
Clinical and Pathologic Features of Congenital Myasthenic Syndromes Caused by 35 Genes—A Comprehensive Review

Abstract
:1. Overview of Congenital Myasthenic Syndromes (CMS)
2. Electrophysiology, Muscle Biopsy, Laboratory Examinations, Differential Diagnosis, Epidemiology, Inheritance, and Therapeutic Perspectives
2.1. Electrophysiological Examinations
2.2. Muscle Biopsy and Creatine Kinase (CK)
2.3. Differential Diagnosis
2.4. Epidemiology
2.5. Inheritance
2.6. Therapeutic Perspectives
3. Physiological Aspects of Neuromuscular Signal Transmission
4. Thirty-Five Genes in 14 Groups of CMS
4.1. Endplate AChR Deficiency (CHRNA1, CHRNB1, CHRND, CHRNE, and RAPSN)
4.1.1. Pathomechanisms
4.1.2. Clinical Features and Therapies
4.2. Escobar Variant of Multiple Pterygium Syndrome (EVMPS, Escobar Syndrome) (CHRNG) and Lethal Form of Multiple Pterygium Syndrome (LMPS)/Fetal Akinesia Deformation Sequence (FADS) (CHRNA1, CHRND, MUSK, RAPSN, DOK7, and SLC18A3)
4.2.1. Pathomechanisms
4.2.2. Clinical Features and Therapies
4.3. Slow-Channel CMS (SCCMS) and Fast-Channel CMS (FCCMS) (CHRNA1, CHRNB1, CHRND, and CHRNE)
4.3.1. Pathomechanisms
4.3.2. Clinical Features and Therapies
4.4. Synaptic CMS (COLQ, LAMB2, and COL13A1)
4.4.1. Pathomechanisms
4.4.2. Clinical Features and Therapies
4.5. Sodium Channel CMS (SCN4A)
4.5.1. Pathomechanisms
4.5.2. Clinical Features and Therapies
4.6. Defective AChR Clustering (AGRN, LRP4, MUSK, and DOK7)
4.6.1. Pathomechanisms
4.6.2. Clinical Features and Therapies
4.7. CMS Caused by Defective Structural Molecule at the NMJ (PLEC)
4.7.1. Pathomechanisms
4.7.2. Clinical Features and Therapies
4.8. CMS Caused by Defective Recycling of ACh (CHAT, SLC18A3, SLC5A7, and PREPL)
4.8.1. Pathomechanisms
4.8.2. Clinical Features and Therapies
4.9. Lambert-Eaton Myasthenic Syndrome (LEMS)-Like CMS (SYT2, SNAP25, VAMP1, UNC13A, RPH3A, and LAMA5)
4.9.1. Pathomechanisms
4.9.2. Clinical Features and Therapies
4.10. Glycosylation-Deficient CMS (GFPT1, DPAGT1, ALG2, ALG14, GMPPB)
4.10.1. Pathomechanisms
4.10.2. Clinical Features and Therapies
4.11. CMS Caused by Defective Nerve Terminal Formation (MYO9A and SLC25A1)
4.11.1. Pathomechanisms
4.11.2. Clinical Features and Therapies
4.12. CMS Caused by Defective Nuclear Membrane Protein (TOR1AIP1)
4.12.1. Pathomechanisms
4.12.2. Clinical Features and Therapies
4.13. CMS Caused by Defective Chromatin Remodeling Protein (CHD8)
4.13.1. Pathomechanisms
4.13.2. Clinical Features and Therapies
4.14. CMS in PURA Syndrome (PURA)
4.14.1. Pathomechanisms
4.14.2. Clinical Features and Therapies
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACh | acetylcholine |
| AChE | acetylcholinesterase |
| AChR | acetylcholine receptor |
| ALG14 | asparagine-linked glycosylation 14 homolog |
| ALG2 | asparagine-linked glycosylation 2 homolog |
| AMC | arthrogryposis multiplex congenita |
| ASD | autism spectrum disorder |
| CaV1.1 | L-type calcium channel |
| CDG | congenital disorder of glycosylation |
| ChAT | choline acetyltransferase |
| CHD8 | chromatin helicase DNA binding protein 8 |
| ChEI | cholinesterase inhibitor |
| ChT | high affinity choline transporter |
| CK | creatine kinase |
| CMAP | compound muscle action potential |
| CMS | congenital myasthenic syndromes |
| CNS | central nervous system |
| ColQ | collagen Q |
| CTD | C-terminal domain of ColQ |
| Ctgf | connective tissue growth factor |
| D2L2AD | combined D-2- and L-2-hydroxyglutaric aciduria |
| DHMN7A | autosomal dominant distal hereditary motor neuropathy type VIIA |
| DHPR | dihydropyridine receptor |
| Dok-7 | docking protein 7 |
| DPAGT1 | dolichyl-phosphate N-acetylglucosamine phosphotransferase 1 |
| EBS | epidermolysis bullosa simplex |
| EPP | endplate potential |
| EVMPS | Escobar variant of multiple pterygium syndrome |
| FADS | fetal akinesia deformation sequence |
| FCCMS | fast-channel congenital myasthenic syndrome |
| Fgf18 | fibroblast growth factor 18 |
| GAP | Rho GTPase-activating protein |
| GFPT1 | glutamine--fructose-6-phosphate transaminase 1 |
| GMPPB | GDP-mannose pyrophosphorylase B |
| HCS | hypotonia-cystinuria syndrome |
| HSP | heparan sulfate proteoglycan |
| HSPBD | HSP-binding domain |
| IBM | inclusion body myositis |
| IDDAM | intellectual developmental disorder with autism and macrocephaly |
| LAP1 | lamin-associated protein 1 |
| LEMS | Lambert-Eaton myasthenic syndrome |
| LGMD17 | autosomal recessive limb-girdle muscular dystrophy 17 |
| LMPS | lethal form of multiple pterygium syndrome |
| LRP4 | low density lipoprotein receptor-related protein 4 |
| MDDG | muscular dystrophy-dystroglycanopathy |
| MG | myasthenia gravis |
| MuSK | muscle-specific receptor tyrosine kinase |
| Myo9a | myosin 9A |
| NaV1.4 | Skeletal muscle voltage-gated sodium channel |
| NCAM | neuronal cell adhesion molecule |
| NIPPV | non-invasive positive pressure ventilation |
| NMJ | neuromuscular junction |
| PRAD | proline-rich attachment domain |
| PREPL | propyl endopeptidase like |
| PURA | purine-rich element-binding protein A |
| R-CMAP | repetitive compound muscle action potential |
| RNS | repetitive nerve stimulation |
| Rspo2 | R-spondin 2 |
| RyR | ryanodine receptor |
| SCCMS | slow-schannel congenital myasthenic syndrome |
| SFEMG | single-fiber electromyography |
| SIDS | sudden infantile death syndrome |
| SNARE | soluble NSF attachment protein receptor |
| SNV | single nucleotide variant |
| SOST2 | sclerosteosis type 2 |
| SR | sarcoplasmic reticulum |
| SSRI | selective serotonin reuptake inhibitor |
| SV2A | synaptic vesicle glycoprotein 2A |
| UDP-GlcNAc | uridine diphosphate N-acetylglucosamine |
| vAChT | vesicular acetylcholine transporter |
| VAMP1 | vesicle-associated membrane protein 1 |
| VGCC | voltage-gated calcium channel |
References
- Ohno, K.; Ohkawara, B.; Ito, M.; Engel, A.G. Molecular Genetics of Congenital Myasthenic Syndromes. In eLS; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014. [Google Scholar] [CrossRef]
- Engel, A.G.; Shen, X.M.; Selcen, D.; Sine, S.M. Congenital myasthenic syndromes: Pathogenesis, diagnosis, and treatment. Lancet Neurol. 2015, 14, 420–434. [Google Scholar] [CrossRef]
- Heintz-Buschart, A.; Pandey, U.; Wicke, T.; Sixel-Doring, F.; Janzen, A.; Sittig-Wiegand, E.; Trenkwalder, C.; Oertel, W.H.; Mollenhauer, B.; Wilmes, P. The nasal and gut microbiome in Parkinson’s disease and idiopathic rapid eye movement sleep behavior disorder. Mov. Disord. 2018, 33, 88–98. [Google Scholar] [CrossRef]
- Byring, R.F.; Pihko, H.; Tsujino, A.; Shen, X.M.; Gustafsson, B.; Hackman, P.; Ohno, K.; Engel, A.G.; Udd, B. Congenital myasthenic syndrome associated with episodic apnea and sudden infant death. Neuromuscul. Disord. 2002, 12, 548–553. [Google Scholar] [CrossRef]
- Mannikko, R.; Wong, L.; Tester, D.J.; Thor, M.G.; Sud, R.; Kullmann, D.M.; Sweeney, M.G.; Leu, C.; Sisodiya, S.M.; FitzPatrick, D.R.; et al. Dysfunction of NaV1.4, a skeletal muscle voltage-gated sodium channel, in sudden infant death syndrome: A case-control study. Lancet 2018, 391, 1483–1492. [Google Scholar] [CrossRef]
- Nicole, S.; Chaouch, A.; Torbergsen, T.; Bauche, S.; de Bruyckere, E.; Fontenille, M.J.; Horn, M.A.; van Ghelue, M.; Loseth, S.; Issop, Y.; et al. Agrin mutations lead to a congenital myasthenic syndrome with distal muscle weakness and atrophy. Brain 2014, 137 Pt 9, 2429–2443. [Google Scholar] [CrossRef]
- Rodriguez Cruz, P.M.; Belaya, K.; Basiri, K.; Sedghi, M.; Farrugia, M.E.; Holton, J.L.; Liu, W.W.; Maxwell, S.; Petty, R.; Walls, T.J.; et al. Clinical features of the myasthenic syndrome arising from mutations in GMPPB. J. Neurol. Neurosurg. Psychiatry 2016, 87, 802–809. [Google Scholar] [CrossRef]
- Ohno, K. Is the serum creatine kinase level elevated in congenital myasthenic syndrome? J. Neurol. Neurosurg. Psychiatry 2016, 87, 801. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, R.G.; Herrmann, D.N.; Bansagi, B.; Hasan, B.A.; Lofra, R.M.; Logigian, E.L.; Sowden, J.E.; Almodovar, J.L.; Littleton, J.T.; Zuchner, S.; et al. Electrophysiologic features of SYT2 mutations causing a treatable neuromuscular syndrome. Neurology 2015, 85, 1964–1971. [Google Scholar] [CrossRef]
- Shen, X.M.; Scola, R.H.; Lorenzoni, P.J.; Kay, C.S.; Werneck, L.C.; Brengman, J.; Selcen, D.; Engel, A.G. Novel synaptobrevin-1 mutation causes fatal congenital myasthenic syndrome. Ann. Clin. Transl. Neurol. 2017, 4, 130–138. [Google Scholar] [CrossRef]
- Engel, A.G.; Selcen, D.; Shen, X.M.; Milone, M.; Harper, C.M. Loss of MUNC13-1 function causes microcephaly, cortical hyperexcitability, and fatal myasthenia. Neurol. Genet. 2016, 2, e105. [Google Scholar] [CrossRef]
- Maselli, R.A.; Vazquez, J.; Schrumpf, L.; Arredondo, J.; Lara, M.; Strober, J.B.; Pytel, P.; Wollmann, R.L.; Ferns, M. Presynaptic congenital myasthenic syndrome with altered synaptic vesicle homeostasis linked to compound heterozygous sequence variants in RPH3A. Mol. Genet. Genom. Med. 2018, 6, 434–440. [Google Scholar] [CrossRef]
- Maselli, R.A.; Arredondo, J.; Vazquez, J.; Chong, J.X.; University of Washington Center for Mendelian, G.; Bamshad, M.J.; Nickerson, D.A.; Lara, M.; Ng, F.; Lo, V.L.; et al. Presynaptic congenital myasthenic syndrome with a homozygous sequence variant in LAMA5 combines myopia, facial tics, and failure of neuromuscular transmission. Am. J. Med. Genet. A 2017, 173, 2240–2245. [Google Scholar] [CrossRef] [PubMed]
- Nicolau, S.; Milone, M. The Electrophysiology of Presynaptic Congenital Myasthenic Syndromes With and Without Facilitation: From Electrodiagnostic Findings to Molecular Mechanisms. Front. Neurol. 2019, 10, 257. [Google Scholar] [CrossRef]
- Shen, X.M.; Selcen, D.; Brengman, J.; Engel, A.G. Mutant SNAP25B causes myasthenia, cortical hyperexcitability, ataxia, and intellectual disability. Neurology 2014, 83, 2247–2255. [Google Scholar] [CrossRef] [PubMed]
- Ohno, K.; Anlar, B.; Ozdirim, E.; Brengman, J.M.; DeBleecker, J.L.; Engel, A.G. Myasthenic syndromes in Turkish kinships due to mutations in the acetylcholine receptor. Ann. Neurol. 1998, 44, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Guergueltcheva, V.; Muller, J.S.; Dusl, M.; Senderek, J.; Oldfors, A.; Lindbergh, C.; Maxwell, S.; Colomer, J.; Mallebrera, C.J.; Nascimento, A.; et al. Congenital myasthenic syndrome with tubular aggregates caused by GFPT1 mutations. J. Neurol. 2012, 259, 838–850. [Google Scholar] [CrossRef]
- Huh, S.Y.; Kim, H.S.; Jang, H.J.; Park, Y.E.; Kim, D.S. Limb-girdle myasthenia with tubular aggregates associated with novel GFPT1 mutations. Muscle Nerve 2012, 46, 600–604. [Google Scholar] [CrossRef]
- Selcen, D.; Shen, X.M.; Milone, M.; Brengman, J.; Ohno, K.; Deymeer, F.; Finkel, R.; Rowin, J.; Engel, A.G. GFPT1-myasthenia: Clinical, structural, and electrophysiologic heterogeneity. Neurology 2013, 81, 370–378. [Google Scholar] [CrossRef]
- Bauche, S.; Vellieux, G.; Sternberg, D.; Fontenille, M.J.; De Bruyckere, E.; Davoine, C.S.; Brochier, G.; Messeant, J.; Wolf, L.; Fardeau, M.; et al. Mutations in GFPT1-related congenital myasthenic syndromes are associated with synaptic morphological defects and underlie a tubular aggregate myopathy with synaptopathy. J. Neurol. 2017, 264, 1791–1803. [Google Scholar] [CrossRef] [PubMed]
- Belaya, K.; Finlayson, S.; Slater, C.R.; Cossins, J.; Liu, W.W.; Maxwell, S.; McGowan, S.J.; Maslau, S.; Twigg, S.R.; Walls, T.J.; et al. Mutations in DPAGT1 cause a limb-girdle congenital myasthenic syndrome with tubular aggregates. Am. J. Hum. Genet. 2012, 91, 193–201. [Google Scholar] [CrossRef]
- Finlayson, S.; Palace, J.; Belaya, K.; Walls, T.J.; Norwood, F.; Burke, G.; Holton, J.L.; Pascual-Pascual, S.I.; Cossins, J.; Beeson, D. Clinical features of congenital myasthenic syndrome due to mutations in DPAGT1. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1119–1125. [Google Scholar] [CrossRef]
- Selcen, D.; Shen, X.M.; Brengman, J.; Li, Y.; Stans, A.A.; Wieben, E.; Engel, A.G. DPAGT1 myasthenia and myopathy: Genetic, phenotypic, and expression studies. Neurology 2014, 82, 1822–1830. [Google Scholar] [CrossRef]
- Cossins, J.; Belaya, K.; Hicks, D.; Salih, M.A.; Finlayson, S.; Carboni, N.; Liu, W.W.; Maxwell, S.; Zoltowska, K.; Farsani, G.T.; et al. Congenital myasthenic syndromes due to mutations in ALG2 and ALG14. Brain 2013, 136 Pt 3, 944–956. [Google Scholar] [CrossRef]
- Belaya, K.; Rodriguez Cruz, P.M.; Liu, W.W.; Maxwell, S.; McGowan, S.; Farrugia, M.E.; Petty, R.; Walls, T.J.; Sedghi, M.; Basiri, K.; et al. Mutations in GMPPB cause congenital myasthenic syndrome and bridge myasthenic disorders with dystroglycanopathies. Brain 2015, 138 Pt 9, 2493–2504. [Google Scholar] [CrossRef] [PubMed]
- Engel, A.G.; Lambert, E.H.; Mulder, D.M.; Torres, C.F.; Sahashi, K.; Bertorini, T.E.; Whitaker, J.N. A newly recognized congenital myasthenic syndrome attributed to a prolonged open time of the acetylcholine-induced ion channel. Ann. Neurol. 1982, 11, 553–569. [Google Scholar] [CrossRef]
- Fidzianska, A.; Ryniewicz, B.; Shen, X.M.; Engel, A.G. IBM-type inclusions in a patient with slow-channel syndrome caused by a mutation in the AChR epsilon subunit. Neuromuscul. Disord. 2005, 15, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Carss, K.J.; Stevens, E.; Foley, A.R.; Cirak, S.; Riemersma, M.; Torelli, S.; Hoischen, A.; Willer, T.; van Scherpenzeel, M.; Moore, S.A.; et al. Mutations in GDP-mannose pyrophosphorylase B cause congenital and limb-girdle muscular dystrophies associated with hypoglycosylation of alpha-dystroglycan. Am. J. Hum. Genet. 2013, 93, 29–41. [Google Scholar] [CrossRef]
- Nicolau, S.; Liewluck, T.; Shen, X.M.; Selcen, D.; Engel, A.G.; Milone, M. A homozygous mutation in GMPPB leads to centronuclear myopathy with combined pre- and postsynaptic defects of neuromuscular transmission. Neuromuscul. Disord. 2019, 29, 614–617. [Google Scholar] [CrossRef] [PubMed]
- Souza, P.V.; Batistella, G.N.; Lino, V.C.; Pinto, W.B.; Annes, M.; Oliveira, A.S. Clinical and genetic basis of congenital myasthenic syndromes. Arq. Neuropsiquiatr. 2016, 74, 750–760. [Google Scholar] [CrossRef]
- Kao, J.C.; Milone, M.; Selcen, D.; Shen, X.M.; Engel, A.G.; Liewluck, T. Congenital myasthenic syndromes in adult neurology clinic: A long road to diagnosis and therapy. Neurology 2018, 91, e1770–e1777. [Google Scholar] [CrossRef]
- Lorenzoni, P.J.; Ducci, R.D.; Arndt, R.C.; Hrysay, N.M.C.; Fustes, O.J.H.; Topf, A.; Lochmuller, H.; Werneck, L.C.; Kay, C.S.K.; Scola, R.H. Congenital myasthenic syndrome in a cohort of patients with ‘double’ seronegative myasthenia gravis. Arq. Neuropsiquiatr. 2022, 80, 69–74. [Google Scholar] [CrossRef]
- Regal, L.; Martensson, E.; Maystadt, I.; Voermans, N.; Lederer, D.; Burlina, A.; Juan Fita, M.J.; Hoogeboom, A.J.M.; Olsson Engman, M.; Hollemans, T.; et al. PREPL deficiency: Delineation of the phenotype and development of a functional blood assay. Genet. Med. 2018, 20, 109–118. [Google Scholar] [CrossRef]
- Niles, K.M.; Blaser, S.; Shannon, P.; Chitayat, D. Fetal arthrogryposis multiplex congenita/fetal akinesia deformation sequence (FADS)-Aetiology, diagnosis, and management. Prenat. Diagn. 2019, 39, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Morgan, N.V.; Brueton, L.A.; Cox, P.; Greally, M.T.; Tolmie, J.; Pasha, S.; Aligianis, I.A.; van Bokhoven, H.; Marton, T.; Al-Gazali, L.; et al. Mutations in the embryonal subunit of the acetylcholine receptor (CHRNG) cause lethal and Escobar variants of multiple pterygium syndrome. Am. J. Hum. Genet. 2006, 79, 390–395. [Google Scholar] [CrossRef]
- Hoffmann, K.; Muller, J.S.; Stricker, S.; Megarbane, A.; Rajab, A.; Lindner, T.H.; Cohen, M.; Chouery, E.; Adaimy, L.; Ghanem, I.; et al. Escobar syndrome is a prenatal myasthenia caused by disruption of the acetylcholine receptor fetal gamma subunit. Am. J. Hum. Genet. 2006, 79, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Choi, I.H.; Lee, J.S.; Yoo, Y.; Kim, N.K.; Choi, M.; Ko, J.M.; Shin, Y.B. Rare cases of congenital arthrogryposis multiplex caused by novel recurrent CHRNG mutations. J. Hum. Genet. 2015, 60, 213–215. [Google Scholar] [CrossRef]
- Bayram, Y.; Karaca, E.; Coban Akdemir, Z.; Yilmaz, E.O.; Tayfun, G.A.; Aydin, H.; Torun, D.; Bozdogan, S.T.; Gezdirici, A.; Isikay, S.; et al. Molecular etiology of arthrogryposis in multiple families of mostly Turkish origin. J. Clin. Investig. 2016, 126, 762–778. [Google Scholar] [CrossRef]
- Vogt, J.; Harrison, B.J.; Spearman, H.; Cossins, J.; Vermeer, S.; ten Cate, L.N.; Morgan, N.V.; Beeson, D.; Maher, E.R. Mutation analysis of CHRNA1, CHRNB1, CHRND, and RAPSN genes in multiple pterygium syndrome/fetal akinesia patients. Am. J. Hum. Genet. 2008, 82, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Winters, L.; Van Hoof, E.; De Catte, L.; Van Den Bogaert, K.; de Ravel, T.; De Waele, L.; Corveleyn, A.; Breckpot, J. Massive parallel sequencing identifies RAPSN and PDHA1 mutations causing fetal akinesia deformation sequence. Eur. J. Paediatr. Neurol. 2017, 21, 745–753. [Google Scholar] [CrossRef]
- Bauche, S.; O’Regan, S.; Azuma, Y.; Laffargue, F.; McMacken, G.; Sternberg, D.; Brochier, G.; Buon, C.; Bouzidi, N.; Topf, A.; et al. Impaired Presynaptic High-Affinity Choline Transporter Causes a Congenital Myasthenic Syndrome with Episodic Apnea. Am. J. Hum. Genet. 2016, 99, 753–761. [Google Scholar] [CrossRef]
- Parr, J.R.; Andrew, M.J.; Finnis, M.; Beeson, D.; Vincent, A.; Jayawant, S. How common is childhood myasthenia? The UK incidence and prevalence of autoimmune and congenital myasthenia. Arch. Dis. Child. 2014, 99, 539–542. [Google Scholar] [CrossRef]
- Mihaylova, V.; Scola, R.H.; Gervini, B.; Lorenzoni, P.J.; Kay, C.K.; Werneck, L.C.; Stucka, R.; Guergueltcheva, V.; von der Hagen, M.; Huebner, A.; et al. Molecular characterisation of congenital myasthenic syndromes in Southern Brazil. J. Neurol. Neurosurg. Psychiatry 2010, 81, 973–977. [Google Scholar] [CrossRef]
- Troha Gergeli, A.; Neubauer, D.; Golli, T.; Butenko, T.; Loboda, T.; Maver, A.; Osredkar, D. Prevalence and genetic subtypes of congenital myasthenic syndromes in the pediatric population of Slovenia. Eur. J. Paediatr. Neurol. 2020, 26, 34–38. [Google Scholar] [CrossRef]
- Natera-de Benito, D.; Topf, A.; Vilchez, J.J.; Gonzalez-Quereda, L.; Dominguez-Carral, J.; Diaz-Manera, J.; Ortez, C.; Bestue, M.; Gallano, P.; Dusl, M.; et al. Molecular characterization of congenital myasthenic syndromes in Spain. Neuromuscul. Disord. 2017, 27, 1087–1098. [Google Scholar] [CrossRef]
- Richard, P.; Gaudon, K.; Andreux, F.; Yasaki, E.; Prioleau, C.; Bauche, S.; Barois, A.; Ioos, C.; Mayer, M.; Routon, M.C.; et al. Possible founder effect of rapsyn N88K mutation and identification of novel rapsyn mutations in congenital myasthenic syndromes. J. Med. Genet. 2003, 40, e81. [Google Scholar] [CrossRef]
- Dunne, V.; Maselli, R.A. Common founder effect of rapsyn N88K studied using intragenic markers. J. Hum. Genet. 2004, 49, 366–369. [Google Scholar] [CrossRef]
- Muller, J.S.; Abicht, A.; Burke, G.; Cossins, J.; Richard, P.; Baumeister, S.K.; Stucka, R.; Eymard, B.; Hantai, D.; Beeson, D.; et al. The congenital myasthenic syndrome mutation RAPSN N88K derives from an ancient Indo-European founder. J. Med. Genet. 2004, 41, e104. [Google Scholar] [CrossRef]
- Ohno, K.; Engel, A.G. Lack of founder haplotype for the rapsyn N88K mutation: N88K is an ancient founder mutation or arises from multiple founders. J. Med. Genet. 2004, 41, e8. [Google Scholar] [CrossRef]
- Abicht, A.; Dusl, M.; Gallenmuller, C.; Guergueltcheva, V.; Schara, U.; Della Marina, A.; Wibbeler, E.; Almaras, S.; Mihaylova, V.; von der Hagen, M.; et al. Congenital myasthenic syndromes: Achievements and limitations of phenotype-guided gene-after-gene sequencing in diagnostic practice: A study of 680 patients. Hum. Mutat. 2012, 33, 1474–1484. [Google Scholar] [CrossRef]
- Croxen, R.; Newland, C.; Betty, M.; Vincent, A.; Newsom-Davis, J.; Beeson, D. Novel functional epsilon-subunit polypeptide generated by a single nucleotide deletion in acetylcholine receptor deficiency congenital myasthenic syndrome. Ann. Neurol. 1999, 46, 639–647. [Google Scholar] [CrossRef]
- Polavarapu, K.; Mathur, A.; Joshi, A.; Nashi, S.; Preethish-Kumar, V.; Bardhan, M.; Sharma, P.; Parveen, S.; Seth, M.; Vengalil, S.; et al. A founder mutation in the GMPPB gene [c.1000G > A (p.Asp334Asn)] causes a mild form of limb-girdle muscular dystrophy/congenital myasthenic syndrome (LGMD/CMS) in South Indian patients. Neurogenetics 2021, 22, 271–285. [Google Scholar] [CrossRef]
- Mroczek, M.; Durmus, H.; Topf, A.; Parman, Y.; Straub, V. Four Individuals with a Homozygous Mutation in Exon 1f of the PLEC Gene and Associated Myasthenic Features. Genes 2020, 11, 716. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, H.M.; Wen, T.; Farrell, A.; Mao, R.; Moore, B.; Boyden, S.E.; Bayrak-Toydemir, P.; Nicholas, T.J.; Rynearson, S.; Holt, C.; et al. Rapid genome sequencing identifies a novel de novo SNAP25 variant for neonatal congenital myasthenic syndrome. Cold Spring Harb. Mol. Case Stud. 2022, 8, a006242. [Google Scholar] [CrossRef]
- Qashqari, H.; McNiven, V.; Gonorazky, H.; Mendoza-Londono, R.; Hassan, A.; Kulkarni, T.; Amburgey, K.; Dowling, J.J. PURA syndrome: Neuromuscular junction manifestations with potential therapeutic implications. Neuromuscul. Disord. 2022, 32, 842–844. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, D.N.; Horvath, R.; Sowden, J.E.; Gonzalez, M.; Sanchez-Mejias, A.; Guan, Z.; Whittaker, R.G.; Almodovar, J.L.; Lane, M.; Bansagi, B.; et al. Synaptotagmin 2 mutations cause an autosomal-dominant form of Lambert-Eaton myasthenic syndrome and nonprogressive motor neuropathy. Am. J. Hum. Genet. 2014, 95, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Montes-Chinea, N.I.; Guan, Z.; Coutts, M.; Vidal, C.; Courel, S.; Rebelo, A.P.; Abreu, L.; Zuchner, S.; Littleton, J.T.; Saporta, M.A. Identification of a new SYT2 variant validates an unusual distal motor neuropathy phenotype. Neurol. Genet. 2018, 4, e282. [Google Scholar] [CrossRef] [PubMed]
- Donkervoort, S.; Mohassel, P.; Laugwitz, L.; Zaki, M.S.; Kamsteeg, E.J.; Maroofian, R.; Chao, K.R.; Verschuuren-Bemelmans, C.C.; Horber, V.; Fock, A.J.M.; et al. Biallelic loss of function variants in SYT2 cause a treatable congenital onset presynaptic myasthenic syndrome. Am. J. Med. Genet. A 2020, 182, 2272–2283. [Google Scholar] [CrossRef]
- Maselli, R.A.; van der Linden, H., Jr.; Ferns, M. Recessive congenital myasthenic syndrome caused by a homozygous mutation in SYT2 altering a highly conserved C-terminal amino acid sequence. Am. J. Med. Genet. A 2020, 182, 1744–1749. [Google Scholar] [CrossRef]
- Maselli, R.A.; Wei, D.T.; Hodgson, T.S.; Sampson, J.B.; Vazquez, J.; Smith, H.L.; Pytel, P.; Ferns, M. Dominant and recessive congenital myasthenic syndromes caused by SYT2 mutations. Muscle Nerve 2021, 64, 219–224. [Google Scholar] [CrossRef]
- Schara, U.; Lochmuller, H. Therapeutic strategies in congenital myasthenic syndromes. Neurotherapeutics 2008, 5, 542–547. [Google Scholar] [CrossRef]
- Wargon, I.; Richard, P.; Kuntzer, T.; Sternberg, D.; Nafissi, S.; Gaudon, K.; Lebail, A.; Bauche, S.; Hantai, D.; Fournier, E.; et al. Long-term follow-up of patients with congenital myasthenic syndrome caused by COLQ mutations. Neuromuscul. Disord. 2012, 22, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Yis, U.; Becker, K.; Kurul, S.H.; Uyanik, G.; Bayram, E.; Haliloglu, G.; Polat, A.I.; Ayanoglu, M.; Okur, D.; Tosun, A.F.; et al. Genetic Landscape of Congenital Myasthenic Syndromes From Turkey: Novel Mutations and Clinical Insights. J. Child Neurol. 2017, 32, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Durmus, H.; Shen, X.M.; Serdaroglu-Oflazer, P.; Kara, B.; Parman-Gulsen, Y.; Ozdemir, C.; Brengman, J.; Deymeer, F.; Engel, A.G. Congenital myasthenic syndromes in Turkey: Clinical clues and prognosis with long term follow-up. Neuromuscul. Disord. 2018, 28, 315–322. [Google Scholar] [CrossRef]
- Maselli, R.A.; Ng, J.J.; Anderson, J.A.; Cagney, O.; Arredondo, J.; Williams, C.; Wessel, H.B.; Abdel-Hamid, H.; Wollmann, R.L. Mutations in LAMB2 causing a severe form of synaptic congenital myasthenic syndrome. J. Med. Genet. 2009, 46, 203–208. [Google Scholar] [CrossRef]
- Ben Ammar, A.; Petit, F.; Alexandri, N.; Gaudon, K.; Bauche, S.; Rouche, A.; Gras, D.; Fournier, E.; Koenig, J.; Stojkovic, T.; et al. Phenotype genotype analysis in 15 patients presenting a congenital myasthenic syndrome due to mutations in DOK7. J. Neurol. 2010, 257, 754–766. [Google Scholar] [CrossRef] [PubMed]
- Lashley, D.; Palace, J.; Jayawant, S.; Robb, S.; Beeson, D. Ephedrine treatment in congenital myasthenic syndrome due to mutations in DOK7. Neurology 2010, 74, 1517–1523. [Google Scholar] [CrossRef] [PubMed]
- Khadilkar, S.; Bhutada, A.; Nallamilli, B.; Hegde, M. Limb girdle weakness responding to salbutamol: An Indian family with DOK7 mutation. Indian Pediatr. 2015, 52, 243–244. [Google Scholar] [CrossRef]
- Lozowska, D.; Ringel, S.P.; Winder, T.L.; Liu, J.; Liewluck, T. Anticholinesterase Therapy Worsening Head Drop and Limb Weakness Due to a Novel DOK7 Mutation. J. Clin. Neuromuscul. Dis. 2015, 17, 72–77. [Google Scholar] [CrossRef]
- Luan, X.; Tian, W.; Cao, L. Limb-girdle congenital myasthenic syndrome in a Chinese family with novel mutations in MUSK gene and literature review. Clin. Neurol. Neurosurg. 2016, 150, 41–45. [Google Scholar] [CrossRef]
- Ohkawara, B.; Cabrera-Serrano, M.; Nakata, T.; Milone, M.; Asai, N.; Ito, K.; Ito, M.; Masuda, A.; Ito, Y.; Engel, A.G.; et al. LRP4 third beta-propeller domain mutations cause novel congenital myasthenia by compromising agrin-mediated MuSK signaling in a position-specific manner. Hum. Mol. Genet. 2014, 23, 1856–1868. [Google Scholar] [CrossRef]
- Khan, M.M.; Lustrino, D.; Silveira, W.A.; Wild, F.; Straka, T.; Issop, Y.; O’Connor, E.; Cox, D.; Reischl, M.; Marquardt, T.; et al. Sympathetic innervation controls homeostasis of neuromuscular junctions in health and disease. Proc. Natl. Acad. Sci. USA 2016, 113, 746–750. [Google Scholar] [CrossRef]
- Guven, A.; Demirci, M.; Anlar, B. Recurrent COLQ mutation in congenital myasthenic syndrome. Pediatr. Neurol. 2012, 46, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Fukudome, T.; Ohno, K.; Brengman, J.M.; Engel, A.G. Quinidine normalizes the open duration of slow-channel mutants of the acetylcholine receptor. Neuroreport 1998, 9, 1907–1911. [Google Scholar] [CrossRef]
- Harper, C.M.; Engel, A.G. Quinidine sulfate therapy for the slow-channel congenital myasthenic syndrome. Ann. Neurol. 1998, 43, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Harper, C.M.; Fukodome, T.; Engel, A.G. Treatment of slow-channel congenital myasthenic syndrome with fluoxetine. Neurology 2003, 60, 1710–1713. [Google Scholar] [CrossRef]
- Visser, A.C.; Laughlin, R.S.; Litchy, W.J.; Benarroch, E.E.; Milone, M. Rapsyn congenital myasthenic syndrome worsened by fluoxetine. Muscle Nerve 2017, 55, 131–135. [Google Scholar] [CrossRef]
- Vidanagamage, A.; Gooneratne, I.K.; Nandasiri, S.; Gunaratne, K.; Fernando, A.; Maxwell, S.; Cossins, J.; Beeson, D.; Chang, T. A rare mutation in the COLQ gene causing congenital myasthenic syndrome with remarkable improvement to fluoxetine: A case report. Neuromuscul. Disord. 2021, 31, 246–248. [Google Scholar] [CrossRef] [PubMed]
- Tsujino, A.; Maertens, C.; Ohno, K.; Shen, X.M.; Fukuda, T.; Harper, C.M.; Cannon, S.C.; Engel, A.G. Myasthenic syndrome caused by mutation of the SCN4A sodium channel. Proc. Natl. Acad. Sci. USA 2003, 100, 7377–7382. [Google Scholar] [CrossRef]
- Berghold, V.M.; Koko, M.; Berutti, R.; Plecko, B. Case report: Novel SCN4A variant associated with a severe congenital myasthenic syndrome/myopathy phenotype. Front. Pediatr. 2022, 10, 944784. [Google Scholar] [CrossRef]
- Habbout, K.; Poulin, H.; Rivier, F.; Giuliano, S.; Sternberg, D.; Fontaine, B.; Eymard, B.; Morales, R.J.; Echenne, B.; King, L.; et al. A recessive Nav1.4 mutation underlies congenital myasthenic syndrome with periodic paralysis. Neurology 2016, 86, 161–169. [Google Scholar] [CrossRef]
- O’Connell, K.; Rooney, T.; Alabaf, S.; Ramdas, S.; Beeson, D.; Palace, J. Pregnancy outcomes in patients with congenital myasthenic syndromes. Muscle Nerve 2022, 66, 345–348. [Google Scholar] [CrossRef] [PubMed]
- Desai, R.C.; Vyas, B.; Earles, C.A.; Littleton, J.T.; Kowalchyck, J.A.; Martin, T.F.; Chapman, E.R. The C2B domain of synaptotagmin is a Ca(2+)-sensing module essential for exocytosis. J. Cell Biol. 2000, 150, 1125–1136. [Google Scholar] [CrossRef]
- Apparsundaram, S.; Ferguson, S.M.; George, A.L., Jr.; Blakely, R.D. Molecular cloning of a human, hemicholinium-3-sensitive choline transporter. Biochem. Biophys. Res. Commun. 2000, 276, 862–867. [Google Scholar] [CrossRef] [PubMed]
- Erickson, J.D.; Varoqui, H.; Schafer, M.K.; Modi, W.; Diebler, M.F.; Weihe, E.; Rand, J.; Eiden, L.E.; Bonner, T.I.; Usdin, T.B. Functional identification of a vesicular acetylcholine transporter and its expression from a “cholinergic” gene locus. J. Biol. Chem. 1994, 269, 21929–21932. [Google Scholar] [CrossRef]
- Milone, M.; Wang, H.L.; Ohno, K.; Prince, R.; Fukudome, T.; Shen, X.M.; Brengman, J.M.; Griggs, R.C.; Sine, S.M.; Engel, A.G. Mode switching kinetics produced by a naturally occurring mutation in the cytoplasmic loop of the human acetylcholine receptor epsilon subunit. Neuron 1998, 20, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Unwin, N. Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J. Mol. Biol. 2005, 346, 967–989. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Stiegler, A.L.; Cameron, T.O.; Hallock, P.T.; Gomez, A.M.; Huang, J.H.; Hubbard, S.R.; Dustin, M.L.; Burden, S.J. Lrp4 is a receptor for Agrin and forms a complex with MuSK. Cell 2008, 135, 334–342. [Google Scholar] [CrossRef]
- Zhang, B.; Luo, S.; Wang, Q.; Suzuki, T.; Xiong, W.C.; Mei, L. LRP4 serves as a coreceptor of agrin. Neuron 2008, 60, 285–297. [Google Scholar] [CrossRef]
- Okada, K.; Inoue, A.; Okada, M.; Murata, Y.; Kakuta, S.; Jigami, T.; Kubo, S.; Shiraishi, H.; Eguchi, K.; Motomura, M.; et al. The muscle protein Dok-7 is essential for neuromuscular synaptogenesis. Science 2006, 312, 1802–1805. [Google Scholar] [CrossRef]
- Borges, L.S.; Yechikhov, S.; Lee, Y.I.; Rudell, J.B.; Friese, M.B.; Burden, S.J.; Ferns, M.J. Identification of a motif in the acetylcholine receptor beta subunit whose phosphorylation regulates rapsyn association and postsynaptic receptor localization. J. Neurosci. 2008, 28, 11468–11476. [Google Scholar] [CrossRef]
- Xing, G.; Jing, H.; Yu, Z.; Chen, P.; Wang, H.; Xiong, W.C.; Mei, L. Membraneless condensates by Rapsn phase separation as a platform for neuromuscular junction formation. Neuron 2021, 109, 1963–1978 e5. [Google Scholar] [CrossRef]
- Wu, H.; Lu, Y.; Shen, C.; Patel, N.; Gan, L.; Xiong, W.C.; Mei, L. Distinct roles of muscle and motoneuron LRP4 in neuromuscular junction formation. Neuron 2012, 75, 94–107. [Google Scholar] [CrossRef] [PubMed]
- Yumoto, N.; Kim, N.; Burden, S.J. Lrp4 is a retrograde signal for presynaptic differentiation at neuromuscular synapses. Nature 2012, 489, 438–442. [Google Scholar] [CrossRef]
- Nakashima, H.; Ohkawara, B.; Ishigaki, S.; Fukudome, T.; Ito, K.; Tsushima, M.; Konishi, H.; Okuno, T.; Yoshimura, T.; Ito, M.; et al. R-spondin 2 promotes acetylcholine receptor clustering at the neuromuscular junction via Lgr5. Sci. Rep. 2016, 6, 28512. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ito, M.; Ohkawara, B.; Masuda, A.; Ohno, K. Differential effects of spinal motor neuron-derived and skeletal muscle-derived Rspo2 on acetylcholine receptor clustering at the neuromuscular junction. Sci. Rep. 2018, 8, 13577. [Google Scholar] [CrossRef]
- Ito, K.; Ohkawara, B.; Yagi, H.; Nakashima, H.; Tsushima, M.; Ota, K.; Konishi, H.; Masuda, A.; Imagama, S.; Kiyama, H.; et al. Lack of Fgf18 causes abnormal clustering of motor nerve terminals at the neuromuscular junction with reduced acetylcholine receptor clusters. Sci. Rep. 2018, 8, 434. [Google Scholar] [CrossRef]
- Ohkawara, B.; Kobayakawa, A.; Kanbara, S.; Hattori, T.; Kubota, S.; Ito, M.; Masuda, A.; Takigawa, M.; Lyons, K.M.; Ishiguro, N.; et al. CTGF/CCN2 facilitates LRP4-mediated formation of the embryonic neuromuscular junction. EMBO Rep. 2020, 21, e48462. [Google Scholar] [CrossRef] [PubMed]
- Ohkawara, B.; Ito, M.; Ohno, K. Secreted Signaling Molecules at the Neuromuscular Junction in Physiology and Pathology. Int. J. Mol. Sci. 2021, 22, 2455. [Google Scholar] [CrossRef]
- Zhang, W.; Coldefy, A.S.; Hubbard, S.R.; Burden, S.J. Agrin binds to the N-terminal region of Lrp4 protein and stimulates association between Lrp4 and the first immunoglobulin-like domain in muscle-specific kinase (MuSK). J. Biol. Chem. 2011, 286, 40624–40630. [Google Scholar] [CrossRef] [Green Version]
- Zong, Y.; Zhang, B.; Gu, S.; Lee, K.; Zhou, J.; Yao, G.; Figueiredo, D.; Perry, K.; Mei, L.; Jin, R. Structural basis of agrin-LRP4-MuSK signaling. Genes Dev. 2012, 26, 247–258. [Google Scholar] [CrossRef]
- Otsuka, K.; Ito, M.; Ohkawara, B.; Masuda, A.; Kawakami, Y.; Sahashi, K.; Nishida, H.; Mabuchi, N.; Takano, A.; Engel, A.G.; et al. Collagen Q and anti-MuSK autoantibody competitively suppress agrin/LRP4/MuSK signaling. Sci. Rep. 2015, 5, 13928. [Google Scholar] [CrossRef]
- Ohno, K.; Quiram, P.A.; Milone, M.; Wang, H.L.; Harper, M.C.; Pruitt, J.N., 2nd; Brengman, J.M.; Pao, L.; Fischbeck, K.H.; Crawford, T.O.; et al. Congenital myasthenic syndromes due to heteroallelic nonsense/missense mutations in the acetylcholine receptor epsilon subunit gene: Identification and functional characterization of six new mutations. Hum. Mol. Genet. 1997, 6, 753–766. [Google Scholar] [CrossRef]
- Ohno, K.; Anlar, B.; Engel, A.G. Congenital myasthenic syndrome caused by a mutation in the Ets-binding site of the promoter region of the acetylcholine receptor epsilon subunit gene. Neuromuscul. Disord. 1999, 9, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Ohno, K.; Milone, M.; Shen, X.M.; Engel, A.G. A frameshifting mutation in CHRNE unmasks skipping of the preceding exon. Hum. Mol. Genet. 2003, 12, 3055–3066. [Google Scholar] [CrossRef]
- Milone, M.; Shen, X.M.; Ohno, K.; Harper, M.C.; Fukudome, T.; Stilling, G.; Brengman, J.M.; Engel, A.G. Unusual congenital myasthenic syndrome (CMS) with endplate (EP) AChR deficiency caused by alpha subunit mutations and a remitting relapsing clinical course. Neurology 1999, 52, A185–A186. [Google Scholar]
- Shen, X.M.; Brengman, J.M.; Sine, S.M.; Engel, A.G. Myasthenic syndrome AChRalpha C-loop mutant disrupts initiation of channel gating. J. Clin. Investig. 2012, 122, 2613–2621. [Google Scholar] [CrossRef] [PubMed]
- Azuma, Y.; Nakata, T.; Tanaka, M.; Shen, X.M.; Ito, M.; Iwata, S.; Okuno, T.; Nomura, Y.; Ando, N.; Ishigaki, K.; et al. Congenital myasthenic syndrome in Japan: Ethnically unique mutations in muscle nicotinic acetylcholine receptor subunits. Neuromuscul. Disord. 2015, 25, 60–69. [Google Scholar] [CrossRef]
- Masuda, A.; Shen, X.M.; Ito, M.; Matsuura, T.; Engel, A.G.; Ohno, K. hnRNP H enhances skipping of a nonfunctional exon P3A in CHRNA1 and a mutation disrupting its binding causes congenital myasthenic syndrome. Hum. Mol. Genet. 2008, 17, 4022–4035. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.A.; Masuda, A.; Ohe, K.; Ito, M.; Hutchinson, D.O.; Mayeda, A.; Engel, A.G.; Ohno, K. HnRNP L and hnRNP LL antagonistically modulate PTB-mediated splicing suppression of CHRNA1 pre-mRNA. Sci. Rep. 2013, 3, 2931. [Google Scholar] [CrossRef]
- Ohno, K.; Rahman, M.A.; Nazim, M.; Nasrin, F.; Lin, Y.; Takeda, J.I.; Masuda, A. Splicing regulation and dysregulation of cholinergic genes expressed at the neuromuscular junction. J. Neurochem. 2017, 142 (Suppl. 2), 64–72. [Google Scholar] [CrossRef] [Green Version]
- Ohno, K.; Engel, A.G.; Shen, X.M.; Selcen, D.; Brengman, J.; Harper, C.M.; Tsujino, A.; Milone, M. Rapsyn mutations in humans cause endplate acetylcholine-receptor deficiency and myasthenic syndrome. Am. J. Hum. Genet. 2002, 70, 875–885. [Google Scholar] [CrossRef]
- Cossins, J.; Burke, G.; Maxwell, S.; Spearman, H.; Man, S.; Kuks, J.; Vincent, A.; Palace, J.; Fuhrer, C.; Beeson, D. Diverse molecular mechanisms involved in AChR deficiency due to rapsyn mutations. Brain 2006, 129 Pt 10, 2773–2783. [Google Scholar] [CrossRef] [PubMed]
- Xing, G.; Jing, H.; Zhang, L.; Cao, Y.; Li, L.; Zhao, K.; Dong, Z.; Chen, W.; Wang, H.; Cao, R.; et al. A mechanism in agrin signaling revealed by a prevalent Rapsyn mutation in congenital myasthenic syndrome. eLife 2019, 8, e49180. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.W.; Wong, K.S.; Leung, H.W.; Law, C.Y. Limb girdle myasthenia with digenic RAPSN and a novel disease gene AK9 mutations. Eur. J. Hum. Genet. 2017, 25, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Milone, M.; Shen, X.M.; Selcen, D.; Ohno, K.; Brengman, J.; Iannaccone, S.T.; Harper, C.M.; Engel, A.G. Myasthenic syndrome due to defects in rapsyn: Clinical and molecular findings in 39 patients. Neurology 2009, 73, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Engel, A.G.; Ohno, K.; Bouzat, C.; Sine, S.M.; Griggs, R.C. End-plate acetylcholine receptor deficiency due to nonsense mutations in the epsilon subunit. Ann. Neurol. 1996, 40, 810–817. [Google Scholar] [CrossRef] [PubMed]
- Burke, G.; Cossins, J.; Maxwell, S.; Owens, G.; Vincent, A.; Robb, S.; Nicolle, M.; Hilton-Jones, D.; Newsom-Davis, J.; Palace, J.; et al. Rapsyn mutations in hereditary myasthenia: Distinct early- and late-onset phenotypes. Neurology 2003, 61, 826–828. [Google Scholar] [CrossRef]
- Dunne, V.; Maselli, R.A. Identification of pathogenic mutations in the human rapsyn gene. J. Hum. Genet. 2003, 48, 204–207. [Google Scholar] [CrossRef]
- Maselli, R.A.; Dunne, V.; Pascual-Pascual, S.I.; Bowe, C.; Agius, M.; Frank, R.; Wollmann, R.L. Rapsyn mutations in myasthenic syndrome due to impaired receptor clustering. Muscle Nerve 2003, 28, 293–301. [Google Scholar] [CrossRef]
- Muller, J.S.; Mildner, G.; Muller-Felber, W.; Schara, U.; Krampfl, K.; Petersen, B.; Petrova, S.; Stucka, R.; Mortier, W.; Bufler, J.; et al. Rapsyn N88K is a frequent cause of congenital myasthenic syndromes in European patients. Neurology 2003, 60, 1805–1810. [Google Scholar] [CrossRef]
- Ohno, K.; Sadeh, M.; Blatt, I.; Brengman, J.M.; Engel, A.G. E-box mutations in the RAPSN promoter region in eight cases with congenital myasthenic syndrome. Hum. Mol. Genet. 2003, 12, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Banwell, B.L.; Ohno, K.; Sieb, J.P.; Engel, A.G. Novel truncating RAPSN mutations causing congenital myasthenic syndrome responsive to 3,4-diaminopyridine. Neuromuscul. Disord. 2004, 14, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Muller, J.S.; Abicht, A.; Christen, H.J.; Stucka, R.; Schara, U.; Mortier, W.; Huebner, A.; Lochmuller, H. A newly identified chromosomal microdeletion of the rapsyn gene causes a congenital myasthenic syndrome. Neuromuscul. Disord. 2004, 14, 744–749. [Google Scholar] [CrossRef]
- Yasaki, E.; Prioleau, C.; Barbier, J.; Richard, P.; Andreux, F.; Leroy, J.P.; Dartevelle, P.; Koenig, J.; Molgo, J.; Fardeau, M.; et al. Electrophysiological and morphological characterization of a case of autosomal recessive congenital myasthenic syndrome with acetylcholine receptor deficiency due to a N88K rapsyn homozygous mutation. Neuromuscul. Disord. 2004, 14, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Ioos, C.; Barois, A.; Richard, P.; Eymard, B.; Hantai, D.; Estournet-Mathiaud, B. Congenital myasthenic syndrome due to rapsyn deficiency: Three cases with arthrogryposis and bulbar symptoms. Neuropediatrics 2004, 35, 246–249. [Google Scholar] [CrossRef]
- Muller, J.S.; Baumeister, S.K.; Rasic, V.M.; Krause, S.; Todorovic, S.; Kugler, K.; Muller-Felber, W.; Abicht, A.; Lochmuller, H. Impaired receptor clustering in congenital myasthenic syndrome with novel RAPSN mutations. Neurology 2006, 67, 1159–1164. [Google Scholar] [CrossRef] [PubMed]
- Maselli, R.; Dris, H.; Schnier, J.; Cockrell, J.; Wollmann, R. Congenital myasthenic syndrome caused by two non-N88K rapsyn mutations. Clin. Genet. 2007, 72, 63–65. [Google Scholar] [CrossRef]
- Gaudon, K.; Penisson-Besnier, I.; Chabrol, B.; Bouhour, F.; Demay, L.; Ben Ammar, A.; Bauche, S.; Vial, C.; Nicolas, G.; Eymard, B.; et al. Multiexon deletions account for 15% of congenital myasthenic syndromes with RAPSN mutations after negative DNA sequencing. J. Med. Genet. 2010, 47, 795–796. [Google Scholar] [CrossRef]
- Brugnoni, R.; Maggi, L.; Canioni, E.; Moroni, I.; Pantaleoni, C.; D’Arrigo, S.; Riva, D.; Cornelio, F.; Bernasconi, P.; Mantegazza, R. Identification of previously unreported mutations in CHRNA1, CHRNE and RAPSN genes in three unrelated Italian patients with congenital myasthenic syndromes. J. Neurol. 2010, 257, 1119–1123. [Google Scholar] [CrossRef]
- Alseth, E.H.; Maniaol, A.H.; Elsais, A.; Nakkestad, H.L.; Tallaksen, C.; Gilhus, N.E.; Skeie, G.O. Investigation for RAPSN and DOK-7 mutations in a cohort of seronegative myasthenia gravis patients. Muscle Nerve 2011, 43, 574–577. [Google Scholar] [CrossRef]
- Leshinsky-Silver, E.; Shapira, D.; Yosovitz, K.; Ginsberg, M.; Lerman-Sagie, T.; Lev, D. A novel mutation in the TPR6 domain of the RAPSN gene associated with congenital myasthenic syndrome. J. Neurol. Sci. 2012, 316, 112–115. [Google Scholar] [CrossRef]
- Lee, H.; Deignan, J.L.; Dorrani, N.; Strom, S.P.; Kantarci, S.; Quintero-Rivera, F.; Das, K.; Toy, T.; Harry, B.; Yourshaw, M.; et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA 2014, 312, 1880–1887. [Google Scholar] [CrossRef] [PubMed]
- Natera-de Benito, D.; Bestue, M.; Vilchez, J.J.; Evangelista, T.; Topf, A.; Garcia-Ribes, A.; Trujillo-Tiebas, M.J.; Garcia-Hoyos, M.; Ortez, C.; Camacho, A.; et al. Long-term follow-up in patients with congenital myasthenic syndrome due to RAPSN mutations. Neuromuscul. Disord. 2016, 26, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Aharoni, S.; Sadeh, M.; Shapira, Y.; Edvardson, S.; Daana, M.; Dor-Wollman, T.; Mimouni-Bloch, A.; Halevy, A.; Cohen, R.; Sagie, L.; et al. Congenital myasthenic syndrome in Israel: Genetic and clinical characterization. Neuromuscul. Disord. 2017, 27, 136–140. [Google Scholar] [CrossRef]
- Estephan, E.P.; Zambon, A.A.; Marchiori, P.E.; da Silva, A.M.S.; Caldas, V.M.; Moreno, C.A.M.; Reed, U.C.; Horvath, R.; Topf, A.; Lochmuller, H.; et al. Clinical variability of early-onset congenital myasthenic syndrome due to biallelic RAPSN mutations in Brazil. Neuromuscul. Disord. 2018, 28, 961–964. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, I.O.; Reynoso, C.; Chavez, G.; Engel, A.G. Congenital myasthenic syndrome due to rapsyn deficiency: A case report with a new mutation and compound heterozygosity. Medwave 2019, 19, e7645. [Google Scholar] [CrossRef]
- Liu, P.; Meng, L.; Normand, E.A.; Xia, F.; Song, X.; Ghazi, A.; Rosenfeld, J.; Magoulas, P.L.; Braxton, A.; Ward, P.; et al. Reanalysis of Clinical Exome Sequencing Data. N. Engl. J. Med. 2019, 380, 2478–2480. [Google Scholar] [CrossRef] [PubMed]
- Westra, D.; Schouten, M.I.; Stunnenberg, B.C.; Kusters, B.; Saris, C.G.J.; Erasmus, C.E.; van Engelen, B.G.; Bulk, S.; Verschuuren-Bemelmans, C.C.; Gerkes, E.H.; et al. Panel-Based Exome Sequencing for Neuromuscular Disorders as a Diagnostic Service. J. Neuromuscul. Dis. 2019, 6, 241–258. [Google Scholar] [CrossRef] [PubMed]
- Estephan, E.P.; Zambon, A.A.; Thompson, R.; Polavarapu, K.; Jomaa, D.; Topf, A.; Helito, P.V.P.; Heise, C.O.; Moreno, C.A.M.; Silva, A.M.S.; et al. Congenital myasthenic syndrome: Correlation between clinical features and molecular diagnosis. Eur. J. Neurol. 2022, 29, 833–842. [Google Scholar] [CrossRef]
- Krenn, M.; Sener, M.; Rath, J.; Zulehner, G.; Keritam, O.; Wagner, M.; Laccone, F.; Iglseder, S.; Marte, S.; Baumgartner, M.; et al. The clinical and molecular landscape of congenital myasthenic syndromes in Austria: A nationwide study. J. Neurol. 2022, 270, 909–916. [Google Scholar] [CrossRef]
- Ozturk, S.; Gulec, A.; Erdogan, M.; Demir, M.; Canpolat, M.; Gumus, H.; Caglayan, A.O.; Dundar, M.; Per, H. Congenital Myasthenic Syndromes in Turkey: Clinical and Molecular Characterization of 16 Cases With Three Novel Mutations. Pediatr. Neurol. 2022, 136, 43–49. [Google Scholar] [CrossRef]
- Saito, M.; Ogasawara, M.; Inaba, Y.; Osawa, Y.; Nishioka, M.; Yamauchi, S.; Atsumi, K.; Takeuchi, S.; Imai, K.; Motobayashi, M.; et al. Successful treatment of congenital myasthenic syndrome caused by a novel compound heterozygous variant in RAPSN. Brain Dev. 2022, 44, 50–55. [Google Scholar] [CrossRef]
- Sadeh, M.; Shen, X.M.; Engel, A.G. Beneficial effect of albuterol in congenital myasthenic syndrome with epsilon-subunit mutations. Muscle Nerve 2011, 44, 289–291. [Google Scholar] [CrossRef] [PubMed]
- Ishigaki, K.; Murakami, T.; Ito, Y.; Yanagisawa, A.; Kodaira, K.; Shishikura, K.; Suzuki, H.; Hirayama, Y.; Osawa, M. Treatment approach to congenital myasthenic syndrome in a patient with acetylcholine receptor deficiency. No To Hattatsu 2009, 41, 37–42. [Google Scholar]
- Erger, F.; Burau, K.; Elsasser, M.; Zimmermann, K.; Moog, U.; Netzer, C. Uniparental isodisomy as a cause of recessive Mendelian disease: A diagnostic pitfall with a quick and easy solution in medium/large NGS analyses. Eur. J. Hum. Genet. 2018, 26, 1392–1395. [Google Scholar] [CrossRef] [PubMed]
- Vogt, J.; Morgan, N.V.; Marton, T.; Maxwell, S.; Harrison, B.J.; Beeson, D.; Maher, E.R. Germline mutation in DOK7 associated with fetal akinesia deformation sequence. J. Med. Genet. 2009, 46, 338–340. [Google Scholar] [CrossRef]
- Michalk, A.; Stricker, S.; Becker, J.; Rupps, R.; Pantzar, T.; Miertus, J.; Botta, G.; Naretto, V.G.; Janetzki, C.; Yaqoob, N.; et al. Acetylcholine receptor pathway mutations explain various fetal akinesia deformation sequence disorders. Am. J. Hum. Genet. 2008, 82, 464–476. [Google Scholar] [CrossRef]
- Radhakrishnan, P.; Shukla, A.; Girisha, K.M.; Nayak, S.S. Biallelic c.1263dupC in DOK7 results in fetal akinesia deformation sequence. Am. J. Med. Genet. A 2020, 182, 804–807. [Google Scholar] [CrossRef] [PubMed]
- Hakonen, A.H.; Polvi, A.; Saloranta, C.; Paetau, A.; Heikkila, P.; Almusa, H.; Ellonen, P.; Jakkula, E.; Saarela, J.; Aittomaki, K. SLC18A3 variants lead to fetal akinesia deformation sequence early in pregnancy. Am. J. Med. Genet. A 2019, 179, 1362–1365. [Google Scholar] [CrossRef]
- Vogt, J.; Morgan, N.V.; Rehal, P.; Faivre, L.; Brueton, L.A.; Becker, K.; Fryns, J.P.; Holder, S.; Islam, L.; Kivuva, E.; et al. CHRNG genotype-phenotype correlations in the multiple pterygium syndromes. J. Med. Genet. 2012, 49, 21–26. [Google Scholar] [CrossRef]
- Al Kaissi, A.; Kenis, V.; Laptiev, S.; Ghachem, M.B.; Klaushofer, K.; Ganger, R.; Grill, F. Is webbing (pterygia) a constant feature in patients with Escobar syndrome? Orthop. Surg. 2013, 5, 297–301. [Google Scholar] [CrossRef]
- Laquerriere, A.; Maluenda, J.; Camus, A.; Fontenas, L.; Dieterich, K.; Nolent, F.; Zhou, J.; Monnier, N.; Latour, P.; Gentil, D.; et al. Mutations in CNTNAP1 and ADCY6 are responsible for severe arthrogryposis multiplex congenita with axoglial defects. Hum. Mol. Genet. 2014, 23, 2279–2289. [Google Scholar] [CrossRef] [PubMed]
- Sung, K.H.; Lee, S.H.; Kim, N.; Cho, T.J. Orthopaedic manifestations and treatment outcome of two siblings with Escobar syndrome and homozygous mutations in the CHRNG gene. J. Pediatr. Orthop. B 2015, 24, 262–267. [Google Scholar] [CrossRef]
- Retterer, K.; Juusola, J.; Cho, M.T.; Vitazka, P.; Millan, F.; Gibellini, F.; Vertino-Bell, A.; Smaoui, N.; Neidich, J.; Monaghan, K.G.; et al. Clinical application of whole-exome sequencing across clinical indications. Genet. Med. 2016, 18, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Abouelhoda, M.; Sobahy, T.; El-Kalioby, M.; Patel, N.; Shamseldin, H.; Monies, D.; Al-Tassan, N.; Ramzan, K.; Imtiaz, F.; Shaheen, R.; et al. Clinical genomics can facilitate countrywide estimation of autosomal recessive disease burden. Genet. Med. 2016, 18, 1244–1249. [Google Scholar] [CrossRef] [PubMed]
- Kariminejad, A.; Almadani, N.; Khoshaeen, A.; Olsson, B.; Moslemi, A.R.; Tajsharghi, H. Truncating CHRNG mutations associated with interfamilial variability of the severity of the Escobar variant of multiple pterygium syndrome. BMC Genet. 2016, 17, 71. [Google Scholar] [CrossRef] [PubMed]
- Monies, D.; Abouelhoda, M.; Assoum, M.; Moghrabi, N.; Rafiullah, R.; Almontashiri, N.; Alowain, M.; Alzaidan, H.; Alsayed, M.; Subhani, S.; et al. Lessons Learned from Large-Scale, First-Tier Clinical Exome Sequencing in a Highly Consanguineous Population. Am. J. Hum. Genet. 2019, 104, 1182–1201. [Google Scholar] [CrossRef]
- Pingel, J.; Andersen, J.D.; Christiansen, S.L.; Borsting, C.; Morling, N.; Lorentzen, J.; Kirk, H.; Doessing, S.; Wong, C.; Nielsen, J.B. Sequence variants in muscle tissue-related genes may determine the severity of muscle contractures in cerebral palsy. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2019, 180, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Pergande, M.; Motameny, S.; Ozdemir, O.; Kreutzer, M.; Wang, H.; Daimaguler, H.S.; Becker, K.; Karakaya, M.; Ehrhardt, H.; Elcioglu, N.; et al. The genomic and clinical landscape of fetal akinesia. Genet. Med. 2020, 22, 511–523. [Google Scholar] [CrossRef]
- Shamseldin, H.E.; Shaheen, R.; Ewida, N.; Bubshait, D.K.; Alkuraya, H.; Almardawi, E.; Howaidi, A.; Sabr, Y.; Abdalla, E.M.; Alfaifi, A.Y.; et al. The morbid genome of ciliopathies: An update. Genet. Med. 2020, 22, 1051–1060. [Google Scholar] [CrossRef]
- Croxen, R.; Hatton, C.; Shelley, C.; Brydson, M.; Chauplannaz, G.; Oosterhuis, H.; Vincent, A.; Newsom-Davis, J.; Colquhoun, D.; Beeson, D. Recessive inheritance and variable penetrance of slow-channel congenital myasthenic syndromes. Neurology 2002, 59, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Croxen, R.; Hatton, C.; Shelley, C.; Brydson, M.; Chauplannaz, G.; Oosterhuis, H.; Vincent, A.; Newsom-Davis, J.; Colquhoun, D.; Beeson, D. Voluntary partial retraction of: Recessive inheritance and variable penetrance of slow-channel congenital myasthenic syndromes. Neurology 2009, 72, 294. [Google Scholar] [PubMed]
- Ohno, K.; Hutchinson, D.O.; Milone, M.; Brengman, J.M.; Bouzat, C.; Sine, S.M.; Engel, A.G. Congenital myasthenic syndrome caused by prolonged acetylcholine receptor channel openings due to a mutation in the M2 domain of the epsilon subunit. Proc. Natl. Acad. Sci. USA 1995, 92, 758–762. [Google Scholar] [CrossRef]
- Shen, X.M.; Okuno, T.; Milone, M.; Otsuka, K.; Takahashi, K.; Komaki, H.; Giles, E.; Ohno, K.; Engel, A.G. Mutations Causing Slow-Channel Myasthenia Reveal That a Valine Ring in the Channel Pore of Muscle AChR is Optimized for Stabilizing Channel Gating. Hum. Mutat. 2016, 37, 1051–1059. [Google Scholar] [CrossRef]
- Groshong, J.S.; Spencer, M.J.; Bhattacharyya, B.J.; Kudryashova, E.; Vohra, B.P.; Zayas, R.; Wollmann, R.L.; Miller, R.J.; Gomez, C.M. Calpain activation impairs neuromuscular transmission in a mouse model of the slow-channel myasthenic syndrome. J. Clin. Investig. 2007, 117, 2903–2912. [Google Scholar] [CrossRef]
- Di Castro, A.; Martinello, K.; Grassi, F.; Eusebi, F.; Engel, A.G. Pathogenic point mutations in a transmembrane domain of the epsilon subunit increase the Ca2+ permeability of the human endplate ACh receptor. J. Physiol. 2007, 579 Pt 3, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Milone, M.; Wang, H.L.; Ohno, K.; Fukudome, T.; Pruitt, J.N.; Bren, N.; Sine, S.M.; Engel, A.G. Slow-channel myasthenic syndrome caused by enhanced activation, desensitization, and agonist binding affinity attributable to mutation in the M2 domain of the acetylcholine receptor alpha subunit. J. Neurosci. 1997, 17, 5651–5665. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; Basta, T.; Teng, J.; Lee, M.; Worrell, B.T.; Stowell, M.H.B.; Hibbs, R.E. Structural mechanism of muscle nicotinic receptor desensitization and block by curare. Nat. Struct. Mol. Biol. 2022, 29, 386–394. [Google Scholar] [CrossRef]
- Ohno, K.; Wang, H.L.; Milone, M.; Bren, N.; Brengman, J.M.; Nakano, S.; Quiram, P.; Pruitt, J.N.; Sine, S.M.; Engel, A.G. Congenital myasthenic syndrome caused by decreased agonist binding affinity due to a mutation in the acetylcholine receptor epsilon subunit. Neuron 1996, 17, 157–170. [Google Scholar] [CrossRef]
- Shen, X.M.; Ohno, K.; Tsujino, A.; Brengman, J.M.; Gingold, M.; Sine, S.M.; Engel, A.G. Mutation causing severe myasthenia reveals functional asymmetry of AChR signature cystine loops in agonist binding and gating. J. Clin. Investig. 2003, 111, 497–505. [Google Scholar] [CrossRef]
- Shen, X.M.; Fukuda, T.; Ohno, K.; Sine, S.M.; Engel, A.G. Congenital myasthenia-related AChR delta subunit mutation interferes with intersubunit communication essential for channel gating. J. Clin. Investig. 2008, 118, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.M.; Brengman, J.M.; Edvardson, S.; Sine, S.M.; Engel, A.G. Highly fatal fast-channel syndrome caused by AChR epsilon subunit mutation at the agonist binding site. Neurology 2012, 79, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.L.; Ohno, K.; Milone, M.; Brengman, J.M.; Evoli, A.; Batocchi, A.P.; Middleton, L.T.; Christodoulou, K.; Engel, A.G.; Sine, S.M. Fundamental gating mechanism of nicotinic receptor channel revealed by mutation causing a congenital myasthenic syndrome. J. Gen. Physiol. 2000, 116, 449–462. [Google Scholar] [CrossRef]
- Shen, X.M.; Ohno, K.; Sine, S.M.; Engel, A.G. Subunit-specific contribution to agonist binding and channel gating revealed by inherited mutation in muscle acetylcholine receptor M3-M4 linker. Brain 2005, 128 Pt 2, 345–355. [Google Scholar] [CrossRef]
- Wang, H.L.; Milone, M.; Ohno, K.; Shen, X.M.; Tsujino, A.; Batocchi, A.P.; Tonali, P.; Brengman, J.; Engel, A.G.; Sine, S.M. Acetylcholine receptor M3 domain: Stereochemical and volume contributions to channel gating. Nat. Neurosci. 1999, 2, 226–233. [Google Scholar] [CrossRef]
- Sine, S.M.; Ohno, K.; Bouzat, C.; Auerbach, A.; Milone, M.; Pruitt, J.N.; Engel, A.G. Mutation of the acetylcholine receptor alpha subunit causes a slow-channel myasthenic syndrome by enhancing agonist binding affinity. Neuron 1995, 15, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Gomez, C.M.; Gammack, J.T. A leucine-to-phenylalanine substitution in the acetylcholine receptor ion channel in a family with the slow-channel syndrome. Neurology 1995, 45, 982–985. [Google Scholar] [CrossRef]
- Engel, A.G.; Ohno, K.; Milone, M.; Wang, H.L.; Nakano, S.; Bouzat, C.; Pruitt, J.N., 2nd; Hutchinson, D.O.; Brengman, J.M.; Bren, N.; et al. New mutations in acetylcholine receptor subunit genes reveal heterogeneity in the slow-channel congenital myasthenic syndrome. Hum. Mol. Genet. 1996, 5, 1217–1227. [Google Scholar] [CrossRef]
- Gomez, C.M.; Maselli, R.; Gammack, J.; Lasalde, J.; Tamamizu, S.; Cornblath, D.R.; Lehar, M.; McNamee, M.; Kuncl, R.W. A beta-subunit mutation in the acetylcholine receptor channel gate causes severe slow-channel syndrome. Ann. Neurol. 1996, 39, 712–723. [Google Scholar] [CrossRef]
- Milone, M.; Ohno, K.; Wang, H.L.; Fukudome, T.; Pruitt, J.N.; Sine, S.M.; Engel, A.G. Novel slow-channel syndrome due to mutation in the acetylcholine receptor (AChR) alpha subunit with increased conductance, nanomolar affinity for acetylcholine, and prolonged open durations of the AChR channel. Ann. Neurol. 1996, 40, 9. [Google Scholar]
- Croxen, R.; Newland, C.; Beeson, D.; Oosterhuis, H.; Chauplannaz, G.; Vincent, A.; Newsom-Davis, J. Mutations in different functional domains of the human muscle acetylcholine receptor alpha subunit in patients with the slow-channel congenital myasthenic syndrome. Hum. Mol. Genet. 1997, 6, 767–774. [Google Scholar] [CrossRef]
- Gomez, C.M.; Maselli, R.; Gundeck, J.E.; Chao, M.; Day, J.W.; Tamamizu, S.; Lasalde, J.A.; McNamee, M.; Wollmann, R.L. Slow-channel transgenic mice: A model of postsynaptic organellar degeneration at the neuromuscular junction. J. Neurosci. 1997, 17, 4170–4179. [Google Scholar] [CrossRef] [Green Version]
- Wintzen, A.R.; Plomp, J.J.; Molenaar, P.C.; van Dijk, J.G.; van Kempen, G.T.; Vos, R.M.; Wokke, J.H.; Vincent, A. Acquired slow-channel syndrome: A form of myasthenia gravis with prolonged open time of the acetylcholine receptor channel. Ann. Neurol. 1998, 44, 657–664. [Google Scholar] [CrossRef]
- Scola, R.H.; Werneck, L.C.; Iwamoto, F.M.; Comerlato, E.A.; Kay, C.K. Acquired slow-channel syndrome. Muscle Nerve 2000, 23, 1582–1585. [Google Scholar] [CrossRef]
- Gomez, C.M.; Maselli, R.A.; Vohra, B.P.; Navedo, M.; Stiles, J.R.; Charnet, P.; Schott, K.; Rojas, L.; Keesey, J.; Verity, A.; et al. Novel delta subunit mutation in slow-channel syndrome causes severe weakness by novel mechanisms. Ann. Neurol. 2002, 51, 102–112. [Google Scholar] [CrossRef]
- Hatton, C.J.; Shelley, C.; Brydson, M.; Beeson, D.; Colquhoun, D. Properties of the human muscle nicotinic receptor, and of the slow-channel myasthenic syndrome mutant epsilonL221F, inferred from maximum likelihood fits. J. Physiol. 2003, 547 Pt 3, 729–760. [Google Scholar] [CrossRef] [PubMed]
- Colomer, J.; Muller, J.S.; Vernet, A.; Nascimento, A.; Pons, M.; Gonzalez, V.; Abicht, A.; Lochmuller, H. Long-term improvement of slow-channel congenital myasthenic syndrome with fluoxetine. Neuromuscul. Disord. 2006, 16, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Lorenzoni, P.J.; Kay, C.S.; Arruda, W.O.; Scola, R.H.; Werneck, L.C. Neurophysiological study in slow-channel congenital myasthenic syndrome: Case report. Arq. Neuropsiquiatr. 2006, 64, 318–321. [Google Scholar] [CrossRef] [PubMed]
- Navedo, M.F.; Lasalde-Dominicci, J.A.; Baez-Pagan, C.A.; Diaz-Perez, L.; Rojas, L.V.; Maselli, R.A.; Staub, J.; Schott, K.; Zayas, R.; Gomez, C.M. Novel beta subunit mutation causes a slow-channel syndrome by enhancing activation and decreasing the rate of agonist dissociation. Mol. Cell Neurosci. 2006, 32, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.M.; Deymeer, F.; Sine, S.M.; Engel, A.G. Slow-channel mutation in acetylcholine receptor alphaM4 domain and its efficient knockdown. Ann. Neurol. 2006, 60, 128–136. [Google Scholar] [CrossRef]
- Outteryck, O.; Richard, P.; Lacour, A.; Fournier, E.; Zephir, H.; Gaudon, K.; Eymard, B.; Hantai, D.; Vermersch, P.; Stojkovic, T. Novel epsilon subunit mutation of the muscle acetylcholine receptor causing a slow-channel congenital myasthenic syndrome. J. Neurol. Neurosurg. Psychiatry 2009, 80, 450–451. [Google Scholar] [CrossRef] [PubMed]
- Chaouch, A.; Muller, J.S.; Guergueltcheva, V.; Dusl, M.; Schara, U.; Rakocevic-Stojanovic, V.; Lindberg, C.; Scola, R.H.; Werneck, L.C.; Colomer, J.; et al. A retrospective clinical study of the treatment of slow-channel congenital myasthenic syndrome. J. Neurol. 2012, 259, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Witoonpanich, R.; Pulkes, T.; Dejthevaporn, C.; Witoonpanich, P.; Yodnopklao, P.; Wetchaphanphesat, S.; Brengman, J.; Engel, A.G. Phenotypic heterogeneity in a large Thai slow-channel congenital myasthenic syndrome kinship: Correction. Neuromuscul. Disord. 2012, 22, 478. [Google Scholar] [CrossRef]
- Tan, J.Z.; Man, Y.; Xiao, F. A Missense Mutation in Epsilon-subunit of Acetylcholine Receptor Causing Autosomal Dominant Slow-channel Congenital Myasthenic Syndrome in a Chinese Family. Chin. Med. J. 2016, 129, 2596–2602. [Google Scholar] [CrossRef] [PubMed]
- Angelini, C.; Lispi, L.; Salvoro, C.; Mostacciuolo, M.L.; Vazza, G. Clinical and genetic characterization of an Italian family with slow-channel syndrome. Neurol. Sci. 2019, 40, 503–507. [Google Scholar] [CrossRef]
- Shen, X.M.; Milone, M.; Wang, H.L.; Banwell, B.; Selcen, D.; Sine, S.M.; Engel, A.G. Slow-channel myasthenia due to novel mutation in M2 domain of AChR delta subunit. Ann. Clin. Transl. Neurol. 2019, 6, 2066–2078. [Google Scholar] [CrossRef]
- Di, L.; Chen, H.; Lu, Y.; Selcen, D.; Engel, A.G.; Da, Y.; Shen, X.M. Determinants of the repetitive-CMAP occurrence and therapy efficacy in slow-channel myasthenia. Neurology 2020, 95, e2781–e2793. [Google Scholar] [CrossRef]
- Gooneratne, I.K.; Nandasiri, S.; Maxwell, S.; Webster, R.; Cossins, J.; Beeson, D.; Gunaratne, K.; Herath, L.; Senanayake, S.; Chang, T. Slow-Channel Congenital Myasthenic Syndrome due to a Novel Mutation in the Acetylcholine Receptor Alpha Subunit in a South Asian: A Case Report. J. Neuromuscul. Dis. 2021, 8, 163–167. [Google Scholar] [CrossRef]
- Huang, K.; Luo, Y.B.; Bi, F.F.; Yang, H. Pharmacological Strategy for Congenital Myasthenic Syndrome with CHRNE Mutations: A Meta-Analysis of Case Reports. Curr. Neuropharmacol. 2021, 19, 718–729. [Google Scholar] [CrossRef]
- Kudryavtsev, D.; Isaeva, A.; Barkova, D.; Spirova, E.; Mukhutdinova, R.; Kasheverov, I.; Tsetlin, V. Point Mutations of Nicotinic Receptor alpha1 Subunit Reveal New Molecular Features of G153S Slow-Channel Myasthenia. Molecules 2021, 26, 1278. [Google Scholar] [CrossRef]
- Santovito, L.S.; Brugnoni, R.; Banfi, P.; Maggi, L. Salbutamol as effective treatment in slow-channel syndrome- first report. Neurol. Sci. 2021, 42, 1611–1612. [Google Scholar] [CrossRef]
- Tawara, N.; Yamashita, S.; Takamatsu, K.; Yamasaki, Y.; Mukaino, A.; Nakane, S.; Farshadyeganeh, P.; Ohno, K.; Ando, Y. Efficacy of salbutamol monotherapy in slow-channel congenital myasthenic syndrome caused by a novel mutation in CHRND. Muscle Nerve 2021, 63, E30–E32. [Google Scholar] [CrossRef]
- Dejthevaporn, C.; Wetchaphanphesat, S.; Pulkes, T.; Rattanasiri, S.; Engel, A.G.; Witoonpanich, R. Treatment of slow-channel congenital myasthenic syndrome in a Thai family with fluoxetine. J. Clin. Neurosci. 2022, 96, 85–89. [Google Scholar] [CrossRef]
- Kinali, M.; Beeson, D.; Pitt, M.C.; Jungbluth, H.; Simonds, A.K.; Aloysius, A.; Cockerill, H.; Davis, T.; Palace, J.; Manzur, A.Y.; et al. Congenital myasthenic syndromes in childhood: Diagnostic and management challenges. J. Neuroimmunol. 2008, 201–202, 6–12. [Google Scholar] [CrossRef]
- Webster, R.G.; Cossins, J.; Lashley, D.; Maxwell, S.; Liu, W.W.; Wickens, J.R.; Martinez-Martinez, P.; de Baets, M.; Beeson, D. A mouse model of the slow channel myasthenic syndrome: Neuromuscular physiology and effects of ephedrine treatment. Exp. Neurol. 2013, 248, 286–298. [Google Scholar] [CrossRef]
- Finlayson, S.; Spillane, J.; Kullmann, D.M.; Howard, R.; Webster, R.; Palace, J.; Beeson, D. Slow channel congenital myasthenic syndrome responsive to a combination of fluoxetine and salbutamol. Muscle Nerve 2013, 47, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Brownlow, S.; Webster, R.; Croxen, R.; Brydson, M.; Neville, B.; Lin, J.P.; Vincent, A.; Newsom-Davis, J.; Beeson, D. Acetylcholine receptor delta subunit mutations underlie a fast-channel myasthenic syndrome and arthrogryposis multiplex congenita. J. Clin. Investig. 2001, 108, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.M.; Ohno, K.; Fukudome, T.; Tsujino, A.; Brengman, J.M.; De Vivo, D.C.; Packer, R.J.; Engel, A.G. Congenital myasthenic syndrome caused by low-expressor fast-channel AChR delta subunit mutation. Neurology 2002, 59, 1881–1888. [Google Scholar] [CrossRef]
- Sine, S.M.; Shen, X.M.; Wang, H.L.; Ohno, K.; Lee, W.Y.; Tsujino, A.; Brengmann, J.; Bren, N.; Vajsar, J.; Engel, A.G. Naturally occurring mutations at the acetylcholine receptor binding site independently alter ACh binding and channel gating. J. Gen. Physiol. 2002, 120, 483–496. [Google Scholar] [CrossRef]
- Sine, S.M.; Wang, H.L.; Ohno, K.; Shen, X.M.; Lee, W.Y.; Engel, A.G. Mechanistic diversity underlying fast channel congenital myasthenic syndromes. Ann. N. Y. Acad. Sci. 2003, 998, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Webster, R.; Brydson, M.; Croxen, R.; Newsom-Davis, J.; Vincent, A.; Beeson, D. Mutation in the AChR ion channel gate underlies a fast channel congenital myasthenic syndrome. Neurology 2004, 62, 1090–1096. [Google Scholar] [CrossRef]
- Palace, J.; Lashley, D.; Bailey, S.; Jayawant, S.; Carr, A.; McConville, J.; Robb, S.; Beeson, D. Clinical features in a series of fast channel congenital myasthenia syndrome. Neuromuscul. Disord. 2012, 22, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Webster, R.; Liu, W.W.; Chaouch, A.; Lochmuller, H.; Beeson, D. Fast-channel congenital myasthenic syndrome with a novel acetylcholine receptor mutation at the alpha-epsilon subunit interface. Neuromuscul. Disord. 2014, 24, 143–147. [Google Scholar] [CrossRef]
- Shen, X.M.; Di, L.; Shen, S.; Zhao, Y.; Neumeyer, A.M.; Selcen, D.; Sine, S.M.; Engel, A.G. A novel fast-channel myasthenia caused by mutation in beta subunit of AChR reveals subunit-specific contribution of the intracellular M1-M2 linker to channel gating. Exp. Neurol. 2020, 331, 113375. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.M.; Brengman, J.M.; Shen, S.; Durmus, H.; Preethish-Kumar, V.; Yuceyar, N.; Vengalil, S.; Nalini, A.; Deymeer, F.; Sine, S.M.; et al. Mutations causing congenital myasthenia reveal principal coupling pathway in the acetylcholine receptor epsilon-subunit. JCI Insight 2018, 3, e97826. [Google Scholar] [CrossRef] [PubMed]
- Deprez, P.; Inestrosa, N.C.; Krejci, E. Two different heparin-binding domains in the triple-helical domain of ColQ, the collagen tail subunit of synaptic acetylcholinesterase. J. Biol. Chem. 2003, 278, 23233–23242. [Google Scholar] [CrossRef]
- Peng, H.B.; Xie, H.; Rossi, S.G.; Rotundo, R.L. Acetylcholinesterase clustering at the neuromuscular junction involves perlecan and dystroglycan. J. Cell Biol. 1999, 145, 911–921. [Google Scholar] [CrossRef]
- Cartaud, A.; Strochlic, L.; Guerra, M.; Blanchard, B.; Lambergeon, M.; Krejci, E.; Cartaud, J.; Legay, C. MuSK is required for anchoring acetylcholinesterase at the neuromuscular junction. J. Cell Biol. 2004, 165, 505–515. [Google Scholar] [CrossRef]
- Kawakami, Y.; Ito, M.; Hirayama, M.; Sahashi, K.; Ohkawara, B.; Masuda, A.; Nishida, H.; Mabuchi, N.; Engel, A.G.; Ohno, K. Anti-MuSK autoantibodies block binding of collagen Q to MuSK. Neurology 2011, 77, 1819–1826. [Google Scholar] [CrossRef]
- Ohno, K.; Brengman, J.; Tsujino, A.; Engel, A.G. Human endplate acetylcholinesterase deficiency caused by mutations in the collagen-like tail subunit (ColQ) of the asymmetric enzyme. Proc. Natl. Acad. Sci. USA 1998, 95, 9654–9659. [Google Scholar] [CrossRef]
- Donger, C.; Krejci, E.; Serradell, A.P.; Eymard, B.; Bon, S.; Nicole, S.; Chateau, D.; Gary, F.; Fardeau, M.; Massoulie, J.; et al. Mutation in the human acetylcholinesterase-associated collagen gene, COLQ, is responsible for congenital myasthenic syndrome with end-plate acetylcholinesterase deficiency (Type Ic). Am. J. Hum. Genet. 1998, 63, 967–975. [Google Scholar] [CrossRef]
- Ohno, K.; Brengman, J.M.; Felice, K.J.; Cornblath, D.R.; Engel, A.G. Congenital end-plate acetylcholinesterase deficiency caused by a nonsense mutation and an A-->G splice-donor-site mutation at position +3 of the collagenlike-tail-subunit gene (COLQ): How does G at position +3 result in aberrant splicing? Am. J. Hum. Genet. 1999, 65, 635–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohno, K.; Engel, A.G.; Brengman, J.M.; Shen, X.-M.; Heidenrich, F.R.; Vincent, A.; Milone, M.; Tan, E.; Demirci, M.; Walsh, P.; et al. The spectrum of mutations causing endplate acetylcholinesterase deficiency. Ann. Neurol. 2000, 47, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Nakata, T.; Ito, M.; Azuma, Y.; Otsuka, K.; Noguchi, Y.; Komaki, H.; Okumura, A.; Shiraishi, K.; Masuda, A.; Natsume, J.; et al. Mutations in the C-terminal domain of ColQ in endplate acetylcholinesterase deficiency compromise ColQ-MuSK interaction. Hum. Mutat. 2013, 34, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.; Krejci, E.; Molgo, J.; Cunningham, J.M.; Massoulie, J.; Sanes, J.R. Genetic analysis of collagen Q: Roles in acetylcholinesterase and butyrylcholinesterase assembly and in synaptic structure and function. J. Cell Biol. 1999, 144, 1349–1360. [Google Scholar] [CrossRef]
- Ito, M.; Suzuki, Y.; Okada, T.; Fukudome, T.; Yoshimura, T.; Masuda, A.; Takeda, S.; Krejci, E.; Ohno, K. Protein-anchoring strategy for delivering acetylcholinesterase to the neuromuscular junction. Mol. Ther. 2012, 20, 1384–1392. [Google Scholar] [CrossRef]
- Bartels, C.F.; Zelinski, T.; Lockridge, O. Mutation at Codon-322 in the Human Acetylcholinesterase (Ache) Gene Accounts for Yt Blood-Group Polymorphism. Am. J. Hum. Genet. 1993, 52, 928–936. [Google Scholar]
- Kimbell, L.M.; Ohno, K.; Engel, A.G.; Rotundo, R.L. C-terminal and heparin-binding domains of collagenic tail subunit are both essential for anchoring acetylcholinesterase at the synapse. J. Biol. Chem. 2004, 279, 10997–11005. [Google Scholar] [CrossRef]
- Rogers, R.S.; Nishimune, H. The role of laminins in the organization and function of neuromuscular junctions. Matrix Biol. 2017, 57–58, 86–105. [Google Scholar] [CrossRef]
- Carlson, S.S.; Valdez, G.; Sanes, J.R. Presynaptic calcium channels and alpha3-integrins are complexed with synaptic cleft laminins, cytoskeletal elements and active zone components. J. Neurochem. 2010, 115, 654–666. [Google Scholar] [CrossRef]
- Chen, J.; Billings, S.E.; Nishimune, H. Calcium channels link the muscle-derived synapse organizer laminin beta2 to Bassoon and CAST/Erc2 to organize presynaptic active zones. J. Neurosci. 2011, 31, 512–525. [Google Scholar] [CrossRef] [PubMed]
- Nishimune, H.; Stanford, J.A.; Mori, Y. Role of exercise in maintaining the integrity of the neuromuscular junction. Muscle Nerve 2014, 49, 315–324. [Google Scholar] [CrossRef]
- Chen, F.; Liu, Y.; Sugiura, Y.; Allen, P.D.; Gregg, R.G.; Lin, W. Neuromuscular synaptic patterning requires the function of skeletal muscle dihydropyridine receptors. Nat. Neurosci. 2011, 14, 570–577. [Google Scholar] [CrossRef] [Green Version]
- Zenker, M.; Aigner, T.; Wendler, O.; Tralau, T.; Muntefering, H.; Fenski, R.; Pitz, S.; Schumacher, V.; Royer-Pokora, B.; Wuhl, E.; et al. Human laminin beta2 deficiency causes congenital nephrosis with mesangial sclerosis and distinct eye abnormalities. Hum. Mol. Genet. 2004, 13, 2625–2632. [Google Scholar] [CrossRef] [PubMed]
- Hasselbacher, K.; Wiggins, R.C.; Matejas, V.; Hinkes, B.G.; Mucha, B.; Hoskins, B.E.; Ozaltin, F.; Nurnberg, G.; Becker, C.; Hangan, D.; et al. Recessive missense mutations in LAMB2 expand the clinical spectrum of LAMB2-associated disorders. Kidney Int. 2006, 70, 1008–1012. [Google Scholar] [CrossRef] [PubMed]
- Noakes, P.G.; Gautam, M.; Mudd, J.; Sanes, J.R.; Merlie, J.P. Aberrant differentiation of neuromuscular junctions in mice lacking s-laminin/laminin beta 2. Nature 1995, 374, 258–262. [Google Scholar] [CrossRef]
- Latvanlehto, A.; Fox, M.A.; Sormunen, R.; Tu, H.; Oikarainen, T.; Koski, A.; Naumenko, N.; Shakirzyanova, A.; Kallio, M.; Ilves, M.; et al. Muscle-derived collagen XIII regulates maturation of the skeletal neuromuscular junction. J. Neurosci. 2010, 30, 12230–12241. [Google Scholar] [CrossRef]
- Logan, C.V.; Cossins, J.; Rodriguez Cruz, P.M.; Parry, D.A.; Maxwell, S.; Martinez-Martinez, P.; Riepsaame, J.; Abdelhamed, Z.A.; Lake, A.V.; Moran, M.; et al. Congenital Myasthenic Syndrome Type 19 Is Caused by Mutations in COL13A1, Encoding the Atypical Non-fibrillar Collagen Type XIII alpha1 Chain. Am. J. Hum. Genet. 2015, 97, 878–885. [Google Scholar] [CrossRef]
- Haronen, H.; Zainul, Z.; Tu, H.; Naumenko, N.; Sormunen, R.; Miinalainen, I.; Shakirzyanova, A.; Oikarainen, T.; Abdullin, A.; Martin, P.; et al. Collagen XIII secures pre- and postsynaptic integrity of the neuromuscular synapse. Hum. Mol. Genet. 2017, 26, 2076–2090. [Google Scholar] [CrossRef]
- Kemppainen, A.V.; Finnila, M.A.; Heikkinen, A.; Haronen, H.; Izzi, V.; Kauppinen, S.; Saarakkala, S.; Pihlajaniemi, T.; Koivunen, J. The CMS19 disease model specifies a pivotal role for collagen XIII in bone homeostasis. Sci. Rep. 2022, 12, 5866. [Google Scholar] [CrossRef]
- Ohno, K.; Brengman, J.; Engel, A. How does an A-to-G splice donor site mutation at position+3 result in aberrant splicing? A lesson learned from a mutation in the COLQ gene. Am. J. Hum. Genet. 1999, 65, A80. [Google Scholar] [CrossRef] [PubMed]
- Shapira, Y.A.; Sadeh, M.E.; Bergtraum, M.P.; Tsujino, A.; Ohno, K.; Shen, X.M.; Brengman, J.; Edwardson, S.; Matoth, I.; Engel, A.G. Three novel COLQ mutations and variation of phenotypic expressivity due to G240X. Neurology 2002, 58, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Ishigaki, K.; Nicolle, D.; Krejci, E.; Leroy, J.P.; Koenig, J.; Fardeau, M.; Eymard, B.; Hantai, D. Two novel mutations in the COLQ gene cause endplate acetylcholinesterase deficiency. Neuromuscul. Disord. 2003, 13, 236–244. [Google Scholar] [CrossRef]
- Muller, J.S.; Petrova, S.; Kiefer, R.; Stucka, R.; Konig, C.; Baumeister, S.K.; Huebner, A.; Lochmuller, H.; Abicht, A. Synaptic congenital myasthenic syndrome in three patients due to a novel missense mutation (T441A) of the COLQ gene. Neuropediatrics 2004, 35, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, F.; Hoppenz, M.; Klaeren, R.; Reimann, J.; Woelfle, J. Novel COLQ mutation 950delC in synaptic congenital myasthenic syndrome and symptomatic heterozygous relatives. Neuromuscul. Disord. 2007, 17, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, V.; Muller, J.S.; Vilchez, J.J.; Salih, M.A.; Kabiraj, M.M.; D’Amico, A.; Bertini, E.; Wolfle, J.; Schreiner, F.; Kurlemann, G.; et al. Clinical and molecular genetic findings in COLQ-mutant congenital myasthenic syndromes. Brain 2008, 131 Pt 3, 747–759. [Google Scholar] [CrossRef] [PubMed]
- Yeung, W.L.; Lam, C.W.; Ng, P.C. Intra-familial variation in clinical manifestations and response to ephedrine in siblings with congenital myasthenic syndrome caused by novel COLQ mutations. Dev. Med. Child Neurol. 2010, 52, e243–e244. [Google Scholar] [CrossRef]
- Duran, G.S.; Uzunhan, T.A.; Ekici, B.; Citak, A.; Aydinli, N.; Caliskan, M. Severe scoliosis in a patient with COLQ mutation and congenital myasthenic syndrome: A clue for diagnosis. Acta Neurol. Belg. 2013, 113, 531–532. [Google Scholar] [CrossRef]
- Arredondo, J.; Lara, M.; Ng, F.; Gochez, D.A.; Lee, D.C.; Logia, S.P.; Nguyen, J.; Maselli, R.A. COOH-terminal collagen Q (COLQ) mutants causing human deficiency of endplate acetylcholinesterase impair the interaction of ColQ with proteins of the basal lamina. Hum. Genet. 2014, 133, 599–616. [Google Scholar] [CrossRef]
- Matlik, H.N.; Milhem, R.M.; Saadeldin, I.Y.; Al-Jaibeji, H.S.; Al-Gazali, L.; Ali, B.R. Clinical and molecular analysis of a novel COLQ missense mutation causing congenital myasthenic syndrome in a Syrian family. Pediatr. Neurol. 2014, 51, 165–169. [Google Scholar] [CrossRef]
- Wang, W.; Wu, Y.; Wang, C.; Jiao, J.; Klein, C.J. Copy number analysis reveals a novel multiexon deletion of the COLQ gene in congenital myasthenia. Neurol. Genet. 2016, 2, e117. [Google Scholar] [CrossRef]
- Al-Muhaizea, M.A.; Al-Mobarak, S.B. COLQ-mutant Congenital Myasthenic Syndrome with Microcephaly: A Unique Case with Literature Review. Transl. Neurosci. 2017, 8, 65–69. [Google Scholar]
- Padmanabha, H.; Saini, A.G.; Sankhyan, N.; Singhi, P. COLQ-Related Congenital Myasthenic Syndrome and Response to Salbutamol Therapy. J. Clin. Neuromuscul. Dis. 2017, 18, 162–163. [Google Scholar] [CrossRef]
- Zhang, Q.L.; Xu, M.J.; Wang, T.L.; Zhu, Z.Q.; Lai, F.; Zheng, X.C. Newly discovered COLQ gene mutation and its clinical features in patients with acetyl cholinesterase deficiency. J. Integr. Neurosci. 2018, 17, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Laforgia, N.; De Cosmo, L.; Palumbo, O.; Ranieri, C.; Sesta, M.; Capodiferro, D.; Pantaleo, A.; Iapicca, P.; Lastella, P.; Capozza, M.; et al. The First Case of Congenital Myasthenic Syndrome Caused by a Large Homozygous Deletion in the C-Terminal Region of COLQ (Collagen Like Tail Subunit of Asymmetric Acetylcholinesterase) Protein. Genes 2020, 11, 1519. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.Q.; Zhang, J.W.; Wang, L.Y.; Xue, P.; An, H.B. Congenital myasthenic syndrome with COLQ gene mutation: Report of a case. Zhonghua Bing Li Xue Za Zhi 2020, 49, 269–271. [Google Scholar]
- Tay, C.G.; Fong, C.Y.; Li, L.; Ganesan, V.; Teh, C.M.; Gan, C.S.; Thong, M.K. Congenital myasthenic syndrome with novel pathogenic variants in the COLQ gene associated with the presence of antibodies to acetylcholine receptors. J. Clin. Neurosci. 2020, 72, 468–471. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Wang, C.; Lin, L.; Yuan, F.; Wang, S.; Wang, Y.; Wang, A.; Wang, C.; Wu, S.; Lan, X.; et al. Mechanisms of Congenital Myasthenia Caused by Three Mutations in the COLQ Gene. Front. Pediatr. 2021, 9, 679342. [Google Scholar] [CrossRef]
- Pallithanam, J.J.; Prabhudesai, S.P.; Naik, N.; Gauns, S. COLQ-Related Congenital Myasthenic Syndrome in a Child from Western India. Neurol. India 2021, 69, 228–229. [Google Scholar]
- Al-Sharif, F.; Alamer, M.F.; Taher, H.O.; Gazzaz, R.Y.; AlRuwaithi, A.O.; Miliany, T.T.; Alrufaihi, M.A.; Al Amer, A.F. Co-occurrence of Glycogen Storage Disease Type 2 and Congenital Myasthenic Syndrome Type 5 in a Pediatric Patient: A Case Report. Cureus 2022, 14, e26345. [Google Scholar] [CrossRef]
- El Kadiri, Y.; Ratbi, I.; Sefiani, A.; Lyahyai, J. Novel copy number variation of COLQ gene in a Moroccan patient with congenital myasthenic syndrome: A case report and review of the literature. BMC Neurol. 2022, 22, 292. [Google Scholar] [CrossRef]
- Yamashita, A.; Muramatsu, Y.; Matsuda, H.; Okamoto, H. General anesthesia for treating scoliosis with congenital myasthenia syndrome: A case report. JA Clin. Rep. 2022, 8, 70. [Google Scholar] [CrossRef] [PubMed]
- Bestue-Cardiel, M.; Saenz de Cabezon-Alvarez, A.; Capablo-Liesa, J.L.; Lopez-Pison, J.; Pena-Segura, J.L.; Martin-Martinez, J.; Engel, A.G. Congenital endplate acetylcholinesterase deficiency responsive to ephedrine. Neurology 2005, 65, 144–146. [Google Scholar] [CrossRef]
- Liewluck, T.; Selcen, D.; Engel, A.G. Beneficial effects of albuterol in congenital endplate acetylcholinesterase deficiency and Dok-7 myasthenia. Muscle Nerve 2011, 44, 789–794. [Google Scholar] [CrossRef]
- Chan, S.H.; Wong, V.C.; Engel, A.G. Neuromuscular junction acetylcholinesterase deficiency responsive to albuterol. Pediatr. Neurol. 2012, 47, 137–140. [Google Scholar] [CrossRef]
- Dusl, M.; Moreno, T.; Munell, F.; Macaya, A.; Gratacos, M.; Abicht, A.; Strom, T.M.; Lochmuller, H.; Senderek, J. Congenital myasthenic syndrome caused by novel COL13A1 mutations. J. Neurol. 2019, 266, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez Cruz, P.M.; Cossins, J.; Estephan, E.P.; Munell, F.; Selby, K.; Hirano, M.; Maroofin, R.; Mehrjardi, M.Y.V.; Chow, G.; Carr, A.; et al. The clinical spectrum of the congenital myasthenic syndrome resulting from COL13A1 mutations. Brain 2019, 142, 1547–1560. [Google Scholar] [CrossRef]
- Kediha, M.I.; Tazir, M.; Sternberg, D.; Eymard, B.; Alipacha, L. Moderate phenotype of a congenital myasthenic syndrome type 19 caused by mutation of the COL13A1 gene: A case report. J. Med. Case Rep. 2022, 16, 134. [Google Scholar] [CrossRef]
- Arnold, W.D.; Feldman, D.H.; Ramirez, S.; He, L.; Kassar, D.; Quick, A.; Klassen, T.L.; Lara, M.; Nguyen, J.; Kissel, J.T.; et al. Defective fast inactivation recovery of Nav 1.4 in congenital myasthenic syndrome. Ann. Neurol. 2015, 77, 840–850. [Google Scholar] [CrossRef] [PubMed]
- Statland, J.M.; Fontaine, B.; Hanna, M.G.; Johnson, N.E.; Kissel, J.T.; Sansone, V.A.; Shieh, P.B.; Tawil, R.N.; Trivedi, J.; Cannon, S.C.; et al. Review of the Diagnosis and Treatment of Periodic Paralysis. Muscle Nerve 2018, 57, 522–530. [Google Scholar] [CrossRef]
- Lerche, H.; Heine, R.; Pika, U.; George, A.L., Jr.; Mitrovic, N.; Browatzki, M.; Weiss, T.; Rivet-Bastide, M.; Franke, C.; Lomonaco, M.; et al. Human sodium channel myotonia: Slowed channel inactivation due to substitutions for a glycine within the III-IV linker. J. Physiol. 1993, 470, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Ptacek, L.J.; George, A.L., Jr.; Barchi, R.L.; Griggs, R.C.; Riggs, J.E.; Robertson, M.; Leppert, M.F. Mutations in an S4 segment of the adult skeletal muscle sodium channel cause paramyotonia congenita. Neuron 1992, 8, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Duan, H.Q.; Li, Q.X.; Luo, Y.B.; Bi, F.F.; Yang, H. Clinicopathological-genetic features of congenital myasthenic syndrome from a Chinese neuromuscular centre. J. Cell Mol. Med. 2022, 26, 3828–3836. [Google Scholar] [CrossRef]
- Ohkawara, B.; Shen, X.; Selcen, D.; Nazim, M.; Bril, V.; Tarnopolsky, M.A.; Brady, L.; Fukami, S.; Amato, A.A.; Yis, U.; et al. Congenital myasthenic syndrome-associated agrin variants affect clustering of acetylcholine receptors in a domain-specific manner. JCI Insight 2020, 5, e132023. [Google Scholar] [CrossRef] [PubMed]
- Leupin, O.; Piters, E.; Halleux, C.; Hu, S.; Kramer, I.; Morvan, F.; Bouwmeester, T.; Schirle, M.; Bueno-Lozano, M.; Fuentes, F.J.; et al. Bone overgrowth-associated mutations in the LRP4 gene impair sclerostin facilitator function. J. Biol. Chem. 2011, 286, 19489–19500. [Google Scholar] [CrossRef]
- Li, Y.; Pawlik, B.; Elcioglu, N.; Aglan, M.; Kayserili, H.; Yigit, G.; Percin, F.; Goodman, F.; Nurnberg, G.; Cenani, A.; et al. LRP4 mutations alter Wnt/beta-catenin signaling and cause limb and kidney malformations in Cenani-Lenz syndrome. Am. J. Hum. Genet. 2010, 86, 696–706. [Google Scholar] [CrossRef] [Green Version]
- Chevessier, F.; Faraut, B.; Ravel-Chapuis, A.; Richard, P.; Gaudon, K.; Bauche, S.; Prioleau, C.; Herbst, R.; Goillot, E.; Ioos, C.; et al. MUSK, a new target for mutations causing congenital myasthenic syndrome. Hum. Mol. Genet. 2004, 13, 3229–3240. [Google Scholar] [CrossRef]
- Maselli, R.A.; Arredondo, J.; Cagney, O.; Ng, J.J.; Anderson, J.A.; Williams, C.; Gerke, B.J.; Soliven, B.; Wollmann, R.L. Mutations in MUSK causing congenital myasthenic syndrome impair MuSK-Dok-7 interaction. Hum. Mol. Genet. 2010, 19, 2370–2379. [Google Scholar] [CrossRef]
- Beeson, D.; Higuchi, O.; Palace, J.; Cossins, J.; Spearman, H.; Maxwell, S.; Newsom-Davis, J.; Burke, G.; Fawcett, P.; Motomura, M.; et al. Dok-7 mutations underlie a neuromuscular junction synaptopathy. Science 2006, 313, 1975–1978. [Google Scholar] [CrossRef]
- Muller, J.S.; Herczegfalvi, A.; Vilchez, J.J.; Colomer, J.; Bachinski, L.L.; Mihaylova, V.; Santos, M.; Schara, U.; Deschauer, M.; Shevell, M.; et al. Phenotypical spectrum of DOK7 mutations in congenital myasthenic syndromes. Brain 2007, 130 Pt 6, 1497–1506. [Google Scholar] [CrossRef]
- Hamuro, J.; Higuchi, O.; Okada, K.; Ueno, M.; Iemura, S.; Natsume, T.; Spearman, H.; Beeson, D.; Yamanashi, Y. Mutations causing DOK7 congenital myasthenia ablate functional motifs in Dok-7. J. Biol. Chem. 2008, 283, 5518–5524. [Google Scholar] [CrossRef] [PubMed]
- Selcen, D.; Milone, M.; Shen, X.M.; Harper, C.M.; Stans, A.A.; Wieben, E.D.; Engel, A.G. Dok-7 myasthenia: Phenotypic and molecular genetic studies in 16 patients. Ann. Neurol. 2008, 64, 71–87. [Google Scholar] [CrossRef]
- Zhang, S.; Ohkawara, B.; Ito, M.; Huang, Z.; Zhao, F.; Nakata, T.; Takeuchi, T.; Sakurai, H.; Komaki, H.; Kamon, M.; et al. A mutation in DOK7 in congenital myasthenic syndrome forms aggresome in cultured cells, and reduces DOK7 expression and MuSK phosphorylation in patient-derived iPS cells. Hum. Mol. Genet. 2022, ddac306. [Google Scholar] [CrossRef] [PubMed]
- Huze, C.; Bauche, S.; Richard, P.; Chevessier, F.; Goillot, E.; Gaudon, K.; Ben Ammar, A.; Chaboud, A.; Grosjean, I.; Lecuyer, H.A.; et al. Identification of an agrin mutation that causes congenital myasthenia and affects synapse function. Am. J. Hum. Genet. 2009, 85, 155–167. [Google Scholar] [CrossRef]
- Maselli, R.A.; Fernandez, J.M.; Arredondo, J.; Navarro, C.; Ngo, M.; Beeson, D.; Cagney, O.; Williams, D.C.; Wollmann, R.L.; Yarov-Yarovoy, V.; et al. LG2 agrin mutation causing severe congenital myasthenic syndrome mimics functional characteristics of non-neural (z-) agrin. Hum. Genet. 2012, 131, 1123–1135. [Google Scholar] [CrossRef]
- Xi, J.; Yan, C.; Liu, W.W.; Qiao, K.; Lin, J.; Tian, X.; Wu, H.; Lu, J.; Wong, L.J.; Beeson, D.; et al. Novel SEA and LG2 Agrin mutations causing congenital Myasthenic syndrome. Orphanet J. Rare Dis. 2017, 12, 182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Dai, Y.; Han, J.N.; Chen, Z.H.; Ling, L.; Pu, C.Q.; Cui, L.Y.; Huang, X.S. A Novel AGRN Mutation Leads to Congenital Myasthenic Syndrome Only Affecting Limb-girdle Muscle. Chin. Med. J. 2017, 130, 2279–2282. [Google Scholar] [PubMed]
- Wu, L.; Brady, L.; Shoffner, J.; Tarnopolsky, M.A. Next-Generation Sequencing to Diagnose Muscular Dystrophy, Rhabdomyolysis, and HyperCKemia. Can. J. Neurol. Sci. 2018, 45, 262–268. [Google Scholar] [CrossRef]
- Gan, S.; Yang, H.; Xiao, T.; Pan, Z.; Wu, L. AGRN Gene Mutation Leads to Congenital Myasthenia Syndromes: A Pediatric Case Report and Literature Review. Neuropediatrics 2020, 51, 364–367. [Google Scholar] [CrossRef]
- Wang, A.; Xiao, Y.; Huang, P.; Liu, L.; Xiong, J.; Li, J.; Mao, D.; Liu, L. Novel NtA and LG1 Mutations in Agrin in a Single Patient Causes Congenital Myasthenic Syndrome. Front. Neurol. 2020, 11, 239. [Google Scholar] [CrossRef]
- Xia, P.; Xie, F.; Zhou, Z.J.; Lv, W. Novel LG1 Mutations in Agrin Causing Congenital Myasthenia Syndrome. Intern. Med. 2022, 61, 887–890. [Google Scholar] [CrossRef]
- Geremek, M.; Dudarewicz, L.; Obersztyn, E.; Paczkowska, M.; Smyk, M.; Sobecka, K.; Nowakowska, B. Null variants in AGRN cause lethal fetal akinesia deformation sequence. Clin. Genet. 2020, 97, 634–638. [Google Scholar] [CrossRef] [PubMed]
- Takata, A.; Miyake, N.; Tsurusaki, Y.; Fukai, R.; Miyatake, S.; Koshimizu, E.; Kushima, I.; Okada, T.; Morikawa, M.; Uno, Y.; et al. Integrative Analyses of De Novo Mutations Provide Deeper Biological Insights into Autism Spectrum Disorder. Cell Rep. 2018, 22, 734–747. [Google Scholar] [CrossRef]
- Previtali, S.C.; Zhao, E.; Lazarevic, D.; Pipitone, G.B.; Fabrizi, G.M.; Manganelli, F.; Mazzeo, A.; Pareyson, D.; Schenone, A.; Taroni, F.; et al. Expanding the spectrum of genes responsible for hereditary motor neuropathies. J. Neurol. Neurosurg. Psychiatry 2019, 90, 1171–1179. [Google Scholar] [CrossRef]
- Mihaylova, V.; Salih, M.A.; Mukhtar, M.M.; Abuzeid, H.A.; El-Sadig, S.M.; von der Hagen, M.; Huebner, A.; Nurnberg, G.; Abicht, A.; Muller, J.S.; et al. Refinement of the clinical phenotype in musk-related congenital myasthenic syndromes. Neurology 2009, 73, 1926–1928. [Google Scholar] [CrossRef] [PubMed]
- Ben Ammar, A.; Soltanzadeh, P.; Bauche, S.; Richard, P.; Goillot, E.; Herbst, R.; Gaudon, K.; Huze, C.; Schaeffer, L.; Yamanashi, Y.; et al. A mutation causes MuSK reduced sensitivity to agrin and congenital myasthenia. PLoS ONE 2013, 8, e53826. [Google Scholar] [CrossRef]
- Maggi, L.; Brugnoni, R.; Scaioli, V.; Winden, T.L.; Morandi, L.; Engel, A.G.; Mantegazza, R.; Bernasconi, P. Marked phenotypic variability in two siblings with congenital myasthenic syndrome due to mutations in MUSK. J. Neurol. 2013, 260, 2894–2896. [Google Scholar] [CrossRef]
- Gallenmuller, C.; Muller-Felber, W.; Dusl, M.; Stucka, R.; Guergueltcheva, V.; Blaschek, A.; von der Hagen, M.; Huebner, A.; Muller, J.S.; Lochmuller, H.; et al. Salbutamol-responsive limb-girdle congenital myasthenic syndrome due to a novel missense mutation and heteroallelic deletion in MUSK. Neuromuscul. Disord. 2014, 24, 31–35. [Google Scholar] [CrossRef]
- Al-Shahoumi, R.; Brady, L.I.; Schwartzentruber, J.; Tarnopolsky, M.A. Two cases of congenital myasthenic syndrome with vocal cord paralysis. Neurology 2015, 84, 1281–1282. [Google Scholar] [CrossRef]
- Giarrana, M.L.; Joset, P.; Sticht, H.; Robb, S.; Steindl, K.; Rauch, A.; Klein, A. A severe congenital myasthenic syndrome with “dropped head” caused by novel MUSK mutations. Muscle Nerve 2015, 52, 668–673. [Google Scholar] [CrossRef]
- Owen, D.; Topf, A.; Preethish-Kumar, V.; Lorenzoni, P.J.; Vroling, B.; Scola, R.H.; Dias-Tosta, E.; Geraldo, A.; Polavarapu, K.; Nashi, S.; et al. Recessive variants of MuSK are associated with late onset CMS and predominant limb girdle weakness. Am. J. Med. Genet. A 2018, 176, 1594–1601. [Google Scholar] [CrossRef]
- Pinto, M.V.; Saw, J.L.; Milone, M. Congenital Vocal Cord Paralysis and Late-Onset Limb-Girdle Weakness in MuSK-Congenital Myasthenic Syndrome. Front. Neurol. 2019, 10, 1300. [Google Scholar] [CrossRef]
- Shen, Y.; Wang, B.; Zheng, X.; Zhang, W.; Wu, H.; Hei, M. A Neonate With MuSK Congenital Myasthenic Syndrome Presenting With Refractory Respiratory Failure. Front. Pediatr. 2020, 8, 166. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez Cruz, P.M.; Cossins, J.; Cheung, J.; Maxwell, S.; Jayawant, S.; Herbst, R.; Waithe, D.; Kornev, A.P.; Palace, J.; Beeson, D. Congenital myasthenic syndrome due to mutations in MUSK suggests that the level of MuSK phosphorylation is crucial for governing synaptic structure. Hum. Mutat. 2020, 41, 619–631. [Google Scholar] [CrossRef]
- Tan-Sindhunata, M.B.; Mathijssen, I.B.; Smit, M.; Baas, F.; de Vries, J.I.; van der Voorn, J.P.; Kluijt, I.; Hagen, M.A.; Blom, E.W.; Sistermans, E.; et al. Identification of a Dutch founder mutation in MUSK causing fetal akinesia deformation sequence. Eur. J. Hum. Genet. 2015, 23, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Wilbe, M.; Ekvall, S.; Eurenius, K.; Ericson, K.; Casar-Borota, O.; Klar, J.; Dahl, N.; Ameur, A.; Anneren, G.; Bondeson, M.L. MuSK: A new target for lethal fetal akinesia deformation sequence (FADS). J. Med. Genet. 2015, 52, 195–202. [Google Scholar] [CrossRef]
- Palace, J.; Lashley, D.; Newsom-Davis, J.; Cossins, J.; Maxwell, S.; Kennett, R.; Jayawant, S.; Yamanashi, Y.; Beeson, D. Clinical features of the DOK7 neuromuscular junction synaptopathy. Brain 2007, 130 Pt 6, 1507–1515. [Google Scholar] [CrossRef]
- Anderson, J.A.; Ng, J.J.; Bowe, C.; McDonald, C.; Richman, D.P.; Wollmann, R.L.; Maselli, R.A. Variable phenotypes associated with mutations in DOK7. Muscle Nerve 2008, 37, 448–456. [Google Scholar] [CrossRef]
- Schara, U.; Barisic, N.; Deschauer, M.; Lindberg, C.; Straub, V.; Strigl-Pill, N.; Wendt, M.; Abicht, A.; Muller, J.S.; Lochmuller, H. Ephedrine therapy in eight patients with congenital myasthenic syndrome due to DOK7 mutations. Neuromuscul. Disord. 2009, 19, 828–832. [Google Scholar] [CrossRef]
- Srour, M.; Bolduc, V.; Guergueltcheva, V.; Lochmuller, H.; Gendron, D.; Shevell, M.I.; Poulin, C.; Mathieu, J.; Bouchard, J.P.; Brais, B. DOK7 mutations presenting as a proximal myopathy in French Canadians. Neuromuscul. Disord. 2010, 20, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Jephson, C.G.; Mills, N.A.; Pitt, M.C.; Beeson, D.; Aloysius, A.; Muntoni, F.; Robb, S.A.; Bailey, C.M. Congenital stridor with feeding difficulty as a presenting symptom of Dok7 congenital myasthenic syndrome. Int. J. Pediatr. Otorhinolaryngol. 2010, 74, 991–994. [Google Scholar] [CrossRef] [PubMed]
- Cossins, J.; Liu, W.W.; Belaya, K.; Maxwell, S.; Oldridge, M.; Lester, T.; Robb, S.; Beeson, D. The spectrum of mutations that underlie the neuromuscular junction synaptopathy in DOK7 congenital myasthenic syndrome. Hum. Mol. Genet. 2012, 21, 3765–3775. [Google Scholar] [CrossRef] [PubMed]
- Irahara, K.; Komaki, H.; Honda, R.; Okumura, A.; Shiraishi, K.; Kobayashi, Y.; Azuma, Y.; Nakata, T.; Ohya, Y.; Sasaki, M. Clinical features of congenital myasthenic syndrome in Japan. No To Hattatsu 2012, 44, 450–454. [Google Scholar]
- Mahjneh, I.; Lochmuller, H.; Muntoni, F.; Abicht, A. DOK7 limb-girdle myasthenic syndrome mimicking congenital muscular dystrophy. Neuromuscul. Disord. 2013, 23, 36–42. [Google Scholar] [CrossRef]
- Burke, G.; Hiscock, A.; Klein, A.; Niks, E.H.; Main, M.; Manzur, A.Y.; Ng, J.; de Vile, C.; Muntoni, F.; Beeson, D.; et al. Salbutamol benefits children with congenital myasthenic syndrome due to DOK7 mutations. Neuromuscul. Disord. 2013, 23, 170–175. [Google Scholar] [CrossRef]
- Lorenzoni, P.J.; Scola, R.H.; Kay, C.S.; Filla, L.; Miranda, A.P.; Pinheiro, J.M.; Chaouch, A.; Lochmuller, H.; Werneck, L.C. Salbutamol therapy in congenital myasthenic syndrome due to DOK7 mutation. J. Neurol. Sci. 2013, 331, 155–157. [Google Scholar] [CrossRef] [PubMed]
- Lorenzoni, P.J.; Scola, R.H.; Kay, C.S.; Lochmuller, H.; Werneck, L.C. Congenital myasthenic syndrome and minicore-like myopathy with DOK7 mutation. Muscle Nerve 2013, 48, 151–152. [Google Scholar] [CrossRef]
- Nishikawa, A.; Mori-Yoshimura, M.; Okamoto, T.; Oya, Y.; Nakata, T.; Ohno, K.; Murata, M. Beneficial effects of 3,4-diaminopyridine in a 26-year-old woman with DOK7 congenital myasthenic syndrome who was originally diagnosed with facioscapulohumeral dystrophy. Rinsho Shinkeigaku 2014, 54, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Witting, N.; Crone, C.; Duno, M.; Vissing, J. Clinical and neurophysiological response to pharmacological treatment of DOK7 congenital myasthenia in an older patient. Clin. Neurol. Neurosurg. 2015, 130, 168–170. [Google Scholar] [CrossRef]
- Bevilacqua, J.A.; Lara, M.; Diaz, J.; Campero, M.; Vazquez, J.; Maselli, R.A. Congenital Myasthenic Syndrome due to DOK7 mutations in a family from Chile. Eur. J. Transl. Myol. 2017, 27, 6832. [Google Scholar] [CrossRef]
- Gaist, D.; Mogensen, J.; Pedersen, E.G.; Schroder, H.D.; Vissing, J.; Andersen, H.; Hertz, J.M. DOK7 congenital myasthenia may be associated with severe mitral valve insufficiency. J. Neurol. Sci. 2017, 379, 217–218. [Google Scholar] [CrossRef]
- Daum, H.; Meiner, V.; Elpeleg, O.; Harel, T.; Collaborating, A. Fetal exome sequencing: Yield and limitations in a tertiary referral center. Ultrasound Obstet. Gynecol. 2019, 53, 80–86. [Google Scholar] [CrossRef]
- Tayade, K.; Salunkhe, M.; Agarwal, A.; Radhakrishnan, D.M.; Srivastava, A.K. DOK7 congenital myasthenic syndrome responsive to oral salbutamol. QJM 2022, 115, 323–324. [Google Scholar] [CrossRef]
- Bastos, P.; Barbosa, R.; Fernandes, M.; Alonso, I. A late-onset congenital myasthenic syndrome due to a heterozygous DOK7 mutation. Neuromuscul. Disord. 2020, 30, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Miyana, K.; Ishiyama, A.; Saito, Y.; Nishino, I. Tulobuterol is a potential therapeutic drug in congenital myasthenic syndrome. Pediatr. Int. 2022, 64, e15115. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.; Cruz, S.; Peres, J.; Santos, L.; Tavares, P.; Basto, J.P.; Salgado, V.; Valverde, A.H. DOK7 myasthenic syndrome with subacute adult onset during pregnancy and partial response to fluoxetine. Neuromuscul. Disord. 2018, 28, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Oury, J.; Zhang, W.; Leloup, N.; Koide, A.; Corrado, A.D.; Ketavarapu, G.; Hattori, T.; Koide, S.; Burden, S.J. Mechanism of disease and therapeutic rescue of Dok7 congenital myasthenia. Nature 2021, 595, 404–408. [Google Scholar] [CrossRef]
- Oh, S.J.; King, P.H.; Schindler, A. Life-Long Steroid Responsive Familial Myopathy With Docking Protein 7 Mutation. J. Clin. Neuromuscul. Dis. 2022, 24, 80–84. [Google Scholar] [CrossRef]
- Smith, F.J.; Eady, R.A.; Leigh, I.M.; McMillan, J.R.; Rugg, E.L.; Kelsell, D.P.; Bryant, S.P.; Spurr, N.K.; Geddes, J.F.; Kirtschig, G.; et al. Plectin deficiency results in muscular dystrophy with epidermolysis bullosa. Nat. Genet. 1996, 13, 450–457. [Google Scholar] [CrossRef]
- Gundesli, H.; Talim, B.; Korkusuz, P.; Balci-Hayta, B.; Cirak, S.; Akarsu, N.A.; Topaloglu, H.; Dincer, P. Mutation in exon 1f of PLEC, leading to disruption of plectin isoform 1f, causes autosomal-recessive limb-girdle muscular dystrophy. Am. J. Hum. Genet. 2010, 87, 834–841. [Google Scholar] [CrossRef]
- Banwell, B.L.; Russel, J.; Fukudome, T.; Shen, X.M.; Stilling, G.; Engel, A.G. Myopathy, myasthenic syndrome, and epidermolysis bullosa simplex due to plectin deficiency. J. Neuropathol. Exp. Neurol. 1999, 58, 832–846. [Google Scholar] [CrossRef] [PubMed]
- Maselli, R.A.; Arredondo, J.; Cagney, O.; Mozaffar, T.; Skinner, S.; Yousif, S.; Davis, R.R.; Gregg, J.P.; Sivak, M.; Konia, T.H.; et al. Congenital myasthenic syndrome associated with epidermolysis bullosa caused by homozygous mutations in PLEC1 and CHRNE. Clin. Genet. 2011, 80, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Selcen, D.; Juel, V.C.; Hobson-Webb, L.D.; Smith, E.C.; Stickler, D.E.; Bite, A.V.; Ohno, K.; Engel, A.G. Myasthenic syndrome caused by plectinopathy. Neurology 2011, 76, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Mihailovska, E.; Raith, M.; Valencia, R.G.; Fischer, I.; Al Banchaabouchi, M.; Herbst, R.; Wiche, G. Neuromuscular synapse integrity requires linkage of acetylcholine receptors to postsynaptic intermediate filament networks via rapsyn-plectin 1f complexes. Mol. Biol. Cell 2014, 25, 4130–4149. [Google Scholar] [CrossRef] [PubMed]
- Fine, J.D.; Stenn, J.; Johnson, L.; Wright, T.; Bock, H.G.; Horiguchi, Y. Autosomal recessive epidermolysis bullosa simplex. Generalized phenotypic features suggestive of junctional or dystrophic epidermolysis bullosa, and association with neuromuscular diseases. Arch. Dermatol. 1989, 125, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Fattahi, Z.; Kahrizi, K.; Nafissi, S.; Fadaee, M.; Abedini, S.S.; Kariminejad, A.; Akbari, M.R.; Najmabadi, H. Report of a patient with limb-girdle muscular dystrophy, ptosis and ophthalmoparesis caused by plectinopathy. Arch. Iran. Med. 2015, 18, 60–64. [Google Scholar]
- Gonzalez Garcia, A.; Tutmaher, M.S.; Upadhyayula, S.R.; Sanchez Russo, R.; Verma, S. Novel PLEC gene variants causing congenital myasthenic syndrome. Muscle Nerve 2019, 60, E40–E43. [Google Scholar] [CrossRef]
- Vahidnezhad, H.; Youssefian, L.; Harvey, N.; Tavasoli, A.R.; Saeidian, A.H.; Sotoudeh, S.; Varghaei, A.; Mahmoudi, H.; Mansouri, P.; Mozafari, N.; et al. Mutation update: The spectra of PLEC sequence variants and related plectinopathies. Hum. Mutat. 2022, 43, 1706–1731. [Google Scholar] [CrossRef]
- Ohno, K.; Tsujino, A.; Shen, X.-M.; Brengman, J.M.; Harper, C.M.; Bajzer, Z.; Udd, B.; Beyring, R.; Robb, S.; Kirkham, F.J.; et al. Choline acetyltransferase mutations cause myasthenic syndrome associated with episodic apnea in humans. Proc. Natl. Acad. Sci. USA 2001, 98, 2017–2022. [Google Scholar] [CrossRef]
- Shen, X.M.; Crawford, T.O.; Brengman, J.; Acsadi, G.; Iannaconne, S.; Karaca, E.; Khoury, C.; Mah, J.K.; Edvardson, S.; Bajzer, Z.; et al. Functional consequences and structural interpretation of mutations of human choline acetyltransferase. Hum. Mutat. 2011, 32, 1259–1267. [Google Scholar] [CrossRef] [PubMed]
- Stankiewicz, P.; Kulkarni, S.; Dharmadhikari, A.V.; Sampath, S.; Bhatt, S.S.; Shaikh, T.H.; Xia, Z.; Pursley, A.N.; Cooper, M.L.; Shinawi, M.; et al. Recurrent deletions and reciprocal duplications of 10q11.21q11.23 including CHAT and SLC18A3 are likely mediated by complex low-copy repeats. Hum. Mutat. 2012, 33, 165–179. [Google Scholar] [CrossRef]
- Schwartz, M.; Sternberg, D.; Whalen, S.; Afenjar, A.; Isapof, A.; Chabrol, B.; Portnoi, M.F.; Heide, S.; Keren, B.; Chantot-Bastaraud, S.; et al. How chromosomal deletions can unmask recessive mutations? Deletions in 10q11.2 associated with CHAT or SLC18A3 mutations lead to congenital myasthenic syndrome. Am. J. Med. Genet. A 2018, 176, 151–155. [Google Scholar] [CrossRef]
- Barwick, K.E.; Wright, J.; Al-Turki, S.; McEntagart, M.M.; Nair, A.; Chioza, B.; Al-Memar, A.; Modarres, H.; Reilly, M.M.; Dick, K.J.; et al. Defective presynaptic choline transport underlies hereditary motor neuropathy. Am. J. Hum. Genet. 2012, 91, 1103–1107. [Google Scholar] [CrossRef] [PubMed]
- Ingram, G.; Barwick, K.E.; Hartley, L.; McEntagart, M.; Crosby, A.H.; Llewelyn, G.; Morris, H.R. Distal hereditary motor neuropathy with vocal cord paresis: From difficulty in choral singing to a molecular genetic diagnosis. Pract. Neurol. 2016, 16, 247–251. [Google Scholar] [CrossRef]
- Wang, H.; Salter, C.G.; Refai, O.; Hardy, H.; Barwick, K.E.S.; Akpulat, U.; Kvarnung, M.; Chioza, B.A.; Harlalka, G.; Taylan, F.; et al. Choline transporter mutations in severe congenital myasthenic syndrome disrupt transporter localization. Brain 2017, 140, 2838–2850. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, S.M.; Bazalakova, M.; Savchenko, V.; Tapia, J.C.; Wright, J.; Blakely, R.D. Lethal impairment of cholinergic neurotransmission in hemicholinium-3-sensitive choline transporter knockout mice. Proc. Natl. Acad. Sci. USA 2004, 101, 8762–8767. [Google Scholar] [CrossRef] [PubMed]
- Lund, D.; Ruggiero, A.M.; Ferguson, S.M.; Wright, J.; English, B.A.; Reisz, P.A.; Whitaker, S.M.; Peltier, A.C.; Blakely, R.D. Motor neuron-specific overexpression of the presynaptic choline transporter: Impact on motor endurance and evoked muscle activity. Neuroscience 2010, 171, 1041–1053. [Google Scholar] [CrossRef]
- English, B.A.; Appalsamy, M.; Diedrich, A.; Ruggiero, A.M.; Lund, D.; Wright, J.; Keller, N.R.; Louderback, K.M.; Robertson, D.; Blakely, R.D. Tachycardia, reduced vagal capacity, and age-dependent ventricular dysfunction arising from diminished expression of the presynaptic choline transporter. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H799–H810. [Google Scholar] [CrossRef]
- Jaeken, J.; Martens, K.; Francois, I.; Eyskens, F.; Lecointre, C.; Derua, R.; Meulemans, S.; Slootstra, J.W.; Waelkens, E.; de Zegher, F.; et al. Deletion of PREPL, a gene encoding a putative serine oligopeptidase, in patients with hypotonia-cystinuria syndrome. Am. J. Hum. Genet. 2006, 78, 38–51. [Google Scholar] [CrossRef]
- Regal, L.; Shen, X.M.; Selcen, D.; Verhille, C.; Meulemans, S.; Creemers, J.W.; Engel, A.G. PREPL deficiency with or without cystinuria causes a novel myasthenic syndrome. Neurology 2014, 82, 1254–1260. [Google Scholar] [CrossRef]
- Barbosa, M.; Lopes, A.; Mota, C.; Martins, E.; Oliveira, J.; Alves, S.; De Bonis, P.; Mota Mdo, C.; Dias, C.; Rodrigues-Santos, P.; et al. Clinical, biochemical and molecular characterization of cystinuria in a cohort of 12 patients. Clin. Genet. 2012, 81, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Maselli, R.A.; Chen, D.; Mo, D.; Bowe, C.; Fenton, G.; Wollmann, R.L. Choline acetyltransferase mutations in myasthenic syndrome due to deficient acetylcholine resynthesis. Muscle Nerve 2003, 27, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.; Abicht, A.; Krampfl, K.; Voss, W.; Stucka, R.; Mildner, G.; Petrova, S.; Schara, U.; Mortier, W.; Bufler, J.; et al. Congenital myasthenic syndrome due to a novel missense mutation in the gene encoding choline acetyltransferase. Neuromuscul. Disord. 2003, 13, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Barisic, N.; Muller, J.S.; Paucic-Kirincic, E.; Gazdik, M.; Lah-Tomulic, K.; Pertl, A.; Sertic, J.; Zurak, N.; Lochmuller, H.; Abicht, A. Clinical variability of CMS-EA (congenital myasthenic syndrome with episodic apnea) due to identical CHAT mutations in two infants. Eur. J. Paediatr. Neurol. 2005, 9, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Mallory, L.A.; Shaw, J.G.; Burgess, S.L.; Estrella, E.; Nurko, S.; Burpee, T.M.; Agus, M.S.; Darras, B.T.; Kunkel, L.M.; Kang, P.B. Congenital myasthenic syndrome with episodic apnea. Pediatr. Neurol. 2009, 41, 42–45. [Google Scholar] [CrossRef]
- Yeung, W.L.; Lam, C.W.; Fung, L.W.; Hon, K.L.; Ng, P.C. Severe congenital myasthenia gravis of the presynaptic type with choline acetyltransferase mutation in a Chinese infant with respiratory failure. Neonatology 2009, 95, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Schara, U.; Christen, H.J.; Durmus, H.; Hietala, M.; Krabetz, K.; Rodolico, C.; Schreiber, G.; Topaloglu, H.; Talim, B.; Voss, W.; et al. Long-term follow-up in patients with congenital myasthenic syndrome due to CHAT mutations. Eur. J. Paediatr. Neurol. 2010, 14, 326–333. [Google Scholar] [CrossRef]
- Dilena, R.; Abicht, A.; Sergi, P.; Comi, G.P.; Di Fonzo, A.; Chidini, G.; Natacci, F.; Barbieri, S.; Lochmuller, H. Congenital myasthenic syndrome due to choline acetyltransferase mutations in infants: Clinical suspicion and comprehensive electrophysiological assessment are important for early diagnosis. J. Child Neurol. 2014, 29, 389–393. [Google Scholar] [CrossRef]
- Arredondo, J.; Lara, M.; Gospe, S.M., Jr.; Mazia, C.G.; Vaccarezza, M.; Garcia-Erro, M.; Bowe, C.M.; Chang, C.H.; Mezei, M.M.; Maselli, R.A. Choline Acetyltransferase Mutations Causing Congenital Myasthenic Syndrome: Molecular Findings and Genotype-Phenotype Correlations. Hum. Mutat. 2015, 36, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.S.; Ambang, T.; Ahmad-Annuar, A.; Rajahram, G.S.; Wong, K.T.; Goh, K.J. Congenital myasthenic syndrome due to novel CHAT mutations in an ethnic kadazandusun family. Muscle Nerve 2016, 53, 822–826. [Google Scholar] [CrossRef]
- Brunelli, L.; Mao, R.; Jenkins, S.M.; Bleyl, S.B.; Dames, S.A.; Miller, C.E.; Ostrander, B.; Tvrdik, T.; Andrews, S.; Flores, J.; et al. A rapid gene sequencing panel strategy to facilitate precision neonatal medicine. Am. J. Med. Genet. A 2017, 173, 1979–1982. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.M.; Fang, F.; Ding, C.H.; Zhang, W.H.; Deng, J.; Chen, C.H.; Wang, X.; Liu, J.; Li, Z.; Jia, X.L.; et al. Clinical and genetic characteristics of congenital myasthenia syndrome with episodic apnea caused by CHAT gene mutation: A report of 2 cases. Zhonghua Er Ke Za Zhi 2018, 56, 216–220. [Google Scholar] [PubMed]
- Zhang, Y.; Cheng, X.; Luo, C.; Lei, M.; Mao, F.; Shi, Z.; Cao, W.; Zhang, J.; Zhang, Q. Congenital Myasthenic Syndrome Caused by a Novel Hemizygous CHAT Mutation. Front. Pediatr. 2020, 8, 185. [Google Scholar] [CrossRef] [PubMed]
- O’Grady, G.L.; Verschuuren, C.; Yuen, M.; Webster, R.; Menezes, M.; Fock, J.M.; Pride, N.; Best, H.A.; Benavides Damm, T.; Turner, C.; et al. Variants in SLC18A3, vesicular acetylcholine transporter, cause congenital myasthenic syndrome. Neurology 2016, 87, 1442–1448. [Google Scholar] [CrossRef]
- Aran, A.; Segel, R.; Kaneshige, K.; Gulsuner, S.; Renbaum, P.; Oliphant, S.; Meirson, T.; Weinberg-Shukron, A.; Hershkovitz, Y.; Zeligson, S.; et al. Vesicular acetylcholine transporter defect underlies devastating congenital myasthenia syndrome. Neurology 2017, 88, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Lamond, A.; Buckley, D.; O’Dea, J.; Turner, L. Variants of SLC18A3 leading to congenital myasthenic syndrome in two children with varying presentations. BMJ Case Rep. 2021, 14, e237799. [Google Scholar] [CrossRef] [PubMed]
- Pardal-Fernandez, J.M.; Carrascosa-Romero, M.C.; Alvarez, S.; Medina-Monzon, M.C.; Caamano, M.B.; de Cabo, C. A new severe mutation in the SLC5A7 gene related to congenital myasthenic syndrome type 20. Neuromuscul. Disord. 2018, 28, 881–884. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.W.; Murrell, J.R.; Nesbitt, A.I.; Pechter, K.B.; Balciuniene, J.; Zhao, X.; Yu, Z.; Denenberg, E.H.; DeChene, E.T.; Wilkens, A.B.; et al. Automated Clinical Exome Reanalysis Reveals Novel Diagnoses. J. Mol. Diagn. 2019, 21, 38–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, S.; Miyake, N.; Tapia, C.; Matsumoto, N. The second point mutation in PREPL: A case report and literature review. J. Hum. Genet. 2018, 63, 677–681. [Google Scholar] [CrossRef]
- Laugwitz, L.; Redler, S.; Buchert, R.; Sturm, M.; Zeile, I.; Schara, U.; Wieczorek, D.; Haack, T.; Distelmaier, F. Isolated PREPL deficiency associated with congenital myasthenic syndrome-22. Klin. Padiatr. 2018, 230, 281–283. [Google Scholar] [CrossRef]
- Kim, M.J.; Yum, M.S.; Seo, G.H.; Lee, Y.; Jang, H.N.; Ko, T.S.; Lee, B.H. Clinical Application of Whole Exome Sequencing to Identify Rare but Remediable Neurologic Disorders. J. Clin. Med. 2020, 9, 3724. [Google Scholar] [CrossRef] [PubMed]
- Shchagina, O.; Bessonova, L.; Bychkov, I.; Beskorovainaya, T.; Poliakov, A. A Family Case of Congenital Myasthenic Syndrome-22 Induced by Different Combinations of Molecular Causes in Siblings. Genes 2020, 11, 821. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wu, B.; Lu, Y.; Ni, Q.; Liu, R.; Zhou, W.; Wang, H. First maternal uniparental disomy for chromosome 2 with PREPL novel frameshift mutation of congenital myasthenic syndrome 22 in an infant. Mol. Genet. Genom. Med. 2020, 8, e1144. [Google Scholar] [CrossRef]
- Yang, Q.; Hua, R.; Qian, J.; Yi, S.; Shen, F.; Zhang, Q.; Li, M.; Yi, S.; Luo, J.; Fan, X. PREPL Deficiency: A Homozygous Splice Site PREPL Mutation in a Patient With Congenital Myasthenic Syndrome and Absence of Ovaries and Hypoplasia of Uterus. Front. Genet. 2020, 11, 198. [Google Scholar] [CrossRef] [PubMed]
- Prior, D.E.; Ghosh, P.S. Congenital Myasthenic Syndrome From a Single Center: Phenotypic and Genotypic features. J. Child Neurol. 2021, 36, 610–617. [Google Scholar] [CrossRef]
- Rizo, J.; Xu, J. The Synaptic Vesicle Release Machinery. Annu. Rev. Biophys. 2015, 44, 339–367. [Google Scholar] [CrossRef] [PubMed]
- Lipstein, N.; Verhoeven-Duif, N.M.; Michelassi, F.E.; Calloway, N.; van Hasselt, P.M.; Pienkowska, K.; van Haaften, G.; van Haelst, M.M.; van Empelen, R.; Cuppen, I.; et al. Synaptic UNC13A protein variant causes increased neurotransmission and dyskinetic movement disorder. J. Clin. Investig. 2017, 127, 1005–1018. [Google Scholar] [CrossRef] [PubMed]
- Salpietro, V.; Lin, W.; Delle Vedove, A.; Storbeck, M.; Liu, Y.; Efthymiou, S.; Manole, A.; Wiethoff, S.; Ye, Q.; Saggar, A.; et al. Homozygous mutations in VAMP1 cause a presynaptic congenital myasthenic syndrome. Ann. Neurol. 2017, 81, 597–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Muhaizea, M.A.; AlQuait, L.; AlRasheed, A.; AlHarbi, S.; Albader, A.A.; AlMass, R.; Albakheet, A.; Alhumaidan, A.; AlRasheed, M.M.; Colak, D.; et al. Pyrostigmine therapy in a patient with VAMP1-related congenital myasthenic syndrome. Neuromuscul. Disord. 2020, 30, 611–615. [Google Scholar] [CrossRef] [PubMed]
- Polavarapu, K.; Vengalil, S.; Preethish-Kumar, V.; Arunachal, G.; Nashi, S.; Mohan, D.; Chawla, T.; Bardhan, M.; Nandeesh, B.; Gupta, P.; et al. Recessive VAMP1 mutations associated with severe congenital myasthenic syndromes—A recognizable clinical phenotype. Eur. J. Paediatr. Neurol. 2021, 31, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Schopperle, W.M.; Murrey, H.; Jaramillo, A.; Dagan, D.; Griffith, L.C.; Levitan, I.B. A dynamically regulated 14-3-3, Slob, and Slowpoke potassium channel complex in Drosophila presynaptic nerve terminals. Neuron 1999, 22, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Schluter, O.M.; Schnell, E.; Verhage, M.; Tzonopoulos, T.; Nicoll, R.A.; Janz, R.; Malenka, R.C.; Geppert, M.; Sudhof, T.C. Rabphilin knock-out mice reveal that rabphilin is not required for rab3 function in regulating neurotransmitter release. J. Neurosci. 1999, 19, 5834–5846. [Google Scholar] [CrossRef]
- Staunton, J.; Ganetzky, B.; Nonet, M.L. Rabphilin potentiates soluble N-ethylmaleimide sensitive factor attachment protein receptor function independently of rab3. J. Neurosci. 2001, 21, 9255–9264. [Google Scholar] [CrossRef] [PubMed]
- Burns, M.E.; Sasaki, T.; Takai, Y.; Augustine, G.J. Rabphilin-3A: A multifunctional regulator of synaptic vesicle traffic. J. Gen. Physiol. 1998, 111, 243–255. [Google Scholar] [CrossRef]
- Miner, J.H.; Cunningham, J.; Sanes, J.R. Roles for laminin in embryogenesis: Exencephaly, syndactyly, and placentopathy in mice lacking the laminin alpha5 chain. J. Cell Biol. 1998, 143, 1713–1723. [Google Scholar] [CrossRef] [PubMed]
- Nishimune, H.; Valdez, G.; Jarad, G.; Moulson, C.L.; Muller, U.; Miner, J.H.; Sanes, J.R. Laminins promote postsynaptic maturation by an autocrine mechanism at the neuromuscular junction. J. Cell Biol. 2008, 182, 1201–1215. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Rush, J.S.; Karaoglu, D.; Krasnewich, D.; Lubinsky, M.S.; Waechter, C.J.; Gilmore, R.; Freeze, H.H. Deficiency of UDP-GlcNAc:Dolichol Phosphate N-Acetylglucosamine-1 Phosphate Transferase (DPAGT1) causes a novel congenital disorder of Glycosylation Type Ij. Hum. Mutat. 2003, 22, 144–150. [Google Scholar] [CrossRef]
- Thiel, C.; Schwarz, M.; Peng, J.; Grzmil, M.; Hasilik, M.; Braulke, T.; Kohlschutter, A.; von Figura, K.; Lehle, L.; Korner, C. A new type of congenital disorders of glycosylation (CDG-Ii) provides new insights into the early steps of dolichol-linked oligosaccharide biosynthesis. J. Biol. Chem. 2003, 278, 22498–22505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senderek, J.; Muller, J.S.; Dusl, M.; Strom, T.M.; Guergueltcheva, V.; Diepolder, I.; Laval, S.H.; Maxwell, S.; Cossins, J.; Krause, S.; et al. Hexosamine biosynthetic pathway mutations cause neuromuscular transmission defect. Am. J. Hum. Genet. 2011, 88, 162–172. [Google Scholar] [CrossRef]
- Zoltowska, K.; Webster, R.; Finlayson, S.; Maxwell, S.; Cossins, J.; Muller, J.; Lochmuller, H.; Beeson, D. Mutations in GFPT1 that underlie limb-girdle congenital myasthenic syndrome result in reduced cell-surface expression of muscle AChR. Hum. Mol. Genet. 2013, 22, 2905–2913. [Google Scholar] [CrossRef]
- Selvam, P.; Arunachal, G.; Danda, S.; Chapla, A.; Sivadasan, A.; Alexander, M.; Thomas, M.M.; Thomas, N.J. Congenital Myasthenic Syndrome: Spectrum of Mutations in an Indian Cohort. J. Clin. Neuromuscul. Dis. 2018, 20, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, C.; Mori-Yoshimura, M.; Noguchi, S.; Endo, Y.; Oya, Y.; Murata, M.; Nishino, I.; Takahashi, Y. Phenotype of a limb-girdle congenital myasthenic syndrome patient carrying a GFPT1 mutation. Brain Dev. 2019, 41, 470–473. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.; Zheng, Y.; Lin, J.; Wu, X.; Yu, Y.; Zhu, M.; Fang, X.; Zhou, M.; Li, X.; Hong, D. Diverse myopathological features in the congenital myasthenia syndrome with GFPT1 mutation. Brain Behav. 2022, 12, e2469. [Google Scholar] [CrossRef] [PubMed]
- Basiri, K.; Belaya, K.; Liu, W.W.; Maxwell, S.; Sedghi, M.; Beeson, D. Clinical features in a large Iranian family with a limb-girdle congenital myasthenic syndrome due to a mutation in DPAGT1. Neuromuscul. Disord. 2013, 23, 469–472. [Google Scholar] [CrossRef]
- Klein, A.; Robb, S.; Rushing, E.; Liu, W.W.; Belaya, K.; Beeson, D. Congenital myasthenic syndrome caused by mutations in DPAGT. Neuromuscul. Disord. 2015, 25, 253–256. [Google Scholar] [CrossRef]
- Bogdanova-Mihaylova, P.; Murphy, R.P.J.; Alexander, M.D.; McHugh, J.C.; Foley, A.R.; Brett, F.; Murphy, S.M. Congenital myasthenic syndrome due to DPAGT1 mutations mimicking congenital myopathy in an Irish family. Eur. J. Neurol. 2018, 25, e22–e23. [Google Scholar] [CrossRef]
- Etzel, J.D.; Neely, K.A.; Ely, A.L. Congenital glycosylation disorder: A novel presentation of coexisting anterior and posterior segment pathology and its implications in pediatric cataract management. J. AAPOS 2019, 23, 297–300. [Google Scholar] [CrossRef]
- Luo, S.; Cai, S.; Maxwell, S.; Yue, D.; Zhu, W.; Qiao, K.; Zhu, Z.; Zhou, L.; Xi, J.; Lu, J.; et al. Novel mutations in the C-terminal region of GMPPB causing limb-girdle muscular dystrophy overlapping with congenital myasthenic syndrome. Neuromuscul. Disord. 2017, 27, 557–564. [Google Scholar] [CrossRef]
- Tian, W.T.; Zhou, H.Y.; Zhan, F.X.; Zhu, Z.Y.; Yang, J.; Chen, S.D.; Luan, X.H.; Cao, L. Lysosomal degradation of GMPPB is associated with limb-girdle muscular dystrophy type 2T. Ann. Clin. Transl. Neurol. 2019, 6, 1062–1071. [Google Scholar] [CrossRef]
- Monies, D.M.; Al-Hindi, H.N.; Al-Muhaizea, M.A.; Jaroudi, D.J.; Al-Younes, B.; Naim, E.A.; Wakil, S.M.; Meyer, B.F.; Bohlega, S. Clinical and pathological heterogeneity of a congenital disorder of glycosylation manifesting as a myasthenic/myopathic syndrome. Neuromuscul. Disord. 2014, 24, 353–359. [Google Scholar] [CrossRef]
- Schorling, D.C.; Rost, S.; Lefeber, D.J.; Brady, L.; Muller, C.R.; Korinthenberg, R.; Tarnopolsky, M.; Bonnemann, C.G.; Rodenburg, R.J.; Bugiani, M.; et al. Early and lethal neurodegeneration with myasthenic and myopathic features: A new ALG14-CDG. Neurology 2017, 89, 657–664. [Google Scholar] [CrossRef]
- Kvarnung, M.; Taylan, F.; Nilsson, D.; Anderlid, B.M.; Malmgren, H.; Lagerstedt-Robinson, K.; Holmberg, E.; Burstedt, M.; Nordenskjold, M.; Nordgren, A.; et al. Genomic screening in rare disorders: New mutations and phenotypes, highlighting ALG14 as a novel cause of severe intellectual disability. Clin. Genet. 2018, 94, 528–537. [Google Scholar] [CrossRef]
- Palombo, F.; Piccolo, B.; Saccani, E.; Fiorini, C.; Capristo, M.; Caporali, L.; Pisani, F.; Carelli, V. A novel ALG14 missense variant in an alive child with myopathy, epilepsy, and progressive cerebral atrophy. Am. J. Med. Genet. A 2021, 185, 1918–1921. [Google Scholar] [CrossRef]
- Katata, Y.; Uneoka, S.; Saijyo, N.; Aihara, Y.; Miyazoe, T.; Koyamaishi, S.; Oikawa, Y.; Ito, Y.; Abe, Y.; Numata-Uematsu, Y.; et al. The longest reported sibling survivors of a severe form of congenital myasthenic syndrome with the ALG14 pathogenic variant. Am. J. Med. Genet. A 2022, 188, 1293–1298. [Google Scholar] [CrossRef]
- Mensch, A.; Cordts, I.; Scholle, L.; Joshi, P.R.; Kleeberg, K.; Emmer, A.; Beck-Woedl, S.; Park, J.; Haack, T.B.; Stoltenburg-Didinger, G.; et al. GFPT1-Associated Congenital Myasthenic Syndrome Mimicking a Glycogen Storage Disease—Diagnostic Pitfalls in Myopathology Solved by Next-Generation-Sequencing. J. Neuromuscul. Dis. 2022, 9, 533–541. [Google Scholar] [CrossRef]
- Siddiqui, S.; Polavarapu, K.; Bardhan, M.; Preethish-Kumar, V.; Joshi, A.; Nashi, S.; Vengalil, S.; Raju, S.; Chawla, T.; Leena, S.; et al. Distinct and Recognisable Muscle MRI Pattern in a Series of Adults Harbouring an Identical GMPPB Gene Mutation. J. Neuromuscul. Dis. 2022, 9, 95–109. [Google Scholar] [CrossRef]
- Johnson, K.; Bertoli, M.; Phillips, L.; Topf, A.; Van den Bergh, P.; Vissing, J.; Witting, N.; Nafissi, S.; Jamal-Omidi, S.; Lusakowska, A.; et al. Detection of variants in dystroglycanopathy-associated genes through the application of targeted whole-exome sequencing analysis to a large cohort of patients with unexplained limb-girdle muscle weakness. Skelet. Muscle 2018, 8, 23. [Google Scholar] [CrossRef]
- Ehrstedt, C.; Liu, W.W.; Frykholm, C.; Beeson, D.; Punga, A.R. Novel pathogenic ALG2 mutation causing congenital myasthenic syndrome: A case report. Neuromuscul. Disord. 2022, 32, 80–83. [Google Scholar] [CrossRef]
- Ma, Y.; Xiong, T.; Lei, G.; Ding, J.; Yang, R.; Li, Z.; Guo, J.; Shen, D. Novel compound heterozygous variants in the GFPT1 gene leading to rare limb-girdle congenital myasthenic syndrome with rimmed vacuoles. Neurol. Sci. 2021, 42, 3485–3490. [Google Scholar] [CrossRef]
- Liao, W.; Elfrink, K.; Bahler, M. Head of myosin IX binds calmodulin and moves processively toward the plus-end of actin filaments. J. Biol. Chem. 2010, 285, 24933–24942. [Google Scholar] [CrossRef]
- O’Connor, E.; Topf, A.; Muller, J.S.; Cox, D.; Evangelista, T.; Colomer, J.; Abicht, A.; Senderek, J.; Hasselmann, O.; Yaramis, A.; et al. Identification of mutations in the MYO9A gene in patients with congenital myasthenic syndrome. Brain 2016, 139 Pt 8, 2143–2153. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, E.; Phan, V.; Cordts, I.; Cairns, G.; Hettwer, S.; Cox, D.; Lochmuller, H.; Roos, A. MYO9A deficiency in motor neurons is associated with reduced neuromuscular agrin secretion. Hum. Mol. Genet. 2018, 27, 1434–1446. [Google Scholar] [CrossRef]
- Li, Q.; Gulati, A.; Lemaire, M.; Nottoli, T.; Bale, A.; Tufro, A. Rho-GTPase Activating Protein myosin MYO9A identified as a novel candidate gene for monogenic focal segmental glomerulosclerosis. Kidney Int. 2021, 99, 1102–1117. [Google Scholar] [CrossRef]
- Nota, B.; Struys, E.A.; Pop, A.; Jansen, E.E.; Fernandez Ojeda, M.R.; Kanhai, W.A.; Kranendijk, M.; van Dooren, S.J.; Bevova, M.R.; Sistermans, E.A.; et al. Deficiency in SLC25A1, encoding the mitochondrial citrate carrier, causes combined D-2- and L-2-hydroxyglutaric aciduria. Am. J. Hum. Genet. 2013, 92, 627–631. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, R.S.; Mayor, J.A.; Wood, D.O. The mitochondrial tricarboxylate transport protein. cDNA cloning, primary structure, and comparison with other mitochondrial transport proteins. J. Biol. Chem. 1993, 268, 13682–13690. [Google Scholar] [CrossRef] [PubMed]
- Chaouch, A.; Porcelli, V.; Cox, D.; Edvardson, S.; Scarcia, P.; De Grassi, A.; Pierri, C.L.; Cossins, J.; Laval, S.H.; Griffin, H.; et al. Mutations in the Mitochondrial Citrate Carrier SLC25A1 are Associated with Impaired Neuromuscular Transmission. J. Neuromuscul. Dis. 2014, 1, 75–90. [Google Scholar] [CrossRef]
- Al-Futaisi, A.; Ahmad, F.; Al-Kasbi, G.; Al-Thihli, K.; Koul, R.; Al-Maawali, A. Missense mutations in SLC25A1 are associated with congenital myasthenic syndrome type 23. Clin. Genet. 2020, 97, 666–667. [Google Scholar] [CrossRef]
- Balaraju, S.; Topf, A.; McMacken, G.; Kumar, V.P.; Pechmann, A.; Roper, H.; Vengalil, S.; Polavarapu, K.; Nashi, S.; Mahajan, N.P.; et al. Congenital myasthenic syndrome with mild intellectual disability caused by a recurrent SLC25A1 variant. Eur. J. Hum. Genet. 2020, 28, 373–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Zhang, M.; Zhang, L.; Shi, Y.; Zhao, L.; Wu, B.; Li, X.; Zhou, S. A case report of an intermediate phenotype between congenital myasthenic syndrome and D-2- and L-2-hydroxyglutaric aciduria due to novel SLC25A1 variants. BMC Neurol. 2020, 20, 278. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, Y.; Bian, Y.; Yao, S.; Liu, P.; Yu, M.; Zhang, W.; Wang, Z.; Yuan, Y. Congenital myasthenic syndrome in China: Genetic and myopathological characterization. Ann. Clin. Transl. Neurol. 2021, 8, 898–907. [Google Scholar] [CrossRef] [PubMed]
- Senior, A.; Gerace, L. Integral membrane proteins specific to the inner nuclear membrane and associated with the nuclear lamina. J. Cell Biol. 1988, 107 Pt 1, 2029–2036. [Google Scholar] [CrossRef]
- Zhao, C.; Brown, R.S.; Chase, A.R.; Eisele, M.R.; Schlieker, C. Regulation of Torsin ATPases by LAP1 and LULL1. Proc. Natl. Acad. Sci. USA 2013, 110, E1545–E1554. [Google Scholar] [CrossRef]
- Kayman-Kurekci, G.; Talim, B.; Korkusuz, P.; Sayar, N.; Sarioglu, T.; Oncel, I.; Sharafi, P.; Gundesli, H.; Balci-Hayta, B.; Purali, N.; et al. Mutation in TOR1AIP1 encoding LAP1B in a form of muscular dystrophy: A novel gene related to nuclear envelopathies. Neuromuscul. Disord. 2014, 24, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Dorboz, I.; Coutelier, M.; Bertrand, A.T.; Caberg, J.H.; Elmaleh-Berges, M.; Laine, J.; Stevanin, G.; Bonne, G.; Boespflug-Tanguy, O.; Servais, L. Severe dystonia, cerebellar atrophy, and cardiomyopathy likely caused by a missense mutation in TOR1AIP1. Orphanet J. Rare Dis. 2014, 9, 174. [Google Scholar] [CrossRef] [PubMed]
- Ghaoui, R.; Benavides, T.; Lek, M.; Waddell, L.B.; Kaur, S.; North, K.N.; MacArthur, D.G.; Clarke, N.F.; Cooper, S.T. TOR1AIP1 as a cause of cardiac failure and recessive limb-girdle muscular dystrophy. Neuromuscul. Disord. 2016, 26, 500–503. [Google Scholar] [CrossRef]
- Cossins, J.; Webster, R.; Maxwell, S.; Rodriguez Cruz, P.M.; Knight, R.; Llewelyn, J.G.; Shin, J.Y.; Palace, J.; Beeson, D. Congenital myasthenic syndrome due to a TOR1AIP1 mutation: A new disease pathway for impaired synaptic transmission. Brain Commun. 2020, 2, fcaa174. [Google Scholar] [CrossRef] [PubMed]
- Malfatti, E.; Catchpool, T.; Nouioua, S.; Sihem, H.; Fournier, E.; Carlier, R.Y.; Cardone, N.; Davis, M.R.; Laing, N.G.; Sternberg, D.; et al. A TOR1AIP1 variant segregating with an early onset limb girdle myasthenia-Support for the role of LAP1 in NMJ function and disease. Neuropathol. Appl. Neurobiol. 2022, 48, e12743. [Google Scholar] [CrossRef]
- Sakamoto, I.; Kishida, S.; Fukui, A.; Kishida, M.; Yamamoto, H.; Hino, S.; Michiue, T.; Takada, S.; Asashima, M.; Kikuchi, A. A novel beta-catenin-binding protein inhibits beta-catenin-dependent Tcf activation and axis formation. J. Biol. Chem. 2000, 275, 32871–32878. [Google Scholar] [CrossRef] [Green Version]
- Thompson, B.A.; Tremblay, V.; Lin, G.; Bochar, D.A. CHD8 is an ATP-dependent chromatin remodeling factor that regulates beta-catenin target genes. Mol. Cell Biol. 2008, 28, 3894–3904. [Google Scholar] [CrossRef]
- Nishiyama, M.; Skoultchi, A.I.; Nakayama, K.I. Histone H1 recruitment by CHD8 is essential for suppression of the Wnt-beta-catenin signaling pathway. Mol. Cell Biol. 2012, 32, 501–512. [Google Scholar] [CrossRef]
- Lee, C.Y.; Petkova, M.; Morales-Gonzalez, S.; Gimber, N.; Schmoranzer, J.; Meisel, A.; Bohmerle, W.; Stenzel, W.; Schuelke, M.; Schwarz, J.M. A spontaneous missense mutation in the chromodomain helicase DNA-binding protein 8 (CHD8) gene: A novel association with congenital myasthenic syndrome. Neuropathol. Appl. Neurobiol. 2020, 46, 588–601. [Google Scholar] [CrossRef] [PubMed]
- Li, X.M.; Dong, X.P.; Luo, S.W.; Zhang, B.; Lee, D.H.; Ting, A.K.; Neiswender, H.; Kim, C.H.; Carpenter-Hyland, E.; Gao, T.M.; et al. Retrograde regulation of motoneuron differentiation by muscle beta-catenin. Nat. Neurosci. 2008, 11, 262–268. [Google Scholar] [CrossRef]
- Latcheva, N.K.; Delaney, T.L.; Viveiros, J.M.; Smith, R.A.; Bernard, K.M.; Harsin, B.; Marenda, D.R.; Liebl, F.L.W. The CHD Protein, Kismet, is Important for the Recycling of Synaptic Vesicles during Endocytosis. Sci. Rep. 2019, 9, 19368. [Google Scholar] [CrossRef]
- McDiarmid, T.A.; Belmadani, M.; Liang, J.; Meili, F.; Mathews, E.A.; Mullen, G.P.; Hendi, A.; Wong, W.R.; Rand, J.B.; Mizumoto, K.; et al. Systematic phenomics analysis of autism-associated genes reveals parallel networks underlying reversible impairments in habituation. Proc. Natl. Acad. Sci. USA 2020, 117, 656–667. [Google Scholar] [CrossRef]
- Bernier, R.; Golzio, C.; Xiong, B.; Stessman, H.A.; Coe, B.P.; Penn, O.; Witherspoon, K.; Gerdts, J.; Baker, C.; Vulto-van Silfhout, A.T.; et al. Disruptive CHD8 mutations define a subtype of autism early in development. Cell 2014, 158, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Spengler, D. Chromatin Remodeler CHD8 in Autism and Brain Development. J. Clin. Med. 2021, 10, 366. [Google Scholar] [CrossRef]
- Lalani, S.R.; Zhang, J.; Schaaf, C.P.; Brown, C.W.; Magoulas, P.; Tsai, A.C.; El-Gharbawy, A.; Wierenga, K.J.; Bartholomew, D.; Fong, C.T.; et al. Mutations in PURA cause profound neonatal hypotonia, seizures, and encephalopathy in 5q31.3 microdeletion syndrome. Am. J. Hum. Genet. 2014, 95, 579–583. [Google Scholar] [CrossRef]
- Hunt, D.; Leventer, R.J.; Simons, C.; Taft, R.; Swoboda, K.J.; Gawne-Cain, M.; the DDD Study; Magee, A.C.; Turnpenny, P.D.; Baralle, D. Whole exome sequencing in family trios reveals de novo mutations in PURA as a cause of severe neurodevelopmental delay and learning disability. J. Med. Genet. 2014, 51, 806–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, A.J.; Bai, R.; Cho, M.T.; Anyane-Yeboa, K.; Ahimaz, P.; Wilson, A.L.; Kendall, F.; Hay, B.; Moss, T.; Nardini, M.; et al. De novo mutations in PURA are associated with hypotonia and developmental delay. Cold Spring Harb. Mol. Case Stud. 2015, 1, a000356. [Google Scholar] [CrossRef]
- Reijnders, M.R.F.; Janowski, R.; Alvi, M.; Self, J.E.; van Essen, T.J.; Vreeburg, M.; Rouhl, R.P.W.; Stevens, S.J.C.; Stegmann, A.P.A.; Schieving, J.; et al. PURA syndrome: Clinical delineation and genotype-phenotype study in 32 individuals with review of published literature. J. Med. Genet. 2018, 55, 104–113. [Google Scholar] [CrossRef]
- Wyrebek, R.; DiBartolomeo, M.; Brooks, S.; Geller, T.; Crenshaw, M.; Iyadurai, S. Hypotonic infant with PURA syndrome-related channelopathy successfully treated with pyridostigmine. Neuromuscul. Disord. 2022, 32, 166–169. [Google Scholar] [CrossRef] [PubMed]





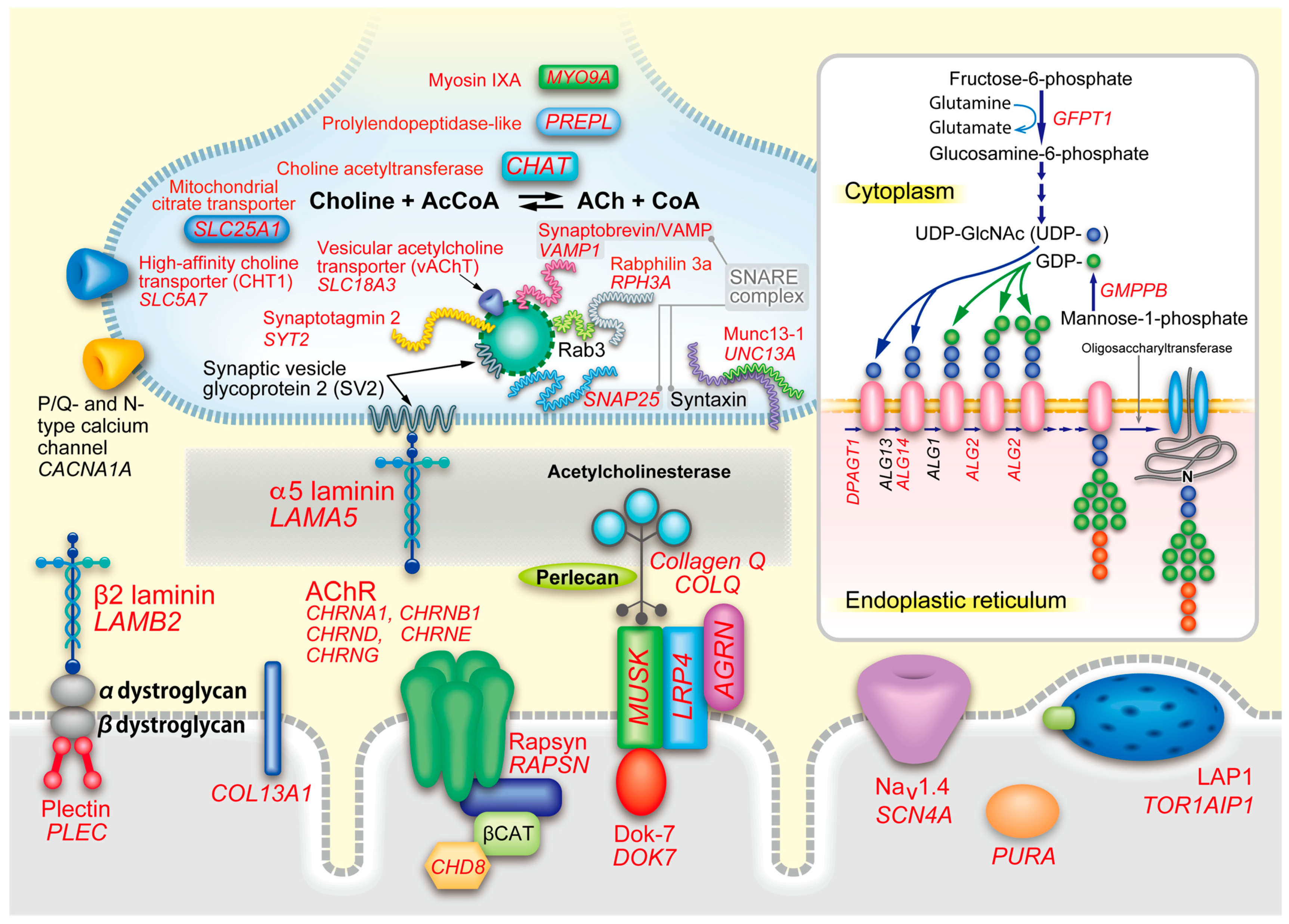{kind=link}
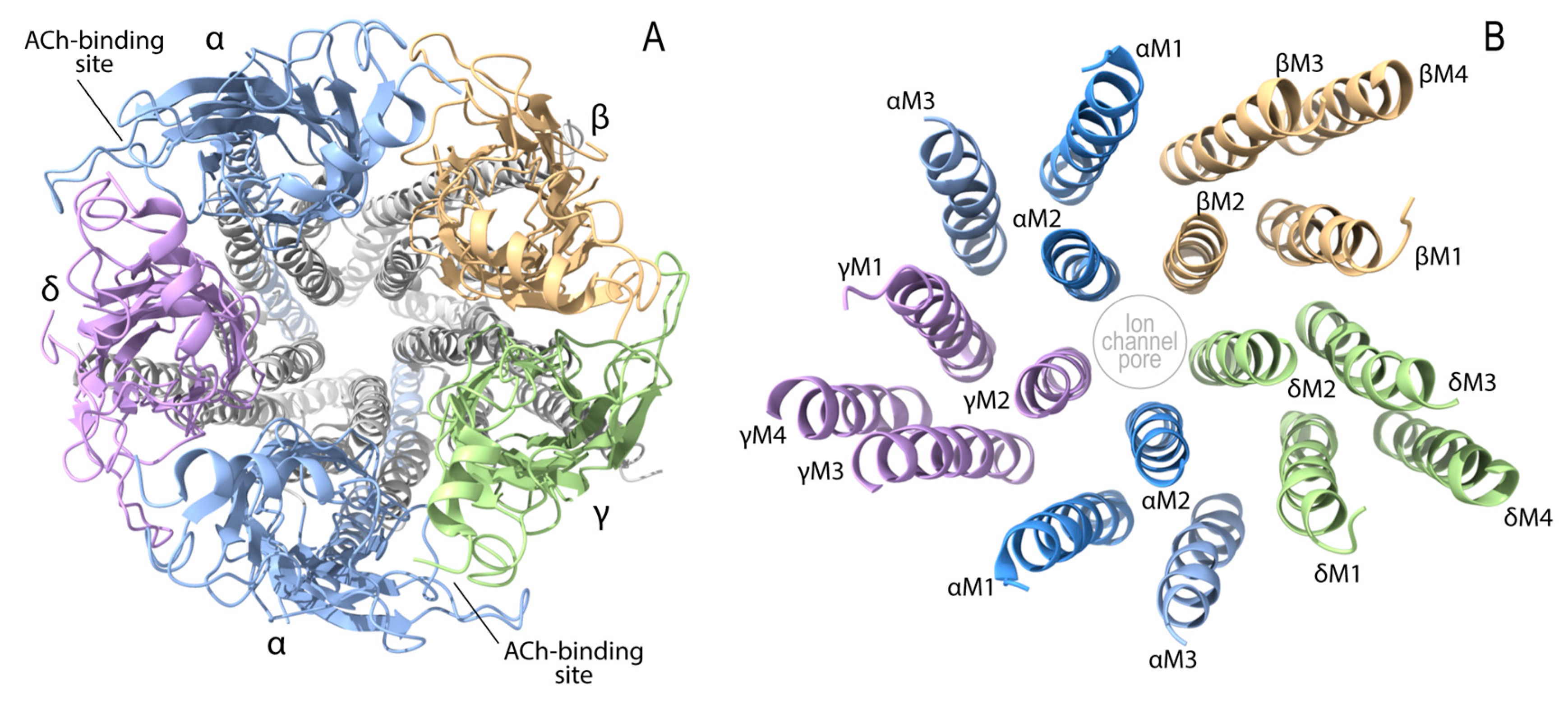{kind=link}
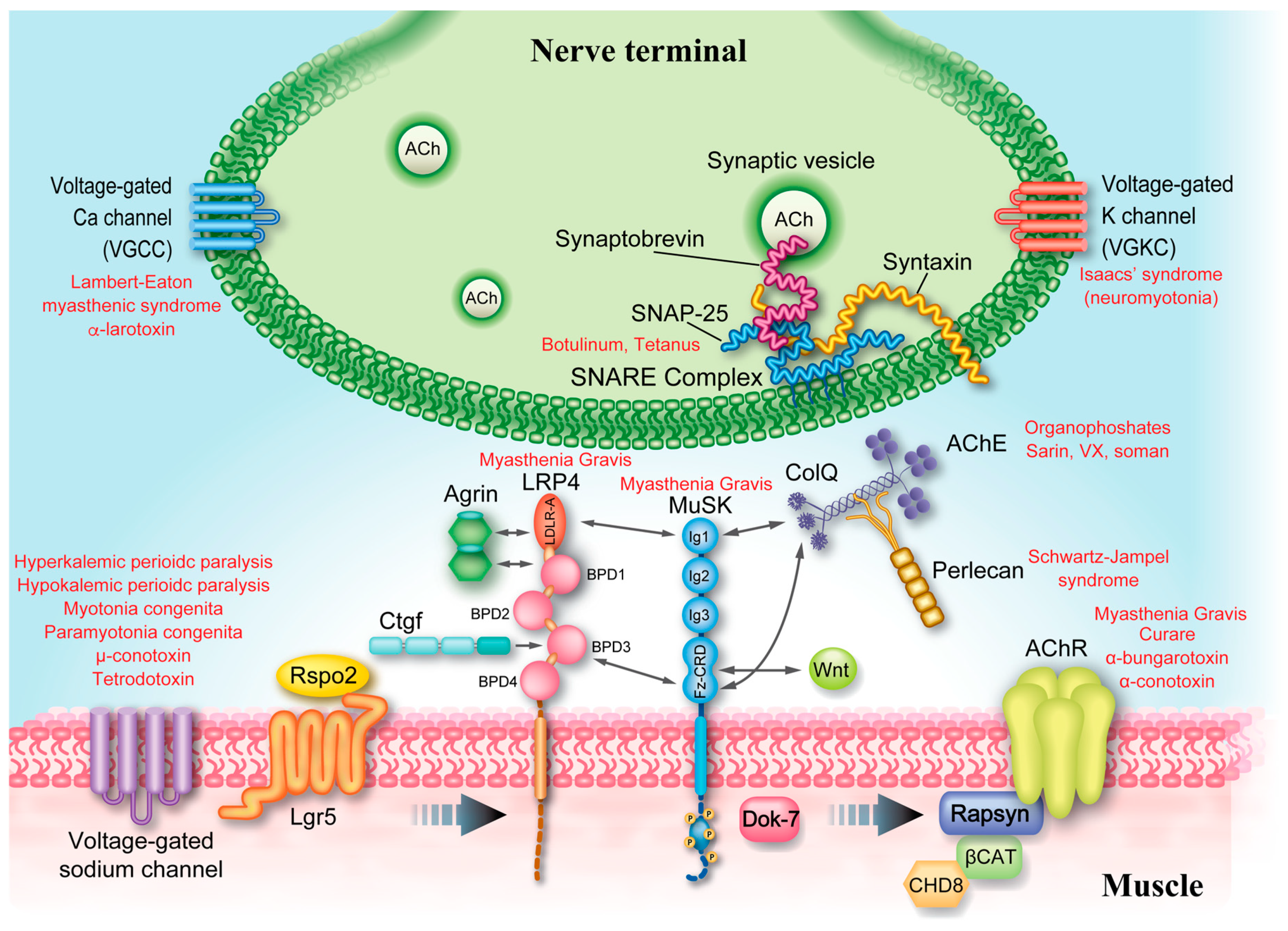{kind=link}
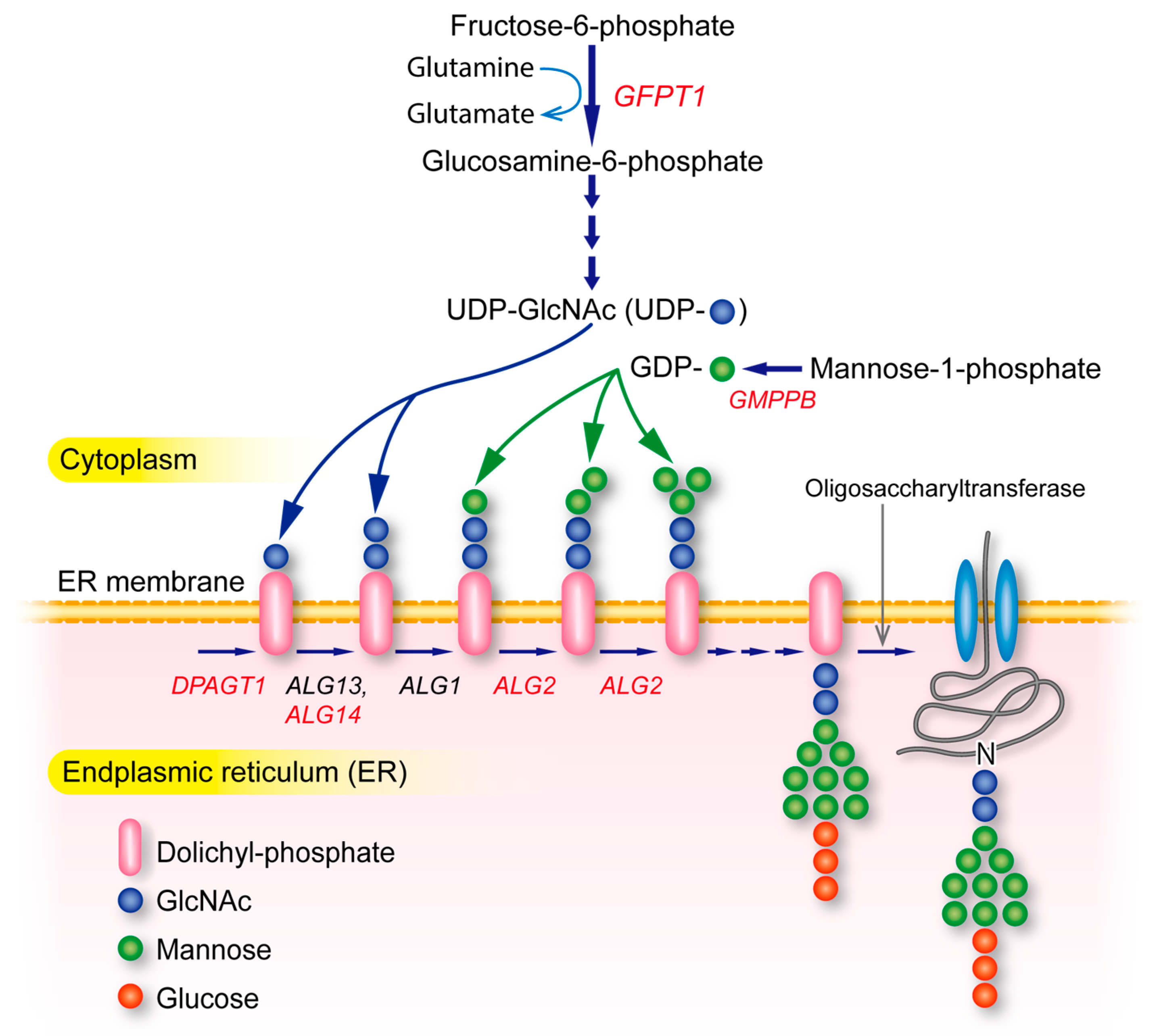{kind=link}
| Section | Phenotype | Gene | OMIM | # a | Inheritance | Low-Frequency RNS | High-Frequency RNS | Treatment | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ChEIs | Ephedrine | Salbutamol (albuterol) | Amifampridine | Quinidine | Fluoxetine | Acetazolamide | |||||||||
| 4.1 | Endplate AChR deficiency | CHRNA1 | - b | AR | decrement | effective | effective | effective | effective | ||||||
| 4.1 | Endplate AChR deficiency | CHRNB1 | CMS2C | - b | AR | decrement | effective | effective | effective | effective | |||||
| 4.1 | Endplate AChR deficiency | CHRND | CMS3C | - b | AR | decrement | effective | effective | effective | effective | |||||
| 4.1 | Endplate AChR deficiency | CHRNE | CMS4C | - b | AR | decrement | effective | effective | effective | effective | |||||
| 4.1 | Endplate AChR deficiency | RAPSN | CMS11 | [38] | AR | decrement | effective | effective | effective | effective | |||||
| 4.2 | Escobar syndrome | CHRNG | 101 | AR | decrement | ||||||||||
| 4.2 | FADS | CHRNA1 | (4) | AR | decrement | ||||||||||
| 4.2 | FADS | CHRND | (6) | AR | decrement | ||||||||||
| 4.2 | FADS | MUSK | (6) | AR | decrement | ||||||||||
| 4.2 | FADS | RAPSN | (8) | AR | decrement | ||||||||||
| 4.2 | FADS | DOK7 | (2) | AR | decrement | ||||||||||
| 4.2 | FADS | SLC18A3 | (1) | AR | decrement | ||||||||||
| 4.3 | SCCMS | CHRNA1 | CMS1A | (14) | AD | decrement, repetitive CMAP | mostly ineffective | effective in some reports | effective in some reports | effective | effective | ||||
| 4.3 | SCCMS | CHRNB1 | CMS2A | (5) | AD | decrement, repetitive CMAP | mostly ineffective | effective in some reports | effective in some reports | effective | effective | ||||
| 4.3 | SCCMS | CHRND | CMS3A | (4) | AD | decrement, repetitive CMAP | mostly ineffective | effective in some reports | effective in some reports | effective | effective | ||||
| 4.3 | SCCMS | CHRNE | CMS4A | (11) | AD/AR | decrement, repetitive CMAP | mostly ineffective | effective in some reports | effective in some reports | effective | effective | ||||
| 4.3 | FCCMS | CHRNA1 | CMS1B | (3) c | AR | decrement | effective | presumably effective, but no report | effective in a report | effective | |||||
| 4.3 | FCCMS | CHRNB1 | CMS2B | (1) c | AR | decrement | effective | presumably effective, but no report | effective in a report | effective | |||||
| 4.3 | FCCMS | CHRND | CMS3B | (1) c | AR | decrement | effective | presumably effective, but no report | effective in a report | effective | |||||
| 4.3 | FCCMS | CHRNE | CMS4B | (6) c | AR | decrement | effective | presumably effective, but no report | effective in a report | effective | |||||
| 4.4 | Endplate AChE deficiency | COLQ | CMS5 | [30] | AR | decrement, repetitive CMAP | contraindication, but effective in some reported patients | effective in some reports | effective in some reports | effective in a report | |||||
| 4.4 | Synaptic CMS | LAMB2 | 1 | AR | decrement | contraindication | effective | ||||||||
| 4.4 | Synaptic CMS | COL13A1 | CMS19 | 41 | AR | decrement | ineffective | effective | effective | ||||||
| 4.5 | Sodium channel CMS | SCN4A | CMS16 | 6 | AR | no decrement | decrement | Effective, ineffective, or marked adverse effects | Slightly effective | effective or ineffective | |||||
| 4.6 | CMS caused by defective AChR clustering | AGRN | CMS8 | [13] | AR | decrement | Ineffective or mildly effective | effective | effective | ineffective or slightly effective | |||||
| 4.6 | CMS caused by defective AChR clustering | MUSK | CMS9 | [15] | AR | decrement | Ineffective or worsened | effective | effective | ||||||
| 4.6 | CMS caused by defective AChR clustering | LRP4 | CMS17 | 1 | AR | decrement | worsened | ||||||||
| 4.6 | CMS caused by defective AChR clustering | DOK7 | CMS10 | [34] | AR | decrement | Combination of ineffective and worsening | effective | effective | effective in some reports | effective in some reports | ||||
| 4.7 | CMS caused by defective structural molecules | PLEC | 22 | AR | decrement | effective or ineffective | effective in some reports | effective or ineffective | |||||||
| 4.8 | CMS caused by defective recycling of ACh | CHAT | CMS6 | [19] | AR | no decrement | decrement in some patients | effective | effective | ||||||
| 4.8 | CMS caused by defective recycling of ACh | SLC18A3 | CMS21 | 7 | AR | decrement at rest or only after isometric muscle contraction | decrement in some patients | effective | effective | effective | |||||
| 4.8 | CMS caused by defective recycling of ACh | SLC5A7 | CMS20 | 12 | AR | decrement at rest or only after isometric muscle contraction | decrement in some patients | effective | effective | ineffective | |||||
| 4.8 | CMS caused by defective recycling of ACh | PREPL | CMS22 | 18 | AR | decrement | decrement in some patients | effective | |||||||
| 4.9 | LEMS-like CMS | SYT2 | CMS7ACMS7B | 2 | AD/AR | decrement | increment | effective | effective | effective | |||||
| 4.9 | LEMS-like CMS | SNAP25 | CMS18 | 2 | AD | decrement | ineffective | effective | |||||||
| 4.9 | LEMS-like CMS | UNC13A | 1 | AR | decrement | increment | minimally effective | minimally effective | |||||||
| 4.9 | LEMS-like CMS | VAMP1 | CMS25 | 9 | AR | decrement | increment | effective | |||||||
| 4.9 | LEMS-like CMS | RPH3A | 1 | AR | no decrement | increment | effective | ||||||||
| 4.9 | LEMS-like CMS | LAMA5 | 1 | AR | decrement | increment | effective | effective | |||||||
| 4.10 | Glycosylation-deficient CMS | GFPT1 | CMS12 | [17] | AR | decrement | effective | effective | |||||||
| 4.10 | Glycosylation-deficient CMS | DPAGT1 | CMS13 | [5] | AR | decrement | effective | effective | effective | ||||||
| 4.10 | Glycosylation-deficient CMS | ALG2 | CMS14 | 9 | AR | decrement | effective or ineffective | effective | effective | ||||||
| 4.10 | Glycosylation-deficient CMS | ALG14 | CMS15 | 12 | AR | decrement | effective | ||||||||
| 4.10 | Glycosylation-deficient CMS | GMPPB | [9] | AR | decrement | effective | effective | ||||||||
| 4.11 | CMS caused by defective nerve terminal formation | MYO9A | CMS24 | 3 | AR | decrement | effective | effective | |||||||
| 4.11 | CMS caused by defective nerve terminal formation | SLC25A1 | CMS23 | 19 | AR | decrement | ineffective in most patients | ineffective in most patients | |||||||
| 4.12 | CMS caused by defective nuclear membrane protein | TOR1AIP1 | 5 | AR | decrement | effective | no additional effect | ||||||||
| 4.13 | CMS caused by defective chromatin remodeling protein | CHD8 | 2 | AR | decrement | ineffective | ineffective | markedly effective | |||||||
| 4.14 | CMS in PURA syndrome | PURA | 3 | AD | decrement, repetitive CMAP | effective in a patient, but not in another patient | effective in a patient | ||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ohno, K.; Ohkawara, B.; Shen, X.-M.; Selcen, D.; Engel, A.G. Clinical and Pathologic Features of Congenital Myasthenic Syndromes Caused by 35 Genes—A Comprehensive Review. Int. J. Mol. Sci. 2023, 24, 3730. https://doi.org/10.3390/ijms24043730
Ohno K, Ohkawara B, Shen X-M, Selcen D, Engel AG. Clinical and Pathologic Features of Congenital Myasthenic Syndromes Caused by 35 Genes—A Comprehensive Review. International Journal of Molecular Sciences. 2023; 24(4):3730. https://doi.org/10.3390/ijms24043730
Chicago/Turabian StyleOhno, Kinji, Bisei Ohkawara, Xin-Ming Shen, Duygu Selcen, and Andrew G. Engel. 2023. "Clinical and Pathologic Features of Congenital Myasthenic Syndromes Caused by 35 Genes—A Comprehensive Review" International Journal of Molecular Sciences 24, no. 4: 3730. https://doi.org/10.3390/ijms24043730