
The Complete Mitochondrial Genome and Phylogenetic Analysis of the Freshwater Shellfish Novaculina chinensis (Bivalvia: Pharidae)

Abstract
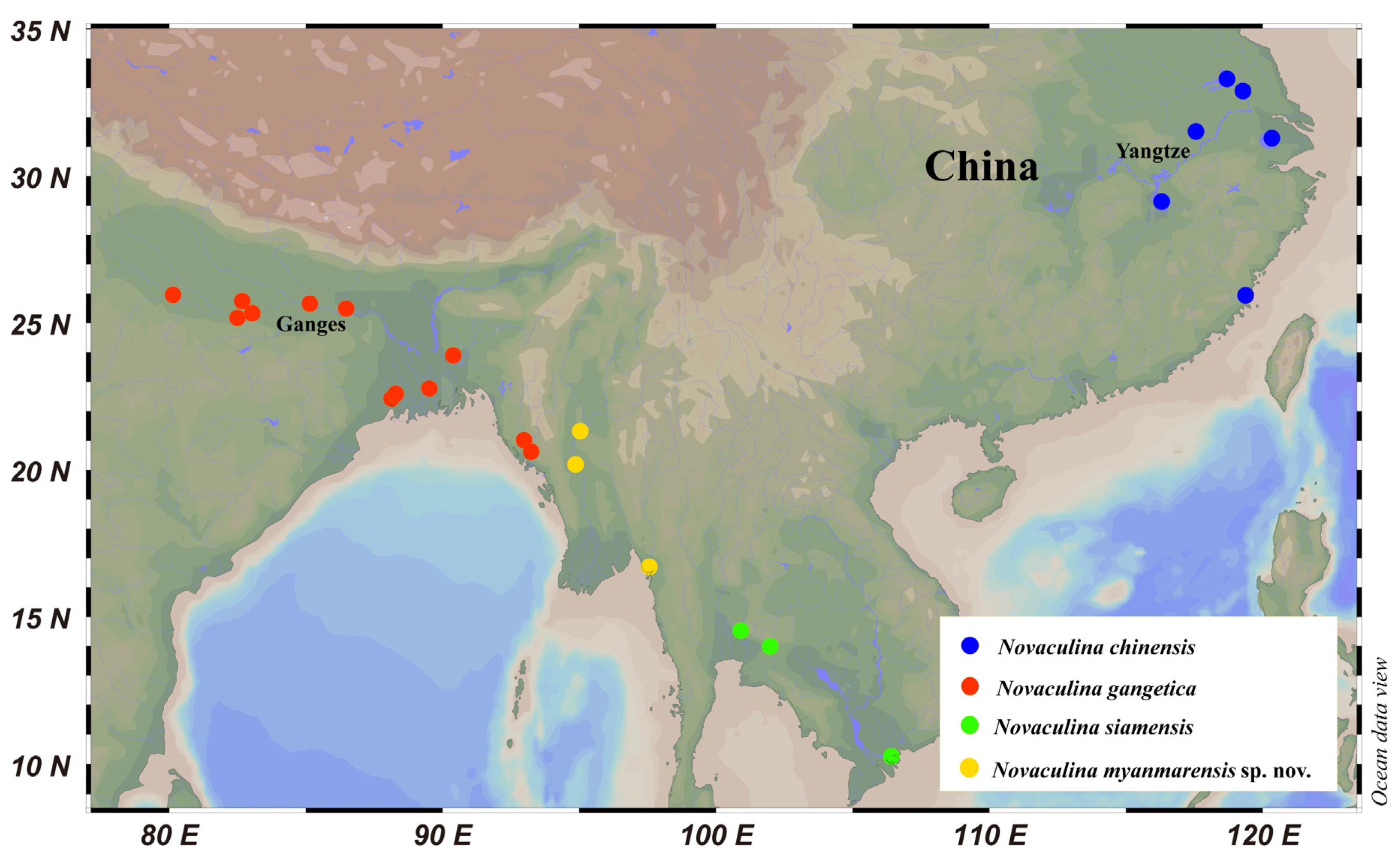
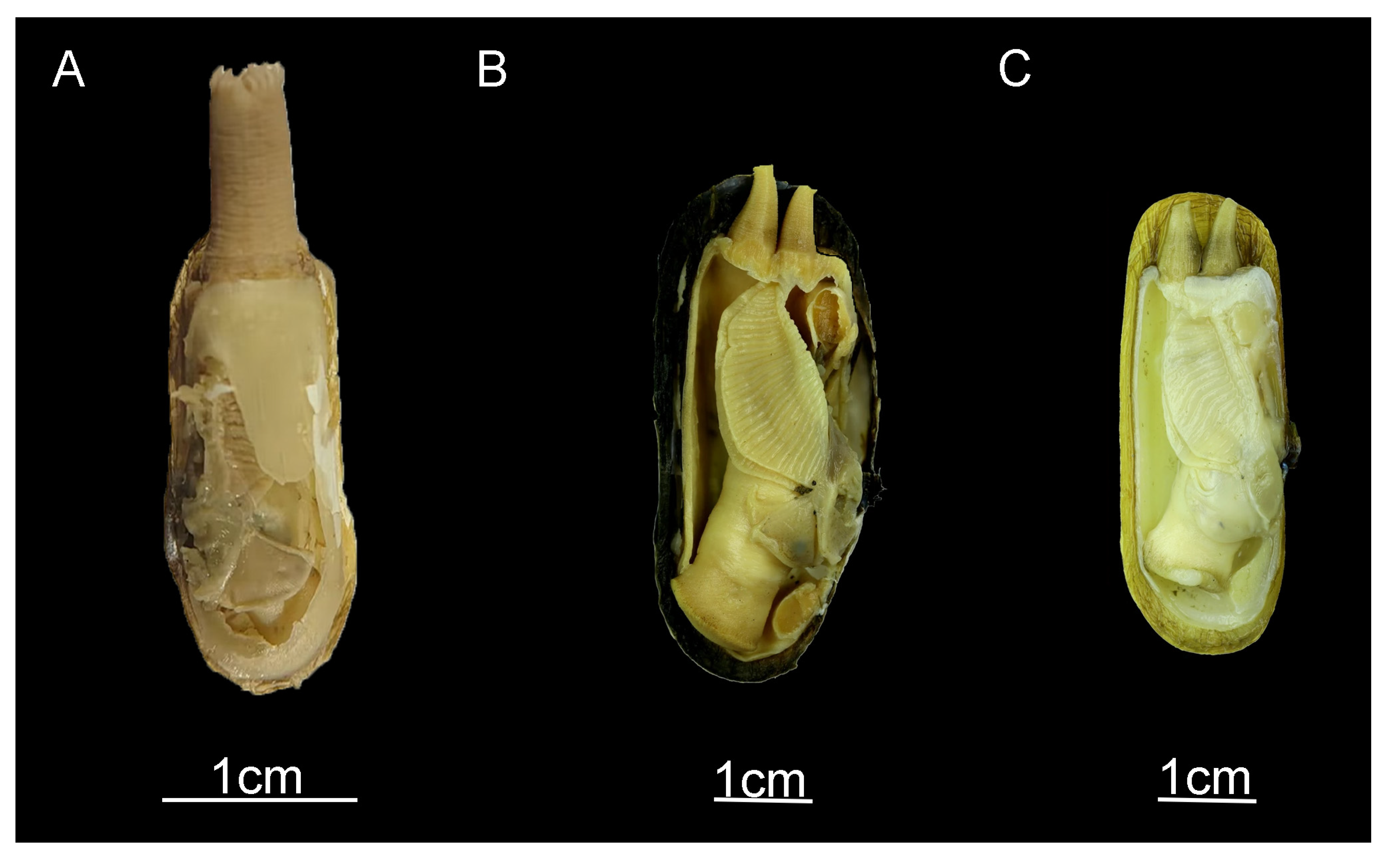
:1. Introduction
2. Results and Discussion
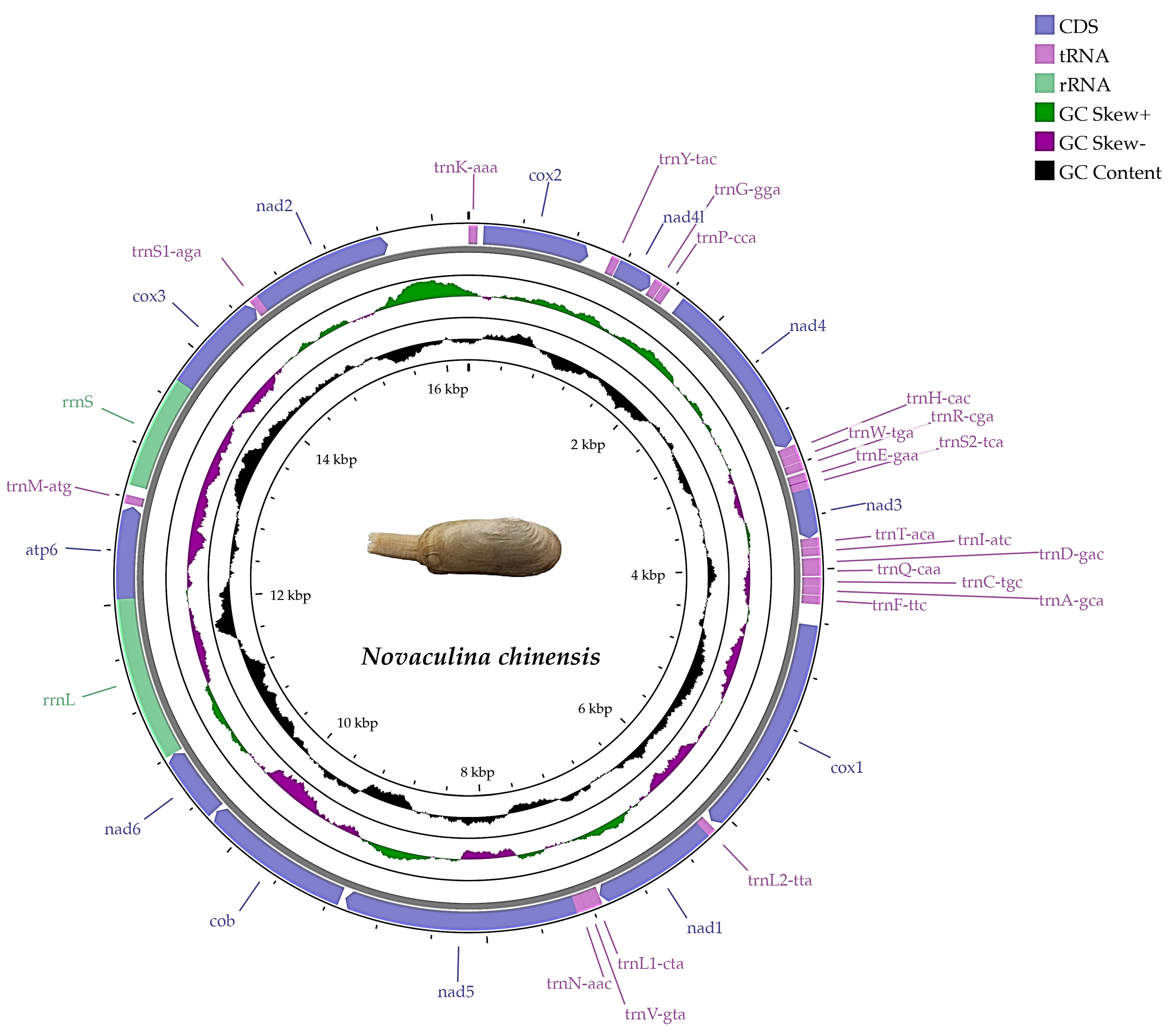
2.1. Genome Features
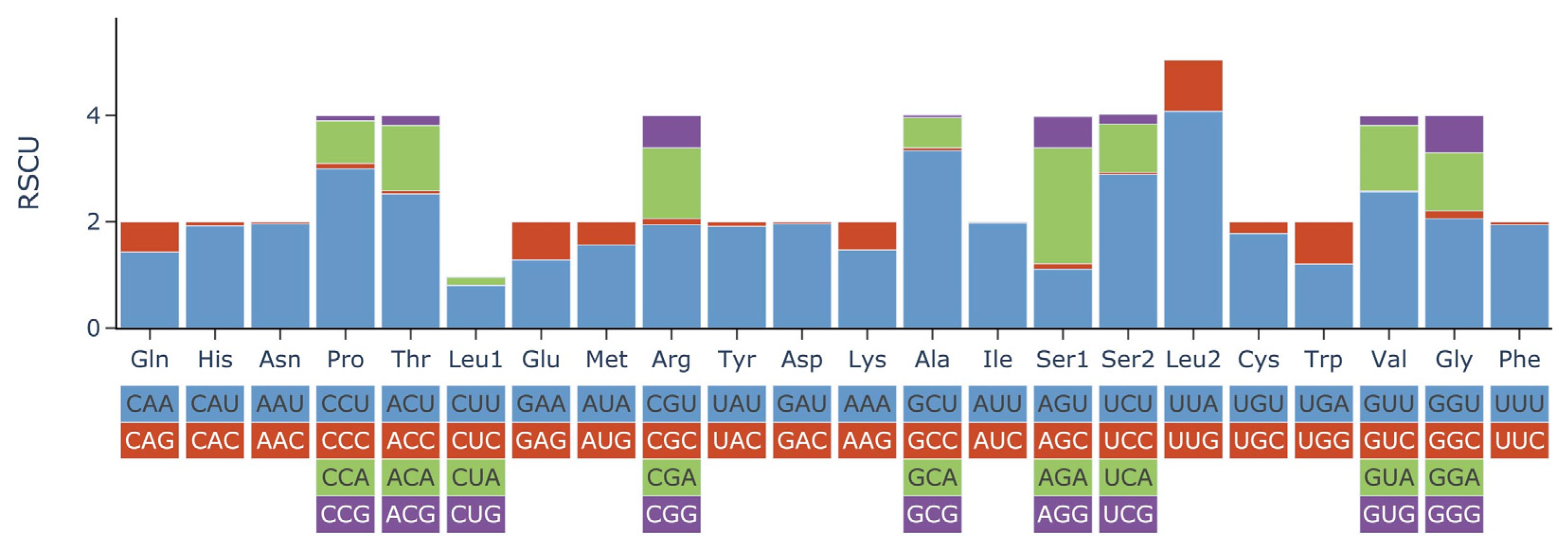
2.2. Protein-Coding Genes, Transfer RNA, and Ribosomal RNA Genes
2.3. KaKs Analysis
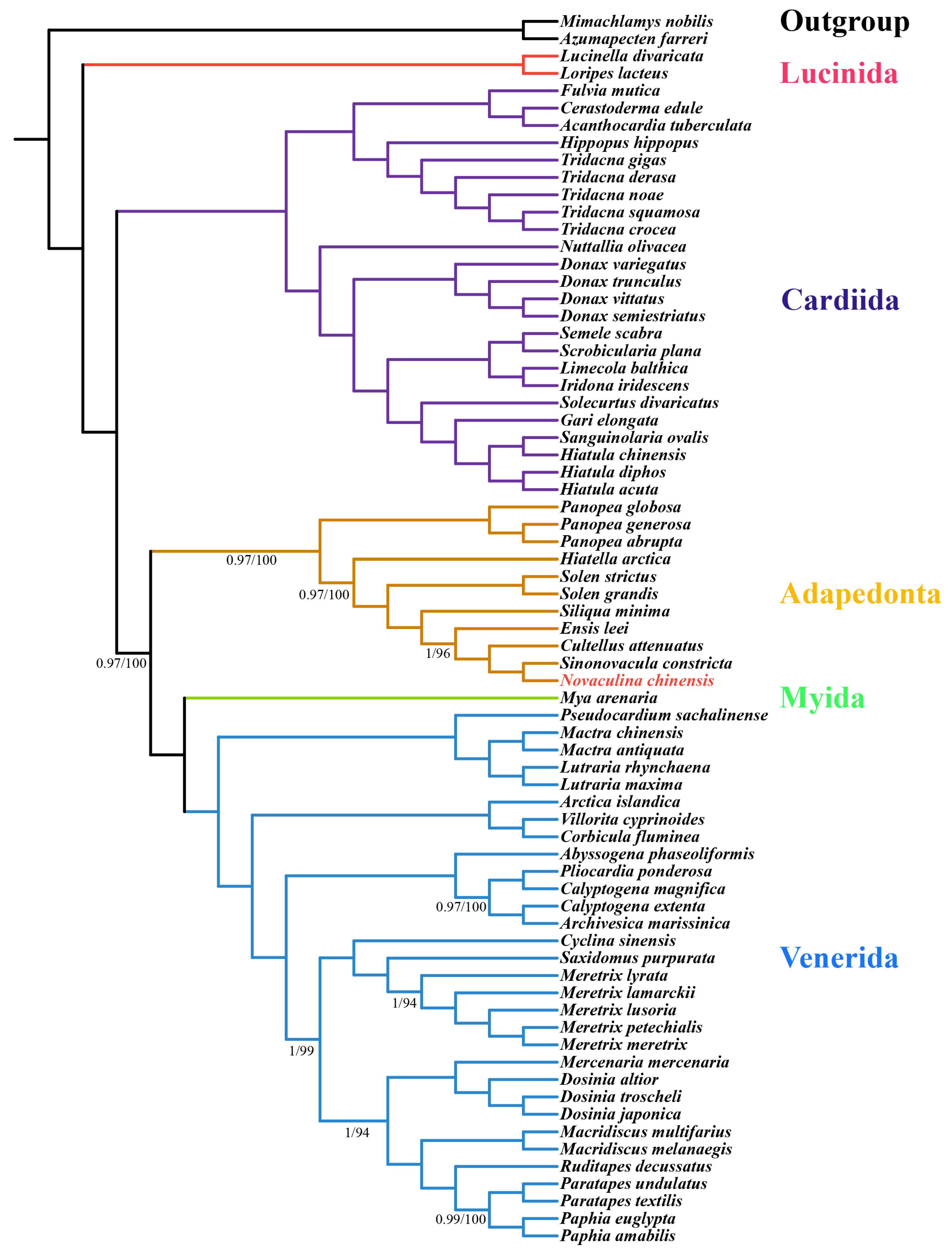
2.4. Analysis of Phylogenetic
3. Materials and Methods
3.1. Sample Collection and DNA Extraction
3.2. Sequencing and Genome Assembly
3.3. Annotation and Sequence Analysis of Mitogenome
3.4. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saeedi, H.; Costello, M.J. The Biology, Ecology, and Societal Importance of Razor Clams. In Encyclopedia of the World’s Biomes; Goldstein, M.I., DellaSala, D.A., Eds.; Elsevier: Oxford, UK, 2020; pp. 494–498. [Google Scholar]
- Wang, Y.; Yang, Y.; Kong, L.; Sasaki, T.; Li, Q. Phylogenomic Resolution of Imparidentia (Mollusca: Bivalvia) Diversification through Mitochondrial Genomes. Mar. Life Sci. Technol. 2023, 5, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Bolotov, I.N.; Vikhrev, I.V.; Lopes-Lima, M.; Lunn, Z.; Chan, N.; Win, T.; Aksenova, O.V.; Gofarov, M.Y.; Kondakov, A.V.; Konopleva, E.S.; et al. Discovery of Novaculina myanmarensis Sp. Nov. (Bivalvia: Pharidae: Pharellinae) Closes the Freshwater Razor Clams Range Disjunction in Southeast Asia. Sci. Rep. 2018, 8, 16325. [Google Scholar] [CrossRef] [PubMed]
- Win, T.; Bolotov, I.N.; Vikhrev, I.V.; Lunn, Z.; Chan, N.; Konopleva, E.S.; Gofarov, M.Y.; Tomilova, A.A.; Kondakov, A.V. Phylogeography and Distribution of the Freshwater Razor Clams Novaculina myanmarensis and N. Gangetica in Myanmar, with Notes on Two Doubtful Nominal Taxa Described as Novaculina Members (Bivalvia: Pharidae). Ecol. Monten. 2021, 40, 59–67. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Zhang, W.Z. A New Species of Freshwater Razor Clam, Novaculina chinensis, from Jiangsu Province, China. Acta Zootaxonomica Sin. 1979, 4, 356–357. [Google Scholar]
- Shu, F.; Zhu, Q.; Zhang, N.; Zhang, X.; Xie, S. Novaculina chinensis Found in Lake Weishan, Shandong Province. Chin. J. Zool. 2013, 48, 278–280. [Google Scholar] [CrossRef]
- Rao, X.; Xu, Y.; Chen, Y.; Lin, G. A Study on Induced Spawning of Novaculina chinensis. J. Fujian Teach. Univ. Nat. Sci. Ed. 2003, 78–81+93. [Google Scholar]
- Jiang, J.; Chen, Y.; Xu, Y.; Huang, J.; Yan, G. A Study on the Seasonal Changes of Novaculina chinensis in Taojiang. J. Fujian Teach. Univ. Nat. Sci. Ed. 1998, 14, 86–91. [Google Scholar]
- Rao, X.; Chen, Y.; Xu, Y. Study on the Morphology of Novaculina Chinensis. J. -Fujian Teach. Univ. Nat. Sci. Ed. 1998, 14, 71–75. [Google Scholar]
- Xiaozhen, R.; Yinshan, C.; Wenlie, C.; Youqin, X.; Lianyun, C. Ultrastructural Studies on Spermatogenesis of Novaculina chinensis. Chin. J. Zool. 2000, 35, 2–5. [Google Scholar]
- Rao, X. Ultrastructures of Novaculina chinensis during Oogenesis. Chin. J. Appl. Environ. Biol. 2001, 7, 248–253. [Google Scholar]
- Bieler, R.; Mikkelsen, P.M.; Collins, T.M.; Glover, E.A.; González, V.L.; Graf, D.L.; Harper, E.M.; Healy, J.; Kawauchi, G.Y.; Sharma, P.P. Investigating the Bivalve Tree of Life–an Exemplar-Based Approach Combining Molecular and Novel Morphological Characters. Invertebr. Syst. 2014, 28, 32–115. [Google Scholar] [CrossRef]
- Ghiselli, F.; Gomes-dos-Santos, A.; Adema, C.M.; Lopes-Lima, M.; Sharbrough, J.; Boore, J.L. Molluscan Mitochondrial Genomes Break the Rules. Philos. Trans. R. Soc. B 2021, 376, 20200159. [Google Scholar] [CrossRef]
- Lavrov, D.V.; Pett, W. Animal Mitochondrial DNA as We Do Not Know It: Mt-Genome Organization and Evolution in Nonbilaterian Lineages. Genome Biol. Evol. 2016, 8, 2896–2913. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Guo, Y.; Yan, C.; Ye, Y.; Yan, X.; Li, J.; Xu, K.; Guo, B.; Lü, Z. Novel Gene Rearrangement in the Mitochondrial Genome of Siliqua minima (Bivalvia, Adapedonta) and Phylogenetic Implications for Imparidentia. PLoS ONE 2021, 16, e0249446. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Xu, Y.; Wang, S.; Zhang, R.; Guo, K.; Xu, W.; Zhao, Z. Complete Mitochondrial Genome Sequence and Phylogenetic Analysis of Procambarus clarkii and Cambaroides dauricus from China. Int. J. Mol. Sci. 2023, 24, 11282. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yu, R.; Ma, P.; Li, C. Complete Mitochondrial Genome of Cultellus attenuatus and Its Phylogenetic Implications. Mol. Biol. Rep. 2022, 49, 8163–8168. [Google Scholar] [CrossRef] [PubMed]
- Zheng, R.; Li, J.; Niu, D. The Complete DNA Sequence of the Mitochondrial Genome of Sinonovacula constricta (Bivalvia: Solecurtidae). Acta Oceanol. Sin. 2010, 29, 88–92. [Google Scholar] [CrossRef]
- Wu, R.-W.; Liu, X.-J.; Wang, S.; Roe, K.J.; Ouyang, S.; Wu, X.-P. Analysis of Mitochondrial Genomes Resolves the Phylogenetic Position of Chinese Freshwater Mussels (Bivalvia, Unionidae). ZooKeys 2019, 812, 23–46. [Google Scholar] [CrossRef]
- Pavan-Kumar, A.; Varshney, S.; Suman, S.; Das, R.; Chaudhari, A.; Krishna, G. Complete Mitochondrial Genome of Freshwater Pearl Mussel Lamellidens marginalis (Lamarck, 1819) and Its Phylogenetic Relation within Unionidae Family. Mol. Biol. Rep. 2022, 49, 9593–9603. [Google Scholar] [CrossRef]
- Xu, M.; Gu, Z.; Huang, J.; Guo, B.; Jiang, L.; Xu, K.; Ye, Y.; Li, J. The Complete Mitochondrial Genome of Mytilisepta virgata (Mollusca: Bivalvia), Novel Gene Rearrangements, and the Phylogenetic Relationships of Mytilidae. Genes 2023, 14, 910. [Google Scholar] [CrossRef]
- Bolotov, I.N.; Aksenova, O.V.; Bakken, T.; Glasby, C.J.; Gofarov, M.Y.; Kondakov, A.V.; Konopleva, E.S.; Lopes-Lima, M.; Lyubas, A.A.; Wang, Y.; et al. Discovery of a Silicate Rock-Boring Organism and Macrobioerosion in Fresh Water. Nat. Commun. 2018, 9, 2882. [Google Scholar] [CrossRef] [PubMed]
- Breton, S.; Stewart, D.T.; Hoeh, W.R. Characterization of a Mitochondrial ORF from the Gender-Associated mtDNAs of Mytilus Spp. (Bivalvia: Mytilidae): Identification of the “Missing” ATPase 8 Gene. Mar. Genom. 2010, 3, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Li, Q.; Kong, L.; Yu, H. Limited Locomotive Ability Relaxed Selective Constraints on Molluscs Mitochondrial Genomes. Sci. Rep. 2017, 7, 10628. [Google Scholar] [CrossRef] [PubMed]
- Sperl, W.; Ješina, P.; Zeman, J.; Mayr, J.A.; Demeirleir, L.; VanCoster, R.; Pickova, A.; Hansikova, H.; Houšt’ková, H.; Krejčík, Z. Deficiency of Mitochondrial ATP Synthase of Nuclear Genetic Origin. Neuromuscul. Disord. 2006, 16, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhang, J.; Xia, J.; Yang, J.; Guo, J.; Deng, Z.; Luo, M. Comparative Characterization of the Complete Mitochondrial Genomes of the Three Apple Snails (Gastropoda: Ampullariidae) and the Phylogenetic Analyses. Int. J. Mol. Sci. 2018, 19, 3646. [Google Scholar] [CrossRef] [PubMed]
- Mayrose, I.; Doron-Faigenboim, A.; Bacharach, E.; Pupko, T. Towards Realistic Codon Models: Among Site Variability and Dependency of Synonymous and Non-Synonymous Rates. Bioinformatics 2007, 23, i319–i327. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Guo, X.; Chen, J.; Li, J.; Zhang, S.; Zheng, S.; Wang, Y.; Peng, Y.; Zhang, K.; Liu, Y. The Complete Mitochondrial Genome of Hemigrapsus sinensis (Brachyura, Grapsoidea, Varunidae) and Its Phylogenetic Position within Grapsoidea. Genes Genom. 2023, 45, 377–391. [Google Scholar] [CrossRef]
- Yang, Z.; Bielawski, J.P. Statistical Methods for Detecting Molecular Adaptation. Trends Ecol. Evol. 2000, 15, 496–503. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, J.; Zhao, X.-Q.; Wang, J.; Wong, G.K.-S.; Yu, J. KaKs_Calculator: Calculating Ka and Ks Through Model Selection and Model Averaging. Genom. Proteom. Bioinform. 2006, 4, 259–263. [Google Scholar] [CrossRef]
- Saccone, C.; De Giorgi, C.; Gissi, C.; Pesole, G.; Reyes, A. Evolutionary Genomics in Metazoa: The Mitochondrial DNA as a Model System. Gene 1999, 238, 195–209. [Google Scholar] [CrossRef]
- Lemer, S.; Bieler, R.; Giribet, G. Resolving the Relationships of Clams and Cockles: Dense Transcriptome Sampling Drastically Improves the Bivalve Tree of Life. Proc. R. Soc. B 2019, 286, 20182684. [Google Scholar] [CrossRef] [PubMed]
- Audino, J.A.; Serb, J.M.; Marian, J.E.A. Phylogeny and Anatomy of Marine Mussels (Bivalvia: Mytilidae) Reveal Convergent Evolution of Siphon Traits. Zool. J. Linn. Soc. 2020, 190, 592–612. [Google Scholar] [CrossRef]
- Sartori, A.F.; Printrakoon, C.; Mikkelsen, P.M.; Bieler, R. Siphonal Structure in the Veneridae (Bivalvia: Heterodonta) with an Assessment of Its Phylogenetic Application and a Review of Venerids of the Gulf of Thailand. Raffles Bull. Zool. 2008, 18, 103–125. [Google Scholar]
- An Analytical Approach to a Classification of the Bivalvia. Philos. Trans. R. Soc. Lond. B 1978, 284, 425–436. [CrossRef]
- Zwarts, L.; Wanink, J. Siphon Size and Burying Depth in Deposit- and Suspension-Feeding Benthic Bivalves. Mar. Biol. 1989, 100, 227–240. [Google Scholar] [CrossRef]
- McLusky, D.S.; Elliott, M. The Feeding and Survival Strategies of Estuarine Molluscs. Feed. Surviv. Srategies Estuar. Org. 1981, 15, 109–121. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de Novo Metazoan Mitochondrial Genome Annotation. Mol. Phylogenetics Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Grant, J.R.; Enns, E.; Marinier, E.; Mandal, A.; Herman, E.K.; Chen, C.; Graham, M.; Van Domselaar, G.; Stothard, P. Proksee: In-Depth Characterization and Visualization of Bacterial Genomes. Nucleic Acids Res. 2023, 51, W484–W492. [Google Scholar] [CrossRef]
- Xiang, C.; Gao, F.; Jakovlić, I.; Lei, H.; Hu, Y.; Zhang, H.; Zou, H.; Wang, G.; Zhang, D. Using PhyloSuite for Molecular Phylogeny and Tree-based Analyses. iMeta 2023, 2, e87. [Google Scholar] [CrossRef]
- Hassanin, A.; Leger, N.; Deutsch, J. Evidence for Multiple Reversals of Asymmetric Mutational Constraints during the Evolution of the Mitochondrial Genome of Metazoa, and Consequences for Phylogenetic Inferences. Syst. Biol. 2005, 54, 277–298. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Talavera, G.; Castresana, J. Improvement of Phylogenies after Removing Divergent and Ambiguously Aligned Blocks from Protein Sequence Alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Strand | Position (Start–End) | Length(bp) | Intergenic_Spacer | Start_Codon | Stop_Codon |
|---|---|---|---|---|---|---|
| trnK-aaa | + | 1–67 | 67 | - | - | - |
| cox2 | + | 113–922 | 810 | 45 | ATG | TAG |
| trnY-tac | + | 1086–1149 | 64 | 163 | - | - |
| nad4l | + | 1157–1444 | 288 | 7 | ATG | TAA |
| trnG-gga | + | 1446–1511 | 66 | 1 | - | - |
| trnP-cca | + | 1518–1582 | 65 | 6 | - | - |
| nad4 | + | 1696–3057 | 1362 | 113 | ATT | TAG |
| trnH-cac | + | 3060–3123 | 64 | 2 | - | - |
| trnW-tga | + | 3126–3192 | 67 | 2 | - | - |
| trnR-cga | + | 3196–3260 | 65 | 3 | - | - |
| trnE-gaa | + | 3279–3345 | 67 | 18 | - | - |
| trnS2-tca | + | 3340–3402 | 63 | −6 | - | - |
| nad3 | + | 3403–3768 | 366 | 0 | ATG | TAG |
| trnT-aca | + | 3768–3833 | 66 | −1 | - | - |
| trnI-atc | + | 3837–3902 | 66 | 3 | - | - |
| trnD-gac | + | 3918–3983 | 66 | 15 | - | - |
| trnQ-caa | + | 3984–4051 | 68 | 0 | - | - |
| trnC-tgc | + | 4062–4126 | 65 | 10 | - | - |
| trnA-gca | + | 4129–4193 | 65 | 2 | - | - |
| trnF-ttc | + | 4199–4262 | 64 | 5 | - | - |
| cox1 | + | 4421–6100 | 1680 | 158 | ATT | TAA |
| trnL2-tta | + | 6128–6192 | 65 | 27 | - | - |
| nad1 | + | 6193–7119 | 927 | 0 | ATG | TAA |
| trnL1-cta | + | 7124–7191 | 68 | 4 | - | - |
| trnV-gta | + | 7192–7255 | 64 | 0 | - | - |
| trnN-aac | + | 7256–7321 | 66 | 0 | - | - |
| nad5 | + | 7322–9082 | 1761 | 0 | ATT | TAA |
| cob | + | 9119–10264 | 1146 | 36 | ATG | TAA |
| nad6 | + | 10277–10807 | 531 | 12 | ATG | TAG |
| rrnL | + | 10808–12039 | 1232 | 0 | - | - |
| atp6 | + | 12040–12738 | 699 | 0 | ATG | TAA |
| trnM-atg | + | 12751–12816 | 66 | 12 | - | - |
| rrnS | + | 12894–13742 | 849 | 77 | - | - |
| cox3 | + | 13741–14529 | 789 | −2 | ATG | TAG |
| trnS1-aga | + | 14529–14595 | 67 | −1 | - | - |
| nad2 | + | 14596–15648 | 1053 | 0 | ATG | TAA |
| Region | Size (bp) | A% | T% | G% | C% | AT% | GC% | AT-Skew | GC-Skew |
|---|---|---|---|---|---|---|---|---|---|
| Mitogenome | 16,262 | 28.15 | 43.45 | 18.86 | 9.54 | 71.60 | 28.40 | −0.214 | 0.328 |
| PCGs | 11,502 | 25.38 | 46.38 | 18.81 | 9.42 | 71.76 | 28.24 | −0.293 | 0.333 |
| tRNAs | 1444 | 32.06 | 36.22 | 18.91 | 12.81 | 68.28 | 31.72 | −0.061 | 0.192 |
| rRNAs | 2212 | 33.95 | 36.53 | 18.63 | 10.90 | 70.48 | 29.52 | −0.04 | 0.26 |
| Codon | Count | RSCU | Codon | Count | RSCU | Codon | Count | RSCU | Codon | Count | RSCU |
|---|---|---|---|---|---|---|---|---|---|---|---|
| UUU (F) | 401 | 1.94 | UCU (S) | 146 | 2.89 | UAU (Y) | 148 | 1.91 | UGU (C) | 73 | 1.78 |
| UUC (F) | 12 | 0.06 | UCC (S) | 2 | 0.04 | UAC (Y) | 7 | 0.09 | UGC (C) | 9 | 0.22 |
| UUA (L) | 360 | 4.08 | UCA (S) | 46 | 0.91 | UAA (*) | 7 | 1.17 | UGA (W) | 58 | 1.2 |
| UUG (L) | 85 | 0.96 | UCG (S) | 9 | 0.18 | UAG (*) | 5 | 0.83 | UGG (W) | 39 | 0.8 |
| CUU (L) | 71 | 0.8 | CCU (P) | 90 | 3 | CAU (H) | 68 | 1.92 | CGU (R) | 32 | 1.94 |
| CUC (L) | 1 | 0.01 | CCC (P) | 3 | 0.1 | CAC (H) | 3 | 0.08 | CGC (R) | 2 | 0.12 |
| CUA (L) | 12 | 0.14 | CCA (P) | 24 | 0.8 | CAA (Q) | 33 | 1.43 | CGA (R) | 22 | 1.33 |
| CUG (L) | 1 | 0.01 | CCG (P) | 3 | 0.1 | CAG (Q) | 13 | 0.57 | CGG (R) | 10 | 0.61 |
| AUU (I) | 255 | 1.98 | ACU (T) | 80 | 2.52 | AAU (N) | 96 | 1.96 | AGU (S) | 56 | 1.11 |
| AUC (I) | 3 | 0.02 | ACC (T) | 2 | 0.06 | AAC (N) | 2 | 0.04 | AGC (S) | 5 | 0.1 |
| AUA (M) | 161 | 1.56 | ACA (T) | 39 | 1.23 | AAA (K) | 73 | 1.47 | AGA (S) | 110 | 2.18 |
| AUG (M) | 46 | 0.44 | ACG (T) | 6 | 0.19 | AAG (K) | 26 | 0.53 | AGG (S) | 30 | 0.59 |
| GUU (V) | 223 | 2.56 | GCU (A) | 141 | 3.34 | GAU (D) | 91 | 1.96 | GGU (G) | 165 | 2.06 |
| GUC (V) | 2 | 0.02 | GCC (A) | 2 | 0.05 | GAC (D) | 2 | 0.04 | GGC (G) | 11 | 0.14 |
| GUA (V) | 107 | 1.23 | GCA (A) | 24 | 0.57 | GAA (E) | 57 | 1.28 | GGA (G) | 88 | 1.1 |
| GUG (V) | 16 | 0.18 | GCG (A) | 2 | 0.05 | GAG (E) | 32 | 0.72 | GGG (G) | 56 | 0.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Z.; Song, Y.; Zheng, Z.; Liu, Y.; Yao, H.; Rao, X.; Lin, G. The Complete Mitochondrial Genome and Phylogenetic Analysis of the Freshwater Shellfish Novaculina chinensis (Bivalvia: Pharidae). Int. J. Mol. Sci. 2024, 25, 67. https://doi.org/10.3390/ijms25010067
Zhou Z, Song Y, Zheng Z, Liu Y, Yao H, Rao X, Lin G. The Complete Mitochondrial Genome and Phylogenetic Analysis of the Freshwater Shellfish Novaculina chinensis (Bivalvia: Pharidae). International Journal of Molecular Sciences. 2024; 25(1):67. https://doi.org/10.3390/ijms25010067
Chicago/Turabian StyleZhou, Ziquan, Yuxin Song, Zewen Zheng, Yunguang Liu, Haiyan Yao, Xiaozhen Rao, and Gang Lin. 2024. "The Complete Mitochondrial Genome and Phylogenetic Analysis of the Freshwater Shellfish Novaculina chinensis (Bivalvia: Pharidae)" International Journal of Molecular Sciences 25, no. 1: 67. https://doi.org/10.3390/ijms25010067