
Novel Compound Inhibitors of HIV-1NL4-3 Vpu

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmids
2.2. Compounds
2.3. Cell Lines
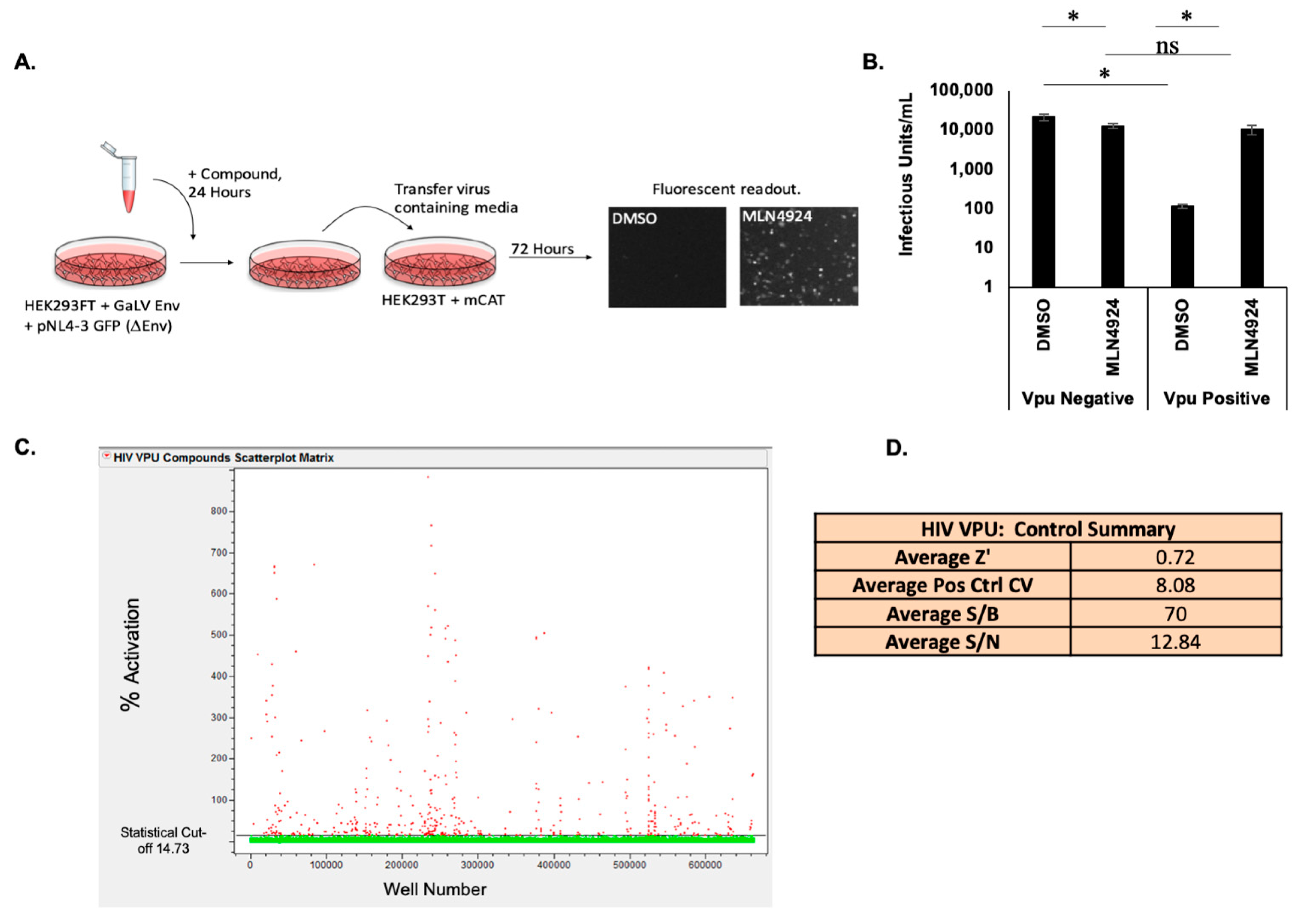
2.4. GaLV Assay/Screen/Flow
2.5. High Throughput Screen
2.6. Cellular Target Counter Screens
2.7. CD4 and Tetherin Surface Labeling in PBMCs
2.8. CD4 Surface Labeling in TZM-GFP
2.9. Flow Cytometry
2.10. 2G12 Surface Labeling
2.11. ADCC
3. Results
3.1. High Throughput Screen for Vpu Inhibitors
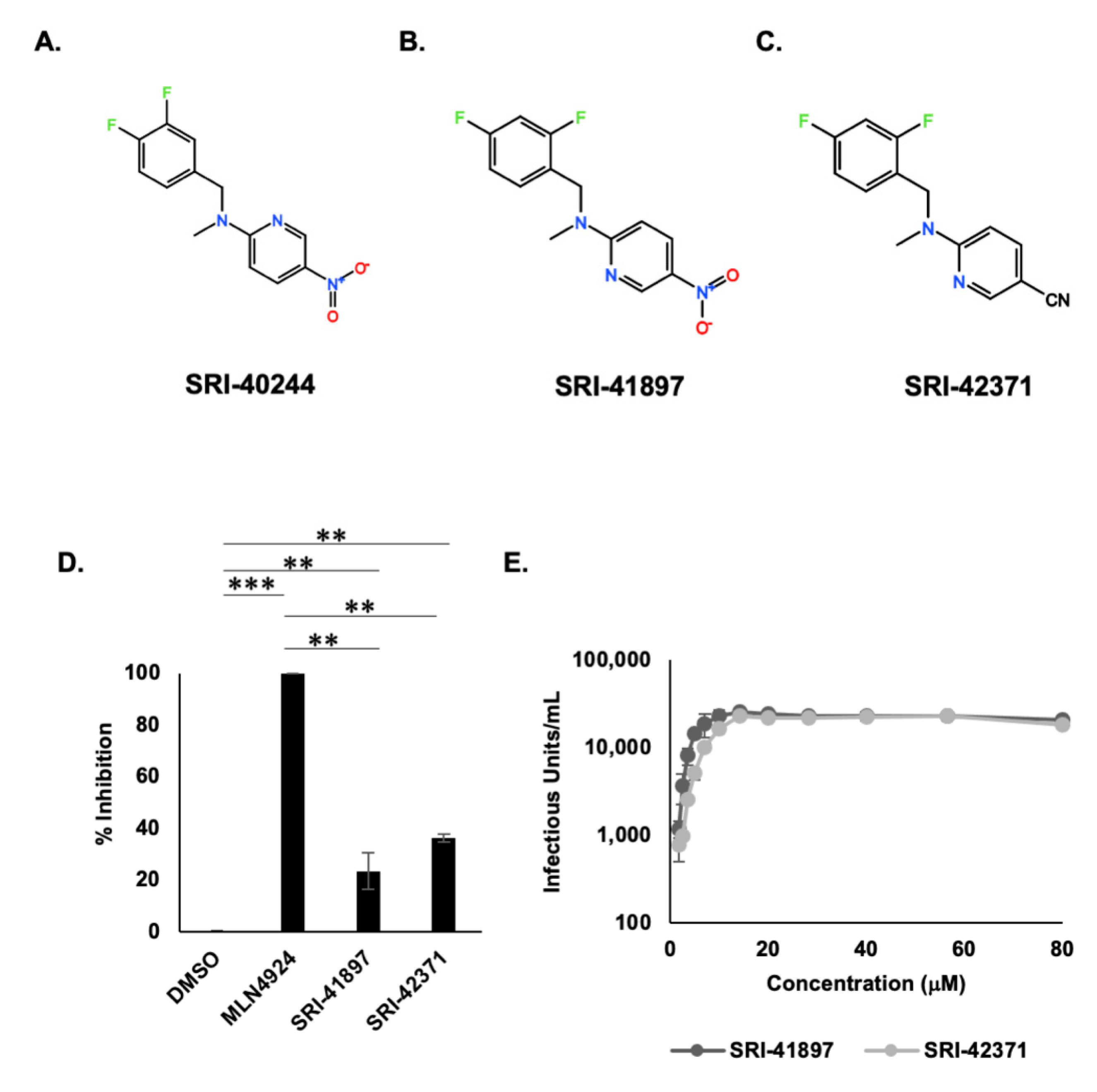
3.2. HTS Reveals Potent Vpu Inhibitors
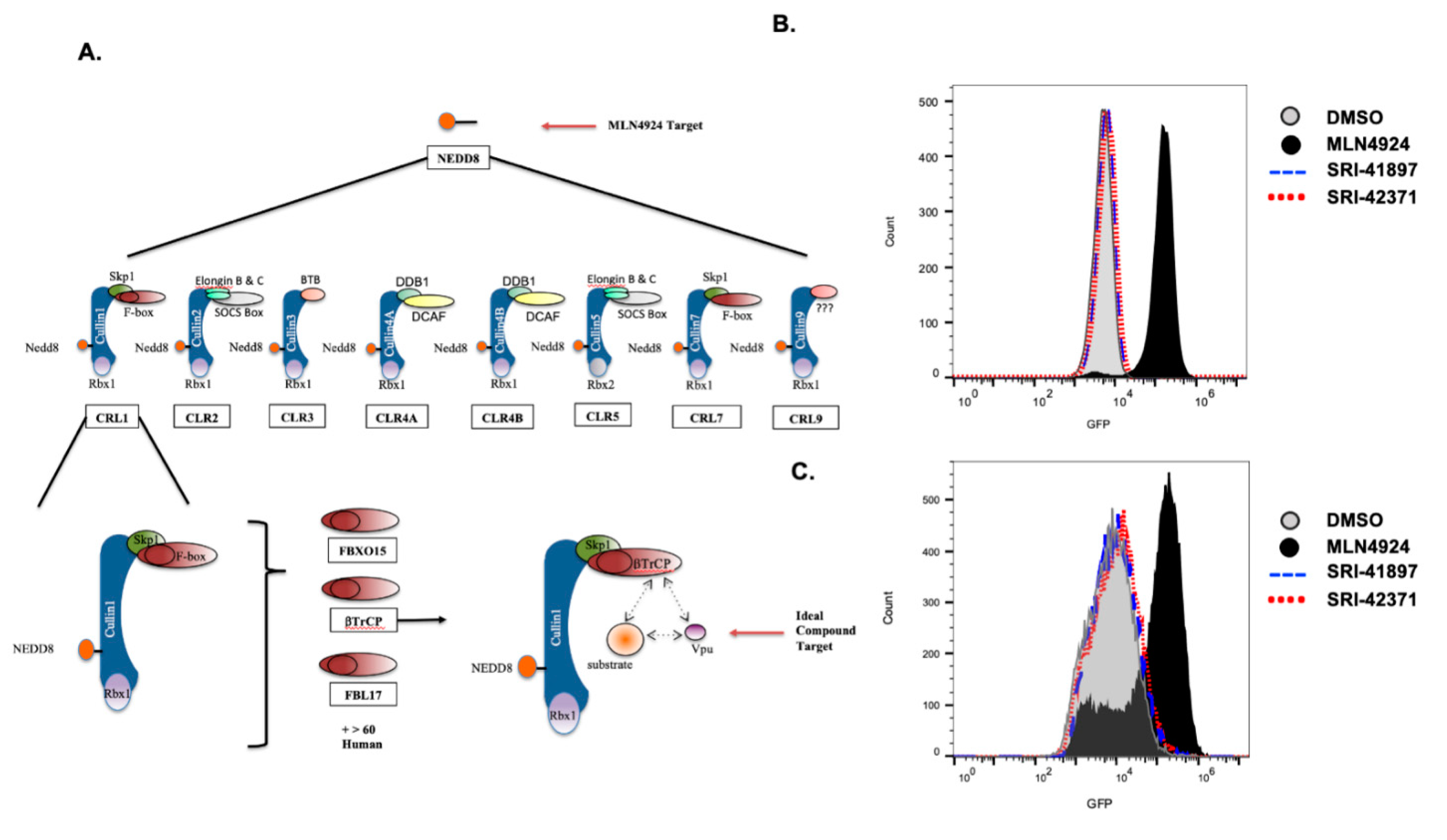
3.3. Vpu- Independent Counter Screens
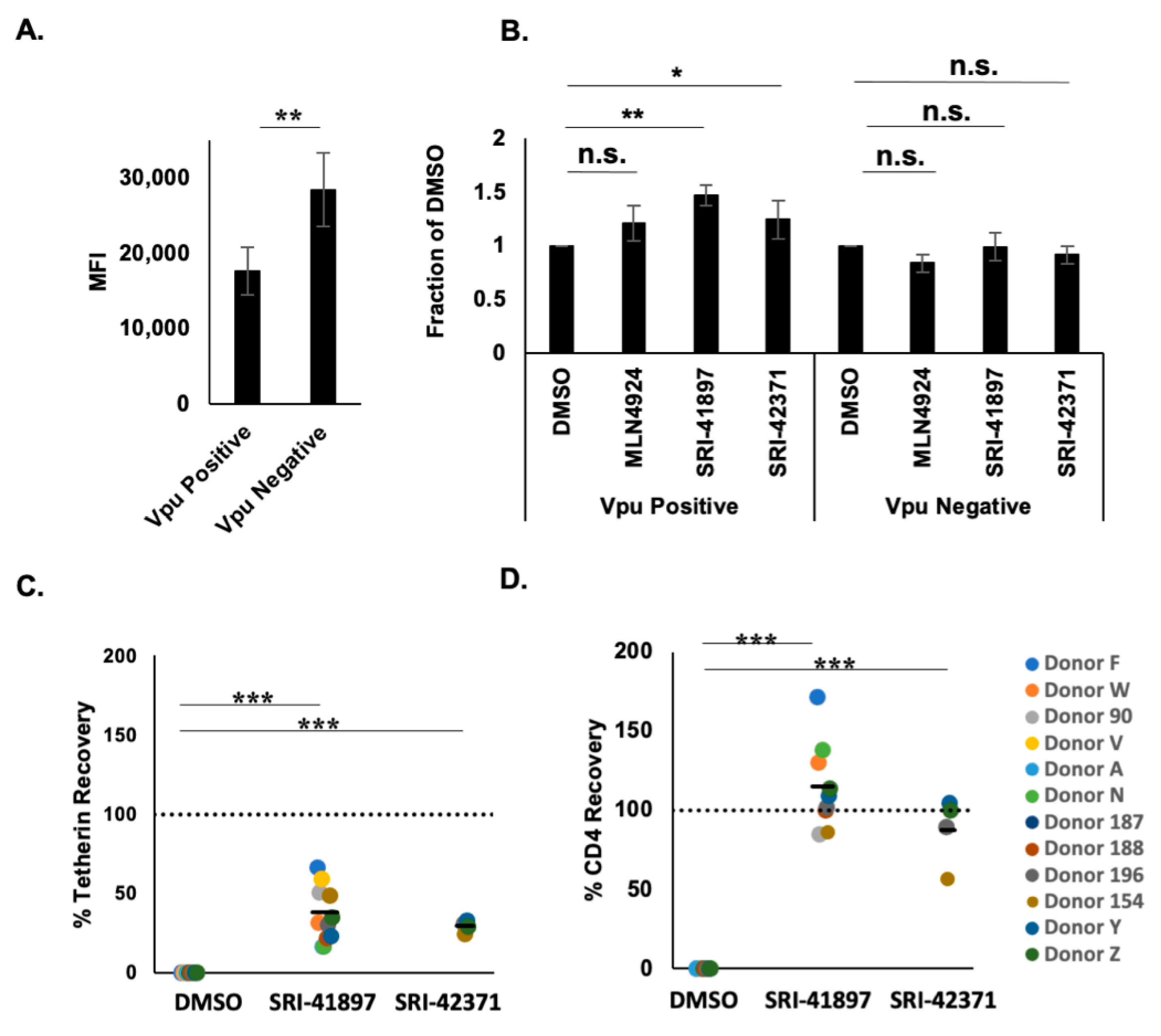
3.4. Compounds Rescue CD4 and BST-2/Tetherin Expression
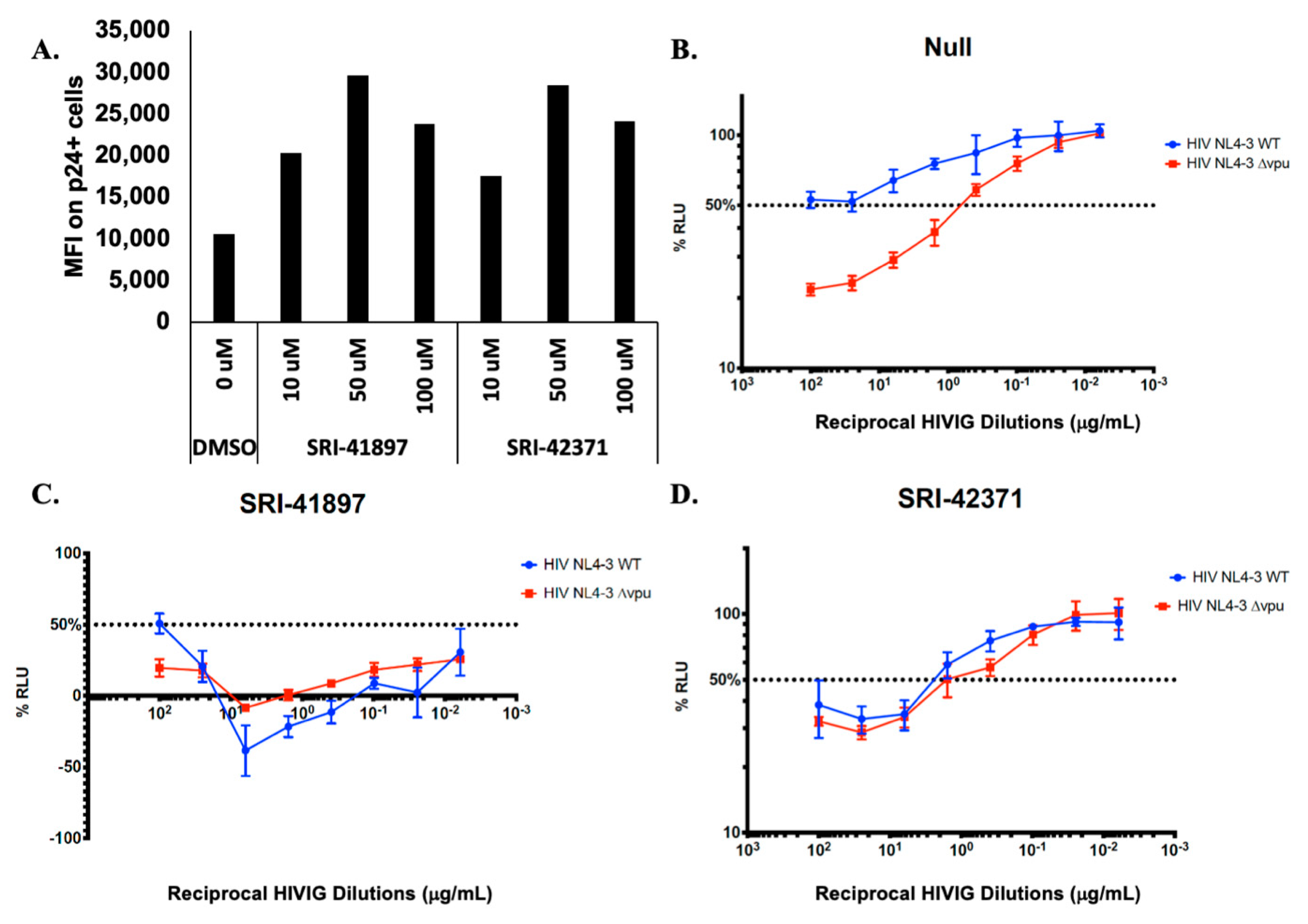
3.5. Compounds Rescue ADCC Response
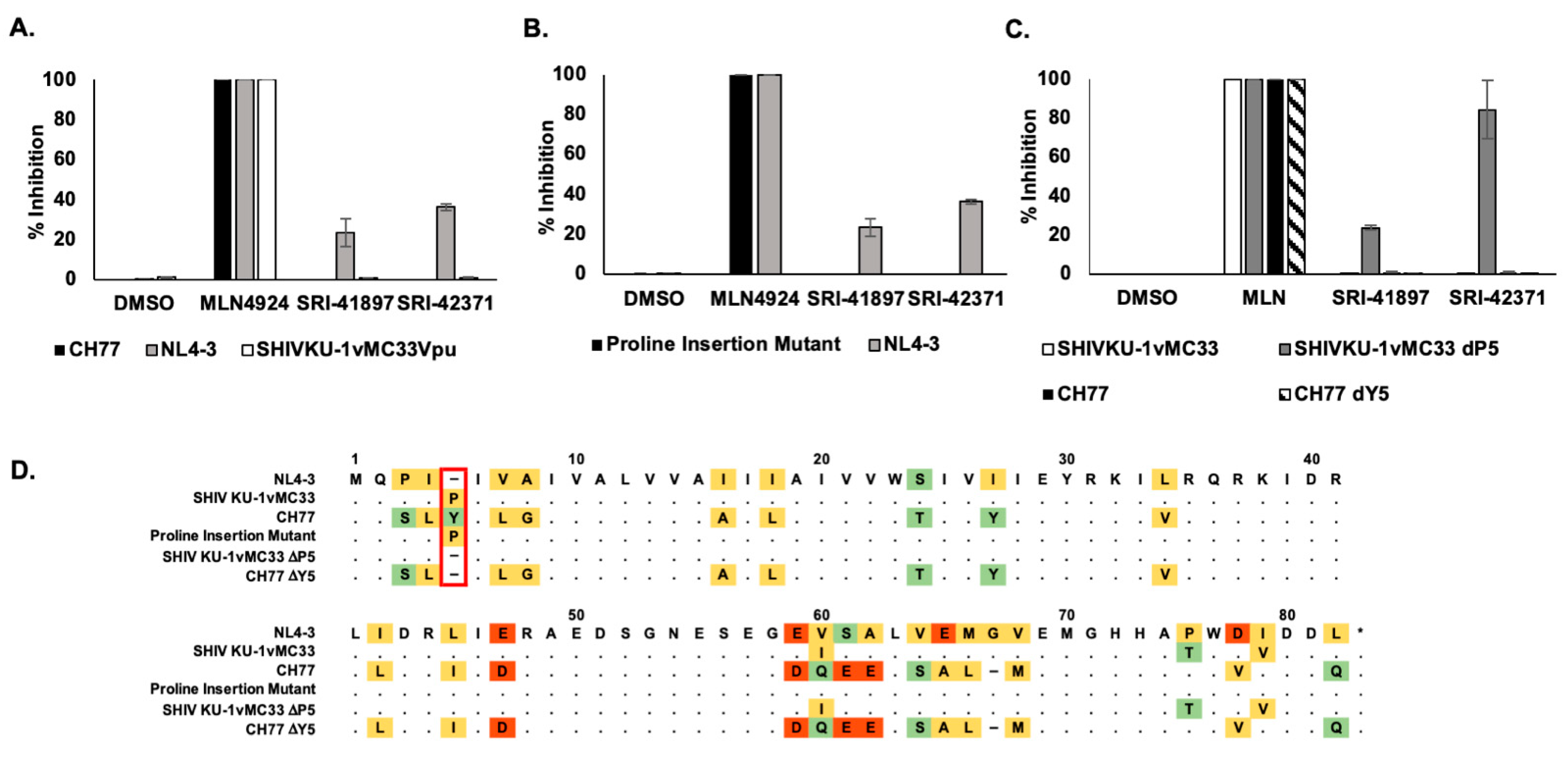
3.6. Compounds Are NL4-3 Strain Specific
4. Discussion
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Harrigan, P.R.; Whaley, M.; Montaner, J.S. Rate of HIV-1 RNA rebound upon stopping antiretroviral therapy. AIDS 1999, 13, F59–F62. [Google Scholar] [CrossRef] [PubMed]
- Killian, M.S.; Roop, J.; Ng, S.; Hecht, F.M.; Levy, J.A. CD8+ cell anti-HIV activity rapidly increases upon discontinuation of early antiretroviral therapy. J. Clin. Immunol. 2009, 29, 311–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crespo, M.; Paradineiro, J.C.; Ribera, E.; Ruiz, I.; Falco, V.; Lopez-Quinones, J.; Ocana, I.; Pahissa, A. A case of multiorgan failure following interruption of antiretroviral treatment. Eur. J. Clin. Microbiol. Infect. Dis. 2004, 23, 63–65. [Google Scholar] [CrossRef] [PubMed]
- Davey, R.T., Jr.; Bhat, N.; Yoder, C.; Chun, T.W.; Metcalf, J.A.; Dewar, R.; Natarajan, V.; Lempicki, R.A.; Adelsberger, J.W.; Miller, K.D.; et al. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc. Natl. Acad. Sci. USA 1999, 96, 15109–15114. [Google Scholar] [CrossRef] [Green Version]
- Hughes, A.J.; Mattson, C.L.; Scheer, S.; Beer, L.; Skarbinski, J. Discontinuation of antiretroviral therapy among adults receiving HIV care in the United States. J. Acquir. Immune Defic. Syndr. 2014, 66, 80–89. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, M.E. Vpu Protein: The Viroporin Encoded by HIV-1. Viruses 2015, 7, 4352–4368. [Google Scholar] [CrossRef] [Green Version]
- Willey, R.L.; Maldarelli, F.; Martin, M.A.; Strebel, K. Human immunodeficiency virus type 1 Vpu protein induces rapid degradation of CD4. J. Virol. 1992, 66, 7193–7200. [Google Scholar] [CrossRef] [Green Version]
- Neil, S.J.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef] [Green Version]
- Van Damme, N.; Goff, D.; Katsura, C.; Jorgenson, R.L.; Mitchell, R.; Johnson, M.C.; Stephens, E.B.; Guatelli, J. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 2008, 3, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Shah, A.H.; Sowrirajan, B.; Davis, Z.B.; Ward, J.P.; Campbell, E.M.; Planelles, V.; Barker, E. Degranulation of natural killer cells following interaction with HIV-1-infected cells is hindered by downmodulation of NTB-A by Vpu. Cell Host Microbe 2010, 8, 397–409. [Google Scholar] [CrossRef] [Green Version]
- Matusali, G.; Potesta, M.; Santoni, A.; Cerboni, C.; Doria, M. The human immunodeficiency virus type 1 Nef and Vpu proteins downregulate the natural killer cell-activating ligand PVR. J. Virol. 2012, 86, 4496–4504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez, P.W.; Famiglietti, M.; Sowrirajan, B.; DePaula-Silva, A.B.; Rodesch, C.; Barker, E.; Bosque, A.; Planelles, V. Downmodulation of CCR7 by HIV-1 Vpu results in impaired migration and chemotactic signaling within CD4(+) T cells. Cell Rep. 2014, 7, 2019–2030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matheson, N.; Wals, K.; Weekes, M.; Rapiteanu, R.; Vigan, R.; Antrobus, R.; Smith, D.; Neil, S.; Lehner, P. Antagonism of aminoacid transport in primary CD4 T cells by HIV-1 Vpu. Lancet 2015, 385, S66. [Google Scholar] [CrossRef]
- Schubert, U.; Henklein, P.; Boldyreff, B.; Wingender, E.; Strebel, K.; Porstmann, T. The human immunodeficiency virus type 1 encoded Vpu protein is phosphorylated by casein kinase-2 (CK-2) at positions Ser52 and Ser56 within a predicted alpha-helix-turn-alpha-helix-motif. J. Mol. Biol. 1994, 236, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Margottin, F.; Bour, S.P.; Durand, H.; Selig, L.; Benichou, S.; Richard, V.; Thomas, D.; Strebel, K.; Benarous, R. A novel human WD protein, h-beta TrCp, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Mol. Cell 1998, 1, 565–574. [Google Scholar] [CrossRef]
- Song, Y.E.; Cyburt, D.; Lucas, T.M.; Gregory, D.A.; Lyddon, T.D.; Johnson, M.C. betaTrCP is Required for HIV-1 Vpu Modulation of CD4, GaLV Env, and BST-2/Tetherin. Viruses 2018, 10, 573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dube, M.; Roy, B.B.; Guiot-Guillain, P.; Binette, J.; Mercier, J.; Chiasson, A.; Cohen, E.A. Antagonism of tetherin restriction of HIV-1 release by Vpu involves binding and sequestration of the restriction factor in a perinuclear compartment. PLoS Pathog. 2010, 6, e1000856. [Google Scholar] [CrossRef] [Green Version]
- Kueck, T.; Foster, T.L.; Weinelt, J.; Sumner, J.C.; Pickering, S.; Neil, S.J. Serine Phosphorylation of HIV-1 Vpu and Its Binding to Tetherin Regulates Interaction with Clathrin Adaptors. PLoS Pathog. 2015, 11, e1005141. [Google Scholar] [CrossRef] [Green Version]
- Vigan, R.; Neil, S.J. Determinants of tetherin antagonism in the transmembrane domain of the human immunodeficiency virus type 1 Vpu protein. J. Virol. 2010, 84, 12958–12970. [Google Scholar] [CrossRef] [Green Version]
- Bolduan, S.; Reif, T.; Schindler, M.; Schubert, U. HIV-1 Vpu mediated downregulation of CD155 requires alanine residues 10, 14 and 18 of the transmembrane domain. Virology 2014, 464–465, 375–384. [Google Scholar] [CrossRef]
- Prevost, J.; Pickering, S.; Mumby, M.J.; Medjahed, H.; Gendron-Lepage, G.; Delgado, G.G.; Dirk, B.S.; Dikeakos, J.D.; Sturzel, C.M.; Sauter, D.; et al. Upregulation of BST-2 by Type I Interferons Reduces the Capacity of Vpu To Protect HIV-1-Infected Cells from NK Cell Responses. mBio 2019, 10, e01113-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schubert, U.; Ferrer-Montiel, A.V.; Oblatt-Montal, M.; Henklein, P.; Strebel, K.; Montal, M. Identification of an ion channel activity of the Vpu transmembrane domain and its involvement in the regulation of virus release from HIV-1-infected cells. FEBS Lett. 1996, 398, 12–18. [Google Scholar] [CrossRef] [Green Version]
- Ewart, G.D.; Nasr, N.; Naif, H.; Cox, G.B.; Cunningham, A.L.; Gage, P.W. Potential new anti-human immunodeficiency virus type 1 compounds depress virus replication in cultured human macrophages. Antimicrob. Agents Chemother. 2004, 48, 2325–2330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forthal, D.N.; Finzi, A. Antibody-dependent cellular cytotoxicity in HIV infection. AIDS 2018, 32, 2439–2451. [Google Scholar] [CrossRef]
- Kramski, M.; Stratov, I.; Kent, S.J. The role of HIV-specific antibody-dependent cellular cytotoxicity in HIV prevention and the influence of the HIV-1 Vpu protein. AIDS 2015, 29, 137–144. [Google Scholar] [CrossRef] [Green Version]
- Veillette, M.; Coutu, M.; Richard, J.; Batraville, L.A.; Dagher, O.; Bernard, N.; Tremblay, C.; Kaufmann, D.E.; Roger, M.; Finzi, A. The HIV-1 gp120 CD4-bound conformation is preferentially targeted by antibody-dependent cellular cytotoxicity-mediating antibodies in sera from HIV-1-infected individuals. J. Virol. 2015, 89, 545–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veillette, M.; Desormeaux, A.; Medjahed, H.; Gharsallah, N.E.; Coutu, M.; Baalwa, J.; Guan, Y.; Lewis, G.; Ferrari, G.; Hahn, B.H.; et al. Interaction with cellular CD4 exposes HIV-1 envelope epitopes targeted by antibody-dependent cell-mediated cytotoxicity. J. Virol. 2014, 88, 2633–2644. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, R.A.; Hamlin, R.E.; Monroe, A.; Moldt, B.; Hotta, M.T.; Rodriguez Caprio, G.; Fierer, D.S.; Simon, V.; Chen, B.K. HIV-1 Vpu antagonism of tetherin inhibits antibody-dependent cellular cytotoxic responses by natural killer cells. J. Virol. 2014, 88, 6031–6046. [Google Scholar] [CrossRef] [Green Version]
- Arias, J.F.; Heyer, L.N.; von Bredow, B.; Weisgrau, K.L.; Moldt, B.; Burton, D.R.; Rakasz, E.G.; Evans, D.T. Tetherin antagonism by Vpu protects HIV-infected cells from antibody-dependent cell-mediated cytotoxicity. Proc. Natl. Acad. Sci. USA 2014, 111, 6425–6430. [Google Scholar] [CrossRef] [Green Version]
- Sato, K.; Misawa, N.; Fukuhara, M.; Iwami, S.; An, D.S.; Ito, M.; Koyanagi, Y. Vpu augments the initial burst phase of HIV-1 propagation and downregulates BST2 and CD4 in humanized mice. J. Virol. 2012, 86, 5000–5013. [Google Scholar] [CrossRef] [Green Version]
- Dave, V.P.; Hajjar, F.; Dieng, M.M.; Haddad, E.; Cohen, E.A. Efficient BST2 antagonism by Vpu is critical for early HIV-1 dissemination in humanized mice. Retrovirology 2013, 10, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khoury, G.; Ewart, G.; Luscombe, C.; Miller, M.; Wilkinson, J. Antiviral efficacy of the novel compound BIT225 against HIV-1 release from human macrophages. Antimicrob. Agents Chemother. 2010, 54, 835–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luscombe, C.A.; Huang, Z.; Murray, M.G.; Miller, M.; Wilkinson, J.; Ewart, G.D. A novel Hepatitis C virus p7 ion channel inhibitor, BIT225, inhibits bovine viral diarrhea virus in vitro and shows synergism with recombinant interferon-alpha-2b and nucleoside analogues. Antivir. Res. 2010, 86, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Luscombe, C.A.; Avihingsanon, A.; Supparatpinyo, K.; Gatechompol, S.; Han, W.M.; Ewart, G.D.; Thomson, A.S.; Miller, M.; Becker, S.; Murphy, R.L. Human immunodeficiency virus type-1 Vpu inhibitor, BIT225, in combination with 3-drug antiretroviral therapy modulates inflammation and immune cells functions. J. Infect. Dis. 2020, 223, 1914–1922. [Google Scholar] [CrossRef] [PubMed]
- Behmard, E.; Abdolmaleki, P.; Taghdir, M. Understanding the inhibitory mechanism of BIT225 drug against p7 viroporin using computational study. Biophys. Chem. 2018, 233, 47–54. [Google Scholar] [CrossRef]
- Kuhl, B.D.; Cheng, V.; Donahue, D.A.; Sloan, R.D.; Liang, C.; Wilkinson, J.; Wainberg, M.A. The HIV-1 Vpu viroporin inhibitor BIT225 does not affect Vpu-mediated tetherin antagonism. PLoS ONE 2011, 6, e27660. [Google Scholar] [CrossRef] [Green Version]
- Tokarev, A.; Stoneham, C.; Lewinski, M.K.; Mukim, A.; Deshmukh, S.; Vollbrecht, T.; Spina, C.A.; Guatelli, J. Pharmacologic Inhibition of Nedd8 Activation Enzyme Exposes CD4-Induced Epitopes within Env on Cells Expressing HIV-1. J. Virol. 2016, 90, 2486–2502. [Google Scholar] [CrossRef] [Green Version]
- Lucas, T.M.; Lyddon, T.D.; Cannon, P.M.; Johnson, M.C. Pseudotyping incompatibility between HIV-1 and gibbon ape leukemia virus Env is modulated by Vpu. J. Virol. 2010, 84, 2666–2674. [Google Scholar] [CrossRef] [Green Version]
- Janaka, S.K.; Lucas, T.M.; Johnson, M.C. Sequences in gibbon ape leukemia virus envelope that confer sensitivity to HIV-1 accessory protein Vpu. J. Virol. 2011, 85, 11945–11954. [Google Scholar] [CrossRef] [Green Version]
- Zufferey, R.; Nagy, D.; Mandel, R.J.; Naldini, L.; Trono, D. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat. Biotechnol. 1997, 15, 871–875. [Google Scholar] [CrossRef]
- Adachi, A.; Gendelman, H.E.; Koenig, S.; Folks, T.; Willey, R.; Rabson, A.; Martin, M.A. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 1986, 59, 284–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chukkapalli, V.; Hogue, I.B.; Boyko, V.; Hu, W.S.; Ono, A. Interaction between the human immunodeficiency virus type 1 Gag matrix domain and phosphatidylinositol-(4,5)-bisphosphate is essential for efficient gag membrane binding. J. Virol. 2008, 82, 2405–2417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salamango, D.J.; McCann, J.L.; Demir, O.; Brown, W.L.; Amaro, R.E.; Harris, R.S. APOBEC3B Nuclear Localization Requires Two Distinct N-Terminal Domain Surfaces. J. Mol. Biol. 2018, 430, 2695–2708. [Google Scholar] [CrossRef] [PubMed]
- Rosa, A.; Chande, A.; Ziglio, S.; De Sanctis, V.; Bertorelli, R.; Goh, S.L.; McCauley, S.M.; Nowosielska, A.; Antonarakis, S.E.; Luban, J.; et al. HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature 2015, 526, 212–217. [Google Scholar] [CrossRef] [Green Version]
- Alpert, M.D.; Heyer, L.N.; Williams, D.E.; Harvey, J.D.; Greenough, T.; Allhorn, M.; Evans, D.T. A novel assay for antibody-dependent cell-mediated cytotoxicity against HIV-1- or SIV-infected cells reveals incomplete overlap with antibodies measured by neutralization and binding assays. J. Virol. 2012, 86, 12039–12052. [Google Scholar] [CrossRef] [Green Version]
- Alpert, M.D.; Rahmberg, A.R.; Neidermyer, W.; Ng, S.K.; Carville, A.; Camp, J.V.; Wilson, R.L.; Piatak, M., Jr.; Mansfield, K.G.; Li, W.; et al. Envelope-modified single-cycle simian immunodeficiency virus selectively enhances antibody responses and partially protects against repeated, low-dose vaginal challenge. J. Virol. 2010, 84, 10748–10764. [Google Scholar] [CrossRef] [Green Version]
- Richard, J.; Veillette, M.; Brassard, N.; Iyer, S.S.; Roger, M.; Martin, L.; Pazgier, M.; Schon, A.; Freire, E.; Routy, J.P.; et al. CD4 mimetics sensitize HIV-1-infected cells to ADCC. Proc. Natl. Acad. Sci. USA 2015, 112, E2687–E2694. [Google Scholar] [CrossRef] [Green Version]
- Soucy, T.A.; Smith, P.G.; Milhollen, M.A.; Berger, A.J.; Gavin, J.M.; Adhikari, S.; Brownell, J.E.; Burke, K.E.; Cardin, D.P.; Critchley, S.; et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009, 458, 732–736. [Google Scholar] [CrossRef]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen 1999, 4, 67–73. [Google Scholar] [CrossRef]
- Mehle, A.; Goncalves, J.; Santa-Marta, M.; McPike, M.; Gabuzda, D. Phosphorylation of a novel SOCS-box regulates assembly of the HIV-1 Vif-Cul5 complex that promotes APOBEC3G degradation. Genes Dev. 2004, 18, 2861–2866. [Google Scholar] [CrossRef] [Green Version]
- Sheehy, A.M.; Gaddis, N.C.; Malim, M.H. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat. Med. 2003, 9, 1404–1407. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Yu, Y.; Liu, B.; Luo, K.; Kong, W.; Mao, P.; Yu, X.F. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 2003, 302, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Mehle, A.; Strack, B.; Ancuta, P.; Zhang, C.; McPike, M.; Gabuzda, D. Vif overcomes the innate antiviral activity of APOBEC3G by promoting its degradation in the ubiquitin-proteasome pathway. J. Biol. Chem. 2004, 279, 7792–7798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frescas, D.; Pagano, M. Deregulated proteolysis by the F-box proteins SKP2 and beta-TrCP: Tipping the scales of cancer. Nat. Rev. Cancer 2008, 8, 438–449. [Google Scholar] [CrossRef] [Green Version]
- Isoda, M.; Kanemori, Y.; Nakajo, N.; Uchida, S.; Yamashita, K.; Ueno, H.; Sagata, N. The extracellular signal-regulated kinase-mitogen-activated protein kinase pathway phosphorylates and targets Cdc25A for SCF beta-TrCP-dependent degradation for cell cycle arrest. Mol. Biol. Cell 2009, 20, 2186–2195. [Google Scholar] [CrossRef] [Green Version]
- Kahrstrom, C.T. Viral pathogenesis: Vpu puts the brakes on ADCC. Nat. Rev. Microbiol. 2014, 12, 397. [Google Scholar] [CrossRef]
- Richard, J.; Prevost, J.; Alsahafi, N.; Ding, S.; Finzi, A. Impact of HIV-1 Envelope Conformation on ADCC Responses. Trends Microbiol. 2018, 26, 253–265. [Google Scholar] [CrossRef]
- Klein, J.S.; Webster, A.; Gnanapragasam, P.N.; Galimidi, R.P.; Bjorkman, P.J. A dimeric form of the HIV-1 antibody 2G12 elicits potent antibody-dependent cellular cytotoxicity. AIDS 2010, 24, 1633–1640. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Finzi, A.; Sodroski, J. The Conformational States of the HIV-1 Envelope Glycoproteins. Trends Microbiol. 2020, 28, 655–667. [Google Scholar] [CrossRef]
- Narayan, S.V.; Mukherjee, S.; Jia, F.; Li, Z.; Wang, C.; Foresman, L.; McCormick-Davis, C.; Stephens, E.B.; Joag, S.V.; Narayan, O. Characterization of a neutralization-escape variant of SHIVKU-1, a virus that causes acquired immune deficiency syndrome in pig-tailed macaques. Virology 1999, 256, 54–63. [Google Scholar] [CrossRef] [Green Version]
- McCormick-Davis, C.; Dalton, S.B.; Hout, D.R.; Singh, D.K.; Berman, N.E.; Yong, C.; Pinson, D.M.; Foresman, L.; Stephens, E.B. A molecular clone of simian-human immunodeficiency virus (DeltavpuSHIV(KU-1bMC33)) with a truncated, non-membrane-bound vpu results in rapid CD4(+) T cell loss and neuro-AIDS in pig-tailed macaques. Virology 2000, 272, 112–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nepali, K.; Lee, H.Y.; Liou, J.P. Nitro-Group-Containing Drugs. J. Med. Chem. 2019, 62, 2851–2893. [Google Scholar] [CrossRef] [PubMed]
- Rerks-Ngarm, S.; Pitisuttithum, P.; Nitayaphan, S.; Kaewkungwal, J.; Chiu, J.; Paris, R.; Premsri, N.; Namwat, C.; de Souza, M.; Adams, E.; et al. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N. Engl. J. Med. 2009, 361, 2209–2220. [Google Scholar] [CrossRef] [PubMed]
- Haynes, B.F.; Gilbert, P.B.; McElrath, M.J.; Zolla-Pazner, S.; Tomaras, G.D.; Alam, S.M.; Evans, D.T.; Montefiori, D.C.; Karnasuta, C.; Sutthent, R.; et al. Immune-correlates analysis of an HIV-1 vaccine efficacy trial. N. Engl. J. Med. 2012, 366, 1275–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambotte, O.; Ferrari, G.; Moog, C.; Yates, N.L.; Liao, H.X.; Parks, R.J.; Hicks, C.B.; Owzar, K.; Tomaras, G.D.; Montefiori, D.C.; et al. Heterogeneous neutralizing antibody and antibody-dependent cell cytotoxicity responses in HIV-1 elite controllers. AIDS 2009, 23, 897–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambotte, O.; Pollara, J.; Boufassa, F.; Moog, C.; Venet, A.; Haynes, B.F.; Delfraissy, J.F.; Saez-Cirion, A.; Ferrari, G. High antibody-dependent cellular cytotoxicity responses are correlated with strong CD8 T cell viral suppressive activity but not with B57 status in HIV-1 elite controllers. PLoS ONE 2013, 8, e74855. [Google Scholar] [CrossRef] [Green Version]
- Mabuka, J.; Nduati, R.; Odem-Davis, K.; Peterson, D.; Overbaugh, J. HIV-specific antibodies capable of ADCC are common in breastmilk and are associated with reduced risk of transmission in women with high viral loads. PLoS Pathog. 2012, 8, e1002739. [Google Scholar] [CrossRef] [Green Version]
- Pham, T.N.; Lukhele, S.; Hajjar, F.; Routy, J.P.; Cohen, E.A. HIV Nef and Vpu protect HIV-infected CD4+ T cells from antibody-mediated cell lysis through down-modulation of CD4 and BST2. Retrovirology 2014, 11, 15. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Lin, E.C.; Das, B.B.; Tian, Y.; Opella, S.J. Structural determination of virus protein U from HIV-1 by NMR in membrane environments. Biochim. Biophys. Acta 2015, 1848, 3007–3018. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Robinson, C.A.; Lyddon, T.D.; Gil, H.M.; Evans, D.T.; Kuzmichev, Y.V.; Richard, J.; Finzi, A.; Welbourn, S.; Rasmussen, L.; Nebane, N.M.; et al. Novel Compound Inhibitors of HIV-1NL4-3 Vpu. Viruses 2022, 14, 817. https://doi.org/10.3390/v14040817
Robinson CA, Lyddon TD, Gil HM, Evans DT, Kuzmichev YV, Richard J, Finzi A, Welbourn S, Rasmussen L, Nebane NM, et al. Novel Compound Inhibitors of HIV-1NL4-3 Vpu. Viruses. 2022; 14(4):817. https://doi.org/10.3390/v14040817
Chicago/Turabian StyleRobinson, Carolyn A., Terri D. Lyddon, Hwi Min Gil, David T. Evans, Yury V. Kuzmichev, Jonathan Richard, Andrés Finzi, Sarah Welbourn, Lynn Rasmussen, N. Miranda Nebane, and et al. 2022. "Novel Compound Inhibitors of HIV-1NL4-3 Vpu" Viruses 14, no. 4: 817. https://doi.org/10.3390/v14040817