

Lipoprotein(a) and Atherosclerotic Cardiovascular Disease: Where Do We Stand?
 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Literature Search
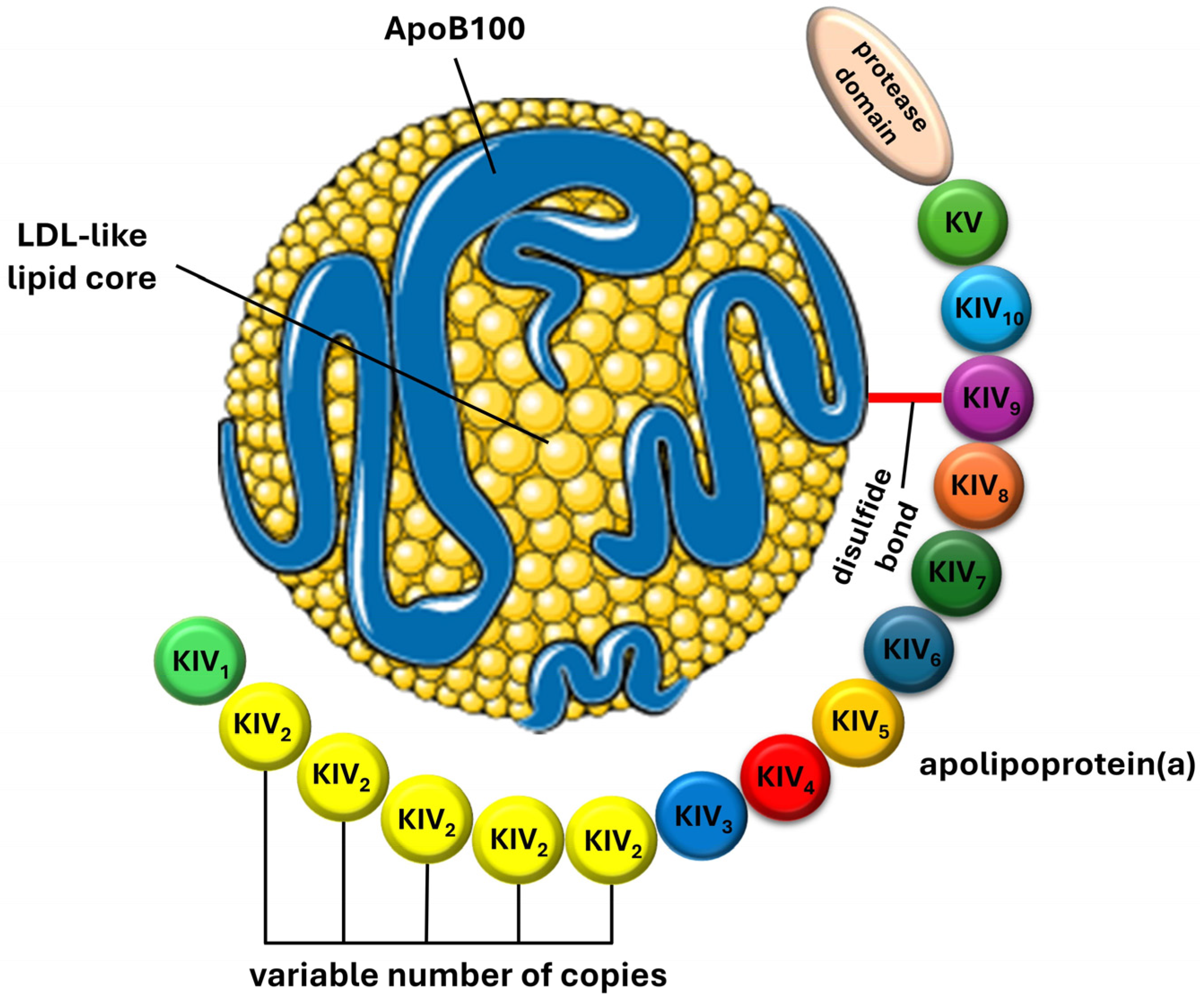
3. Lipoprotein(a): Structure, Synthesis, and Metabolism
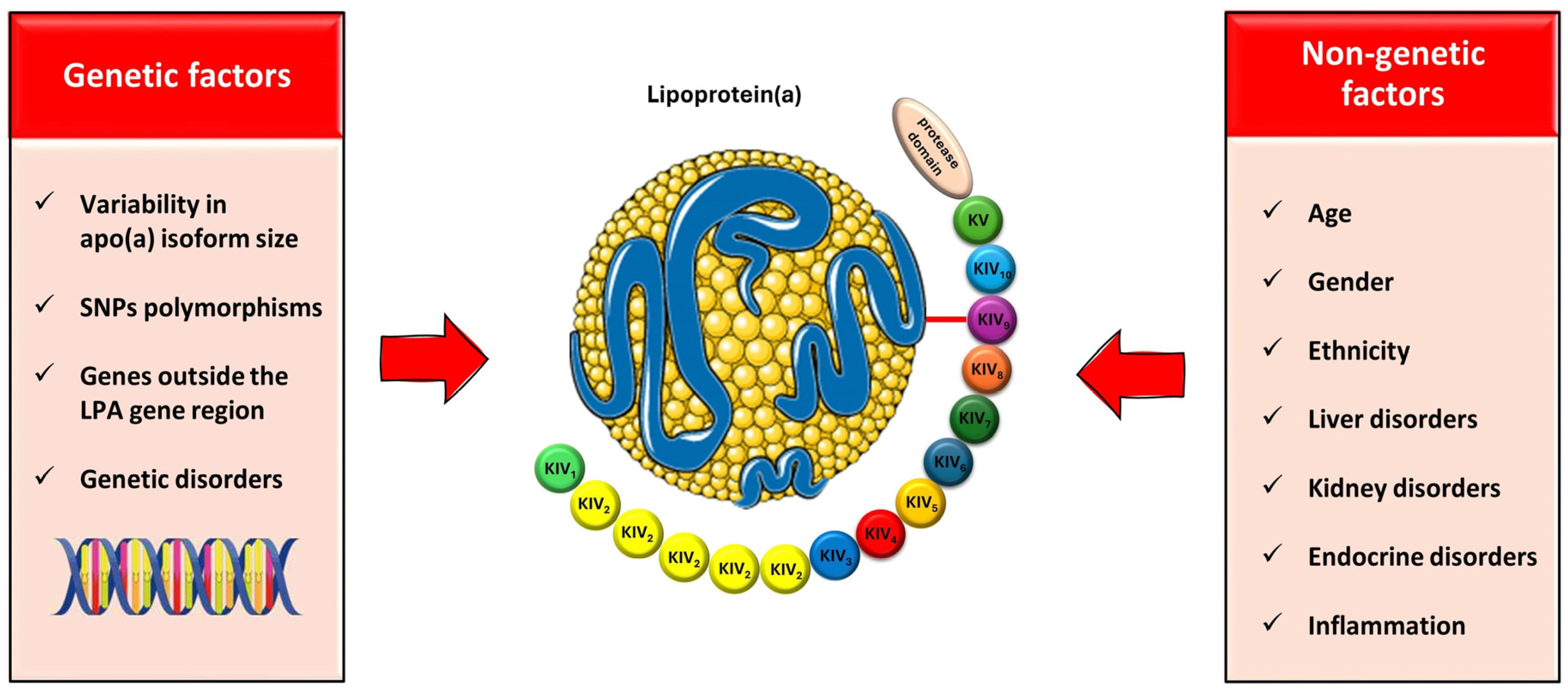
4. Factors Affecting the Lp(a) Levels: Genetics and beyond
4.1. Genetics
4.2. Beyond Genetics
4.2.1. Age, Gender, and Ethnicity
4.2.2. Liver and Kidney Disorders
4.2.3. Hormones
4.2.4. The Role of Inflammation
5. Lp(a) Measurement and Reporting: Current Knowledge and Concerns
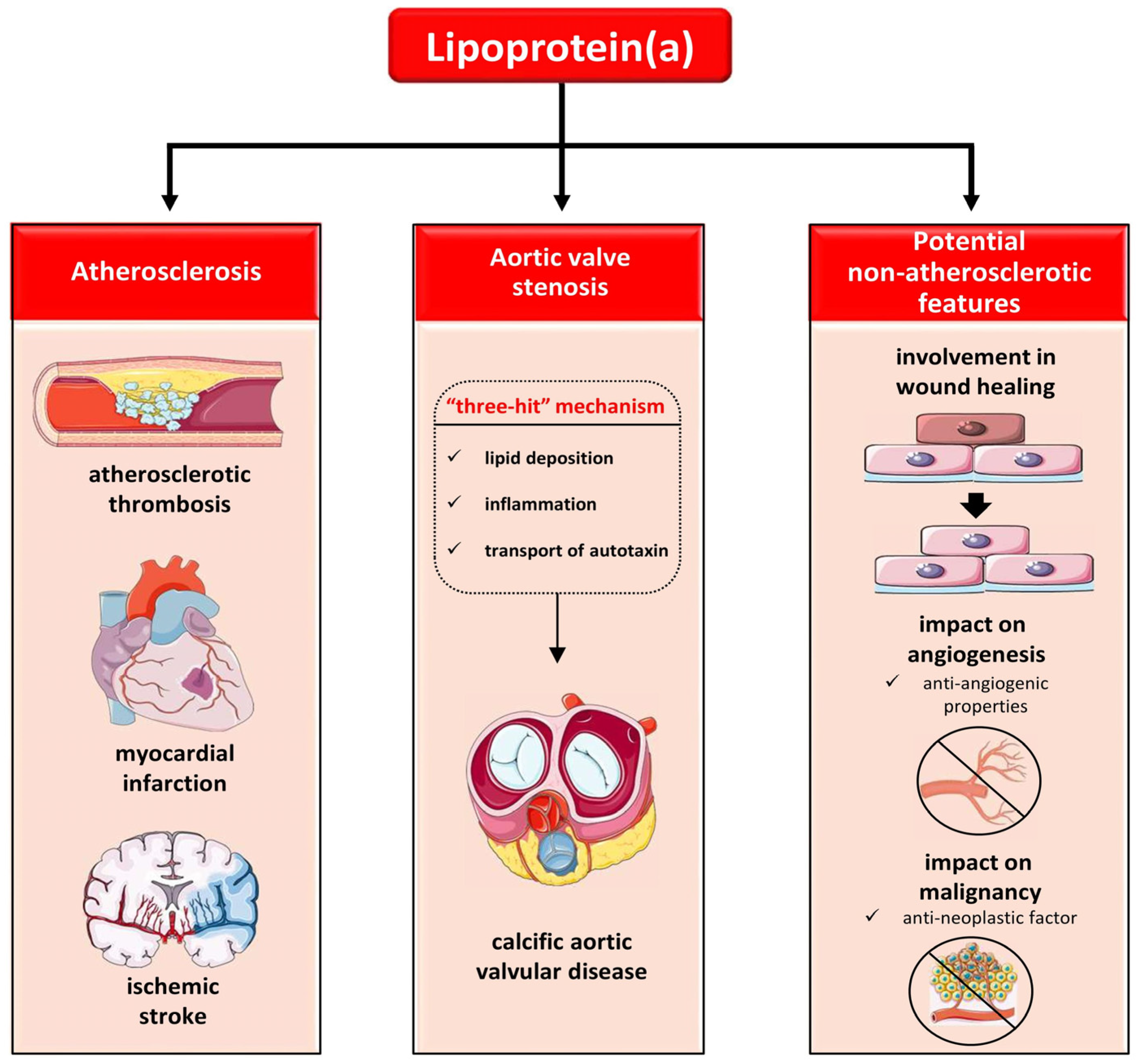
6. The Role of Lp(a) in Atherosclerosis
7. The Role of Lp(a) in Cardiovascular Disease: Focusing on Atherothrombosis
7.1. The Role of Lp(a) in Atherosclerotic Cardiovascular Disease (ASCVD)
7.2. The Role of Lp(a) in Calcific Aortic Vascular Disease (CAVD)
7.3. The Role of Lp(a) in Thrombosis
8. The Role of Lp(a) beyond Atherosclerosis
9. The Impact of Lipid-modifying Interventions on Lp(a) Levels: From Traditional to Novel Agents
9.1. Fasting and Lifestyle Modifications
9.2. Traditional Lipid-Lowering Therapies
9.2.1. Statins
9.2.2. Ezetimibe
9.2.3. Fibrates
9.2.4. Niacin
9.3. Novel Hypolipidemic Agents
9.3.1. Mipomersen
9.3.2. PCSK9 Inhibition
Monoclonal Antibodies against PCSK9
Inclisiran
9.3.3. Bempedoic Acid
9.3.4. CETP Inhibitors
9.4. Lipoprotein Apheresis
10. Molecular Lp(a)-targeting Therapies: In the Heart of the Problem
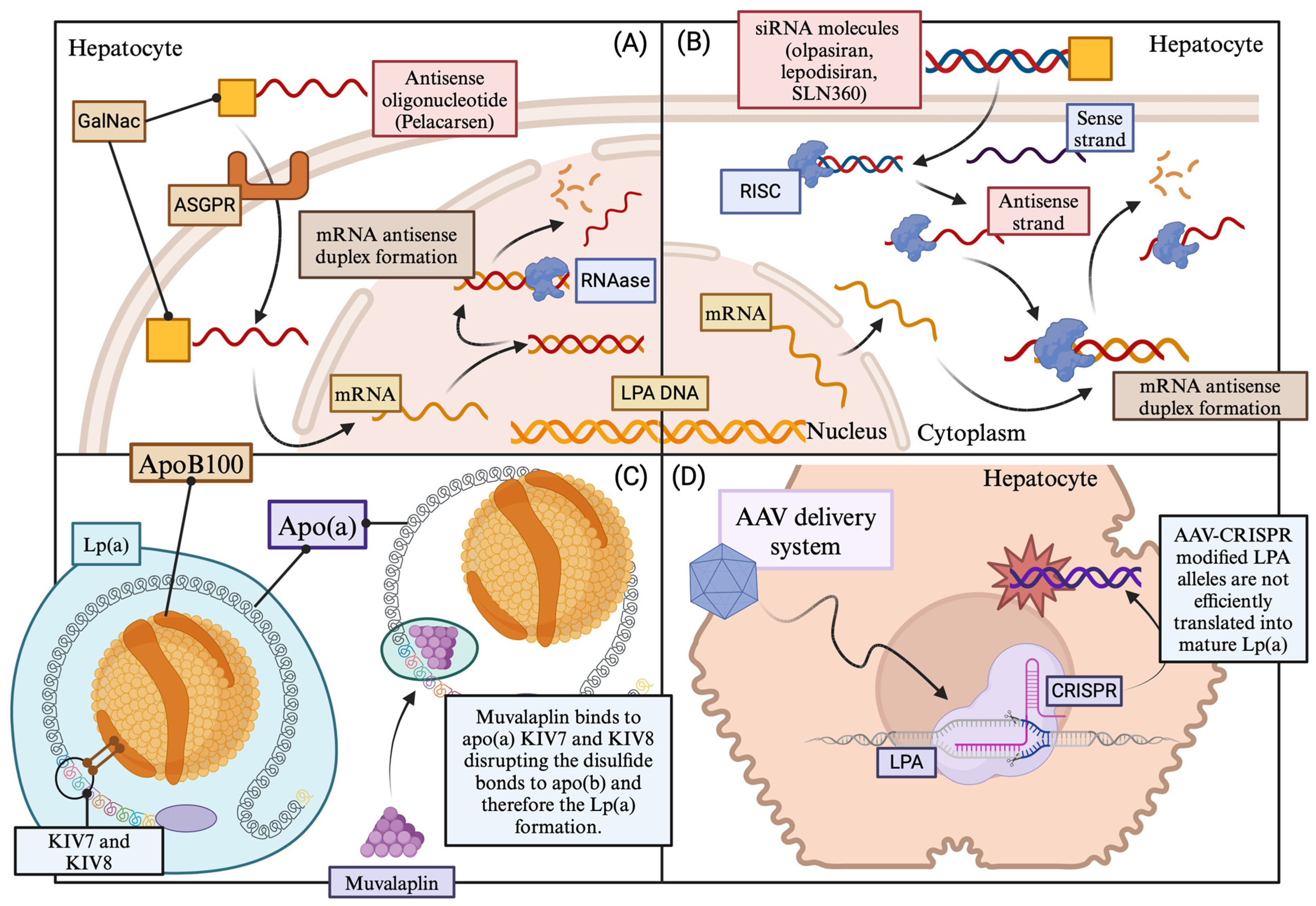
10.1. Pelacarsen
10.2. Olpasiran
10.3. SLN360
10.4. Lepodisiran
10.5. Muvalaplin
10.6. CRISPR/Cas9 Lp(a) Genome Editing
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Gaubatz, J.W.; Heideman, C.; Gotto, A.M., Jr.; Morrisett, J.D.; Dahlen, G.H. Human plasma lipoprotein [a]. Structural properties. J. Biol. Chem. 1983, 258, 4582–4589. [Google Scholar] [CrossRef] [PubMed]
- Lampsas, S.; Xenou, M.; Oikonomou, E.; Pantelidis, P.; Lysandrou, A.; Sarantos, S.; Goliopoulou, A.; Kalogeras, K.; Tsigkou, V.; Kalpis, A.; et al. Lipoprotein(a) in Atherosclerotic Diseases: From Pathophysiology to Diagnosis and Treatment. Molecules 2023, 28, 969. [Google Scholar] [CrossRef] [PubMed]
- Berg, K. A new serum type system in man-The LP system. Acta Pathol. Microbiol. Scand. 1963, 59, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Kamstrup, P.R. Lipoprotein(a) and Cardiovascular Disease. Clin. Chem. 2021, 67, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Kronenberg, F.; Mora, S.; Stroes, E.S.G.; Ference, B.A.; Arsenault, B.J.; Berglund, L.; Dweck, M.R.; Koschinsky, M.; Lambert, G.; Mach, F.; et al. Lipoprotein(a) in atherosclerotic cardiovascular disease and aortic stenosis: A European Atherosclerosis Society consensus statement. Eur. Heart J. 2022, 43, 3925–3946. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S.; Marcovina, S.M. Ancestry, Lipoprotein(a), and Cardiovascular Risk Thresholds: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2022, 80, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Wolski, K.; Cho, L.; Nicholls, S.J.; Kastelein, J.; Leitersdorf, E.; Landmesser, U.; Blaha, M.; Lincoff, A.M.; Morishita, R.; et al. Lipoprotein(a) levels in a global population with established atherosclerotic cardiovascular disease. Open Heart 2022, 9, e002060. [Google Scholar] [CrossRef] [PubMed]
- Kosmas, C.E.; Bousvarou, M.D.; Papakonstantinou, E.J.; Tsamoulis, D.; Koulopoulos, A.; Echavarria Uceta, R.; Guzman, E.; Rallidis, L.S. Novel Pharmacological Therapies for the Management of Hyperlipoproteinemia(a). Int. J. Mol. Sci. 2023, 24, 13622. [Google Scholar] [CrossRef]
- Tselepis, A.D. Treatment of Lp(a): Is It the Future or Are We Ready Today? Curr. Atheroscler. Rep. 2023, 25, 679–689. [Google Scholar] [CrossRef]
- Gupta, K.; Hinkamp, C.; Andrews, T.; Meloche, C.; Minhas, A.M.K.; Slipczuk, L.; Vaughan, E.; Habib, F.Z.; Sheikh, S.; Kalra, D.; et al. Highlights of Cardiovascular Disease Prevention Studies Presented at the 2023 European Society of Cardiology Congress. Curr. Atheroscler. Rep. 2023, 25, 965–978. [Google Scholar] [CrossRef]
- Ruscica, M.; Sirtori, C.R.; Corsini, A.; Watts, G.F.; Sahebkar, A. Lipoprotein(a): Knowns, unknowns and uncertainties. Pharmacol. Res. 2021, 173, 105812. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.; Noureen, A.; Kronenberg, F.; Utermann, G. Structure, function, and genetics of lipoprotein (a). J. Lipid Res. 2016, 57, 1339–1359. [Google Scholar] [CrossRef] [PubMed]
- Guevara, J., Jr.; Jan, A.Y.; Knapp, R.; Tulinsky, A.; Morrisett, J.D. Comparison of ligand-binding sites of modeled apo[a] kringle-like sequences in human lipoprotein[a]. Arterioscler. Thromb. 1993, 13, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Santonastaso, A.; Maggi, M.; De Jonge, H.; Scotti, C. High resolution structure of human apolipoprotein (a) kringle IV type 2: Beyond the lysine binding site. J. Lipid Res. 2020, 61, 1687–1696. [Google Scholar] [CrossRef] [PubMed]
- Coassin, S.; Kronenberg, F. Lipoprotein(a) beyond the kringle IV repeat polymorphism: The complexity of genetic variation in the LPA gene. Atherosclerosis 2022, 349, 17–35. [Google Scholar] [CrossRef] [PubMed]
- Trieu, V.N.; McConathy, W.J. A two-step model for lipoprotein(a) formation. J. Biol. Chem. 1995, 270, 15471–15474. [Google Scholar] [CrossRef] [PubMed]
- Chemello, K.; Chan, D.C.; Lambert, G.; Watts, G.F. Recent advances in demystifying the metabolism of lipoprotein(a). Atherosclerosis 2022, 349, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Dieplinger, H.; Utermann, G. The seventh myth of lipoprotein(a): Where and how is it assembled? Curr. Opin. Lipidol. 1999, 10, 275–283. [Google Scholar] [CrossRef]
- Youssef, A.; Clark, J.R.; Marcovina, S.M.; Boffa, M.B.; Koschinsky, M.L. Apo(a) and ApoB Interact Noncovalently Within Hepatocytes: Implications for Regulation of Lp(a) Levels by Modulation of ApoB Secretion. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 289–304. [Google Scholar] [CrossRef]
- Kostner, K.M.; Kostner, G.M. Lipoprotein (a): A historical appraisal. J. Lipid Res. 2017, 58, 1–14. [Google Scholar] [CrossRef]
- McCormick, S.P. Lipoprotein(a): Biology and clinical importance. Clin. Biochem. Rev. 2004, 25, 69–80. [Google Scholar] [PubMed]
- McCormick, S.P.A.; Schneider, W.J. Lipoprotein(a) catabolism: A case of multiple receptors. Pathology 2019, 51, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Pasławska, A.; Tomasik, P.J. Lipoprotein(a)-60 Years Later—What Do We Know? Cells 2023, 12, 2472. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Redpath, G.M.; Williams, M.J.; McCormick, S.P. Recycling of Apolipoprotein(a) After PlgRKT-Mediated Endocytosis of Lipoprotein(a). Circ. Res. 2017, 120, 1091–1102. [Google Scholar] [CrossRef] [PubMed]
- Rader, D.J.; Cain, W.; Ikewaki, K.; Talley, G.; Zech, L.A.; Usher, D.; Brewer, H.B., Jr. The inverse association of plasma lipoprotein(a) concentrations with apolipoprotein(a) isoform size is not due to differences in Lp(a) catabolism but to differences in production rate. J. Clin. Investig. 1994, 93, 2758–2763. [Google Scholar] [CrossRef] [PubMed]
- Fogacci, F.; Di Micoli, V.; Avagimyan, A.; Giovannini, M.; Imbalzano, E.; Cicero, A.F.G. Assessment of Apolipoprotein(a) Isoform Size Using Phenotypic and Genotypic Methods. Int. J. Mol. Sci. 2023, 24, 13886. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C.; Watts, G.F.; Coll, B.; Wasserman, S.M.; Marcovina, S.M.; Barrett, P.H.R. Lipoprotein(a) Particle Production as a Determinant of Plasma Lipoprotein(a) Concentration Across Varying Apolipoprotein(a) Isoform Sizes and Background Cholesterol-Lowering Therapy. J. Am. Heart. Assoc. 2019, 8, e011781. [Google Scholar] [CrossRef]
- White, A.L.; Guerra, B.; Wang, J.; Lanford, R.E. Presecretory degradation of apolipoprotein [a] is mediated by the proteasome pathway. J. Lipid Res. 1999, 40, 275–286. [Google Scholar] [CrossRef]
- Frank, S.L.; Klisak, I.; Sparkes, R.S.; Mohandas, T.; Tomlinson, J.E.; McLean, J.W.; Lawn, R.M.; Lusis, A.J. The apolipoprotein(a) gene resides on human chromosome 6q26-27, in close proximity to the homologous gene for plasminogen. Hum. Genet. 1988, 79, 352–356. [Google Scholar] [CrossRef]
- Rubin, J.; Paultre, F.; Tuck, C.H.; Holleran, S.; Reed, R.G.; Pearson, T.A.; Thomas, C.M.; Ramakrishnan, R.; Berglund, L. Apolipoprotein [a] genotype influences isoform dominance pattern differently in African Americans and Caucasians. J. Lipid Res. 2002, 43, 234–244. [Google Scholar] [CrossRef]
- Enkhmaa, B.; Anuurad, E.; Berglund, L. Lipoprotein (a): Impact by ethnicity and environmental and medical conditions. J. Lipid Res. 2016, 57, 1111–1125. [Google Scholar] [CrossRef] [PubMed]
- Mack, S.; Coassin, S.; Rueedi, R.; Yousri, N.A.; Seppälä, I.; Gieger, C.; Schönherr, S.; Forer, L.; Erhart, G.; Marques-Vidal, P.; et al. A genome-wide association meta-analysis on lipoprotein (a) concentrations adjusted for apolipoprotein (a) isoforms. J. Lipid Res. 2017, 58, 1834–1844. [Google Scholar] [CrossRef] [PubMed]
- Schachtl-Riess, J.F.; Kheirkhah, A.; Grüneis, R.; Di Maio, S.; Schoenherr, S.; Streiter, G.; Losso, J.L.; Paulweber, B.; Eckardt, K.U.; Köttgen, A.; et al. Frequent LPA KIV-2 Variants Lower Lipoprotein(a) Concentrations and Protect Against Coronary Artery Disease. J. Am. Coll. Cardiol. 2021, 78, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Coassin, S.; Erhart, G.; Weissensteiner, H.; de Araújo, M.E.G.; Lamina, C.; Schönherr, S.; Forer, L.; Haun, M.; Losso, J.L.; Köttgen, A.; et al. A novel but frequent variant in LPA KIV-2 is associated with a pronounced Lp(a) and cardiovascular risk reduction. Eur. Heart. J. 2017, 38, 1823–1831. [Google Scholar] [CrossRef] [PubMed]
- Said, M.A.; Yeung, M.W.; van de Vegte, Y.J.; Benjamins, J.W.; Dullaart, R.P.F.; Ruotsalainen, S.; Ripatti, S.; Natarajan, P.; Juarez-Orozco, L.E.; Verweij, N.; et al. Genome-Wide Association Study and Identification of a Protective Missense Variant on Lipoprotein(a) Concentration: Protective Missense Variant on Lipoprotein(a) Concentration-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1792–1800. [Google Scholar] [CrossRef]
- Sticchi, E.; Magi, A.; Kamstrup, P.R.; Marcucci, R.; Prisco, D.; Martinelli, I.; Mannucci, P.M.; Abbate, R.; Giusti, B. Apolipoprotein(a) Kringle-IV Type 2 Copy Number Variation Is Associated with Venous Thromboembolism. PLoS ONE 2016, 11, e0149427. [Google Scholar] [CrossRef]
- Clarke, R.; Peden, J.F.; Hopewell, J.C.; Kyriakou, T.; Goel, A.; Heath, S.C.; Parish, S.; Barlera, S.; Franzosi, M.G.; Rust, S.; et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N. Engl. J. Med. 2009, 361, 2518–2528. [Google Scholar] [CrossRef]
- Morgan, B.M.; Brown, A.N.; Deo, N.; Harrop, T.W.R.; Taiaroa, G.; Mace, P.D.; Wilbanks, S.M.; Merriman, T.R.; Williams, M.J.A.; McCormick, S.P.A. Nonsynonymous SNPs in LPA homologous to plasminogen deficiency mutants represent novel null apo(a) alleles. J. Lipid Res. 2020, 61, 432–444. [Google Scholar] [CrossRef]
- Zabaneh, D.; Kumari, M.; Sandhu, M.; Wareham, N.; Wainwright, N.; Papamarkou, T.; Hopewell, J.; Clarke, R.; Li, K.; Palmen, J.; et al. Meta analysis of candidate gene variants outside the LPA locus with Lp(a) plasma levels in 14,500 participants of six White European cohorts. Atherosclerosis 2011, 217, 447–451. [Google Scholar] [CrossRef]
- Hoekstra, M.; Chen, H.Y.; Rong, J.; Dufresne, L.; Yao, J.; Guo, X.; Tsai, M.Y.; Tsimikas, S.; Post, W.S.; Vasan, R.S.; et al. Genome-Wide Association Study Highlights APOH as a Novel Locus for Lipoprotein(a) Levels-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 458–464. [Google Scholar] [CrossRef]
- Kronenberg, F.; Utermann, G. Lipoprotein(a): Resurrected by genetics. J. Intern. Med. 2013, 273, 6–30. [Google Scholar] [CrossRef] [PubMed]
- Loh, W.J.; Chan, D.C.; Mata, P.; Watts, G.F. Familial Hypercholesterolemia and Elevated Lipoprotein(a): Cascade Testing and Other Implications for Contextual Models of Care. Front. Genet. 2022, 13, 905941. [Google Scholar] [CrossRef] [PubMed]
- de Boer, L.M.; Hof, M.H.; Wiegman, A.; Stroobants, A.K.; Kastelein, J.J.P.; Hutten, B.A. Lipoprotein(a) levels from childhood to adulthood: Data in nearly 3,000 children who visited a pediatric lipid clinic. Atherosclerosis 2022, 349, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Deshotels, M.R.; Sun, C.; Nambi, V.; Virani, S.S.; Matsushita, K.; Yu, B.; Ballantyne, C.M.; Hoogeveen, R.C. Temporal Trends in Lipoprotein(a) Concentrations: The Atherosclerosis Risk in Communities Study. J. Am. Heart Assoc. 2022, 11, e026762. [Google Scholar] [CrossRef] [PubMed]
- Welsh, P.; Welsh, C.; Celis-Morales, C.A.; Brown, R.; Ho, F.K.; Ferguson, L.D.; Mark, P.B.; Lewsey, J.; Gray, S.R.; Lyall, D.M.; et al. Lipoprotein(a) and cardiovascular disease: Prediction, attributable risk fraction, and estimating benefits from novel interventions. Eur. J. Prev. Cardiol. 2022, 28, 1991–2000. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Wang, M.; Pirruccello, J.P.; Ellinor, P.T.; Ng, K.; Kathiresan, S.; Khera, A.V. Lp(a) (Lipoprotein[a]) Concentrations and Incident Atherosclerotic Cardiovascular Disease: New Insights from a Large National Biobank. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 465–474. [Google Scholar] [CrossRef]
- Virani, S.S.; Brautbar, A.; Davis, B.C.; Nambi, V.; Hoogeveen, R.C.; Sharrett, A.R.; Coresh, J.; Mosley, T.H.; Morrisett, J.D.; Catellier, D.J.; et al. Associations between lipoprotein(a) levels and cardiovascular outcomes in black and white subjects: The Atherosclerosis Risk in Communities (ARIC) Study. Circulation 2012, 125, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.; Cao, J.; Steffen, B.T.; Post, W.S.; Stein, J.H.; Tattersall, M.C.; Kaufman, J.D.; McConnell, J.P.; Hoefner, D.M.; Warnick, R.; et al. Race is a key variable in assigning lipoprotein(a) cutoff values for coronary heart disease risk assessment: The Multi-Ethnic Study of Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 996–1001. [Google Scholar] [CrossRef]
- Tsimikas, S.; Clopton, P.; Brilakis, E.S.; Marcovina, S.M.; Khera, A.; Miller, E.R.; de Lemos, J.A.; Witztum, J.L. Relationship of oxidized phospholipids on apolipoprotein B-100 particles to race/ethnicity, apolipoprotein(a) isoform size, and cardiovascular risk factors: Results from the Dallas Heart Study. Circulation 2009, 119, 1711–1719. [Google Scholar] [CrossRef]
- Jenner, J.L.; Ordovas, J.M.; Lamon-Fava, S.; Schaefer, M.M.; Wilson, P.W.; Castelli, W.P.; Schaefer, E.J. Effects of age, sex, and menopausal status on plasma lipoprotein(a) levels. The Framingham Offspring Study. Circulation 1993, 87, 1135–1141. [Google Scholar] [CrossRef]
- Cobbaert, C.; Kesteloot, H. Serum lipoprotein(a) levels in racially different populations. Am. J. Epidemiol. 1992, 136, 441–449. [Google Scholar] [CrossRef]
- Erhart, G.; Lamina, C.; Lehtimäki, T.; Marques-Vidal, P.; Kähönen, M.; Vollenweider, P.; Raitakari, O.T.; Waeber, G.; Thorand, B.; Strauch, K.; et al. Genetic Factors Explain a Major Fraction of the 50% Lower Lipoprotein(a) Concentrations in Finns. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1230–1241. [Google Scholar] [CrossRef]
- Varvel, S.; McConnell, J.P.; Tsimikas, S. Prevalence of Elevated Lp(a) Mass Levels and Patient Thresholds in 532 359 Patients in the United States. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2239–2245. [Google Scholar] [CrossRef]
- Derby, C.A.; Crawford, S.L.; Pasternak, R.C.; Sowers, M.; Sternfeld, B.; Matthews, K.A. Lipid changes during the menopause transition in relation to age and weight: The Study of Women’s Health Across the Nation. Am. J. Epidemiol. 2009, 169, 1352–1361. [Google Scholar] [CrossRef]
- Meroni, M.; Longo, M.; Lombardi, R.; Paolini, E.; Macchi, C.; Corsini, A.; Sirtori, C.R.; Fracanzani, A.L.; Ruscica, M.; Dongiovanni, P. Low Lipoprotein(a) Levels Predict Hepatic Fibrosis in Patients with Nonalcoholic Fatty Liver Disease. Hepatol. Commun. 2022, 6, 535–549. [Google Scholar] [CrossRef]
- Kraft, H.G.; Menzel, H.J.; Hoppichler, F.; Vogel, W.; Utermann, G. Changes of genetic apolipoprotein phenotypes caused by liver transplantation. Implications for apolipoprotein synthesis. J. Clin. Investig. 1989, 83, 137–142. [Google Scholar] [CrossRef]
- Hopewell, J.C.; Haynes, R.; Baigent, C. The role of lipoprotein (a) in chronic kidney disease. J. Lipid Res. 2018, 59, 577–585. [Google Scholar] [CrossRef]
- Kronenberg, F.; König, P.; Lhotta, K.; Ofner, D.; Sandholzer, C.; Margreiter, R.; Dosch, E.; Utermann, G.; Dieplinger, H. Apolipoprotein(a) phenotype-associated decrease in lipoprotein(a) plasma concentrations after renal transplantation. Arterioscler. Thromb. 1994, 14, 1399–1404. [Google Scholar] [CrossRef]
- Enkhmaa, B.; Berglund, L. Non-genetic influences on lipoprotein(a) concentrations. Atherosclerosis 2022, 349, 53–62. [Google Scholar] [CrossRef]
- Simony, S.B.; Mortensen, M.B.; Langsted, A.; Afzal, S.; Kamstrup, P.R.; Nordestgaard, B.G. Sex differences of lipoprotein(a) levels and associated risk of morbidity and mortality by age: The Copenhagen General Population Study. Atherosclerosis 2022, 355, 76–82. [Google Scholar] [CrossRef]
- Aljawini, N.; Aldakhil, L.O.; Habib, S.S. High-Risk Lipoprotein(a) Levels in Saudi Women and Its Relationship to Menopause and Adiposity. Nutrients 2023, 15, 693. [Google Scholar] [CrossRef]
- Anagnostis, P.; Antza, C.; Trakatelli, C.; Lambrinoudaki, I.; Goulis, D.G.; Kotsis, V. The effect of menopause on lipoprotein (a) concentrations: A systematic review and meta-analysis. Maturitas 2023, 167, 39–45. [Google Scholar] [CrossRef]
- Denti, L.; Pasolini, G.; Cortellini, P.; Ferretti, S.; Sanfelici, L.; Ablondi, F.; Valenti, G. Effects of androgen suppression by gonadotropin-releasing hormone agonist and flutamide on lipid metabolism in men with prostate cancer: Focus on lipoprotein(a). Clin. Chem. 1996, 42, 1176–1181. [Google Scholar] [CrossRef]
- Fogacci, F.; Borghi, C.; Davinelli, S.; Scapagnini, G.; Cicero, A.F.G. Impact of anti-oestrogen therapy on lipoprotein(a) in postmenopausal women: A systematic review and meta-analysis of double-blind placebo-controlled clinical studies. Endocrine 2023, 80, 292–302. [Google Scholar] [CrossRef]
- Kotwal, A.; Cortes, T.; Genere, N.; Hamidi, O.; Jasim, S.; Newman, C.B.; Prokop, L.J.; Murad, M.H.; Alahdab, F. Treatment of Thyroid Dysfunction and Serum Lipids: A Systematic Review and Meta-analysis. J. Clin. Endocrinol. Metab. 2020, 105, dgaa672. [Google Scholar] [CrossRef]
- Koutsogianni, A.D.; Liamis, G.; Liberopoulos, E.; Adamidis, P.S.; Florentin, M. Effects of Lipid-Modifying and Other Drugs on Lipoprotein(a) Levels-Potent Clinical Implications. Pharmaceuticals 2023, 16, 750. [Google Scholar] [CrossRef]
- Fernández-Ruiz, I. Menopausal hormone therapy does not lower Lp(a)-associated CHD risk. Nat. Rev. Cardiol. 2022, 19, 351. [Google Scholar] [CrossRef]
- Ramharack, R.; Barkalow, D.; Spahr, M.A. Dominant negative effect of TGF-beta1 and TNF-alpha on basal and IL-6-induced lipoprotein(a) and apolipoprotein(a) mRNA expression in primary monkey hepatocyte cultures. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 984–990. [Google Scholar] [CrossRef]
- Makris, A.; Barkas, F.; Sfikakis, P.P.; Liberopoulos, E.; Filippatos, T.D.; Ray, K.K.; Agouridis, A.P. Lipoprotein(a), Interleukin-6 inhibitors, and atherosclerotic cardiovascular disease: Is there an association? Atheroscler. Plus 2023, 54, 1–6. [Google Scholar] [CrossRef]
- Missala, I.; Kassner, U.; Steinhagen-Thiessen, E. A Systematic Literature Review of the Association of Lipoprotein(a) and Autoimmune Diseases and Atherosclerosis. Int. J. Rheumatol. 2012, 2012, 480784. [Google Scholar] [CrossRef]
- Ziogos, E.; Vavuranakis, M.A.; Harb, T.; Foran, P.L.; Blaha, M.J.; Jones, S.R.; Lai, S.; Gerstenblith, G.; Leucker, T.M. Lipoprotein(a) concentrations in acute myocardial infarction patients are not indicative of levels at six month follow-up. Eur. Heart J. Open 2023, 3, oead035. [Google Scholar] [CrossRef]
- Dzobo, K.E.; Kraaijenhof, J.M.; Stroes, E.S.G.; Nurmohamed, N.S.; Kroon, J. Lipoprotein(a): An underestimated inflammatory mastermind. Atherosclerosis 2022, 349, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Schultz, O.; Oberhauser, F.; Saech, J.; Rubbert-Roth, A.; Hahn, M.; Krone, W.; Laudes, M. Effects of inhibition of interleukin-6 signalling on insulin sensitivity and lipoprotein (a) levels in human subjects with rheumatoid diseases. PLoS ONE 2010, 5, e14328. [Google Scholar] [CrossRef] [PubMed]
- Moriarty, P.M.; Gorby, L.K.; Stroes, E.S.; Kastelein, J.P.; Davidson, M.; Tsimikas, S. Lipoprotein(a) and Its Potential Association with Thrombosis and Inflammation in COVID-19: A Testable Hypothesis. Curr. Atheroscler. Rep. 2020, 22, 48. [Google Scholar] [CrossRef] [PubMed]
- Mooser, V.; Berger, M.M.; Tappy, L.; Cayeux, C.; Marcovina, S.M.; Darioli, R.; Nicod, P.; Chioléro, R. Major reduction in plasma Lp(a) levels during sepsis and burns. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1137–1142. [Google Scholar] [CrossRef] [PubMed]
- Cegla, J.; France, M.; Marcovina, S.M.; Neely, R.D.G. Lp(a): When and how to measure it. Ann. Clin. Biochem. 2021, 58, 16–21. [Google Scholar] [CrossRef]
- Kronenberg, F. Lipoprotein(a) measurement issues: Are we making a mountain out of a molehill? Atherosclerosis 2022, 349, 123–135. [Google Scholar] [CrossRef]
- Scharnagl, H.; Stojakovic, T.; Dieplinger, B.; Dieplinger, H.; Erhart, G.; Kostner, G.M.; Herrmann, M.; März, W.; Grammer, T.B. Comparison of lipoprotein (a) serum concentrations measured by six commercially available immunoassays. Atherosclerosis 2019, 289, 206–213. [Google Scholar] [CrossRef]
- Marcovina, S.M.; Albers, J.J.; Scanu, A.M.; Kennedy, H.; Giaculli, F.; Berg, K.; Couderc, R.; Dati, F.; Rifai, N.; Sakurabayashi, I.; et al. Use of a reference material proposed by the International Federation of Clinical Chemistry and Laboratory Medicine to evaluate analytical methods for the determination of plasma lipoprotein(a). Clin Chem. 2000, 46, 1956–1967. [Google Scholar] [CrossRef]
- Marcovina, S.M.; Clouet-Foraison, N.; Koschinsky, M.L.; Lowenthal, M.S.; Orquillas, A.; Boffa, M.B.; Hoofnagle, A.N.; Vaisar, T. Development of an LC-MS/MS Proposed Candidate Reference Method for the Standardization of Analytical Methods to Measure Lipoprotein(a). Clin. Chem. 2021, 67, 490–499. [Google Scholar] [CrossRef]
- Tsimikas, S.; Fazio, S.; Ferdinand, K.C.; Ginsberg, H.N.; Koschinsky, M.L.; Marcovina, S.M.; Moriarty, P.M.; Rader, D.J.; Remaley, A.T.; Reyes-Soffer, G.; et al. NHLBI Working Group Recommendations to Reduce Lipoprotein(a)-Mediated Risk of Cardiovascular Disease and Aortic Stenosis. J. Am. Coll. Cardiol. 2018, 71, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Marcovina, S.M.; Albers, J.J.; Gabel, B.; Koschinsky, M.L.; Gaur, V.P. Effect of the number of apolipoprotein(a) kringle 4 domains on immunochemical measurements of lipoprotein(a). Clin. Chem. 1995, 41, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Soffer, G.; Ginsberg, H.N.; Berglund, L.; Duell, P.B.; Heffron, S.P.; Kamstrup, P.R.; Lloyd-Jones, D.M.; Marcovina, S.M.; Yeang, C.; Koschinsky, M.L.; et al. Lipoprotein(a): A Genetically Determined, Causal, and Prevalent Risk Factor for Atherosclerotic Cardiovascular Disease: A Scientific Statement From the American Heart Association. Arterioscler. Thromb. Vasc. Biol. 2022, 42, e48–e60. [Google Scholar] [CrossRef] [PubMed]
- Cegla, J.; Neely, R.D.G.; France, M.; Ferns, G.; Byrne, C.D.; Halcox, J.; Datta, D.; Capps, N.; Shoulders, C.; Qureshi, N.; et al. HEART UK consensus statement on Lipoprotein(a): A call to action. Atherosclerosis 2019, 291, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Nordestgaard, B.G.; Tybjaerg-Hansen, A.; Lewis, B. Influx in vivo of low density, intermediate density, and very low density lipoproteins into aortic intimas of genetically hyperlipidemic rabbits. Roles of plasma concentrations, extent of aortic lesion, and lipoprotein particle size as determinants. Arterioscler. Thromb. 1992, 12, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Miles, L.A.; Fless, G.M.; Scanu, A.M.; Baynham, P.; Sebald, M.T.; Skocir, P.; Curtiss, L.K.; Levin, E.G.; Hoover-Plow, J.L.; Plow, E.F. Interaction of Lp(a) with plasminogen binding sites on cells. Thromb. Haemost. 1995, 73, 458–465. [Google Scholar] [CrossRef]
- Anglés-Cano, E.; Rojas, G. Apolipoprotein(a): Structure-function relationship at the lysine-binding site and plasminogen activator cleavage site. Biol. Chem. 2002, 383, 93–99. [Google Scholar] [CrossRef]
- van der Hoek, Y.Y.; Sangrar, W.; Côté, G.P.; Kastelein, J.J.; Koschinsky, M.L. Binding of recombinant apolipoprotein(a) to extracellular matrix proteins. Arterioscler. Thromb. 1994, 14, 1792–1798. [Google Scholar] [CrossRef]
- Rath, M.; Niendorf, A.; Reblin, T.; Dietel, M.; Krebber, H.J.; Beisiegel, U. Detection and quantification of lipoprotein(a) in the arterial wall of 107 coronary bypass patients. Arteriosclerosis 1989, 9, 579–592. [Google Scholar] [CrossRef]
- Kreuzer, J.; Lloyd, M.B.; Bok, D.; Fless, G.M.; Scanu, A.M.; Lusis, A.J.; Haberland, M.E. Lipoprotein (a) displays increased accumulation compared with low-density lipoprotein in the murine arterial wall. Chem. Phys. Lipids 1994, 67–68, 175–190. [Google Scholar] [CrossRef]
- Falcone, D.J.; Salisbury, B.G. Fibronectin stimulates macrophage uptake of low density lipoprotein-heparin-collagen complexes. Arteriosclerosis 1988, 8, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.; Khan, S.; Tam, S.p.; Koschinsky, M.; Taylor, P.; Yacoub, M. Expression of adhesion molecules by lp(a): A potential novel mechanism for its atherogenicity. FASEB J. 1998, 12, 1765–1776. [Google Scholar] [CrossRef] [PubMed]
- Labudovic, D.; Kostovska, I.; Tosheska Trajkovska, K.; Cekovska, S.; Brezovska Kavrakova, J.; Topuzovska, S. Lipoprotein(a)—Link between Atherogenesis and Thrombosis. Prague Med. Rep. 2019, 120, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Klezovitch, O.; Edelstein, C.; Scanu, A.M. Stimulation of interleukin-8 production in human THP-1 macrophages by apolipoprotein(a). Evidence for a critical involvement of elements in its C-terminal domain. J. Biol. Chem. 2001, 276, 46864–46899. [Google Scholar] [CrossRef] [PubMed]
- Baggiolini, M.; Walz, A.; Kunkel, S.L. Neutrophil-activating peptide-1/interleukin 8, a novel cytokine that activates neutrophils. J. Clin. Investig. 1989, 84, 1045–1049. [Google Scholar] [CrossRef] [PubMed]
- van der Valk, F.M.; Bekkering, S.; Kroon, J.; Yeang, C.; Van den Bossche, J.; van Buul, J.D.; Ravandi, A.; Nederveen, A.J.; Verberne, H.J.; Scipione, C.; et al. Oxidized Phospholipids on Lipoprotein(a) Elicit Arterial Wall Inflammation and an Inflammatory Monocyte Response in Humans. Circulation 2016, 134, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Oikonomou, E.; Tsaplaris, P.; Anastasiou, A.; Xenou, M.; Lampsas, S.; Siasos, G.; Pantelidis, P.; Theofilis, P.; Tsatsaragkou, A.; Katsarou, O.; et al. Interleukin-1 in Coronary Artery Disease. Curr. Top. Med. Chem. 2022, 22, 2368–2389. [Google Scholar] [CrossRef] [PubMed]
- Kobiyama, K.; Ley, K. Atherosclerosis. Circ. Res. 2018, 123, 1118–1120. [Google Scholar] [CrossRef]
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2010, 11, 155–161. [Google Scholar] [CrossRef]
- Berglund, L.; Kim, K.; Zhang, W.; Prakash, N.; Truax, K.; Anuurad, E.; Enkhmaa, B. Lp(a)-Associated Oxidized Phospholipids in Healthy Black and White Participants in Relation to apo(a) Size, Age, and Family Structure. J. Am. Heart. Assoc. 2021, 10, e020158. [Google Scholar] [CrossRef]
- Koschinsky, M.L.; Boffa, M.B. Oxidized phospholipid modification of lipoprotein(a): Epidemiology, biochemistry and pathophysiology. Atherosclerosis 2022, 349, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Bergmark, C.; Dewan, A.; Orsoni, A.; Merki, E.; Miller, E.R.; Shin, M.J.; Binder, C.J.; Hörkkö, S.; Krauss, R.M.; Chapman, M.J.; et al. A novel function of lipoprotein [a] as a preferential carrier of oxidized phospholipids in human plasma. J. Lipid Res. 2008, 49, 2230–2239. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S.; Witztum, J.L. The role of oxidized phospholipids in mediating lipoprotein(a) atherogenicity. Curr. Opin. Lipidol. 2008, 19, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Edelstein, C.; Shapiro, S.D.; Klezovitch, O.; Scanu, A.M. Macrophage metalloelastase, MMP-12, cleaves human apolipoprotein(a) in the linker region between kringles IV-4 and IV-5. Potential relevance to lipoprotein(a) biology. J. Biol. Chem. 1999, 274, 10019–10023. [Google Scholar] [CrossRef]
- Edelstein, C.; Italia, J.A.; Klezovitch, O.; Scanu, A.M. Functional and metabolic differences between elastase-generated fragments of human lipoprotein[a] and apolipoprotein[a]. J. Lipid Res. 1996, 37, 1786–1801. [Google Scholar] [CrossRef]
- Oikonomou, E.; Theofilis, P.; Lampsas, S.; Katsarou, O.; Kalogeras, K.; Marinos, G.; Tsatsaragkou, A.; Anastasiou, A.; Lysandrou, A.; Gounaridi, M.I.; et al. Current Concepts and Future Applications of Non-Invasive Functional and Anatomical Evaluation of Coronary Artery Disease. Life 2022, 12, 1803. [Google Scholar] [CrossRef] [PubMed]
- Nordestgaard, B.G.; Langsted, A. Lipoprotein (a) as a cause of cardiovascular disease: Insights from epidemiology, genetics, and biology. J. Lipid Res. 2016, 57, 1953–1975. [Google Scholar] [CrossRef]
- Langsted, A.; Nordestgaard, B.G.; Kamstrup, P.R. Elevated Lipoprotein(a) and Risk of Ischemic Stroke. J. Am. Coll. Cardiol. 2019, 74, 54–66. [Google Scholar] [CrossRef]
- Kamstrup, P.R.; Nordestgaard, B.G. Elevated Lipoprotein(a) Levels, LPA Risk Genotypes, and Increased Risk of Heart Failure in the General Population. JACC Heart Fail. 2016, 4, 78–87. [Google Scholar] [CrossRef]
- Bennet, A.; Di Angelantonio, E.; Erqou, S.; Eiriksdottir, G.; Sigurdsson, G.; Woodward, M.; Rumley, A.; Lowe, G.D.; Danesh, J.; Gudnason, V. Lipoprotein(a) levels and risk of future coronary heart disease: Large-scale prospective data. Arch. Intern. Med. 2008, 168, 598–608. [Google Scholar] [CrossRef]
- Zhu, L.; Zheng, J.; Gao, B.; Jin, X.; He, Y.; Zhou, L.; Huang, J. The correlation between lipoprotein(a) elevations and the risk of recurrent cardiovascular events in CAD patients with different LDL-C levels. BMC Cardiovasc. Disord. 2022, 22, 171. [Google Scholar] [CrossRef] [PubMed]
- Leistner, D.M.; Laguna-Fernandez, A.; Haghikia, A.; Abdelwahed, Y.S.; Schatz, A.S.; Erbay, A.; Roehle, R.; Fonseca, A.F.; Ferber, P.; Landmesser, U. Impact of elevated lipoprotein(a) on coronary artery disease phenotype and severity. Eur. J. Prev. Cardiol. 2024, zwae007. [Google Scholar] [CrossRef] [PubMed]
- Erqou, S.; Thompson, A.; Di Angelantonio, E.; Saleheen, D.; Kaptoge, S.; Marcovina, S.; Danesh, J. Apolipoprotein(a) isoforms and the risk of vascular disease: Systematic review of 40 studies involving 58,000 participants. J. Am. Coll. Cardiol. 2010, 55, 2160–2167. [Google Scholar] [CrossRef] [PubMed]
- Orfanos, P.; Fonseca, A.F.; Hu, X.; Gautam, R.; Montgomery, G.; Studer, R.; Kaur, J.; Saxena, N.; Kaushik, N. Burden of elevated lipoprotein(a) among patients with atherosclerotic cardiovascular disease: Evidence from a systematic literature review and feasibility assessment of meta-analysis. PLoS ONE 2023, 18, e0294250. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Stampfer, M.J.; Rifai, N. Novel risk factors for systemic atherosclerosis: A comparison of C-reactive protein, fibrinogen, homocysteine, lipoprotein(a), and standard cholesterol screening as predictors of peripheral arterial disease. JAMA 2001, 285, 2481–2485. [Google Scholar] [CrossRef] [PubMed]
- Yi, C.; Junyi, G.; Fengju, L.; Qing, Z.; Jie, C. Association between lipoprotein(a) and peripheral arterial disease in coronary artery bypass grafting patients. Clin. Cardiol. 2023, 46, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.H.; Hegele, R.A.; Hackam, D.G.; Koschinsky, M.L.; Huff, M.W.; Spence, J.D. Lipoprotein(a) is associated differentially with carotid stenosis, occlusion, and total plaque area. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1851–1856. [Google Scholar] [CrossRef]
- Sharma, K.; Blaha, M.J.; Blumenthal, R.S.; Musunuru, K. Clinical and research applications of carotid intima-media thickness. Am. J. Cardiol. 2009, 103, 1316–1320. [Google Scholar] [CrossRef]
- Kabłak-Ziembicka, A.; Przewłocki, T. Clinical Significance of Carotid Intima-Media Complex and Carotid Plaque Assessment by Ultrasound for the Prediction of Adverse Cardiovascular Events in Primary and Secondary Care Patients. J. Clin. Med. 2021, 10, 4628. [Google Scholar] [CrossRef]
- França, V.; Gomes, É.I.L.; de Campos, E.V.S.; Zago, V.H.S.; Nunes, V.S.; de Faria, E.C. Relationship between lipoprotein (a) and subclinical carotid atherosclerosis in asymptomatic individuals. Clinics 2022, 77, 100107. [Google Scholar] [CrossRef]
- Lin, L.; Deng, K.Q.; Chen, Z.; Lei, F.; Qin, J.J.; Huang, X.; Sun, T.; Zhang, X.; Hu, Y.; Zhang, P.; et al. Lipoprotein(a) distribution and its association with carotid arteriopathy in the Chinese population. Atherosclerosis 2023, 372, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.T.; Würtz, P.; Havulinna, A.S.; Palta, P.; Tukiainen, T.; Rehnström, K.; Esko, T.; Mägi, R.; Inouye, M.; Lappalainen, T.; et al. Distribution and medical impact of loss-of-function variants in the Finnish founder population. PLoS Genet. 2014, 10, e1004494. [Google Scholar] [CrossRef] [PubMed]
- Paré, G.; Çaku, A.; McQueen, M.; Anand, S.S.; Enas, E.; Clarke, R.; Boffa, M.B.; Koschinsky, M.; Wang, X.; Yusuf, S.; et al. Lipoprotein(a) Levels and the Risk of Myocardial Infarction Among 7 Ethnic Groups. Circulation 2019, 139, 1472–1482. [Google Scholar] [CrossRef] [PubMed]
- Kamstrup, P.R.; Benn, M.; Tybjaerg-Hansen, A.; Nordestgaard, B.G. Extreme lipoprotein(a) levels and risk of myocardial infarction in the general population: The Copenhagen City Heart Study. Circulation 2008, 117, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart. J. 2020, 41, 111–188. [Google Scholar] [CrossRef] [PubMed]
- Khera, A.V.; Everett, B.M.; Caulfield, M.P.; Hantash, F.M.; Wohlgemuth, J.; Ridker, P.M.; Mora, S. Lipoprotein(a) concentrations, rosuvastatin therapy, and residual vascular risk: An analysis from the JUPITER Trial (Justification for the Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin). Circulation 2014, 129, 635–642. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, M.L.; Fazio, S.; Giugliano, R.P.; Stroes, E.S.G.; Kanevsky, E.; Gouni-Berthold, I.; Im, K.; Lira Pineda, A.; Wasserman, S.M.; Češka, R.; et al. Lipoprotein(a), PCSK9 Inhibition, and Cardiovascular Risk. Circulation 2019, 139, 1483–1492. [Google Scholar] [CrossRef] [PubMed]
- Bittner, V.A.; Szarek, M.; Aylward, P.E.; Bhatt, D.L.; Diaz, R.; Edelberg, J.M.; Fras, Z.; Goodman, S.G.; Halvorsen, S.; Hanotin, C.; et al. Effect of Alirocumab on Lipoprotein(a) and Cardiovascular Risk After Acute Coronary Syndrome. J. Am. Coll. Cardiol. 2020, 75, 133–144. [Google Scholar] [CrossRef]
- Zhang, W.; Speiser, J.L.; Ye, F.; Tsai, M.Y.; Cainzos-Achirica, M.; Nasir, K.; Herrington, D.M.; Shapiro, M.D. High-Sensitivity C-Reactive Protein Modifies the Cardiovascular Risk of Lipoprotein(a): Multi-Ethnic Study of Atherosclerosis. J. Am. Coll. Cardiol. 2021, 78, 1083–1094. [Google Scholar] [CrossRef]
- Alushi, B.; Curini, L.; Christopher, M.R.; Grubitzch, H.; Landmesser, U.; Amedei, A.; Lauten, A. Calcific Aortic Valve Disease-Natural History and Future Therapeutic Strategies. Front. Pharmacol. 2020, 11, 685. [Google Scholar] [CrossRef]
- Tsamoulis, D.; Siountri, I.; Rallidis, L.S. Lipoprotein(a): Its Association with Calcific Aortic Valve Stenosis, the Emerging RNA-Related Treatments and the Hope for a New Era in “Treating” Aortic Valve Calcification. J. Cardiovasc. Dev. Dis. 2023, 10, 96. [Google Scholar] [CrossRef] [PubMed]
- Stewart, B.F.; Siscovick, D.; Lind, B.K.; Gardin, J.M.; Gottdiener, J.S.; Smith, V.E.; Kitzman, D.W.; Otto, C.M. Clinical factors associated with calcific aortic valve disease. Cardiovascular Health Study. J. Am. Coll. Cardiol. 1997, 29, 630–634. [Google Scholar] [CrossRef] [PubMed]
- Thanassoulis, G.; Campbell, C.Y.; Owens, D.S.; Smith, J.G.; Smith, A.V.; Peloso, G.M.; Kerr, K.F.; Pechlivanis, S.; Budoff, M.J.; Harris, T.B.; et al. Genetic associations with valvular calcification and aortic stenosis. N. Engl. J. Med. 2013, 368, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Arsenault, B.J.; Boekholdt, S.M.; Dubé, M.P.; Rhéaume, E.; Wareham, N.J.; Khaw, K.T.; Sandhu, M.S.; Tardif, J.C. Lipoprotein(a) levels, genotype, and incident aortic valve stenosis: A prospective Mendelian randomization study and replication in a case-control cohort. Circ. Cardiovasc. Genet. 2014, 7, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Kamstrup, P.R.; Hung, M.Y.; Witztum, J.L.; Tsimikas, S.; Nordestgaard, B.G. Oxidized Phospholipids and Risk of Calcific Aortic Valve Disease: The Copenhagen General Population Study. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1570–1578. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Yu, Y.; Xi, R.; Li, J.; Lai, R.; Wang, T.; Fan, Y.; Zhang, Z.; Xu, H.; Ju, J. Association Between Lipoprotein(a) and Calcific Aortic Valve Disease: A Systematic Review and Meta-Analysis. Front. Cardiovasc. Med. 2022, 9, 877140. [Google Scholar] [CrossRef] [PubMed]
- Pantelidis, P.; Oikonomou, E.; Lampsas, S.; Zakynthinos, G.E.; Lysandrou, A.; Kalogeras, K.; Katsianos, E.; Theofilis, P.; Siasos, G.; Vavuranakis, M.A.; et al. Lipoprotein(a) and calcific aortic valve disease initiation and progression: A systematic review and meta-analysis. Cardiovasc. Res. 2023, 119, 1641–1655. [Google Scholar] [CrossRef]
- Després, A.A.; Perrot, N.; Poulin, A.; Tastet, L.; Shen, M.; Chen, H.Y.; Bourgeois, R.; Trottier, M.; Tessier, M.; Guimond, J.; et al. Lipoprotein(a), Oxidized Phospholipids, and Aortic Valve Microcalcification Assessed by 18F-Sodium Fluoride Positron Emission Tomography and Computed Tomography. CJC Open 2019, 1, 131–140. [Google Scholar] [CrossRef]
- Zheng, K.H.; Tsimikas, S.; Pawade, T.; Kroon, J.; Jenkins, W.S.A.; Doris, M.K.; White, A.C.; Timmers, N.K.L.M.; Hjortnaes, J.; Rogers, M.A.; et al. Lipoprotein(a) and Oxidized Phospholipids Promote Valve Calcification in Patients with Aortic Stenosis. J. Am. Coll. Cardiol. 2019, 73, 2150–2162. [Google Scholar] [CrossRef]
- Kaiser, Y.; Singh, S.S.; Zheng, K.H.; Verbeek, R.; Kavousi, M.; Pinto, S.J.; Vernooij, M.W.; Sijbrands, E.J.G.; Boekholdt, S.M.; de Rijke, Y.B.; et al. Lipoprotein(a) is robustly associated with aortic valve calcium. Heart 2021, 107, 1422–1428. [Google Scholar] [CrossRef]
- Emdin, C.A.; Khera, A.V.; Natarajan, P.; Klarin, D.; Won, H.H.; Peloso, G.M.; Stitziel, N.O.; Nomura, A.; Zekavat, S.M.; Bick, A.G.; et al. Phenotypic Characterization of Genetically Lowered Human Lipoprotein(a) Levels. J. Am. Coll. Cardiol. 2016, 68, 2761–2772. [Google Scholar] [CrossRef] [PubMed]
- Boffa, M.B.; Koschinsky, M.L. Lipoprotein (a): Truly a direct prothrombotic factor in cardiovascular disease? J. Lipid Res. 2016, 57, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Boffa, M.B.; Marar, T.T.; Yeang, C.; Viney, N.J.; Xia, S.; Witztum, J.L.; Koschinsky, M.L.; Tsimikas, S. Potent reduction of plasma lipoprotein (a) with an antisense oligonucleotide in human subjects does not affect ex vivo fibrinolysis. J. Lipid Res. 2019, 60, 2082–2089. [Google Scholar] [CrossRef] [PubMed]
- Anglés-Cano, E.; de la Peña Díaz, A.; Loyau, S. Inhibition of fibrinolysis by lipoprotein(a). Ann. N. Y. Acad. Sci. 2001, 936, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Boffa, M.B. Beyond fibrinolysis: The confounding role of Lp(a) in thrombosis. Atherosclerosis 2022, 349, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Koschinsky, M.L.; Stroes, E.S.G.; Kronenberg, F. Daring to dream: Targeting lipoprotein(a) as a causal and risk-enhancing factor. Pharmacol. Res. 2023, 194, 106843. [Google Scholar] [CrossRef] [PubMed]
- Yano, Y.; Shimokawa, K.; Okada, Y.; Noma, A. Immunolocalization of lipoprotein(a) in wounded tissues. J. Histochem. Cytochem. 1997, 45, 559–568. [Google Scholar] [CrossRef]
- Wilson, D.P.; Jacobson, T.A.; Jones, P.H.; Koschinsky, M.L.; McNeal, C.J.; Nordestgaard, B.G.; Orringer, C.E. Use of Lipoprotein(a) in clinical practice: A biomarker whose time has come. A scientific statement from the National Lipid Association. J. Clin. Lipidol. 2019, 13, 374–392. [Google Scholar] [CrossRef]
- Jawi, M.M.; Frohlich, J.; Chan, S.Y. Lipoprotein(a) the Insurgent: A New Insight into the Structure, Function, Metabolism, Pathogenicity, and Medications Affecting Lipoprotein(a) Molecule. J. Lipids 2020, 2020, 3491764. [Google Scholar] [CrossRef]
- Kim, J.S.; Chang, J.H.; Yu, H.K.; Ahn, J.H.; Yum, J.S.; Lee, S.K.; Jung, K.H.; Park, D.H.; Yoon, Y.; Byun, S.M.; et al. Inhibition of angiogenesis and angiogenesis-dependent tumor growth by the cryptic kringle fragments of human apolipoprotein(a). J. Biol. Chem. 2003, 278, 29000–29008. [Google Scholar] [CrossRef]
- Schulter, V.; Koolwijk, P.; Peters, E.; Frank, S.; Hrzenjak, A.; Graier, W.F.; van Hinsbergh, V.W.; Kostner, G.M. Impact of apolipoprotein(a) on in vitro angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Kalaivani, V.; Jaleel, A. Apolipoprotein(a), an enigmatic anti-angiogenic glycoprotein in human plasma: A curse or cure? Pharmacol. Res. 2020, 158, 104858. [Google Scholar] [CrossRef] [PubMed]
- Kostner, K.M.; Kostner, G.M. Therapy of hyper-Lp(a). Handb. Exp. Pharmacol. 2005, 170, 519–536. [Google Scholar] [CrossRef]
- Law, H.G.; Khan, M.A.; Zhang, W.; Bang, H.; Rood, J.; Most, M.; Lefevre, M.; Berglund, L.; Enkhmaa, B. Reducing saturated fat intake lowers LDL-C but increases Lp(a) levels in African Americans: The GET-READI feeding trial. J. Lipid Res. 2023, 64, 100420. [Google Scholar] [CrossRef] [PubMed]
- Law, H.G.; Meyers, F.J.; Berglund, L.; Enkhmaa, B. Lipoprotein(a) and diet-a challenge for a role of saturated fat in cardiovascular disease risk reduction? Am. J. Clin. Nutr. 2023, 118, 23–26. [Google Scholar] [CrossRef] [PubMed]
- Enkhmaa, B.; Petersen, K.S.; Kris-Etherton, P.M.; Berglund, L. Diet and Lp(a): Does Dietary Change Modify Residual Cardiovascular Risk Conferred by Lp(a)? Nutrients 2020, 12, 2024. [Google Scholar] [CrossRef] [PubMed]
- Ebbeling, C.B.; Knapp, A.; Johnson, A.; Wong, J.M.W.; Greco, K.F.; Ma, C.; Mora, S.; Ludwig, D.S. Effects of a low-carbohydrate diet on insulin-resistant dyslipoproteinemia-a randomized controlled feeding trial. Am. J. Clin. Nutr. 2022, 115, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Fogacci, F.; Di Micoli, V.; Sabouret, P.; Giovannini, M.; Cicero, A.F.G. Lifestyle and Lipoprotein(a) Levels: Does a Specific Counseling Make Sense? J. Clin. Med. 2024, 13, 751. [Google Scholar] [CrossRef]
- Stancu, C.; Sima, A. Statins: Mechanism of action and effects. J. Cell. Mol. Med. 2001, 5, 378–387. [Google Scholar] [CrossRef]
- van Wissen, S.; Smilde, T.J.; Trip, M.D.; de Boo, T.; Kastelein, J.J.; Stalenhoef, A.F. Long term statin treatment reduces lipoprotein(a) concentrations in heterozygous familial hypercholesterolaemia. Heart 2003, 89, 893–896. [Google Scholar] [CrossRef]
- Ridker, P.M.; Danielson, E.; Fonseca, F.A.; Genest, J.; Gotto, A.M., Jr.; Kastelein, J.J.; Koenig, W.; Libby, P.; Lorenzatti, A.J.; MacFadyen, J.G.; et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N. Engl. J. Med. 2008, 359, 2195–2207. [Google Scholar] [CrossRef] [PubMed]
- Arsenault, B.J.; Petrides, F.; Tabet, F.; Bao, W.; Hovingh, G.K.; Boekholdt, S.M.; Ramin-Mangata, S.; Meilhac, O.; DeMicco, D.; Rye, K.A.; et al. Effect of atorvastatin, cholesterol ester transfer protein inhibition, and diabetes mellitus on circulating proprotein subtilisin kexin type 9 and lipoprotein(a) levels in patients at high cardiovascular risk. J. Clin. Lipidol. 2018, 12, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Willeit, P.; Ridker, P.; Nestel, P.J.; Simes, J.; Tonkin, A.M.; Pedersen, T.R.; Schwartz, G.G.; Olsson, A.G.; Colhoun, H.M.; Kronenberg, F.; et al. Baseline and on-statin treatment lipoprotein(a) levels for prediction of cardiovascular events: Individual patient-data meta-analysis of statin outcome trials. Lancet 2018, 392, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- de Boer, L.M.; Oorthuys, A.O.J.; Wiegman, A.; Langendam, M.W.; Kroon, J.; Spijker, R.; Zwinderman, A.H.; Hutten, B.A. Statin therapy and lipoprotein(a) levels: A systematic review and meta-analysis. Eur. J. Prev. Cardiol. 2022, 29, 779–792. [Google Scholar] [CrossRef]
- Tsimikas, S.; Gordts, P.L.S.M.; Nora, C.; Yeang, C.; Witztum, J.L. Statin therapy increases lipoprotein(a) levels. Eur. Heart J. 2020, 41, 2275–2284. [Google Scholar] [CrossRef] [PubMed]
- Yahya, R.; Berk, K.; Verhoeven, A.; Bos, S.; van der Zee, L.; Touw, J.; Erhart, G.; Kronenberg, F.; Timman, R.; Sijbrands, E.; et al. Statin treatment increases lipoprotein(a) levels in subjects with low molecular weight apolipoprotein(a) phenotype. Atherosclerosis 2019, 289, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Scanu, A.M.; Hinman, J. Issues concerning the monitoring of statin therapy in hypercholesterolemic subjects with high plasma lipoprotein(a) levels. Lipids 2002, 37, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.; Sheehan, V.; Gurk-Turner, C. Ezetimibe (Zetia): A new type of lipid-lowering agent. Bayl. Univ. Med. Cent. Proc. 2003, 16, 354–358. [Google Scholar] [CrossRef]
- Lee, Y.J.; Cho, J.Y.; You, S.C.; Lee, Y.H.; Yun, K.H.; Cho, Y.H.; Shin, W.Y.; Im, S.W.; Kang, W.C.; Park, Y.; et al. Moderate-intensity statin with ezetimibe vs. high-intensity statin in patients with diabetes and atherosclerotic cardiovascular disease in the RACING trial. Eur. Heart J. 2023, 44, 972–983. [Google Scholar] [CrossRef]
- Awad, K.; Mikhailidis, D.P.; Katsiki, N.; Muntner, P.; Banach, M.; Lipid and Blood Pressure Meta-Analysis Collaboration (LBPMC) Group. Effect of Ezetimibe Monotherapy on Plasma Lipoprotein(a) Concentrations in Patients with Primary Hypercholesterolemia: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Drugs 2018, 78, 453–462. [Google Scholar] [CrossRef]
- Sahebkar, A.; Simental-Mendía, L.E.; Pirro, M.; Banach, M.; Watts, G.F.; Sirtori, C.; Al-Rasadi, K.; Atkin, S.L. Impact of ezetimibe on plasma lipoprotein(a) concentrations as monotherapy or in combination with statins: A systematic review and meta-analysis of randomized controlled trials. Sci. Rep. 2018, 8, 17887. [Google Scholar] [CrossRef] [PubMed]
- Staels, B.; Dallongeville, J.; Auwerx, J.; Schoonjans, K.; Leitersdorf, E.; Fruchart, J.C. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation 1998, 98, 2088–2093. [Google Scholar] [CrossRef]
- McConathy, W.J.; Trieu, V.N.; Klör, H.U.; Corder, C.N. Lp(a) and plasma triglyceride-rich lipoproteins. Klin. Wochenschr. 1990, 68, 117–1179. [Google Scholar] [PubMed]
- Schwartz, G.G.; Ballantyne, C.M. Existing and emerging strategies to lower Lipoprotein(a). Atherosclerosis 2022, 349, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Sahebkar, A.; Simental-Mendía, L.E.; Watts, G.F.; Serban, M.C.; Banach, M.; Lipid and Blood Pressure Meta-analysis Collaboration (LBPMC) Group. Comparison of the effects of fibrates versus statins on plasma lipoprotein(a) concentrations: A systematic review and meta-analysis of head-to-head randomized controlled trials. BMC Med. 2017, 15, 22. [Google Scholar] [CrossRef] [PubMed]
- Scanu, A.M.; Bamba, R. Niacin and lipoprotein(a): Facts, uncertainties, and clinical considerations. Am. J. Cardiol. 2008, 101, 44B–47B. [Google Scholar] [CrossRef] [PubMed]
- Sahebkar, A.; Reiner, Ž.; Simental-Mendía, L.E.; Ferretti, G.; Cicero, A.F. Effect of extended-release niacin on plasma lipoprotein(a) levels: A systematic review and meta-analysis of randomized placebo-controlled trials. Metabolism 2016, 65, 1664–1678. [Google Scholar] [CrossRef]
- Woudberg, N.J.; Pedretti, S.; Lecour, S.; Schulz, R.; Vuilleumier, N.; James, R.W.; Frias, M.A. Pharmacological Intervention to Modulate HDL: What Do We Target? Front. Pharmacol. 2018, 8, 989. [Google Scholar] [CrossRef]
- Chennamsetty, I.; Kostner, K.M.; Claudel, T.; Vinod, M.; Frank, S.; Weiss, T.S.; Trauner, M.; Kostner, G.M. Nicotinic acid inhibits hepatic APOA gene expression: Studies in humans and in transgenic mice. J. Lipid Res. 2012, 53, 2405–2412. [Google Scholar] [CrossRef]
- HPS2-THRIVE Collaborative Group; Landray, M.J.; Haynes, R.; Hopewell, J.C.; Parish, S.; Aung, T.; Tomson, J.; Wallendszus, K.; Craig, M.; Jiang, L.; et al. Effects of extended-release niacin with laropiprant in high-risk patients. N. Engl. J. Med. 2014, 371, 203–212. [Google Scholar] [CrossRef]
- AIM-HIGH Investigators; Boden, W.E.; Probstfield, J.L.; Anderson, T.; Chaitman, B.R.; Desvignes-Nickens, P.; Koprowicz, K.; McBride, R.; Teo, K.; Weintraub, W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N. Engl. J. Med. 2011, 365, 2255–2267. [Google Scholar] [CrossRef] [PubMed]
- Bell, D.A.; Hooper, A.J.; Burnett, J.R. Mipomersen, an antisense apolipoprotein B synthesis inhibitor. Expert Opin. Investig. Drugs 2011, 20, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.; Goldberg, T. Mipomersen (kynamro): A novel antisense oligonucleotide inhibitor for the management of homozygous familial hypercholesterolemia. Pharm. Ther. 2014, 39, 119–122. [Google Scholar]
- Santos, R.D.; Raal, F.J.; Catapano, A.L.; Witztum, J.L.; Steinhagen-Thiessen, E.; Tsimikas, S. Mipomersen, an antisense oligonucleotide to apolipoprotein B-100, reduces lipoprotein(a) in various populations with hypercholesterolemia: Results of 4 phase III trials. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Panta, R.; Dahal, K.; Kunwar, S. Efficacy and safety of mipomersen in treatment of dyslipidemia: A meta-analysis of randomized controlled trials. J. Clin. Lipidol. 2015, 9, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Fogacci, F.; Ferri, N.; Toth, P.P.; Ruscica, M.; Corsini, A.; Cicero, A.F.G. Efficacy and Safety of Mipomersen: A Systematic Review and Meta-Analysis of Randomized Clinical Trials. Drugs 2019, 79, 751–766. [Google Scholar] [CrossRef] [PubMed]
- Reeskamp, L.F.; Kastelein, J.J.P.; Moriarty, P.M.; Duell, P.B.; Catapano, A.L.; Santos, R.D.; Ballantyne, C.M. Safety and efficacy of mipomersen in patients with heterozygous familial hypercholesterolemia. Atherosclerosis 2019, 280, 109–117. [Google Scholar] [CrossRef]
- Nandakumar, R.; Matveyenko, A.; Thomas, T.; Pavlyha, M.; Ngai, C.; Holleran, S.; Ramakrishnan, R.; Ginsberg, H.N.; Karmally, W.; Marcovina, S.M.; et al. Effects of mipomersen, an apolipoprotein B100 antisense, on lipoprotein (a) metabolism in healthy subjects. J. Lipid Res. 2018, 59, 2397–2402. [Google Scholar] [CrossRef]
- Chambergo-Michilot, D.; Alur, A.; Kulkarni, S.; Agarwala, A. Mipomersen in Familial Hypercholesterolemia: An Update on Health-Related Quality of Life and Patient-Reported Outcomes. Vasc. Health Risk Manag. 2022, 18, 73–80. [Google Scholar] [CrossRef]
- Sjouke, B.; Balak, D.M.; Beuers, U.; Ratziu, V.; Stroes, E.S. Is mipomersen ready for clinical implementation? A transatlantic dilemma. Curr. Opin. Lipidol. 2013, 24, 301–306. [Google Scholar] [CrossRef]
- Alhamadani, F.; Zhang, K.; Parikh, R.; Wu, H.; Rasmussen, T.P.; Bahal, R.; Zhong, X.B.; Manautou, J.E. Adverse Drug Reactions and Toxicity of the Food and Drug Administration-Approved Antisense Oligonucleotide Drugs. Drug Metab. Dispos. 2022, 50, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Lo Surdo, P.; Bottomley, M.J.; Calzetta, A.; Settembre, E.C.; Cirillo, A.; Pandit, S.; Ni, Y.G.; Hubbard, B.; Sitlani, A.; Carfí, A. Mechanistic implications for LDL receptor degradation from the PCSK9/LDLR structure at neutral pH. EMBO Rep. 2011, 12, 1300–1305. [Google Scholar] [CrossRef] [PubMed]
- Leren, T.P. Mutations in the PCSK9 gene in Norwegian subjects with autosomal dominant hypercholesterolemia. Clin. Genet. 2004, 65, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.; Pertsemlidis, A.; Kotowski, I.K.; Graham, R.; Garcia, C.K.; Hobbs, H.H. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat. Genet. 2005, 37, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Langsted, A.; Nordestgaard, B.G.; Benn, M.; Tybjærg-Hansen, A.; Kamstrup, P.R. PCSK9 R46L Loss-of-Function Mutation Reduces Lipoprotein(a), LDL Cholesterol, and Risk of Aortic Valve Stenosis. J. Clin. Endocrinol. Metab. 2016, 101, 3281–3287. [Google Scholar] [CrossRef] [PubMed]
- Hummelgaard, S.; Vilstrup, J.P.; Gustafsen, C.; Glerup, S.; Weyer, K. Targeting PCSK9 to tackle cardiovascular disease. Pharmacol. Ther. 2023, 249, 108480. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Wright, R.S.; Kallend, D.; Koenig, W.; Leiter, L.A.; Raal, F.J.; Bisch, J.A.; Richardson, T.; Jaros, M.; Wijngaard, P.L.J.; et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N. Engl. J. Med. 2020, 382, 1507–1519. [Google Scholar] [CrossRef] [PubMed]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Edelberg, J.M.; Goodman, S.G.; Hanotin, C.; Harrington, R.A.; et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N. Engl. J. Med. 2018, 379, 2097–2107. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Szarek, M.; Bittner, V.A.; Diaz, R.; Goodman, S.G.; Jukema, J.W.; Landmesser, U.; López-Jaramillo, P.; Manvelian, G.; Pordy, R.; et al. Lipoprotein(a) and Benefit of PCSK9 Inhibition in Patients with Nominally Controlled LDL Cholesterol. J. Am. Coll. Cardiol. 2021, 78, 421–433. [Google Scholar] [CrossRef]
- Szarek, M.; Bittner, V.A.; Aylward, P.; Baccara-Dinet, M.; Bhatt, D.L.; Diaz, R.; Fras, Z.; Goodman, S.G.; Halvorsen, S.; Harrington, R.A.; et al. Lipoprotein(a) lowering by alirocumab reduces the total burden of cardiovascular events independent of low-density lipoprotein cholesterol lowering: ODYSSEY OUTCOMES trial. Eur. Heart J. 2020, 41, 4245–4255. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bittner, V.A.; Diaz, R.; Goodman, S.G.; Kim, Y.U.; Jukema, J.W.; Pordy, R.; Roe, M.T.; et al. Peripheral Artery Disease and Venous Thromboembolic Events After Acute Coronary Syndrome: Role of Lipoprotein(a) and Modification by Alirocumab: Prespecified Analysis of the ODYSSEY OUTCOMES Randomized Clinical Trial. Circulation 2020, 141, 1608–1617. [Google Scholar] [CrossRef]
- Cao, Y.X.; Liu, H.H.; Li, S.; Li, J.J. A Meta-Analysis of the Effect of PCSK9-Monoclonal Antibodies on Circulating Lipoprotein (a) Levels. Am. J. Cardiovasc. Drugs 2019, 19, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Farmakis, I.; Doundoulakis, I.; Pagiantza, A.; Zafeiropoulos, S.; Antza, C.; Karvounis, H.; Giannakoulas, G. Lipoprotein(a) Reduction with Proprotein Convertase Subtilisin/Kexin Type 9 Inhibitors: A Systematic Review and Meta-analysis. J. Cardiovasc. Pharmacol. 2021, 77, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Suo, Y.; Yang, L.; Zhang, X.; Yu, Q.; Zeng, M.; Zhang, W.; Jiang, X.; Wang, Y. Effect of PCSK9 Inhibitor on Blood Lipid Levels in Patients with High and Very-High CVD Risk: A Systematic Review and Meta-Analysis. Cardiol. Res. Pract. 2022, 2022, 8729003. [Google Scholar] [CrossRef] [PubMed]
- Katsiki, N.; Vrablik, M.; Banach, M.; Gouni-Berthold, I. Inclisiran, Low-Density Lipoprotein Cholesterol and Lipoprotein (a). Pharmaceuticals 2023, 16, 577. [Google Scholar] [CrossRef]
- Fitzgerald, K.; White, S.; Borodovsky, A.; Bettencourt, B.R.; Strahs, A.; Clausen, V.; Wijngaard, P.; Horton, J.D.; Taubel, J.; Brooks, A.; et al. A Highly Durable RNAi Therapeutic Inhibitor of PCSK9. N. Engl. J. Med. 2017, 376, 41–51. [Google Scholar] [CrossRef]
- Ray, K.K.; Landmesser, U.; Leiter, L.A.; Kallend, D.; Dufour, R.; Karakas, M.; Hall, T.; Troquay, R.P.; Turner, T.; Visseren, F.L.; et al. Inclisiran in Patients at High Cardiovascular Risk with Elevated LDL Cholesterol. N. Engl. J. Med. 2017, 376, 1430–1440. [Google Scholar] [CrossRef]
- Ray, K.K.; Stoekenbroek, R.M.; Kallend, D.; Leiter, L.A.; Landmesser, U.; Wright, R.S.; Wijngaard, P.; Kastelein, J.J.P. Effect of an siRNA Therapeutic Targeting PCSK9 on Atherogenic Lipoproteins: Prespecified Secondary End Points in ORION 1. Circulation 2018, 138, 1304–1316. [Google Scholar] [CrossRef]
- Ray, K.K.; Troquay, R.P.T.; Visseren, F.L.J.; Leiter, L.A.; Wright, R.S.; Vikarunnessa, S.; Talloczy, Z.; Zang, X.; Maheux, P.; Lesogor, A.; et al. Long-term efficacy and safety of inclisiran in patients with high cardiovascular risk and elevated LDL cholesterol (ORION-3): Results from the 4-year open-label extension of the ORION-1 trial. Lancet Diabetes Endocrinol. 2023, 11, 109–119. [Google Scholar] [CrossRef]
- Raal, F.J.; Kallend, D.; Ray, K.K.; Turner, T.; Koenig, W.; Wright, R.S.; Wijngaard, P.L.J.; Curcio, D.; Jaros, M.J.; Leiter, L.A.; et al. Inclisiran for the Treatment of Heterozygous Familial Hypercholesterolemia. N. Engl. J. Med. 2020, 382, 1520–1530. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Kallend, D.; Leiter, L.A.; Raal, F.J.; Koenig, W.; Jaros, M.J.; Schwartz, G.G.; Landmesser, U.; Garcia Conde, L.; Wright, R.S.; et al. Effect of inclisiran on lipids in primary prevention: The ORION-11 trial. Eur. Heart J. 2022, 43, 5047–5057. [Google Scholar] [CrossRef] [PubMed]
- Ballantyne, C.M.; Bays, H.; Catapano, A.L.; Goldberg, A.; Ray, K.K.; Saseen, J.J. Role of Bempedoic Acid in Clinical Practice. Cardiovasc. Drugs Ther. 2021, 35, 853–864. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Lincoff, A.M.; Brennan, D.; Ray, K.K.; Mason, D.; Kastelein, J.J.P.; Thompson, P.D.; Libby, P.; Cho, L.; Plutzky, J.; et al. Bempedoic Acid and Cardiovascular Outcomes in Statin-Intolerant Patients. N. Engl. J. Med. 2023, 388, 1353–1364. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Lei, L.; Ray, K.K.; Ballantyne, C.M.; Bradwin, G.; Rifai, N. Effects of bempedoic acid on CRP, IL-6, fibrinogen and lipoprotein(a) in patients with residual inflammatory risk: A secondary analysis of the CLEAR harmony trial. J. Clin. Lipidol. 2023, 17, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Rubino, J.; MacDougall, D.E.; Sterling, L.R.; Kelly, S.E.; McKenney, J.M.; Lalwani, N.D. Lipid lowering with bempedoic acid added to a proprotein convertase subtilisin/kexin type 9 inhibitor therapy: A randomized, controlled trial. J. Clin. Lipidol. 2021, 15, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.J.; Sniderman, A.D.; Ditmarsch, M.; Dicklin, M.R.; Nicholls, S.J.; Davidson, M.H.; Kastelein, J.J.P. Cholesteryl Ester Transfer Protein Inhibition Reduces Major Adverse Cardiovascular Events by Lowering Apolipoprotein B Levels. Int. J. Mol. Sci. 2022, 23, 9417. [Google Scholar] [CrossRef]
- Ferri, N.; Corsini, A.; Sirtori, C.R.; Ruscica, M. Present therapeutic role of cholesteryl ester transfer protein inhibitors. Pharmacol. Res. 2018, 128, 29–41. [Google Scholar] [CrossRef]
- Xue, H.; Zhang, M.; Liu, J.; Wang, J.; Ren, G. Structure-based mechanism and inhibition of cholesteryl ester transfer protein. Curr. Atheroscler. Rep. 2023, 25, 155–166. [Google Scholar] [CrossRef]
- Bagdade, J.; Barter, P.; Quiroga, C.; Alaupovic, P. Effects of Torcetrapib and Statin Treatment on ApoC-III and Apoprotein-Defined Lipoprotein Subclasses (from the ILLUMINATE Trial). Am. J. Cardiol. 2017, 119, 1753–1756. [Google Scholar] [CrossRef]
- Ballantyne, C.M.; Shah, S.; Sapre, A.; Ashraf, T.B.; Tobias, S.C.; Sahin, T.; Ye, P.; Dong, Y.; Sheu, W.H.; Kang, D.H.; et al. A Multiregional, Randomized Evaluation of the Lipid-Modifying Efficacy and Tolerability of Anacetrapib Added to Ongoing Statin Therapy in Patients with Hypercholesterolemia or Low High-Density Lipoprotein Cholesterol. Am. J. Cardiol. 2017, 120, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.; Zhou, H.; Karmally, W.; Ramakrishnan, R.; Holleran, S.; Liu, Y.; Jumes, P.; Wagner, J.A.; Hubbard, B.; Previs, S.F.; et al. CETP (Cholesteryl Ester Transfer Protein) Inhibition with Anacetrapib Decreases Production of Lipoprotein(a) in Mildly Hypercholesterolemic Subjects. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1770–1775. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhang, Q.; Wang, Y.; Gao, P.; Chen, D. The effect and safety of anacetrapib in the treatment of dyslipidemia: A systematic review and meta-analysis. Postgrad. Med. 2018, 130, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.G.; Ballantyne, C.M.; Barter, P.J.; Kallend, D.; Leiter, L.A.; Leitersdorf, E.; McMurray, J.J.V.; Nicholls, S.J.; Olsson, A.G.; Shah, P.K.; et al. Association of Lipoprotein(a) With Risk of Recurrent Ischemic Events Following Acute Coronary Syndrome: Analysis of the dal-Outcomes Randomized Clinical Trial. JAMA Cardiol. 2018, 3, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, S.J.; Ruotolo, G.; Brewer, H.B.; Wang, M.D.; Liu, L.; Willey, M.B.; Deeg, M.A.; Krueger, K.A.; Nissen, S.E. Evacetrapib alone or in combination with statins lowers lipoprotein(a) and total and small LDL particle concentrations in mildly hypercholesterolemic patients. J. Clin. Lipidol. 2016, 10, 519–527.e4. [Google Scholar] [CrossRef] [PubMed]
- Handhle, A.; Viljoen, A.; Wierzbicki, A.S. Elevated Lipoprotein(a): Background, Current Insights and Future Potential Therapies. Vasc. Health Risk Manag. 2021, 17, 527–542. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, S.J.; Ditmarsch, M.; Kastelein, J.J.; Rigby, S.P.; Kling, D.; Curcio, D.L.; Alp, N.J.; Davidson, M.H. Lipid lowering effects of the CETP inhibitor obicetrapib in combination with high-intensity statins: A randomized phase 2 trial. Nat. Med. 2022, 28, 1672–1678. [Google Scholar] [CrossRef]
- Armitage, J.; Holmes, M.V.; Preiss, D. Cholesteryl Ester Transfer Protein Inhibition for Preventing Cardiovascular Events: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 73, 477–487. [Google Scholar] [CrossRef]
- Taheri, H.; Filion, K.B.; Windle, S.B.; Reynier, P.; Eisenberg, M.J. Cholesteryl Ester Transfer Protein Inhibitors and Cardiovascular Outcomes: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Cardiology 2020, 145, 236–250. [Google Scholar] [CrossRef]
- Furtado, J.D.; Ruotolo, G.; Nicholls, S.J.; Dullea, R.; Carvajal-Gonzalez, S.; Sacks, F.M. Pharmacological Inhibition of CETP (Cholesteryl Ester Transfer Protein) Increases HDL (High-Density Lipoprotein) That Contains ApoC3 and Other HDL Subspecies Associated with Higher Risk of Coronary Heart Disease. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 227–237. [Google Scholar] [CrossRef]
- Kounatidis, D.; Vallianou, N.G.; Poulaki, A.; Evangelopoulos, A.; Panagopoulos, F.; Stratigou, T.; Geladari, E.; Karampela, I.; Dalamaga, M. ApoB100 and Atherosclerosis: What’s New in the 21st Century? Metabolites 2024, 14, 123. [Google Scholar] [CrossRef] [PubMed]
- Marlęga-Linert, J.; Gąsecka, A.; van der Pol, E.; Kuchta, A.; Filipiak, K.J.; Fijałkowski, M.; Gruchała, M.; Nieuwland, R.; Mickiewicz, A. Lipoprotein apheresis affects the concentration of extracellular vesicles in patients with elevated lipoprotein (a). Sci. Rep. 2024, 14, 2762. [Google Scholar] [CrossRef] [PubMed]
- Thompson, G.; Parhofer, K.G. Current Role of Lipoprotein Apheresis. Curr. Atheroscler. Rep. 2019, 21, 26. [Google Scholar] [CrossRef] [PubMed]
- Roeseler, E.; Julius, U.; Heigl, F.; Spitthoever, R.; Heutling, D.; Breitenberger, P.; Leebmann, J.; Lehmacher, W.; Kamstrup, P.R.; Nordestgaard, B.G.; et al. Lipoprotein Apheresis for Lipoprotein(a)-Associated Cardiovascular Disease: Prospective 5 Years of Follow-Up and Apolipoprotein(a) Characterization. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2019–2027. [Google Scholar] [CrossRef] [PubMed]
- Bigazzi, F.; Sbrana, F.; Berretti, D.; Maria Grazia, Z.; Zambon, S.; Fabris, A.; Fonda, M.; Vigna, G.B.; D’Alessandri, G.; Passalacqua, S.; et al. Reduced incidence of cardiovascular events in hyper-Lp(a) patients on lipoprotein apheresis. The G.I.L.A. (Gruppo Interdisciplinare Aferesi Lipoproteica) pilot study. Transfus. Apher. Sci. 2018, 57, 661–664. [Google Scholar] [CrossRef] [PubMed]
- Jain, P. Traditional and novel non-statin lipid-lowering drugs. Indian Heart J. 2023, S0019-4832, 00176-1. [Google Scholar] [CrossRef]
- Sosnowska, B.; Surma, S.; Banach, M. Targeted Treatment against Lipoprotein (a): The Coming Breakthrough in Lipid Lowering Therapy. Pharmaceuticals 2022, 15, 1573. [Google Scholar] [CrossRef]
- Karwatowska-Prokopczuk, E.; Clouet-Foraison, N.; Xia, S.L.; Viney, N.J.; Witztum, J.L.; Marcovina, S.M.; Tsimikas, S. Prevalence and influence of LPA gene variants and isoform size on the Lp(a)-lowering effect of pelacarsen. Atherosclerosis 2021, 324, 102–108. [Google Scholar] [CrossRef]
- Karwatowska-Prokopczuk, E.; Lesogor, A.; Yan, J.H.; Hurh, E.; Hoenlinger, A.; Margolskee, A.; Xia, S.; Tsimikas, S. Efficacy and safety of pelacarsen in lowering Lp(a) in healthy Japanese subjects. J. Clin. Lipidol. 2023, 17, 181–188. [Google Scholar] [CrossRef]
- Sarzani, R.; Spannella, F.; Di Pentima, C.; Giulietti, F.; Landolfo, M.; Allevi, M. Molecular Therapies in Cardiovascular Diseases: Small Interfering RNA in Atherosclerosis, Heart Failure, and Hypertension. Int. J. Mol. Sci. 2023, 25, 328. [Google Scholar] [CrossRef]
- Koren, M.J.; Moriarty, P.M.; Baum, S.J.; Neutel, J.; Hernandez-Illas, M.; Weintraub, H.S.; Florio, M.; Kassahun, H.; Melquist, S.; Varrieur, T.; et al. Preclinical development and phase 1 trial of a novel siRNA targeting lipoprotein(a). Nat. Med. 2022, 28, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Sohn, W.; Winkle, P.; Neutel, J.; Wu, Y.; Jabari, F.; Terrio, C.; Varrieur, T.; Wang, J.; Hellawell, J. Pharmacokinetics, Pharmacodynamics, and Tolerability of Olpasiran in Healthy Japanese and Non-Japanese Participants: Results from a Phase I, Single-dose, Open-label Study. Clin. Ther. 2022, 44, 1237–1247. [Google Scholar] [CrossRef]
- O’Donoghue, M.L.; López, J.A.G.; Knusel, B.; Gencer, B.; Wang, H.; Wu, Y.; Kassahun, H.; Sabatine, M.S. Study design and rationale for the Olpasiran trials of Cardiovascular Events And lipoproteiN(a) reduction-DOSE finding study (OCEAN(a)-DOSE). Am. Heart. J. 2022, 251, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Rider, D.A.; Eisermann, M.; Löffler, K.; Aleku, M.; Swerdlow, D.I.; Dames, S.; Hauptmann, J.; Morrison, E.; Lindholm, M.W.; Schubert, S.; et al. Pre-clinical assessment of SLN360, a novel siRNA targeting LPA, developed to address elevated lipoprotein (a) in cardiovascular disease. Atherosclerosis 2022, 349, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Rider, D.; Chivers, S.; Aretz, J.; Eisermann, M.; Löffler, K.; Hauptmann, J.; Morrison, E.; Campion, G. Preclinical Toxicological Assessment of A Novel siRNA, SLN360, Targeting Elevated Lipoprotein (a) in Cardiovascular Disease. Toxicol. Sci. 2022, 189, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Wolski, K.; Balog, C.; Swerdlow, D.I.; Scrimgeour, A.C.; Rambaran, C.; Wilson, R.J.; Boyce, M.; Ray, K.K.; Cho, L.; et al. Single Ascending Dose Study of a Short Interfering RNA Targeting Lipoprotein(a) Production in Individuals with Elevated Plasma Lipoprotein(a) Levels. JAMA 2022, 327, 1679–1687. [Google Scholar] [CrossRef] [PubMed]
- Nurmohamed, N.S.; Kraaijenhof, J.M.; Stroes, E.S.G. Lp(a): A New Pathway to Target? Curr. Atheroscler. Rep. 2022, 24, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Cho, L. Recent lipoprotein(a) trials. Curr. Opin. Lipidol. 2022, 33, 301–308. [Google Scholar] [CrossRef]
- Nissen, S.E.; Linnebjerg, H.; Shen, X.; Wolski, K.; Ma, X.; Lim, S.; Michael, L.F.; Ruotolo, G.; Gribble, G.; Navar, A.M.; et al. Lepodisiran, an Extended-Duration Short Interfering RNA Targeting Lipoprotein(a): A Randomized Dose-Ascending Clinical Trial. JAMA 2023, 330, 2075–2083. [Google Scholar] [CrossRef]
- Hooper, A.J.; Fernando, P.M.S.; Burnett, J.R. Potential of muvalaplin as a lipoprotein(a) inhibitor. Expert Opin. Investig. Drugs 2024, 33, 5–7. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Nissen, S.E.; Fleming, C.; Urva, S.; Suico, J.; Berg, P.H.; Linnebjerg, H.; Ruotolo, G.; Turner, P.K.; Michael, L.F. Muvalaplin, an Oral Small Molecule Inhibitor of Lipoprotein(a) Formation: A Randomized Clinical Trial. JAMA 2023, 330, 1042–1053. [Google Scholar] [CrossRef]
- Xu, Y.; Li, Z. CRISPR-Cas systems: Overview, innovations and applications in human disease research and gene therapy. Comput. Struct. Biotechnol. J. 2020, 18, 2401–2415. [Google Scholar] [CrossRef] [PubMed]
- Naso, M.F.; Tomkowicz, B.; Perry, W.L., 3rd; Strohl, W.R. Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. BioDrugs 2017, 31, 317–334. [Google Scholar] [CrossRef]
- Doerfler, A.M.; Park, S.H.; Assini, J.M.; Youssef, A.; Saxena, L.; Yaseen, A.B.; De Giorgi, M.; Chuecos, M.; Hurley, A.E.; Li, A.; et al. LPA disruption with AAV-CRISPR potently lowers plasma apo(a) in transgenic mouse model: A proof-of-concept study. Mol Ther Methods Clin. Dev. 2022, 27, 337–351. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Agent | Author/Study/Year | Impact on Lp(a) | Remarks |
|---|---|---|---|
| Atorvastatin | Arsenault et al., Clinical Trial/2018 [162] | Dose-dependent ↑ of Lp(a) with ↑ atorvastatin doses | T2DM was associated with ↓ Lp(a) levels |
| Ezetimibe | Sahebkar et al., Systematic review and meta-analysis of randomized controlled trials/ 2018 [171] | No significant possible reduction (0–5%) | The result was confirmed both when ezetimibe was used as monotherapy and in combination with a statin |
| Fenofibrate | Ko et al., Prospective case-control study/ 2005 [174] | Possible ↑ of Lp(a) levels in patients with hypertriglyceridemia | 1. Changes in Lp(a) levels were negatively correlated with changes in triglyceride levels 2. Liver function influenced the effect of fenofibrate on Lp(a) |
| Niacin | Sahebkar et al., Systematic review and meta-analysis of randomized placebo-controlled trials/2016 [177] | Significant ↓ of Lp(a) levels (WMD: −22.90%, 95% CI: −27.32, −18.48, p < 0.001). | No significant association between the changes in plasma concentrations of Lp(a) with niacin dose, treatment duration, and percentage change in plasma HDL-C concentrations |
| Mipomersen | Reeskamp et al., Randomized Controlled Trial/ 2019 [187] | Significant ↓ of Lp(a) by 27.7% | 1. Additional significant ↓ in LDL-C and ApoB 2. Limited tolerability and ↑ hepatic transaminase levels in 21% of patients |
| Bempedoic acid | Ridker et al., Secondary biomarker analysis of the randomized placebo-controlled multi-center CLEAR Harmony trial /2023 [215] | Median percent change of Lp(a) levels from baseline to 12 weeks was 2.4% | The pattern of lipid lowering and inflammation inhibition with bempedoic acid was almost identical to what is observed with statin therapy |
| Obicetrapib | Nicholls et al., Clinical trial/2022 [227] | Significant ↓ of Lp(a) by 33.8% and 56.5% with doses of 5 mg and 10 mg, respectively | 1. Significant ↓ in LDL-C, non-HDL-C, and ApoB 2. Significant ↑ in HDL-C 3. The most prevalent adverse events were gastrointestinal disorders (primarily nausea) and nervous system disorders (primarily headache). |
| Evolocumab | O’Donoghue, Randomized controlled trial/ 2019 [127] | Median ↓ of Lp(a) at 48 weeks by 26.9% (6.2−46.7%) | Patients with higher baseline Lp(a) levels experienced greater absolute ↓ in Lp(a) and tended to derive greater coronary benefit |
| Alirocumab | Szarek et al., Randomized controlled trial/ 2020 [201] | Median Lp(a) ↓ in Lp(a) −5.0 [−13.6, 0] mg/dL | 1. ↓ of Lp(a) independently predicted lower risk of total cardiovascular events 2. Each 5 mg/dL ↓ in Lp(a) predicted a 2.5% relative ↓ in cardiovascular events |
| Inclisiran | Ray et al., Controlled clinical trial/ 2023 [210] | ↓ Lp(a) of 6.3% and 14.3% in the inclisiran-only arm and switching arm, respectively | Twice-yearly inclisiran provided sustained ↓ in LDL-C and PCSK9 concentrations and was well tolerated over 4 years in the extension study |
| Authors, Year, Agent | Study’s Characteristics | Results |
|---|---|---|
| Karwatowska-Prokopczuk et al., 2023 Pelacarsen [239] | Randomized double-blind, placebo-controlled study: 1. 29 healthy Japanese individuals 2. Effect on serum Lp(a): - SAD: 20 mg, 40 mg, 80 mg - MD: 80 mg monthly (4 doses) | 1. Safety and tolerability 2. No serious side effects 3. In the MD cohort: mean serum peak levels of pelacarsen at 4 h 4. In the SAD cohort: max. ↓ of Lp(a) by 73.7% at 80 mg 5. In the MD cohort: max. ↓ of Lp(a) by 106.2% at day 85 |
| Sohn et al., 2022, Olpasiran [242] | Phase I, open-label, parallel-design clinical study enrolling 27 subjects (Japanese/non-Japanese) - Japanese: received olpasiran at 3 mg, 9 mg, 75 mg, 225 mg - Non-Japanese: received olpasiran at 75 mg | 1. Safety and tolerability 2. No serious side effects 3. ↓ of Lp(a) as early as day 4 4. ↓ of Lp(a) in a dose-dependent way 5. Mean percentage ↓ of Lp(a) from the baseline: 56–99% 6. Max. ↓ of Lp(a) at day 57 7. The magnitude and durability of ↓ of Lp(a) were similar between 2 groups |
| Nissen et al. 2022, SLN360 [246] | Phase 1 clinical trial: 1. 32 adults with Lp(a) ≥ 150 nmol/L with no history of CVD 2. Effect on serum Lp(a): SLN360 vs. placebo at 30 mg, 100 mg, 300 mg, 600 mg | 1. Safety and tolerability 2. No significant side effects 3. Dose-dependent ↓ Lp(a) 4. Median Lp(a) concentrations were ≥70% and ≥80% below baseline at day 150 following administration of the 300 mg and 600 mg SLN360 doses, respectively. 5. ↓ of Lp(a) persisted for at least 150 days after administration |
| Nissen et al. 2023, Lepodisiran [249] | Phase 1 clinical trial: 1. 48 subjects with Lp(a) ≥ 30 mg/dL without CVD. 2. Effect on serum Lp(a): Lepodisiran vs. placebo at 4 mg, 12 mg, 32 mg, 96 mg, 304 mg, and 608 mg 3. Excluded from the study: - Age < 18 years old - Women of reproductive age - Smoking, alcoholism - Chronic renal failure or liver disease | 1. Safety and tolerability 2. Side effects: - Most common: transient pain at the injection site - ↑ SGOT, SGPT, CPK (mild) 3. Peak lepodisiran’s levels at 10.5 h 4. Undetectable levels of lepodisiran by 2 days 5. Dose-dependent ↓ of Lp(a) with ↓ 97% reduction of Lp(a) in the 608- mg dose group 6. The treatment effect lasted longer in the 608 mg dose group |
| Nicholls et al. 2023, Muvalaplin [251] | Phase 1 randomized placebo-controlled human study: 1. 114 healthy individuals aged 18–69 years old with BMI ≤ 30 kg/m2: - 1st part of the study: The effect of 1 dose (1–800 mg) of muvalaplin vs. placebo on serum Lp(a) - 2nd part of the study: The effect of 1 dose (30–800 mg) of muvalaplin vs. placebo taken over 14 days in subjects with Lp(a) ≥ 30 mg/dL | 1. Safe and well-tolerated for up to 14 days of prescription 2. No significant side effects 3. Peak serum levels of muvalaplin 2–5 days post-administration 4. Rapid ↓ of Lp(a) within 1 day 5. Max. ↓ of Lp(a) 63–65% for doses ≥ 100 mg on days 14 and 15 6. Sustained ↓ in Lp(a) levels for up to 50 days, particularly for doses ≥ 300 mg 7. No significant changes in PLG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsioulos, G.; Kounatidis, D.; Vallianou, N.G.; Poulaki, A.; Kotsi, E.; Christodoulatos, G.S.; Tsilingiris, D.; Karampela, I.; Skourtis, A.; Dalamaga, M. Lipoprotein(a) and Atherosclerotic Cardiovascular Disease: Where Do We Stand? Int. J. Mol. Sci. 2024, 25, 3537. https://doi.org/10.3390/ijms25063537
Tsioulos G, Kounatidis D, Vallianou NG, Poulaki A, Kotsi E, Christodoulatos GS, Tsilingiris D, Karampela I, Skourtis A, Dalamaga M. Lipoprotein(a) and Atherosclerotic Cardiovascular Disease: Where Do We Stand? International Journal of Molecular Sciences. 2024; 25(6):3537. https://doi.org/10.3390/ijms25063537
Chicago/Turabian StyleTsioulos, Georgios, Dimitris Kounatidis, Natalia G. Vallianou, Aikaterini Poulaki, Evangelia Kotsi, Gerasimos Socrates Christodoulatos, Dimitrios Tsilingiris, Irene Karampela, Alexandros Skourtis, and Maria Dalamaga. 2024. "Lipoprotein(a) and Atherosclerotic Cardiovascular Disease: Where Do We Stand?" International Journal of Molecular Sciences 25, no. 6: 3537. https://doi.org/10.3390/ijms25063537