
- Anunciación-Llunell, A.; Muñoz, C.; Roggenbuck, D.; Frasca, S.; Pardos-Gea, J.; Esteve-Valverde, E.; Alijotas-Reig, J.; Miró-Mur, F. Differences in Antiphospholipid Antibody Profile between Patients with Obstetric and Thrombotic Antiphospholipid Syndrome. Int. J. Mol. Sci. 2022, 23, 12819. [Google Scholar] [CrossRef]
- Stanisavljevic, N.; Stojanovich, L.; Djokovic, A.; Todic, B.; Dopsaj, V.; Saponjski, J.; Saponjski, D.; Markovic, O.; Belizna, C.; Zdravkovic, M.; et al. Asymmetric Dimethylarginine Is a Marker of Endothelial Dysfunction in Thrombotic Antiphospholipid Syndrome Patients. Int. J. Mol. Sci. 2022, 23, 12309. [Google Scholar] [CrossRef]
- Sammaritano, L.R. Antiphospholipid syndrome. Best Pract. Res. Clin. Rheumatol. 2020, 34, 101463. [Google Scholar] [CrossRef] [PubMed]
- Duarte-García, A.; Pham, M.M.; Crowson, C.S.; Amin, S.; Moder, K.G.; Pruthi, R.K.; Warrington, K.J.; Matteson, E.L. The Epidemiology of Antiphospholipid Syndrome: A Population-Based Study. Arthritis Rheumatol. 2019, 71, 1545–1552. [Google Scholar] [CrossRef]
- Pons-Estel, G.J.; Andreoli, L.; Scanzi, F.; Cervera, R.; Tincani, A. The antiphospholipid syndrome in patients with systemic lupus erythematosus. J. Autoimmun. 2017, 76, 10–20. [Google Scholar] [CrossRef]
- Cervera, R.; Piette, J.C.; Font, J.; Khamashta, M.A.; Shoenfeld, Y.; Camps, M.T.; Jacobsen, S.; La-kos, G.; Tincani, A.; Kontopou-lou-Griva, I.; et al. Antiphospholipid syndrome: Clinical and immunologic manifestations and patterns of disease expression in a cohort of 1,000 patients. Arthritis Rheum. 2002, 46, 1019–1027. [Google Scholar] [CrossRef]
- Negrini, S.; Pappalardo, F.; Murdaca, G.; Indiveri, F.; Puppo, F. The antiphospholipid syndrome: From pathophysiology to treatment. Clin. Exp. Med. 2017, 17, 257–267. [Google Scholar] [CrossRef]
- Ruaro, B.; Sulli, A.; Alessandri, E.; Fraternali-Orcioni, G.; Cutolo, M. Kikuchi-Fujimoto’s disease associated with systemic lupus erythematous: Difficult case report and literature review. Lupus 2014, 23, 939–944. [Google Scholar] [CrossRef]
- Reshetnyak, T.; Cheldieva, F.; Cherkasova, M.; Lila, A.; Nasonov, E. IgA Antiphospholipid Antibodies in Antiphospholipid Syndrome and Systemic Lupus Erythematosus. Int. J. Mol. Sci. 2022, 23, 9432. [Google Scholar] [CrossRef] [PubMed]
- Ruaro, B.; Confalonieri, P.; Santagiuliana, M.; Wade, B.; Baratella, E.; Kodric, M.; Berria, M.; Jaber, M.; Torregiani, C.; Bruni, C.; et al. Correlation between Potential Risk Factors and Pulmonary Embolism in Sarcoidosis Patients Timely Treated. J. Clin. Med. 2021, 10, 2462. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Puerta, J.A.; Cervera, R. Diagnosis and classification of the antiphospholipid syndrome. J. Autoimmun. 2014, 48–49, 20–25. [Google Scholar] [CrossRef]
- Cervera, R. Antiphospholipid syndrome. Thromb. Res. 2017, 151, S43–S47. [Google Scholar] [CrossRef] [PubMed]
- Meroni, P.L.; Borghi, M.O.; Grossi, C.; Chighizola, C.B.; Durigutto, P.; Tedesco, F. Obstetric and vascular antiphospholipid syndrome: Same antibodies but different diseases? Nat. Rev. Rheumatol. 2018, 14, 433–440. [Google Scholar] [CrossRef]
- Solano, C.; Lamuño, M.; Vargas, A.; Amezcua-Guerra, L.M. Comparison of the 1999 Sapporo and 2006 revised criteria for the classification of the antiphospholipid syndrome. Clin. Exp. Rheumatol. 2009, 27, 914–919. [Google Scholar]
- Pires da Rosa, G.; Bettencourt, P.; Rodríguez-Pintó, I.; Cervera, R.; Espinosa, G. “Non-criteria” antiphospholipid syndrome: A nomenclature proposal. Autoimmun. Rev. 2020, 19, 102689. [Google Scholar] [CrossRef]
- Chaturvedi, S.; Braunstein, E.M.; Brodsky, R.A. Antiphospholipid syndrome: Complement activation, complement gene mutations, and therapeutic implications. J. Thromb. Haemost. 2021, 19, 607–616. [Google Scholar] [CrossRef]
- Miyakis, S.; Lockshin, M.D.; Atsumi, T.; Branch, D.W.; Brey, R.L.; Cervera, R.; Derksen, R.H.W.M.; De Groot, P.G.; Koike, T.; Meroni, P.L.; et al. International classification consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J. Thromb. Haemost. 2006, 4, 295–306. [Google Scholar] [CrossRef]
- Cervera, R.; Khamashta, M.A.; Shoenfeld, Y.; Camps, M.T.; Jacobsen, S.; Kiss, E.; Zeher, M.M.; Tincani, A.; Kontopoulou-Griva, I.; Galeazzi, M.; et al. Euro-Phospholipid Project Group (European Forum on Antiphospholipid Antibodies). Morbidity and mortality in the antiphospholipid syndrome during a 5-year period: A multicentre proprospective study. Ann. Rheum. 2009, 68, 1428–1432. [Google Scholar] [CrossRef]
- Alijotas-Reig, J.; Esteve-Valverde, E.; Ferrer-Oliveras, R.; Sáez-Comet, L.; Lefkou, E.; Mekinian, A.; Belizna, C.; Ruffatti, A.; Tinca-ni, A.; Marozio, L.; et al. EUROAPS Study Group. The European Registry on Obstetric Antiphospholipid Syndrome (EUROAPS): A survey of 1000 consecutive cases. Autoimmun. Rev. 2019, 18, 406–414. [Google Scholar] [CrossRef] [Green Version]
- Practice Committee of the American Society for Reproductive Medicine. Evaluation and treatment of recurrent pregnancy loss: A committee opinion. Fertil Steril. 2012, 98, 1103–1111. [Google Scholar] [CrossRef]
- Saccone, G.; Berghella, V.; Maruotti, G.M.; Ghi, T.; Rizzo, G.; Simonazzi, G.; Rizzo, N.; Facchi-netti, F.; Dall’Asta, A.; Visentin, S.; et al. Antiphospholipid antibody profile based obstetric outcomes of primary antiphospholipid syndrome. The PREGNANTS study. Am. J. Obstet. Gynecol. 2017, 216, 525.e1–525.e12. [Google Scholar] [CrossRef]
- Walter, I.J.; Klein Haneveld, M.J.; Lely, A.T.; Bloemenkamp, K.W.M.; Limper, M.; Kooiman, J. Pregnancy outcome predictors in antiphospholipid syndrome: A systematic review and meta-analysis. Autoimmun. Rev. 2021, 20, 102901. [Google Scholar] [CrossRef]
- Marchetti, T.; de Moerloose, P.; Gris, J.C. Antiphospholipid antibodies and the risk of severe and non-severe pre-eclampsia: The NOHA case-control study. J. Thromb. Haemost. 2016, 14, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Canti, V.; Del Rosso, S.; Tonello, M.; Lucianò, R.; Hoxha, A.; Coletto, L.A.; Tessitore, I.V.; Rosa, S.; Manfredi, A.A.; Castiglioni, M.T.; et al. Antiphosphatidylserine/prothrombin Antibodies in Antiphospholipid Syndrome with Intrauterine Growth Restriction and Preeclampsia. J. Rheumatol. 2018, 45, 1263–1272. [Google Scholar] [CrossRef]
- Pleguezuelo, D.E.; Cabrera-Marante, O.; Abad, M.; Rodriguez-Frias, E.A.; Naranjo, L.; Vazquez, A.; Villar, O.; Gil-Etayo, F.J.; Serrano, M.; Perez-Rivilla, A.; et al. Anti-Phosphatidylserine/Prothrombin Antibodies in Healthy Women with Unexplained Recurrent Pregnancy Loss. J. Clin. Med. 2021, 10, 2094. [Google Scholar] [CrossRef] [PubMed]
- Atsumi, T.; Amengual, O.; Koike, T. Antiphospholipid Syndrome: Pathogenesis. In Systemic Lupus Erythematosus; Academic Press: Cambridge, MA, USA, 2011. [Google Scholar]
- Willis, R.; Nigel Harris, E.; Pierangeli, S.S. Pathogenesis of the antiphospholipid syndrome. In Seminars in Thrombosis and Hemostasis; Thieme Medical Publishers: New York, NY, USA, 2012; pp. 305–321. [Google Scholar]
- Schreiber, K.; Sciascia, S.; de Groot, P.G.; Devreese, K.; Jacobsen, S.; Ruiz-Irastorza, G.; Salmon, J.E.; Shoenfeld, Y.; Shovman, O.; Hunt, B.J. Antiphospholipid syndrome. Nat. Rev. Dis. Prim. 2018, 4, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Cariou, R.; Tobelem, G.; Bellucci, S.; Soria, J.; Soria, C.; Maclouf, J.; Caen, J. Effect of lupus anticoagulant on antithrombotic properties of endothelial cells. Inhibition of thrombomodulin-dependent protein C activation. Thromb. Haemost. 1988, 60, 054–058. [Google Scholar]
- Marciniak, E.; Romond, E.H. Impaired catalytic function of activated protein C: A new in vitro manifestation of lupus anticoagulant. Blood 1989, 74, 2426–2432. [Google Scholar] [CrossRef]
- Ames, P.R.; Tommasino, C.; Iannaccone, L.; Brillante, M.; Cimino, R.; Brancaccio, V. Coagulation activation and fibrinolytic imbalance in subjects with idiopathic antiphospholipid antibodies. A crucial role for acquired free protein S deficiency. Thromb. Haemost. 1996, 76, 190–194. [Google Scholar] [CrossRef]
- Ginsberg, J.; Demers, C.; Brill-Edwards, P.; Bona, R.; Johnston, M.; Wong, A. Acquired free protein S deficiency is associated with antiphospholipid antibodies and increased thrombin generation in patients with systemic lupus erythematosus. Am. J. Med. 1995, 98, 379–383. [Google Scholar] [CrossRef]
- Forastiero, R.R.; Martinuzzo, M.E.; Lu, L.; Broze, G.J. Autoimmune antiphospholipid antibodies impair the inhibition of activated factor X by protein Z/protein Z-dependent protease inhibitor. J. Thromb. Haemost. 2003, 1, 1764–1770. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Iverson, G.M.; Qi, J.C.; Cockerill, K.A.; Linnik, M.D.; Konecny, P. Beta 2-glycoprotein I binds factor XI and inhibits its activation by thrombin and factor XIIa: Loss of inhibition by clipped beta 2-glycoprotein I. Proc. Natl. Acad. Sci. USA 2004, 101, 3939–3944. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Y.; Reddel, S.W.; Herzog, H.; Wang, Y.X.; Brighton, T.; France, M.P. Impaired thrombin generation in b2-glycoprotein I null mice. J. Biol. Chem. 2001, 276, 13817–13821. [Google Scholar] [CrossRef] [PubMed]
- Jurado, M.; Paramo, J.A.; Gutierrez-Pimentel, M.; Rocha, E. Fibrinolytic potential and antiphospholipid antibodies in systemic lupus erythematosus and other connective tissue disorders. Thromb. Haemost. 1992, 68, 516–520. [Google Scholar] [CrossRef] [PubMed]
- Etingin, O.R.; Hajjar, D.P.; Hajjar, K.A.; Harpel, P.C.; Nachman, R.L. Lipoprotein (a) regulates plasminogen activator inhibitor-1 expression in endothelial cells. J. Biol. Chem. 1991, 266, 2459–2465. [Google Scholar] [CrossRef]
- Atsumi, T.; Khamashta, M.A.; Andujar, C.; Leandro, M.J.; Amengual, O.; Ames, P.R. Elevated plasma lipoprotein(a) level and its association with impaired fibrinolysis in patients with antiphospholipid syndrome. J. Rheumatol. 1998, 25, 69–73. [Google Scholar]
- Yamazaki, M.; Asakura, H.; Jokaji, H.; Saito, M.; Uotani, C.; Kumabashiri, I. Plasma levels of lipoprotein(a) are elevated in patients with the antiphospholipid antibody syndrome. Thromb. Haemost. 1994, 71, 424–427. [Google Scholar] [CrossRef]
- Cugno, M.; Cabibbe, M.; Galli, M.; Meroni, P.L.; Caccia, S.; Russo, R. Antibodies to tissue-type plasminogen activator (tPA) in patients with antiphospholipid syndrome: Evidence of interaction between the antibodies and the catalytic domain of tPA in 2 patients. Blood 2004, 103, 2121–2126. [Google Scholar] [CrossRef]
- Kornberg, A.; Blank, M.; Kaufman, S.; Shoenfeld, Y. Induction of tissue factor-like activity in monocytes by anti-cardiolipin antibodies. J. Immunol. 1994, 153, 1328–1332. [Google Scholar] [CrossRef]
- Zhang, J.; McCrae, K.R. Annexin A2 mediates endothelial cell activation by antiphospholipid/anti-beta2 glycoprotein I antibodies. Blood 2005, 1964–1969. [Google Scholar] [CrossRef]
- Moestrup, S.K.; Schousboe, I.; Jacobsen, C.; Leheste, J.R.; Christensen, E.I.; Willnow, T.E. Beta2-glycoprotein-I (apolipoprotein H) and beta2-glycoprotein-Iephospholipid complex harbor a recognition site for the endocytic receptor megalin. J. Clin. Investig. 1998, 102, 902–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutters, B.C.; Derksen, R.H.; Tekelenburg, W.L.; Lenting, P.J.; Arnout, J.; de Groot, P.G. Dimers of beta 2-glycoprotein I increase platelet deposition to collagen via interaction with phospholipids and the apolipoprotein E receptor 20. J. Biol. Chem. 2003, 278, 33831–33838. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Giannakopoulos, B.; Yan, Z.; Yu, P.; Berndt, M.C.; Andrews, R.K. Anti-beta2-glycoprotein I antibodies in complex with beta2-glycoprotein I can activate platelets in a dysregulated manner via glycoprotein Ib-IX-V. Arthritis Rheum. 2006, 54, 2558–2567. [Google Scholar] [CrossRef]
- De Wolf, F.; Carreras, L.O.; Moerman, P.; Vermylen, J.; Van Assche, A.; Renaer, M. Decidual vasculopathy and extensive placental infarction in a patient with repeated thromboembolic accidents, recurrent fetal loss, and a lupus anticoagulant. Am. J. Obstet. Gynecol. 1982, 142, 829–834. [Google Scholar] [CrossRef]
- Lyden, T.W.; Vogt, E.; Ng, A.K.; Johnson, P.M.; Rote, N.S. Monoclonal antiphospholipid antibody reactivity against human placental trophoblast. J. Reprod. Immunol. 1992, 22, 1–14. [Google Scholar] [CrossRef]
- Viall, C.A.; Chamley, L.W. Histopathology in the placentae of women with antiphospholipid antibodies: A systematic review of the literature. Autoimmun. Rev. 2015, 14, 446–471. [Google Scholar] [CrossRef]
- Di Simone, N.; Meroni, P.L.; de Papa, N.; Raschi, E.; Caliandro, D.; De Carolis, C.S.; Khamashta, M.A.; Atsumi, T.; Hughes, G.R.; Balestrieri, G.; et al. Antiphospholipid antibodies affect trophoblast gonadotropin secretion and invasiveness by binding directly and through adhered beta2-glycoprotein I. Arthritis Rheum. 2000, 43, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Meroni, P.L.; Borghi, M.O.; Raschi, E.; Tedesco, F. Pathogenesis of antiphospholipid syndrome: Understanding the antibodies. Nat. Rev. Rheumatol. 2011, 7, 330–339. [Google Scholar] [CrossRef]
- Pierangeli, S.; Chen, P.; Raschi, E.; Scurati, S.; Grossi, C.; Borghi, M.; Palomo, I.; Harris, E.; Meroni, P. Antiphospholipid antibodies and the antiphospholipid syndrome: Pathogenic mechanisms. Semin. Thromb. Hemost. 2008, 34, 236–250. [Google Scholar] [CrossRef]
- Meroni, P.L.; Raschi, E.; Grossi, C.; Pregnolato, F.; Trespidi, L.; Acaia, B.; Borghi, M.O. Obstetric and vascular APS: Same autoantibodies but different diseases? Lupus 2012, 21, 708–710. [Google Scholar] [CrossRef]
- D’Ippolito, S.; Meroni, P.L.; Koike, T.; Veglia, M.; Scambia, G.; Di Simone, N. Obstetric antiphospholipid syndrome: A recent classify cation for an old defined disorder. Autoimmun. Rev. 2014, 13, 901–908. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, J.A. Immune recognition at the maternal-fetal interface: Overview. Am. J. Reprod. Immunol. 1992, 28, 127–131. [Google Scholar] [CrossRef]
- Matsuura, E.; Igarashi, Y.; Yasuda, T.; Triplett, D.A.; Koike, T. Anticardiolipin antibodies recognize beta 2-glycoprotein I structure altered by interacting with an oxygen modified solid phase surface. J. Exp. Med. 1994, 179, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Di Simone, N.; Di Nicuolo, F.; D’Ippolito, S.; Castellani, R.; Tersigni, C.; Caruso, A.; Meroni, P.; Marana, R. Antiphospholipid antibodies affect human endometrial angiogenesis. Biol. Reprod. 2010, 83, 212–219. [Google Scholar] [CrossRef]
- Borghi, M.O.; Raschi, E.; Broggini, V.; Grossi, C.; D’Amelio, F.; Chen, P. Antiphospholipid antibodies reactivity with human decidual cells: An additional mechanism of pregnancy complications in APS and a potential target for innovative therapeutic intervention. Ann. Rheum. Dis. 2009, 68, 216–221. [Google Scholar]
- Di Simone, N.; Raschi, E.; Testoni, C.; Castellani, R.; D’Asta, M.; Shi, T.; Krilis, S.A.; Caruso, A.; Meroni, P.L. Pathogenic role of anti β2-glycoprotein I antibodies in antiphospholipid-associated fetal loss: Characterisation of β2-glycoprotein I binding to trophoblast cells and functional effects of anti β2-glycoprotein I antibodies in vitro. Ann. Rheum. Dis. 2005, 64, 462–467. [Google Scholar] [CrossRef]
- Rand, J.H.; Wu, X.-X.; Lapinski, R.; Van Heerde, W.L.; Reutelingsperger, C.P.; Chen, P.P.; Ortel, T.L. Detection of antibody mediated reduction of annexin A5 anticoagulant activity in plasmas of patients with the antiphospholipid syndrome. Blood 2004, 104, 2783–2790. [Google Scholar] [CrossRef]
- Di Simone, N.; Ferrazzani, S.; Castellani, R.; De Carolis, S.; Mancuso, S.; Caruso, A. Heparin and low-dose aspirin restore placental human chorionic gonadotrophin secretion abolished by antiphospholipid antibody-containing sera. Hum. Reprod. 1997, 12, 2061–2065. [Google Scholar] [CrossRef] [PubMed]
- Di Simone, N.; Caliandro, D.; Castellani, R.; Ferrazzani, S.; De Carolis, S.; Caruso, A. Low-molecular weight heparin restores in-vitro trophoblast invasiveness and differentiation in presence of immunoglobulin G fractions obtained from patients with antiphospholipid syndrome. Hum. Reprod. 1999, 14, 489–495. [Google Scholar] [CrossRef]
- Di Simone, N.; Marana, R.; Castellani, R.; Di Nicuolo, F.; D’Alessio, M.C.; Raschi, E.; Borghi, M.O.; Chen, P.P.; Sanguinetti, M.; Caruso, A.; et al. Decreased expression of heparin-binding epidermal growth factor-like growth factor as a newly identified patho-genic mechanism of antiphospholipid-mediated defective placentation. Arthritis Rheum. 2010, 62, 1504–1512. [Google Scholar] [CrossRef]
- Berman, J.; Girardi, G.; Salmon, J.E. TNF-alpha is a critical effector and a target for therapy in antiphospholipid anti-body-induced pregnancy loss. J. Immunol. 2005, 174, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Martinez de la Torre, Y.; Buracchi, C.; Borroni, E.M.; Dupor, J.; Bonecchi, R.; Nebuloni, M.; Pasqualini, F.; Doni, A.; Lauri, E.; Agostinis, C.; et al. Protection against inflammation and autoantibody-caused fetal loss by the chemokine decoy receptor. Proc. Natl. Acad. Sci. USA 2007, 104, 2319–2324. [Google Scholar] [CrossRef] [Green Version]
- Thiam, H.R.; Wong, S.L.; Wagner, D.D.; Waterman, C.M. Cellular Mechanisms of NETosis. Annu. Rev. Cell Dev. Biol. 2020, 36, 191–218. [Google Scholar] [CrossRef] [PubMed]
- de Bont, C.M.; Boelens, W.C.; Pruijn, G.J.M. NETosis, complement, and coagulation: A triangular relationship. Cell Mol. Immunol. 2019, 16, 19–27. [Google Scholar] [CrossRef]
- Cahilog, Z.; Zhao, H.; Wu, L.; Alam, A.; Eguchi, S.; Weng, H.; Ma, D. The Role of Neutrophil NETosis in Organ Injury: Novel Inflammatory Cell Death Mechanisms. Inflammation 2020, 43, 2021–2032. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Yalavarthi, S.; Kanthi, Y.; Mazza, L.F.; Elfline, M.A.; Luke, C.E.; Pinsky, D.J.; Henke, P.K.; Knight, J.S. In vivo role of neutrophil extracellular traps in antiphospholipid anti-body-mediated venous thrombosis. Arthritis Rheumatol. 2017, 69, 665–667. [Google Scholar] [CrossRef] [PubMed]
- Yalavarthi, S.; Gould, T.J.; Rao, A.N.; Mazza, L.F.; Morris, A.E.; Núñez-Álvarez, C.; Hernández-Ramírez, D.; Bockenstedt, P.L.; Liaw, P.C.; Cabral, A.R.; et al. Release of neutrophil extra-cellular traps by neutrophils stimulated with antiphospholipid antibodies: A newly identified mechanism of thrombosis in the antiphospholipid syndrome. Arthritis Rheumatol. 2015, 67, 2990–3003. [Google Scholar] [CrossRef]
- Gladigau, G.; Haselmayer, P.; Scharrer, I.; Munder, M.; Prinz, N.; Lackner, K.; Schild, H.; Stein, P.; Radsak, M.P. A role for Toll-like receptor mediated signals in neutrophils in the patho-genesis of the anti-phospholipid syndrome. PLoS ONE 2012, 7, e42176. [Google Scholar] [CrossRef]
- Marder, W.; Knight, J.S.; Kaplan, M.J.; Somers, E.C.; Zhang, X.; O’Dell, A.A.; Padmanabhan, V.; Lieberman, R.W. Placental histology and neutrophil extracellular traps in lupus and pre-eclampsia pregnancies. Lupus Sci. Med. 2015, 3, e000134. [Google Scholar] [CrossRef] [PubMed]
- Lackner, K.J.; Müller-Calleja, N. Pathogenesis of antiphospholipid syndrome: Recent insights and emerging concepts. Expert Rev. Clin. Immunol. 2018, 15, 199–209. [Google Scholar] [CrossRef]
- Chaturvedi, S.; Alluri, R.; McCrae, K.R. Extracellular vesicles in the antiphospholipid syndrome. Semin. Thromb. Hemost. 2018, 44, 493–504. [Google Scholar]
- Wu, M.; Barnard, J.; Kundu, S.; McCrae, K.R. A novel pathway of cellular activation mediated by antiphospholipid anti-body-induced extracellular vesicles. J. Thromb. Haemost. 2015, 13, 1928–1940. [Google Scholar] [CrossRef] [PubMed]
- Tong, M.; Johansson, C.; Xiao, F.; Stone, P.R.; James, J.L.; Chen, Q.; Cree, L.M.; Chamley, L.W. Antiphospholipid antibodies increase the levels of mitochondrial DNA in placental extracellular vesicles: Alarming for preeclampsia. Sci. Rep. 2017, 7, 16556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotyla, P.J.; Islam, M.A. MicroRNA (miRNA): A New Dimension in the Pathogenesis of Antiphospholipid Syndrome (APS). Int. J. Mol. Sci. 2020, 21, 2076. [Google Scholar] [CrossRef]
- Teruel, R.; Pérez-Sánchez, C.; Corral, J.; Herranz, M.T.; Pérez-Andreu, V.; Saiz, E.; García-Barberá, N.; Martínez-Martínez, I.; Roldán, V.; Vicente, V.; et al. Identification of miRNAs as potential modulators of tissue factor expression in patients with systemic lupus erythematosus and antiphospholipid syndrome. J. Thromb. Haemost. 2011, 9, 1985–1992. [Google Scholar] [CrossRef]
- Pérez-Sánchez, C.; Aguirre, M.A.; Ruiz-Limón, P.; Barbarroja, N.; Jiménez-Gómez, Y.; de La Rosa, I.A.; Rodriguez-Ariza, A.; Collantes-Estévez, E.; Segui, P.; Velasco, F.; et al. Atherothrombosis-associated microRNAs in antiphospholipid syndrome and systemic lupus erythematosus patients. Sci. Rep. 2016, 6, 31375. [Google Scholar] [CrossRef]
- Pérez-Sánchez, C.; la Rosa, I.A.-D.; Aguirre, M.; Luque-Tévar, M.; Ruiz-Limón, P.; Barbarroja, N.; Jiménez-Gómez, Y.; Ábalos-Aguilera, M.C.; Collantes-Estévez, E.; Segui, P.; et al. Circulating microRNAs as biomarkers of disease and typification of the atherothrombotic status in antiphospholipid syndrome. Haematologica 2018, 103, 908–918. [Google Scholar] [CrossRef]
- Alijotas-Reig, J.; Esteve-Valverde, E.; Anunciación-Llunell, A.; Marques-Soares, J.; Pardos-Gea, J.; Miró-Mur, F. Pathogenesis, Diagnosis and Management of Obstetric Antiphospholipid Syndrome: A Comprehensive Review. J. Clin. Med. 2022, 11, 675. [Google Scholar] [CrossRef] [PubMed]
- Hamulyák, E.N.; Scheres, L.J.; Marijnen, M.C.; Goddijn, M.; Middeldorp, S. Aspirin or heparin or both for improving pregnancy outcomes in women with persistent antiphospholipid antibodies and recurrent pregnancy loss. Cochrane Database Syst. Rev. 2020, 5, CD012852. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, B.; Burmester, G.R.; Dörner, T. Heparin or aspirin or both in the treatment of recurrent abortions in women with antiphospholipid antibody (syndrome). Curr. Opin. Rheumatol. 2011, 23, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Girardi, G.; Redecha, P.; Salmon, J.E. Heparin prevents antiphospholipid antibody-induced fetal loss by inhibiting complement activation. Nat Med. 2004, 10, 1222–1226. [Google Scholar] [CrossRef] [PubMed]
- D’Ippolito, S.; Marana, R.; Di Nicuolo, F.; Castellani, R.; Veglia, M.; Stinson, J.; Scambia, G.; Di Simone, N. Effect of Low Molecular Weight Heparins (LMWHs) on antiphospholipid Antibodies (aPL)-mediated inhibition of endometrial angiogenesis. PLoS ONE 2012, 7, e29660. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| 1. Vascular thrombosis | ≥1 clinical episode of thrombosis (arterial or venous), in any tissue or organ. |
| 2. Pregnancy morbidity |
|
| 3. Laboratory criteria |
|
| Subclassification of APS patients (according to aPL positivity) |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Ippolito, S.; Barbaro, G.; Paciullo, C.; Tersigni, C.; Scambia, G.; Di Simone, N. Antiphospholipid Syndrome in Pregnancy: New and Old Pathogenetic Mechanisms. Int. J. Mol. Sci. 2023, 24, 3195. https://doi.org/10.3390/ijms24043195
D’Ippolito S, Barbaro G, Paciullo C, Tersigni C, Scambia G, Di Simone N. Antiphospholipid Syndrome in Pregnancy: New and Old Pathogenetic Mechanisms. International Journal of Molecular Sciences. 2023; 24(4):3195. https://doi.org/10.3390/ijms24043195
Chicago/Turabian StyleD’Ippolito, Silvia, Greta Barbaro, Carmela Paciullo, Chiara Tersigni, Giovanni Scambia, and Nicoletta Di Simone. 2023. "Antiphospholipid Syndrome in Pregnancy: New and Old Pathogenetic Mechanisms" International Journal of Molecular Sciences 24, no. 4: 3195. https://doi.org/10.3390/ijms24043195