
An Insight into Pathophysiological Features and Therapeutic Advances on Ependymoma

 , ,
, ,  ,
,  ,
,  , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods
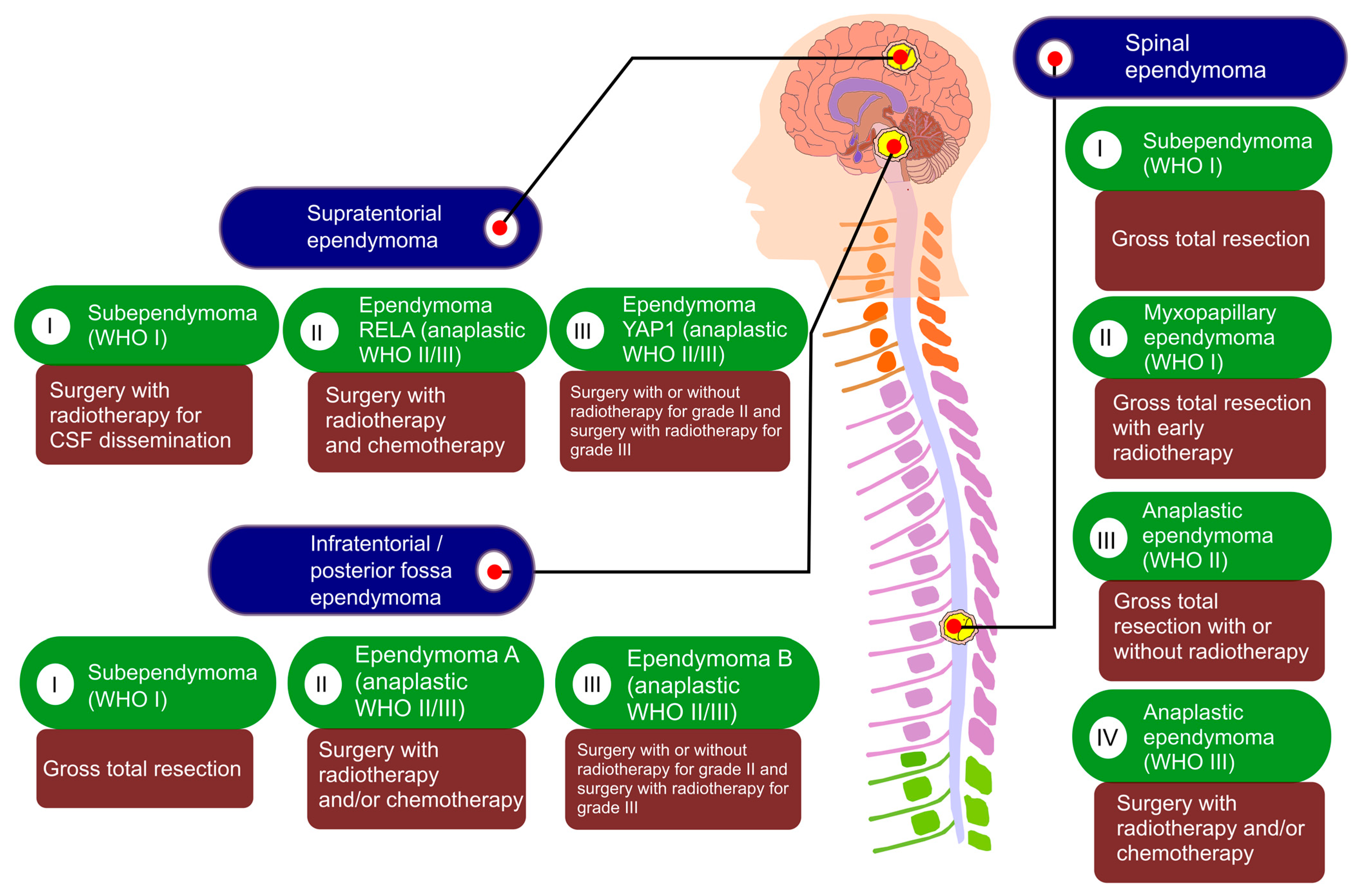
3. Histopathological Classification of Ependymoma
4. A Molecular Classification of Ependymomas Using DNA Methylation Profiling
5. Metabolism of Brain Tumor
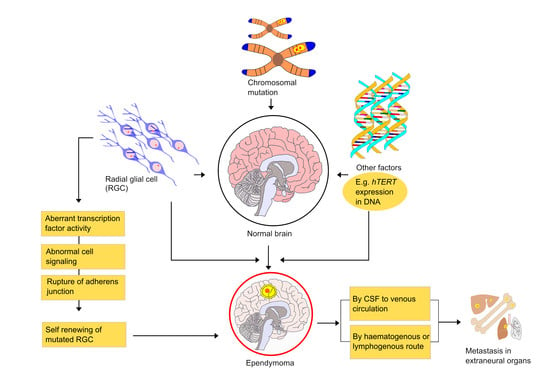
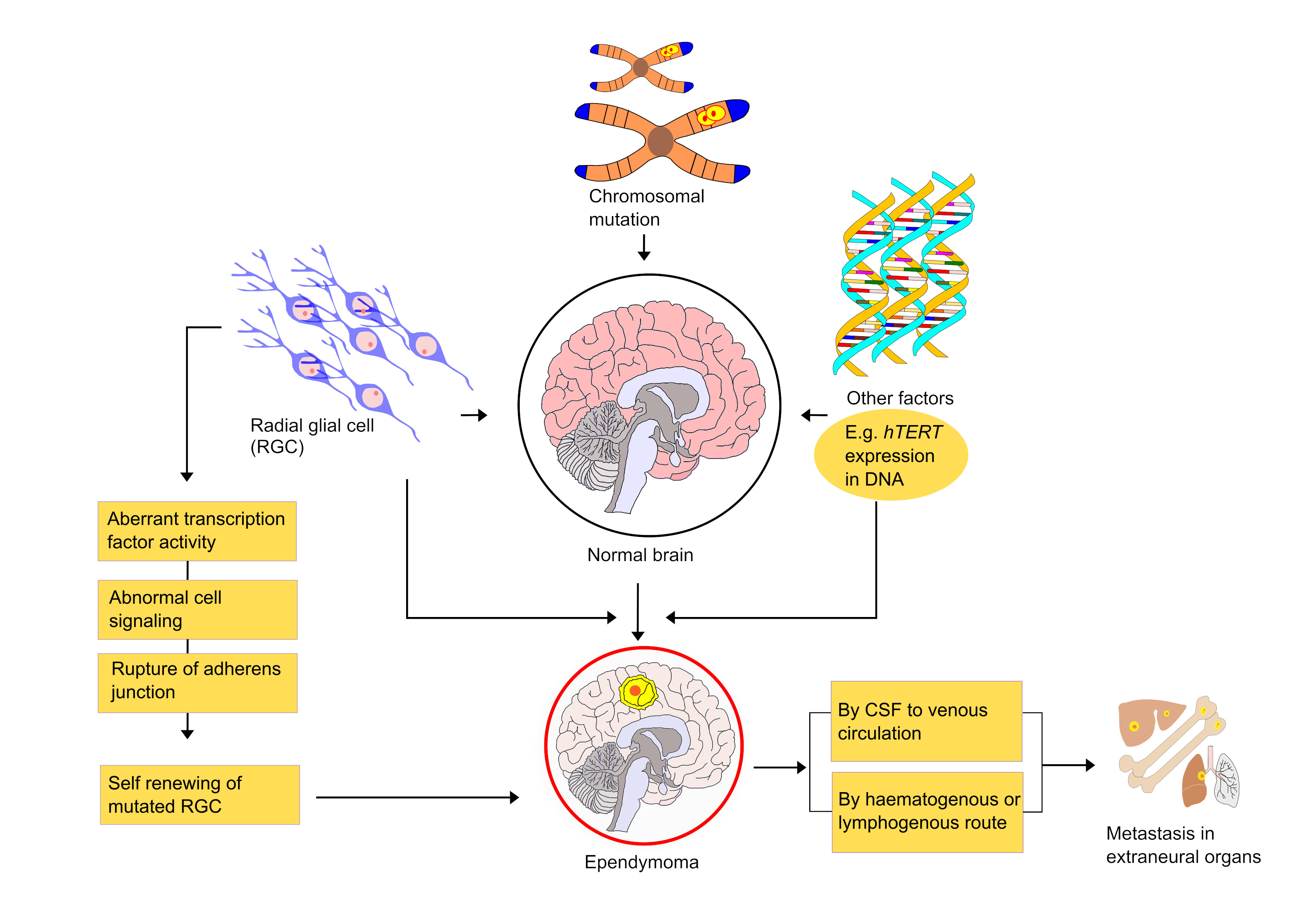
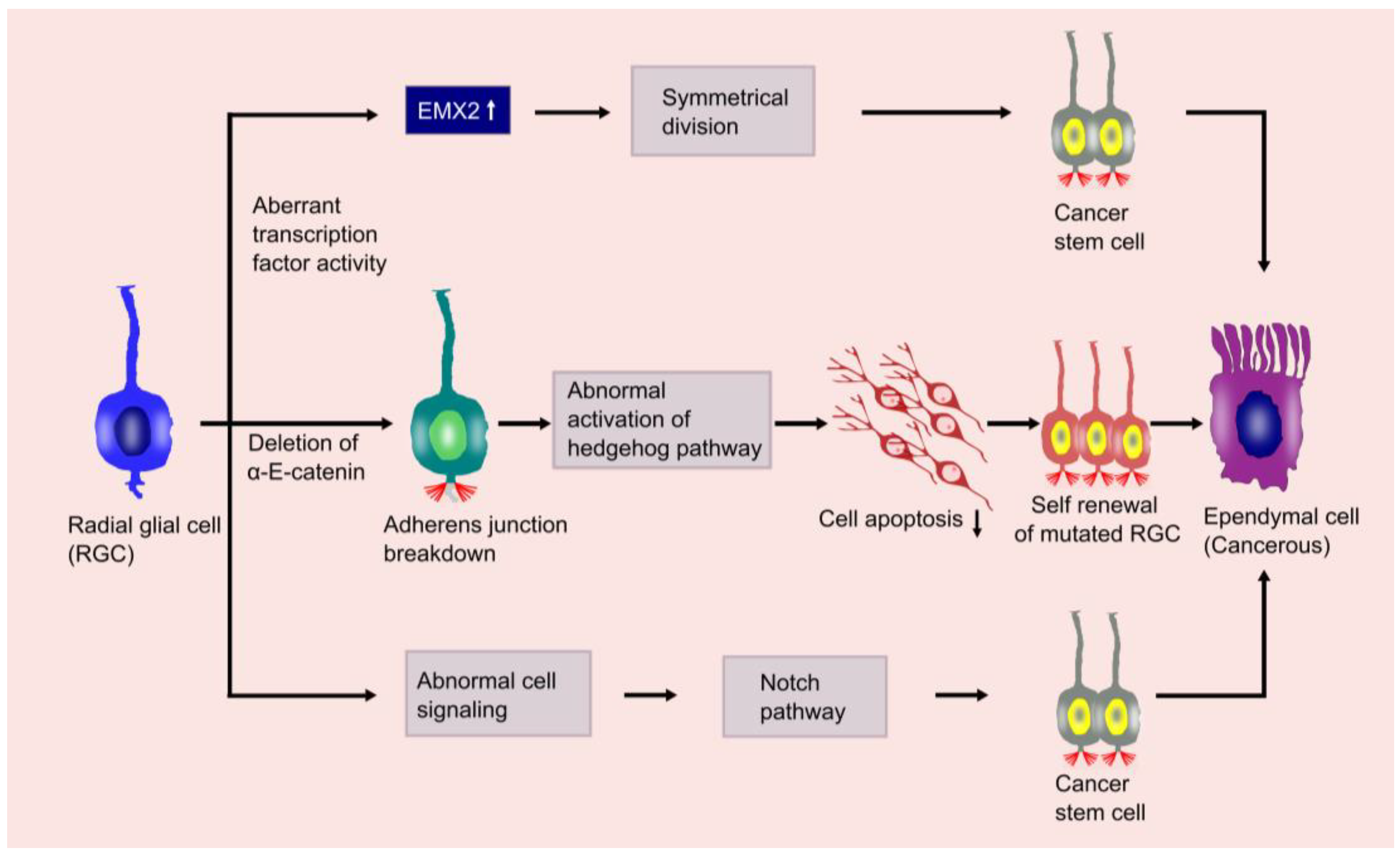
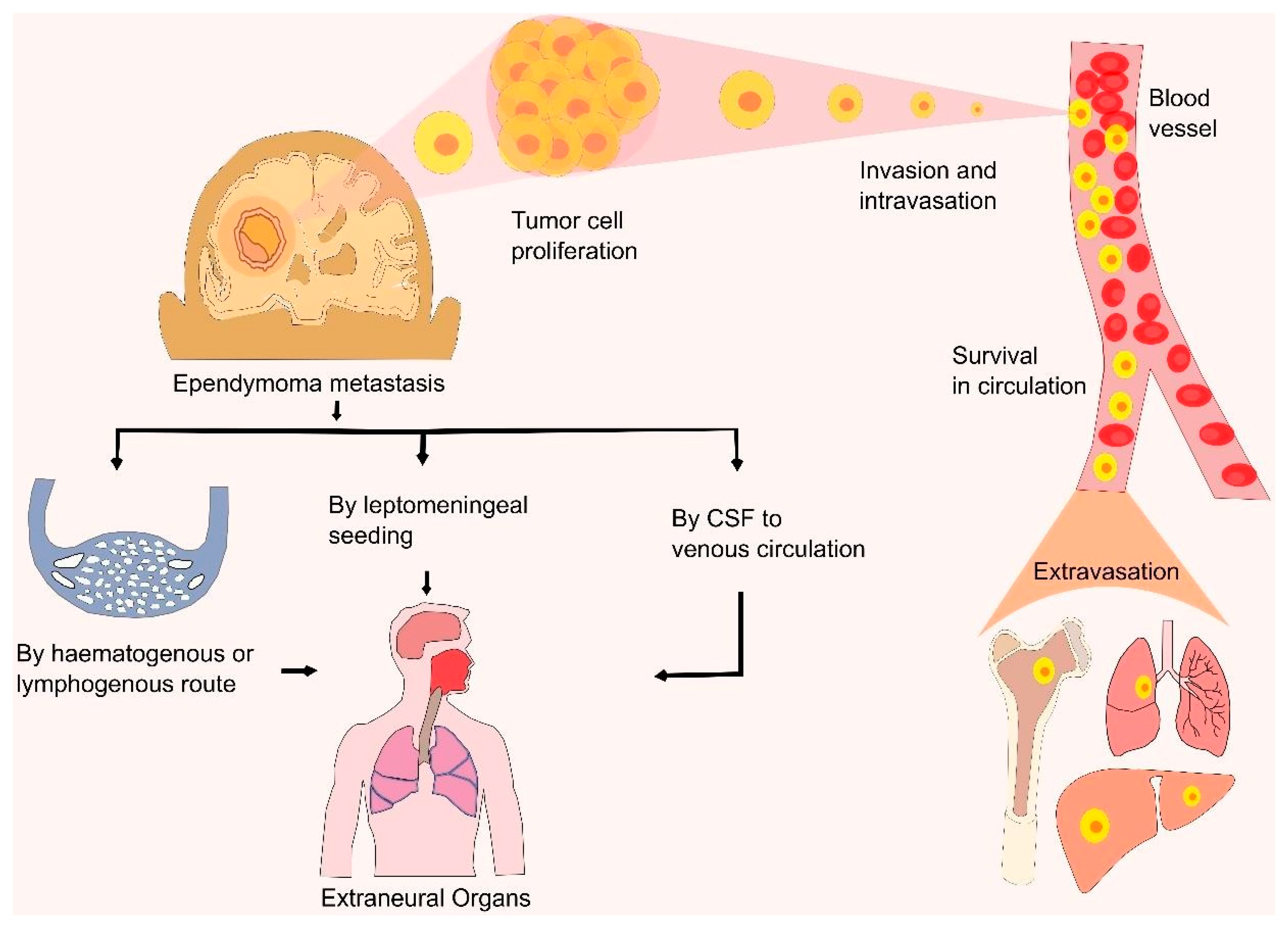
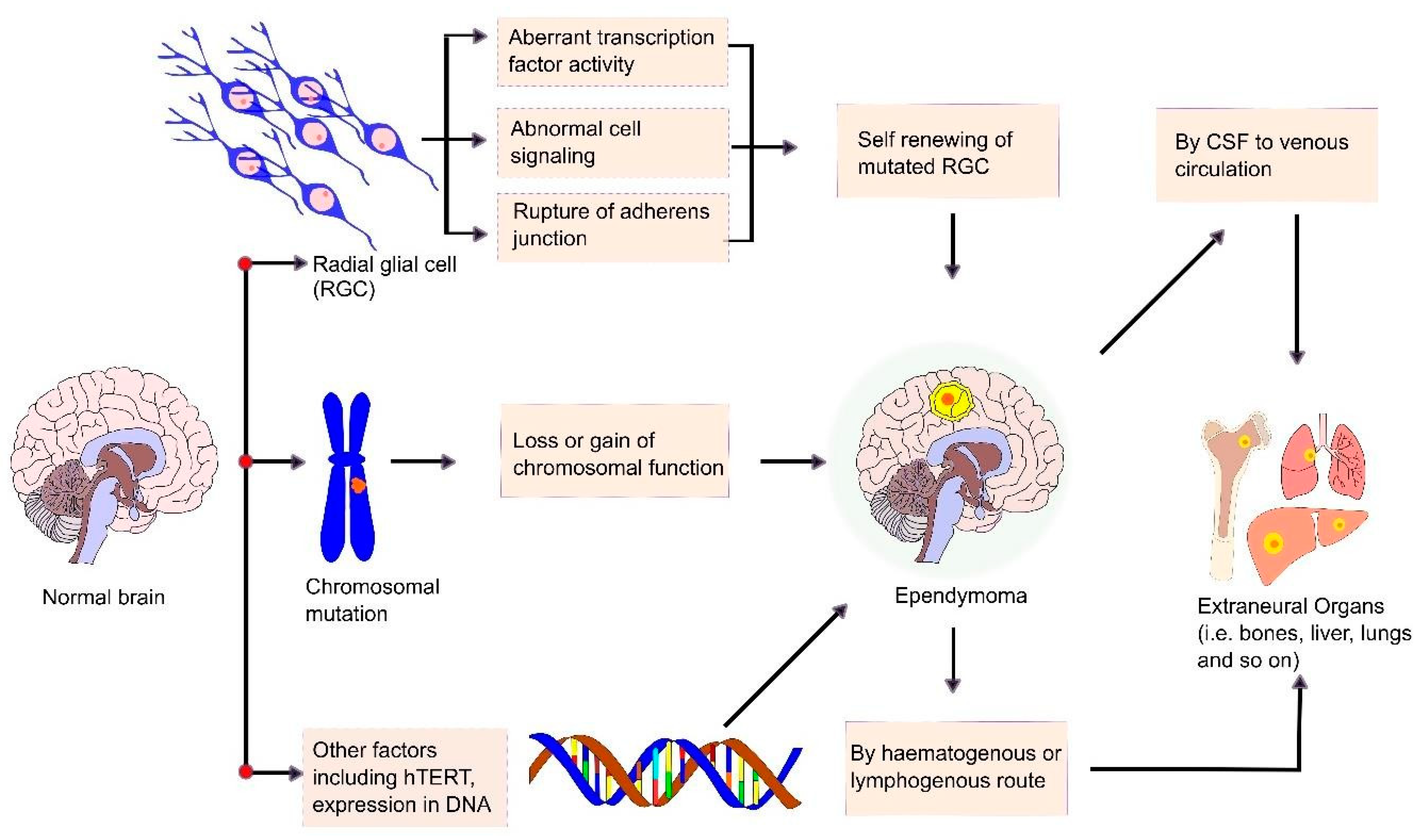
6. Mechanism and Metastasis of Ependymoma
7. Signs and Symptoms
8. Diagnosis
9. Therapeutic Advances
9.1. Surgery
9.2. Radiotherapy
9.3. Chemotherapy
10. Ongoing Trials
11. Limitations, Future Prospective, and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jiménez, A.J.; Domínguez-Pinos, M.-D.; Guerra, M.M.; Fernández-Llebrez, P.; Pérez-Fígares, J.-M. Structure and function of the ependymal barrier and diseases associated with ependyma disruption. Tissue Barriers 2014, 2, e28426. [Google Scholar] [CrossRef]
- Wright, B.L.C.; Lai, J.T.F.; Sinclair, A.J. Cerebrospinal fluid and lumbar puncture: A practical review. J. Neurol. 2012, 259, 1530–1545. [Google Scholar] [CrossRef] [PubMed]
- Worthington, W.C.; Cathcart, R.S.; Cooper, P.; Goldring, I.; Klein, M. Ependymal Cilia: Distribution and Activity in the Adult Human Brain. Science 1963, 139, 221–222. [Google Scholar] [CrossRef]
- Oliver, C.; González, C.A.; Alvial, G.; Flores, C.A.; Rodríguez, E.M.; Bátiz, L.F. Disruption of CDH2/N-Cadherin–Based Adherens Junctions Leads to Apoptosis of Ependymal Cells and Denudation of Brain Ventricular Walls. J. Neuropathol. Exp. Neurol. 2013, 72, 846–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAllister, J.P. Pathophysiology of congenital and neonatal hydrocephalus. Semin. Fetal Neonatal Med. 2012, 17, 285–294. [Google Scholar] [CrossRef]
- Maksoud, Y.A.; Hahn, Y.S.; Engelhard, H. Intracranial ependymoma. Neurosurg. Focus 2002, 13, 1–5. [Google Scholar] [CrossRef]
- Elsamadicy, A.A.; Koo, A.B.; David, W.B.; Lee, V.; Zogg, C.K.; Kundishora, A.J.; Hong, C.S.; DeSpenza, T.; Reeves, B.C.; Kahle, K.T.; et al. Comparison of epidemiology, treatments, and outcomes in pediatric versus adult ependymoma. Neuro Oncol. Adv. 2020, 2, vdaa019. [Google Scholar] [CrossRef] [Green Version]
- Merchant, T.E. Current Clinical Challenges in Childhood Ependymoma: A Focused Review. J. Clin. Oncol. 2017, 35, 2364–2369. [Google Scholar] [CrossRef] [Green Version]
- Neumann, J.E.; Spohn, M.; Obrecht, D.; Mynarek, M.; Thomas, C.; Hasselblatt, M.; Dorostkar, M.M.; Wefers, A.K.; Frank, S.; Monoranu, C.-M.; et al. Molecular characterization of histopathological ependymoma variants. Acta Neuropathol. 2019, 139, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Pajtler, K.W.; Mack, S.C.; Ramaswamy, V.; Smith, C.A.; Witt, H.; Smith, A.; Hansford, J.R.; Von Hoff, K.; Wright, K.D.; Hwang, E.; et al. The current consensus on the clinical management of intracranial ependymoma and its distinct molecular variants. Acta Neuropathol. 2017, 133, 5–12. [Google Scholar] [CrossRef]
- Wu, J.; Armstrong, T.; Gilbert, M.R. Biology and management of ependymomas. Neuro Oncol. 2016, 18, 902–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furie, D.M.; Provenzale, J.M. Supratentorial Ependymomas and Subependymomas. J. Comput. Assist. Tomogr. 1995, 19, 518–526. [Google Scholar] [CrossRef]
- Landau, E.; Boop, F.A.; Conklin, H.M.; Wu, S.; Xiong, X.; Merchant, T.E. Supratentorial Ependymoma: Disease Control, Complications, and Functional Outcomes After Irradiation. Int. J. Radiat. Oncol. 2013, 85, e193–e199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.-Y.; Chang, K.-H.; Yu, I.K.; Kim, K.H.; Kwon, B.J.; Han, M.H.; Kim, I.-O. Intracranial and Spinal Ependymomas: Review of MR Images in 61 Patients. Korean J. Radiol. 2002, 3, 219–228. [Google Scholar] [CrossRef] [Green Version]
- Reni, M.; Gatta, G.; Mazza, E.; Vecht, C. Ependymoma. Crit. Rev. Oncol. Hematol. 2007, 63, 81–89. [Google Scholar] [CrossRef]
- Gerstner, E.R.; Pajtler, K.W. Ependymoma. Seminar. Neurolog. 2018, 38, 104–111. [Google Scholar] [CrossRef]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO Classification of Tumours of the Central Nervous System. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.D.; Poppleton, H.; Fuller, C.; Su, X.; Liu, Y.; Jensen, P.; Magdaleno, S.; Dalton, J.; Calabrese, C.; Board, J. Radial glia cells are candidate stem cells of ependymoma. Cancer Cell 2005, 8, 323–335. [Google Scholar] [CrossRef] [Green Version]
- Rezai, A.R.; Woo, H.H.; Lee, M.; Cohen, H.; Zagzag, D.; Epstein, F.J. Disseminated ependymomas of the central nervous system. J. Neurosurg. 1996, 85, 618–624. [Google Scholar] [CrossRef] [Green Version]
- Brody, A.S.; Frush, D.; Huda, W.; Brent, R.L.; American Academy of Pediatrics Section on Radiology. Radiation Risk to Children From Computed Tomography. Pediatrics 2007, 120, 677–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jabeen, S.; Konar, S.K.; Prasad, C.; Mahadevan, A.; Beniwal, M.; Sadashiva, N.; Santosh, V.; Saini, J. Conventional and Advanced Magnetic Resonance Imaging Features of Supratentorial Extraventricular Ependymomas. J. Comput. Assist. Tomogr. 2020, 44, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Klawinski, D.; Indelicato, D.J.; Hossain, J.; Sandler, E. Surveillance imaging in pediatric ependymoma. Pediatr. Blood Cancer 2020, 67, e28622. [Google Scholar] [CrossRef]
- Gupta, K.; Salunke, P. Understanding Ependymoma Oncogenesis: An Update on Recent Molecular Advances and Current Perspectives. Mol. Neurobiol. 2015, 54, 15–21. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Mehrjardi, M.Z.; Mirzaei, S.; Haghighatkhah, H.R. The many faces of primary cauda equina myxopapillary ependymoma: Clinicoradiological manifestations of two cases and review of the literature. Rom. Neurosurg. 2017, 31, 385–390. [Google Scholar] [CrossRef]
- Sonneland, P.R.L.; Scheithauer, B.W.; Onofrio, B.M. Myxopapillary ependymoma. A clinicopathologic and immunocytochemical study of 77 cases. Cancer 1985, 56, 883–893. [Google Scholar] [CrossRef]
- Ilhan, A.; Furtner, J.; Birner, P.; Roessler, K.; Marosi, C.; Preusser, M. Myxopapillary Ependymoma With Pleuropulmonary Metastases and High Plasma Glial Fibrillary Acidic Protein Levels. J. Clin. Oncol. 2011, 29, e756–e757. [Google Scholar] [CrossRef]
- Eng, L.F.; Ghirnikar, R.S.; Lee, Y.L. Glial Fibrillary Acidic Protein: GFAP-Thirty-One Years (1969–2000). Neurochem. Res. 2000, 25, 1439–1451. [Google Scholar] [CrossRef]
- Orakcioglu, B.; Schramm, P.; Kohlhof, P.; Aschoff, A.; Unterberg, A.; Halatsch, M.-E. Characteristics of thoracolumbar intramedullary subependymomas. J. Neurosurg. Spine 2009, 10, 54–59. [Google Scholar] [CrossRef]
- Godfraind, C. Classification and controversies in pathology of ependymomas. Child’s Nerv. Syst. 2009, 25, 1185–1193. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.B.; Smirniotopoulos, J.G.; Horkanyne-Szakaly, I. From the Radiologic Pathology Archives: Intraventricular Neoplasms: Radiologic-Pathologic Correlation. Radiography 2013, 33, 21–43. [Google Scholar] [CrossRef]
- Louis, D.; Ohgaki, H.; Wiestler, O.; Cavenee, W.; Figarella-Branger, D.; Reifenberger, G.; von Deimling, A. WHO Classification and Grading of Tumours of the Central Nervous System. WHO Classification of Tumours of the Central Nervous System, 4th ed.; Revised; International Agency for Research Centre: Lyon, France, 2016; pp. 12–13. [Google Scholar]
- Ostrom, Q.T.; Gittleman, H.; Fulop, J.; Liu, M.; Blanda, R.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008–2012. Neuro Oncol. 2015, 17, iv1–iv62. [Google Scholar] [CrossRef]
- Schiffer, D.; Giordana, M.T.; Migheli, A.; Palma, L.; Pollo, B.; Soffietti, R.; Tribolo, A. Histologic prognostic factors in ependymoma. Child’s Nerv. Syst. 1991, 7, 177–182. [Google Scholar] [CrossRef]
- Ellison, D.; Love, S.; Chimelli, L.M.C.; Harding, B.; Lowe, J.S.; Vinters, H.V.; Brandner, S.; Yong, W.H. Neuropathology E-Book: A Reference Text of CNS Pathology; Elsevier Health Sciences: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Fouladi, M.; Helton, K.; Dalton, J.; Gilger, E.; Gajjar, A.; Merchant, T.; Kun, L.; Newsham, I.; Burger, P.; Fuller, C. Clear cell ependymoma: A clinicopathologic and radiographic analysis of 10 patients. Cancer 2003, 98, 2232–2244. [Google Scholar] [CrossRef]
- Figarella-Branger, D.; Lechapt-Zalcman, E.; Tabouret, E.; Jünger, S.; de Paula, A.M.; Bouvier, C.; Colin, C.; Jouvet, A.; Forest, F.; Andreiuolo, F. Supratentorial clear cell ependymomas with branching capillaries demonstrate characteristic clinicopathological features and pathological activation of nuclear factor-kappaB signaling. Neuro Oncol. 2016, 18, 919–927. [Google Scholar] [CrossRef] [Green Version]
- Mobley, B.; Kalani, M.-Y.S.; Harsh IV, G.R.; Edwards, M.S.; Vogel, H. Papillary tumor of the spinal cord: Report of 2 cases. Am. J. Surg. Pathol. 2009, 33, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, X.; Zhang, Z.; Chen, Y. Tanycytic ependymoma arising from the right lateral ventricle: A case report and review of the literature. Neuropathology 2008, 28, 427–432. [Google Scholar] [CrossRef]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; International Agency for Research on Cancer. WHO Classification of Tumours of the Central Nervous System; International Agency for Research on Cancer: Lyon, France, 2016. [Google Scholar]
- Pajtler, K.W.; Witt, H.; Sill, M.; Jones, D.T.W.; Hovestadt, V.; Kratochwil, F.; Wani, K.; Tatevossian, R.; Punchihewa, C.; Johann, P.; et al. Molecular Classification of Ependymal Tumors across All CNS Compartments, Histopathological Grades, and Age Groups. Cancer Cell 2015, 27, 728–743. [Google Scholar] [CrossRef] [Green Version]
- Parker, M.; Mohankumar, K.M.; Punchihewa, C.; Weinlich, R.; Dalton, J.D.; Li, Y.; Lee, R.; Tatevossian, R.G.; Phoenix, T.N.; Thiruvenkatam, R. C11orf95–RELA fusions drive oncogenic NF-κB signalling in ependymoma. Nature 2014, 506, 451–455. [Google Scholar] [CrossRef] [Green Version]
- Elsharkawy, A.E.; Abuamona, R.; Bergmann, M.; Salem, S.; Gafumbegete, E.; Röttger, E. Cortical Anaplastic Ependymoma with Significant Desmoplasia: A Case Report and Literature Review. Case Rep. Oncol. Med. 2013, 2013, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Jünger, S.T.; Andreiuolo, F.; Mynarek, M.; Dörner, E.; Mühlen, A.Z.; Rutkowski, S.; Von Bueren, A.O.; Pietsch, T. Ependymomas in infancy: Underlying genetic alterations, histological features, and clinical outcome. Child’s Nerv. Syst. 2020, 36, 2693–2700. [Google Scholar] [CrossRef]
- Mørk, S.J.; Rubinstein, L.J. Ependymoblastoma. A reappraisal of a rare embryonal tumor. Cancer 1985, 55, 1536–1542. [Google Scholar] [CrossRef]
- Foundation, C. Ependymoma Statistics. Available online: https://www.cern-foundation.org/education/ependymoma-basics/ependymoma-statistics (accessed on 3 June 2021).
- Hovestadt, V.; Jones, D.T.W.; Picelli, S.; Wang, W.; Kool, M.; Northcott, P.A.; Sultan, M.; Stachurski, K.; Ryzhova, M.; Warnatz, H.-J.; et al. Decoding the regulatory landscape of medulloblastoma using DNA methylation sequencing. Nat. Cell Biol. 2014, 510, 537–541. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.A.; Wright, K.D.; Poppleton, H.; Mohankumar, K.M.; Finkelstein, D.; Pounds, S.B.; Rand, V.; Leary, S.E.S.; White, E.; Eden, C.; et al. Cross-species genomics matches driver mutations and cell compartments to model ependymoma. Nat. Cell Biol. 2010, 466, 632–636. [Google Scholar] [CrossRef] [PubMed]
- Witt, H.; Mack, S.C.; Ryzhova, M.; Bender, S.; Sill, M.; Isserlin, R.; Benner, A.; Hielscher, T.; Milde, T.; Remke, M.; et al. Delineation of Two Clinically and Molecularly Distinct Subgroups of Posterior Fossa Ependymoma. Cancer Cell 2011, 20, 143–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wani, K.; For the Collaborative Ependymoma Research Network; Armstrong, T.; Vera-Bolanos, E.; Raghunathan, A.; Ellison, D.; Gilbertson, R.; Vaillant, B.; Goldman, S.; Packer, R.J.; et al. A prognostic gene expression signature in infratentorial ependymoma. Acta Neuropathol. 2012, 123, 727–738. [Google Scholar] [CrossRef] [Green Version]
- Ellison, D.W.; Aldape, K.D.; Capper, D.; Fouladi, M.; Gilbert, M.R.; Gilbertson, R.J.; Hawkins, C.; Merchant, T.E.; Pajtler, K.W.; Venneti, S.; et al. cIMPACT-NOW update 7: Advancing the molecular classification of ependymal tumors. Brain Pathol. 2020, 30, 863–866. [Google Scholar] [CrossRef]
- Witt, H.; Gramatzki, D.; Hentschel, B.; Pajtler, K.W.; Felsberg, J.; Schackert, G.; Löffler, M.; Capper, D.; Sahm, F.; Sill, M.; et al. DNA methylation-based classification of ependymomas in adulthood: Implications for diagnosis and treatment. Neuro Oncol. 2018, 20, 1616–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, C.; Thierfelder, F.; Träger, M.; Soschinski, P.; Müther, M.; Edelmann, D.; Förster, A.; Geiler, C.; Kim, H.-Y.; Filipski, K.; et al. TERT promoter mutation and chromosome 6 loss define a high-risk subtype of ependymoma evolving from posterior fossa subependymoma. Acta Neuropathol. 2021, 141, 959–970. [Google Scholar] [CrossRef]
- Ellison, D.W.; Kocak, M.; Figarella-Branger, D.; Felice, G.; Catherine, G.; Pietsch, T.; Frappaz, D.; Massimino, M.; Grill, J.; Boyett, J.M.; et al. Histopathological grading of pediatric ependymoma: Reproducibility and clinical relevance in European trial cohorts. J. Negat. Results Biomed. 2011, 10, 7. [Google Scholar] [CrossRef] [PubMed]
- Griesinger, A.M.; Witt, D.A.; Grob, S.T.; Georgio Westover, S.R.; Donson, A.M.; Sanford, B.; Mulcahy Levy, J.M.; Wong, R.; Moreira, D.C.; DeSisto, J.A. NF-κB upregulation through epigenetic silencing of LDOC1 drives tumor biology and specific immunophenotype in Group A ependymoma. Neuro Oncol. 2017, 19, 1350–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Sun, T.; Kobayashi, K.; Gao, P.; Griffin, J.D. Identification of a Family of Mastermind-Like Transcriptional Coactivators for Mammalian Notch Receptors. Mol. Cell. Biol. 2002, 22, 7688–7700. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Fong, K.-W.; Tang, M.; Han, X.; Gong, Z.; Ma, W.; Hebert, M.; Songyang, Z.; Chen, J. Fam118B, a newly identified component of Cajal bodies, is required for Cajal body formation, snRNP biogenesis and cell viability. J. Cell Sci. 2014, 127, 2029–2039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLendon, R.E.; Halperin, E.C. Is the long-term survival of patients with intracranial glioblastoma multiforme overstated? Cancer: Interdiscipl. Int. J. Am. Cancer Soc. 2003, 98, 1745–1748. [Google Scholar]
- Greene, A.E.; Todorova, M.T.; Seyfried, T.N. Perspectives on the metabolic management of epilepsy through dietary reduction of glucose and elevation of ketone bodies. J. Neurochem. 2003, 86, 529–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donald, D.; Clarke, L.S. Basic Neurochemistry, 6th ed.; Siegel, G.J., Agranoff, B.W., Albers, R.W., Fisher, S.K., Uhler, M.D., Eds.; Lippincott-Raven: Philadelphia, PA, USA, 1999; pp. 645–680. [Google Scholar]
- Mantis, J.G.; Centeno, N.A.; Todorova, M.T.; McGowan, R.; Seyfried, T.N. Management of multifactorial idiopathic epilepsy in EL mice with caloric restriction and the ketogenic diet: Role of glucose and ketone bodies. Nutr. Metab. 2004, 1, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, A.A.M. Cerebral ketone body metabolism. J. Inherit. Metab. Dis. 2005, 28, 109–121. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Sanderson, T.M.; El-Abbadi, M.M.; McGowan, R.; Mukherjee, P. Role of glucose and ketone bodies in the metabolic control of experimental brain cancer. Br. J. Cancer 2003, 89, 1375–1382. [Google Scholar] [CrossRef] [Green Version]
- Tisdale, M.J. Biology of Cachexia. J. Natl. Cancer Inst. 1997, 89, 1763–1773. [Google Scholar] [CrossRef]
- Kashiwaya, Y.; Takeshima, T.; Mori, N.; Nakashima, K.; Clarke, K.; Veech, R.L. d-β-Hydroxybutyrate protects neurons in models of Alzheimer’s and Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 5440–5444. [Google Scholar] [CrossRef] [Green Version]
- Stafstrom, C.E.; Bough, K.J. The Ketogenic Diet for the Treatment of Epilepsy: A Challenge for Nutritional Neuroscientists. Nutr. Neurosci. 2003, 6, 67–79. [Google Scholar] [CrossRef]
- Mukherjee, P.; Abate, L.E.; Seyfried, T.N. Antiangiogenic and Proapoptotic Effects of Dietary Restriction on Experimental Mouse and Human Brain Tumors. Clin. Cancer Res. 2004, 10, 5622–5629. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, P.; El-Abbadi, M.M.; Kasperzyk, J.L.; Ranes, M.K.; Seyfried, T.N. Dietary restriction reduces angiogenesis and growth in an orthotopic mouse brain tumour model. Br. J. Cancer 2002, 86, 1615–1621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, H.Y.; Kim, H.J.; Kim, K.W.; Choi, J.S.; Yu, B.P. Molecular inflammation hypothesis of aging based on the anti-aging mechanism of calorie restriction. Microsc. Res. Tech. 2002, 59, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias, D.J.; Rosen, B.R.; Lev, M.H. Dynamic Magnetic Resonance Perfusion Imaging of Brain Tumors. Oncology 2004, 9, 528–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barry, D.S.; Pakan, J.M.; McDermott, K.W. Radial glial cells: Key organisers in CNS development. Int. J. Biochem. Cell Biol. 2014, 46, 76–79. [Google Scholar] [CrossRef]
- Poppleton, H.; Gilbertson, R.J. Stem cells of ependymoma. Br. J. Cancer 2006, 96, 6–10. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.J.; Jillette, N.; Li, X.-N.; Cheng, A.W.; Lau, C.C. Correction to: C11orf95-RELA reprograms 3D epigenome in supratentorial ependymoma. Acta Neuropathol. 2020, 140, 961–962. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, S.; Liu, S.-T. Cell division symmetry control and cancer stem cells. AIMS Mol. Sci. 2020, 7, 82–101. [Google Scholar] [CrossRef]
- Galli, R.; Fiocco, R.; De Filippis, L.; Muzio, L.; Gritti, A.; Mercurio, S.; Broccoli, V.; Pellegrini, M.; Mallamaci, A.; Vescovi, A.L. Emx2regulates the proliferation of stem cells of the adult mammalian central nervous system. Developement 2002, 129, 1633–1644. [Google Scholar] [CrossRef]
- Götz, M.; Huttner, W.B. The cell biology of neurogenesis. Nat. Rev. Mol. Cell Biol. 2005, 6, 777–788. [Google Scholar] [CrossRef]
- Cavallaro, U.; Christofori, G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat. Rev. Cancer 2004, 4, 118–132. [Google Scholar] [CrossRef] [PubMed]
- Lien, W.-H.; Klezovitch, O.; Fernandez, T.E.; Delrow, J.; Vasioukhin, V. αE-catenin controls cerebral cortical size by regulating the hedgehog signaling pathway. Science 2006, 311, 1609–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Liu, Y.; Gao, R.; Yu, H.; Sun, T. HDAC6 inhibition induces glioma stem cells differentiation and enhances cellular radiation sensitivity through the SHH/Gli1 signaling pathway. Cancer Lett. 2018, 415, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Mack, S.C.; Taylor, M.D. Molecular genetics of ependymoma. Chin. J. Cancer 2011, 30, 669–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, S.J. Notch signalling: A simple pathway becomes complex. Nat. Rev. Mol. Cell Biol. 2006, 7, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Palm, T.; Figarella-Branger, D.; Chapon, F.; Lacroix, C.; Scaravilli, F.; Ellison, D.W.; Salmon, I.; Vikkula, M.; Godfraind, C. Expression profiling of ependymomas unravels localization and tumor grade-specific tumorigenesis. Cancer 2009, 115, 3955–3968. [Google Scholar] [CrossRef]
- Magalhães, T.D.A.; Cruzeiro, G.A.V.; Sousa, G.; Da Silva, K.R.; Lira, R.C.P.; Scrideli, C.A.; Tone, L.G.; Valera, E.T.; Borges, K.S. Notch pathway in ependymoma RELA-fused subgroup: Upregulation and association with cancer stem cells markers expression. Cancer Gene Ther. 2020, 27, 509–512. [Google Scholar] [CrossRef] [Green Version]
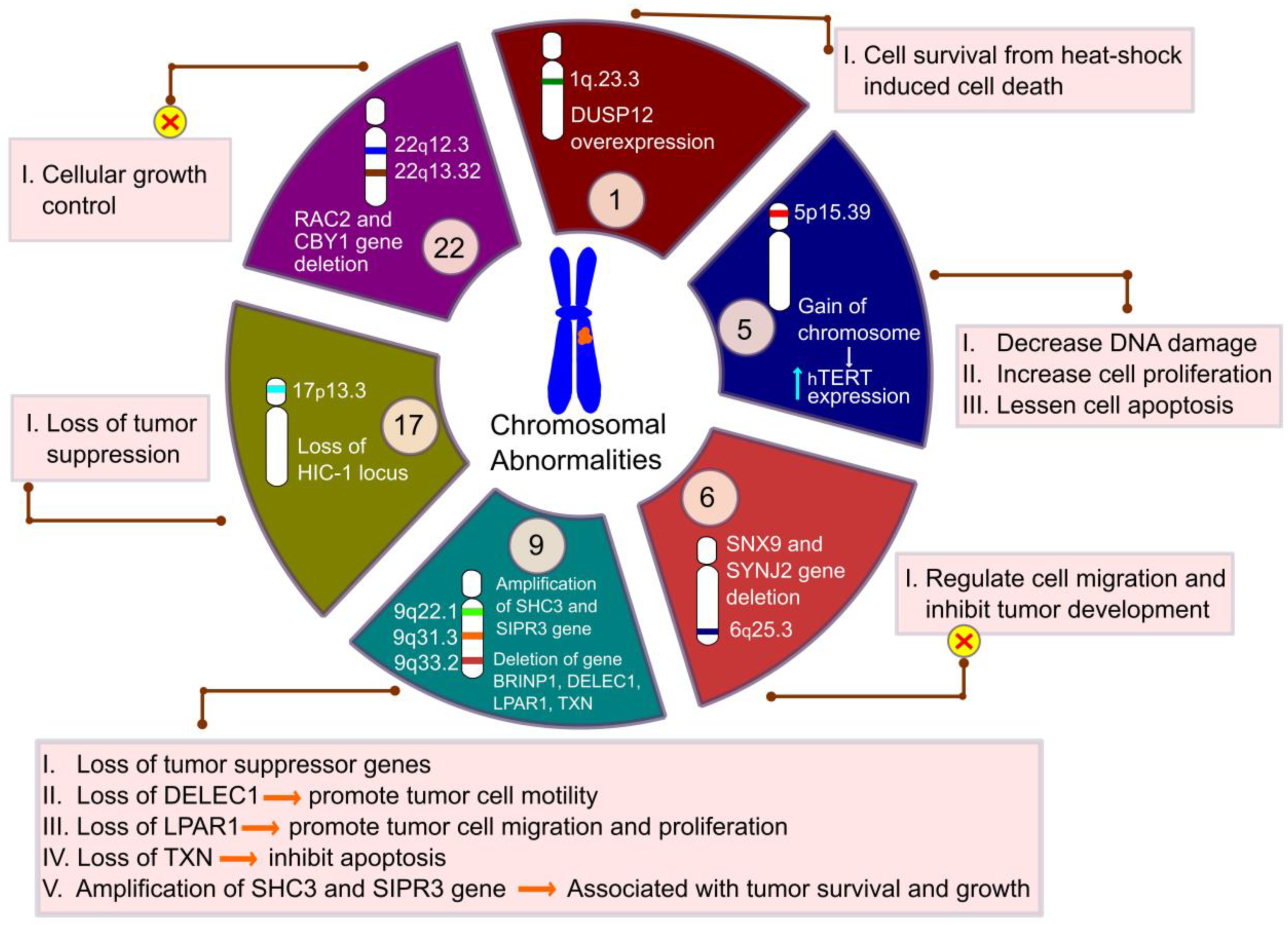
- Karakoula, K.; Suarez-Merino, B.; Ward, S.; Phipps, K.P.; Harkness, W.; Hayward, R.; Thompson, D.; Jacques, T.S.; Harding, B.; Beck, J. Real-time quantitative PCR analysis of pediatric ependymomas identifies novel candidate genes including TPR at 1q25 and CHIBBY at 22q12-qGenes. Chromos. Cancer 2008, 47, 1005–1022. [Google Scholar] [CrossRef]
- Rand, V.; on behalf of the Children’s Cancer and Leukaemia Group Biological Studies Committee; Prebble, E.; Ridley, L.; Howard, M.; Wei, W.; Brundler, M.-A.; Fee, B.E.; Riggins, G.J.; Coyle, B.; et al. Investigation of chromosome 1q reveals differential expression of members of the S100 family in clinical subgroups of intracranial paediatric ependymoma. Br. J. Cancer 2008, 99, 1136–1143. [Google Scholar] [CrossRef] [Green Version]
- Mendrzyk, F.; Korshunov, A.; Benner, A.; Toedt, G.; Pfister, S.; Radlwimmer, B.; Lichter, P. Identification of Gains on 1q and Epidermal Growth Factor Receptor Overexpression as Independent Prognostic Markers in Intracranial Ependymoma. Clin. Cancer Res. 2006, 12, 2070–2079. [Google Scholar] [CrossRef] [Green Version]
- Sharda, P.R.; Bonham, C.A.; Mucaki, E.J.; Butt, Z.; Vacratsis, P.O. The dual-specificity phosphatase hYVH1 interacts with Hsp70 and prevents heat-shock-induced cell death. Biochem. J. 2009, 418, 391–401. [Google Scholar] [CrossRef] [Green Version]
- Monoranu, C.-M.; Huang, B.; Zangen, I.L.-V.; Rutkowski, S.; Vince, G.H.; Gerber, N.U.; Puppe, B.; Roggendorf, W. Correlation between 6q25.3 deletion status and survival in pediatric intracranial ependymomas. Cancer Genet. Cytogenet. 2008, 182, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Tabori, U.; Ma, J.; Carter, M.; Zielenska, M.; Rutka, J.; Bouffet, E.; Bartels, U.; Malkin, D.; Hawkins, C. Human Telomere Reverse Transcriptase Expression Predicts Progression and Survival in Pediatric Intracranial Ependymoma. J. Clin. Oncol. 2006, 24, 1522–1528. [Google Scholar] [CrossRef]
- Ridley, L.; Rahman, R.; Brundler, M.-A.; Ellison, D.; Lowe, J.; Robson, K.; Prebble, E.; Luckett, I.; Gilbertson, R.J.; Parkes, S.; et al. Multifactorial analysis of predictors of outcome in pediatric intracranial ependymoma. Neuro Oncol. 2008, 10, 675–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waha, A.; Koch, A.; Hartmann, W.; Mack, H.; Schramm, J.; Sörensen, N.; Berthold, F.; Wiestler, O.D.; Pietsch, T.; Waha, A. Analysis ofHIC-1 methylation and transcription in human ependymomas. Int. J. Cancer 2004, 110, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Schneider, D.; Monoranu, C.-M.; Huang, B.; Rutkowski, S.; Gerber, N.U.; Krauss, J.; Puppe, B.; Roggendorf, W. Pediatric supratentorial ependymomas show more frequent deletions on chromosome 9 than infratentorial ependymomas: A microsatellite analysis. Cancer Genet. Cytogenet. 2009, 191, 90–96. [Google Scholar] [CrossRef]
- Magrassi, L.; Marziliano, N.; Inzani, F.; Cassini, P.; Chiaranda, I.; Skrap, M.; Pizzolito, S.; Arienta, C.; Arbustini, E. EDG3 and SHC3 on chromosome 9q22 are co-amplified in human ependymomas. Cancer Lett. 2010, 290, 36–42. [Google Scholar] [CrossRef]
- Magrassi, L.; Conti, L.; Lanterna, A.; Zuccato, C.; Marchionni, M.; Cassini, P.; Arienta, C.; Cattaneo, E. Shc3 Affects Human High-Grade Astrocytomas Survival. Oncogene 2005, 24, 5198–5206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.-H.; Chung, C.K.; Ohn, J.H.; Kim, C.H. The Similarities and Differences between Intracranial and Spinal Ependymomas: A Review from a Genetic Research Perspective. J. Korean Neurosurg. Soc. 2016, 59, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Barton, V.N.; Donson, A.M.; Kleinschmidt-DeMasters, B.K.; Birks, D.K.; Handler, M.H.; Foreman, N.K. Unique Molecular Characteristics of Pediatric Myxopapillary Ependymoma. Brain Pathol. 2010, 20, 560–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, S.; Gu, W.; Shou, J.; Xiong, J.; Liu, X.; Sun, B.; Yang, D.; Xie, R. The Molecular Feature of HOX Gene Family in the Intramedullary Spinal Tumors. Spine 2017, 42, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Paço, A.; Freitas, R. HOXgenes as transcriptional and epigenetic regulators during tumorigenesis and their value as therapeutic targets. Epigenomics 2019, 11, 1539–1552. [Google Scholar] [CrossRef] [PubMed]
- Moreno, L.; Popov, S.; Jury, A.; Al Sarraj, S.; Jones, C.; Zacharoulis, S. Role of platelet derived growth factor receptor (PDGFR) over-expression and angiogenesis in ependymoma. J. Neuro Oncol. 2012, 111, 169–176. [Google Scholar] [CrossRef]
- Suarez-Merino, B.; Hubank, M.; Revesz, T.; Harkness, W.; Hayward, R.; Thompson, D.; Darling, J.L.; Thomas, D.G.; Warr, T.J. Microarray analysis of pediatric ependymoma identifies a cluster of 112 candidate genes including four transcripts at 22q12.1-q13.3. Neuro Oncol. 2005, 7, 20–31. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Bergami, P.; Barbero, G. The emerging role of Wnt5a in the promotion of a pro-inflammatory and immunosuppressive tumor microenvironment. Cancer Metastasis Rev. 2020, 39, 933–952. [Google Scholar] [CrossRef]
- Hung, C.-S.; Huang, C.-Y.; Lee, C.-H.; Chen, W.-Y.; Huang, M.-T.; Wei, P.-L.; Chang, Y.-J. IGFBP2 plays an important role in heat shock protein 27-mediated cancer progression and metastasis. Oncotarget 2017, 8, 54978–54992. [Google Scholar] [CrossRef] [Green Version]
- Almeida, A.; Zhu, X.X.; Vogt, N.; Tyagi, R.; Muleris, M.; Dutrillaux, A.-M.; Dutrillaux, B.; Ross, D.; Malfoy, B.; Hanash, S. GAC1, a new member of the leucine-rich repeat superfamily on chromosome band 1q32.1, is amplified and overexpressed in malignant gliomas. Oncogene 1998, 16, 2997–3002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vastrad, B.; Vastrad, C.; Godavarthi, A.; Chandrashekar, R. Molecular mechanisms underlying gliomas and glioblastoma pathogenesis revealed by bioinformatics analysis of microarray data. Med. Oncol. 2017, 34, 182. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Chen, K.; Wu, W.; Zhou, Y.; Zhu, J.; Wu, G.; Cao, L.; Zhang, X.; Guan, H.; Yang, Y.; et al. Overexpression of HOXC10 promotes angiogenesis in human glioma via interaction with PRMT5 and upregulation of VEGFA expression. Theranostics 2018, 8, 5143–5158. [Google Scholar] [CrossRef]
- Zhou, Y.; Shu, C.; Huang, Y. Fibronectin promotes cervical cancer tumorigenesis through activating FAK signaling pathway. J. Cell. Biochem. 2019, 120, 10988–10997. [Google Scholar] [CrossRef] [PubMed]
- Aruga, J.; Millen, K.J. ZIC1 Function in Normal Cerebellar Development and Human Developmental Pathology. Adv. Exp. Med. Biol. 2018, 1046, 249–268. [Google Scholar] [CrossRef]
- Yurt, A.; Selçuki, M.; Ertürk, A.R.; Küpelioglu, A. Large Supratentorial Cortical Ependymoma in a Child. Clin. Med. Res. 2010, 8, 25–27. [Google Scholar] [CrossRef] [Green Version]
- Weiss, L. A metastasizing ependymoma of the cauda equina. Cancer 1955, 8, 161–171. [Google Scholar] [CrossRef]
- Wight, D.G.D.; Holley, K.J.; Finbow, J.A.H.; Griffiths, D.F.; Jasani, B.; Newman, G.R.; Williams, E.D. Metastasizing ependymoma of the cauda equina. J. Clin. Pathol. 1973, 26, 929–935. [Google Scholar] [CrossRef] [Green Version]
- Perrin, E.V. Extracranial Metastases from Intracranial Gliomata. Report of Two Cases in Children. Am. J. Clin. Pathol. 1958, 30, 244–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouffet, E.; Hawkins, C.E.; Ballourah, W.; Taylor, M.; Bartels, U.K.; Schoenhoff, N.; Tsangaris, E.; Huang, A.; Kulkarni, A.; Mabbot, D.J.; et al. Survival Benefit for Pediatric Patients With Recurrent Ependymoma Treated With Reirradiation. Int. J. Radiat. Oncol. 2012, 83, 1541–1548. [Google Scholar] [CrossRef] [PubMed]
- Zacharoulis, S.; Ji, L.; Pollack, I.F.; Duffner, P.; Geyer, R.; Grill, J.; Schild, S.; Jaing, T.-H.; Massimino, M.; Finlay, J.; et al. Metastatic ependymoma: A multi-institutional retrospective analysis of prognostic factors. Pediatr. Blood Cancer 2008, 50, 231–235. [Google Scholar] [CrossRef]
- Umbach, G.; El Ahmadieh, T.Y.; Plitt, A.R.; Aoun, S.G.; Neeley, O.J.; Lyon, K.A.; Fonkem, E.; Raisanen, J.M.; Bishop, J.A.; Wardak, Z.; et al. Extraneural metastatic anaplastic ependymoma: A systematic review and a report of metastases to bilateral parotid glands. Neuro Oncol. Pract. 2019, 7, 218–227. [Google Scholar] [CrossRef]
- Zhu, F.; Ding, J.; Li, Y.; Mao, D.; He, X.; Chen, W.; Lou, L.; Ding, Z. Benign ependymoma with extensive intracranial and spinal cerebrospinal fluid dissemination: Case report and literature review. Br. J. Neurosurg. 2017, 33, 290–293. [Google Scholar] [CrossRef]
- Diaz-Aguilar, D.; Terterov, S.; Tucker, A.M.; Sedighim, S.; Scharnweber, R.; Wang, S.; Merna, C.; Rahman, S. Simultaneous cerebrospinal fluid and hematologic metastases in a high-grade ependymoma. Surg. Neurol. Int. 2018, 9, 93. [Google Scholar] [CrossRef]
- Wang, M.; Wang, H.; Zhou, Y.; Zhan, R.; Wan, S. Myxopapillary Ependymoma in the Third Ventricle Area and Sacral Canal: Dropped or Retrograde Metastasis?—Case Report. Neurol. Med. Chir. 2013, 53, 237–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benesch, M.; Mynarek, M.; Witt, H.; Warmuth-Metz, M.; Pietsch, T.; Bison, B.; Pfister, S.M.; Pajtler, K.W.; Kool, M.; Schüller, U.; et al. Newly Diagnosed Metastatic Intracranial Ependymoma in Children: Frequency, Molecular Characteristics, Treatment, and Outcome in the Prospective HIT Series. Oncology 2019, 24, e921–e929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborti, S.; Govindan, A.; Alapatt, J.P.; Radhakrishnan, M.; Santosh, V. Primary myxopapillary ependymoma of the fourth ventricle with cartilaginous metaplasia: A case report and review of the literature. Brain Tumor Pathol. 2012, 29, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Ngo, T.P.; Dufton, J.; Stern, P.J.; Islam, O. Myxopapillary ependymoma as a cause of back pain in a young male—A case report. J. Can. Chiropr. Assoc. 2013, 57, 150–155. [Google Scholar] [PubMed]
- Bagley, C.A.; Kothbauer, K.F.; Wilson, S.; Bookland, M.J.; Epstein, F.J.; Jallo, G.I. Resection of myxopapillary ependymomas in children. J. Neurosurg. Pediatr. 2007, 106, 261–267. [Google Scholar] [CrossRef] [Green Version]
- Sayegh, E.T.; Aranda, D.; Kim, J.M.; Oh, T.; Parsa, A.T.; Oh, M.C. Prognosis by tumor location in adults with intracranial ependymomas. J. Clin. Neurosci. 2014, 21, 2096–2101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toescu, S.M.; Aquilina, K. Current and Emerging Methods of Management of Ependymoma. Curr. Oncol. Rep. 2019, 21, 78. [Google Scholar] [CrossRef]
- Roncaroli, F.; Consales, A.; Fioravanti, A.; Cenacchi, G. Supratentorial Cortical Ependymoma: Report of Three Cases. Neurosurgery 2005, 57, E192. [Google Scholar] [CrossRef]
- Applegate, G.L.; Marymont, M.H. Intracranial ependymomas: A review: Neuro-oncology. Cancer Investig. 1998, 16, 588–593. [Google Scholar] [CrossRef] [PubMed]
- Rudà, R.; Reifenberger, G.; Frappaz, D.; Pfister, S.M.; Laprie, A.; Santarius, T.; Roth, P.; Tonn, J.C.; Soffietti, R.; Weller, M.; et al. EANO guidelines for the diagnosis and treatment of ependymal tumors. Neuro Oncol. 2018, 20, 445–456. [Google Scholar] [CrossRef] [Green Version]
- Yuh, E.L.; Barkovich, A.J.; Gupta, N. Imaging of ependymomas: MRI and CT. Child’s Nerv. Syst. 2009, 25, 1203–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowak, J.; Seidel, C.; Pietsch, T.; Alkonyi, B.; Fuss, T.L.; Friedrich, C.; Von Hoff, K.; Rutkowski, S.; Warmuth-Metz, M. Systematic comparison of MRI findings in pediatric ependymoblastoma with ependymoma and CNS primitive neuroectodermal tumor not otherwise specified. Neuro Oncol. 2015, 17, 1157–1165. [Google Scholar] [CrossRef] [Green Version]
- Vitanza, N.A.; Partap, S. Pediatric Ependymoma. J. Child Neurol. 2016, 31, 1354–1366. [Google Scholar] [CrossRef]
- Jain, A.; Amin, A.G.; Jain, P.; Burger, P.; Jallo, G.I.; Lim, M.; Bettegowda, C. Subependymoma: Clinical features and surgical outcomes. Neurol. Res. 2012, 34, 677–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandão, L.A.; Poussaint, T.Y. Posterior Fossa Tumors. Neuroimaging Clin. N. Am. 2017, 27, 1–37. [Google Scholar] [CrossRef]
- Panigrahy, A.; Nelson, M.D.; Blüml, S. Magnetic resonance spectroscopy in pediatric neuroradiology: Clinical and research applications. Pediatr. Radiol. 2009, 40, 3–30. [Google Scholar] [CrossRef] [PubMed]
- Jaremko, J.; Jans, L.; Coleman, L.; Ditchfield, M.; Jaremko, J.; Jans, L.; Coleman, L.; Ditchfield, M. Value and Limitations of Diffusion-Weighted Imaging in Grading and Diagnosis of Pediatric Posterior Fossa Tumors. Am. J. Neuroradiol. 2010, 31, 1613–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uematsu, Y.; Hirano, A.; Llena, J.F. Electron microscopic observations of blood vessels in ependymoma. No Shinkei Geka 1988, 16, 1235–1242. [Google Scholar] [PubMed]
- Maj, E.; Szemplińska, B.; Szeszkowski, W.; Prokopienko, M.; Cieszanowski, A.; Marchel, A.; Rowiński, O. Role of Diffusion Tensor Imaging Parameters in the Characterization and Differentiation of Infiltrating and Non-Infiltrating Spinal Cord Tumors. Clin. Neuroradiol. 2020, 30, 739–747. [Google Scholar] [CrossRef] [Green Version]
- Lefton, D.R.; Pinto, R.S.; Martin, S.W. MRI features of intracranial and spinal ependymomas. Pediatr. Neurosurg. 1998, 28, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Khatua, S.; Magnum, R.; Bertrand, K.C.; Zaky, W.; McCall, D.; Mack, S.C. Pediatric ependymoma: Current treatment and newer therapeutic insights. Futur. Oncol. 2018, 14, 3175–3186. [Google Scholar] [CrossRef]
- Mueller, S.; Chang, S. Pediatric brain tumors: Current treatment strategies and future therapeutic approaches. Neurotherapy 2009, 6, 570–586. [Google Scholar] [CrossRef] [Green Version]
- Delgado-López, P.D.; Corrales-García, E.M.; Alonso-García, E.; García-Leal, R.; González-Rodrigálvarez, R.; Araus-Galdós, E.; Martín-Alonso, J. Central nervous system ependymoma: Clinical implications of the new molecular classification, treatment guidelines and controversial issues. Clin. Transl. Oncol. 2019, 21, 1450–1463. [Google Scholar] [CrossRef] [PubMed]
- Zacharoulis, S.; Moreno, L. Ependymoma: An Update. J. Child Neurol. 2009, 24, 1431–1438. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.R.; Ruda, R.; Soffietti, R. Ependymomas in Adults. Curr. Neurol. Neurosci. Rep. 2010, 10, 240–247. [Google Scholar] [CrossRef]
- Shim, K.-W.; Kim, D.-S.; Choi, J.-U. The history of ependymoma management. Child’s Nerv. Syst. 2009, 25, 1167–1183. [Google Scholar] [CrossRef]
- Tsai, C.J.; Wang, Y.; Allen, P.K.; Mahajan, A.; McCutcheon, I.E.; Rao, G.; Rhines, L.D.; Tatsui, C.E.; Armstrong, T.; Maor, M.H.; et al. Outcomes After Surgery and Radiotherapy for Spinal Myxopapillary Ependymoma. Neurosurgery 2014, 75, 205–214. [Google Scholar] [CrossRef] [Green Version]
- Pesce, A.; Palmieri, M.; Armocida, D.; Frati, A.; Miscusi, M.; Raco, A. Spinal Myxopapillary Ependymoma: The Sapienza University Experience and Comprehensive Literature Review Concerning the Clinical Course of 1602 Patients. World Neurosurg. 2019, 129, 245–253. [Google Scholar] [CrossRef]
- Gupta, S.; Haresh, K.P.; Gandhi, A.K.; Mallick, S.; Benson, R.; Sharma, D.N.; Julka, P.K.; Rath, G.K. Prognostic factors and survival outcomes of intracranial ependymoma treated with multimodality approach. Indian J. Med. Paediatr. Oncol. 2017, 38, 420–426. [Google Scholar] [CrossRef]
- Jung, T.-Y.; Jung, S.; Kook, H.; Baek, H.-J. Treatment Decisions of World Health Organization Grade II and III Ependymomas in Molecular Era. J. Korean Neurosurg. Soc. 2018, 61, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Wright, K.D.; Gajjar, A. Current Treatment Options for Pediatric and Adult Patients With Ependymoma. Curr. Treat. Options Oncol. 2012, 13, 465–477. [Google Scholar] [CrossRef]
- Hoffman, K.E.; Yock, T.I. Radiation Therapy for Pediatric Central Nervous System Tumors. J. Child Neurol. 2009, 24, 1387–1396. [Google Scholar] [CrossRef] [PubMed]
- Khatua, S.; Ramaswamy, V.; Bouffet, E. Current therapy and the evolving molecular landscape of paediatric ependymoma. Eur. J. Cancer 2017, 70, 34–41. [Google Scholar] [CrossRef]
- Metellus, P.; Barrie, M.; Figarella-Branger, D.; Chinot, O.; Giorgi, R.; Gouvernet, J.; Jouvet, A.; Guyotat, J. Multicentric French study on adult intracranial ependymomas: Prognostic factors analysis and therapeutic considerations from a cohort of 152 patients. Brain 2007, 130, 1338–1349. [Google Scholar] [CrossRef] [Green Version]
- Perilongo, G.; Massimino, M.; Sotti, G.; Belfontali, T.; Masiero, L.; Rigobello, L.; Garrè, L.; Carli, M.; Lombardi, F.; Solero, C.; et al. Analyses of prognostic factors in a retrospective review of 92 children with ependymoma: Italian Pediatric Neuro-Oncology Group. Med. Pediatr. Oncol. 1997, 29, 79–85. [Google Scholar] [CrossRef]
- Paulino, A.C. The local field in infratentorial ependymoma: Does the entire posterior fossa need to be treated? Int. J. Radiat. Oncol. 2001, 49, 757–761. [Google Scholar] [CrossRef]
- Paulino, A.C.; Wen, B.-C. The significance of radiotherapy treatment duration in intracranial ependymoma. Int. J. Radiat. Oncol. 2000, 47, 585–589. [Google Scholar] [CrossRef]
- Thorp, N.; Gandola, L. Management of Ependymoma in Children, Adolescents and Young Adults. Clin. Oncol. 2019, 31, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Merchant, T.E.; Li, C.; Xiong, X.; Kun, L.E.; Boop, F.A.; Sanford, R.A. Conformal radiotherapy after surgery for paediatric ependymoma: A prospective study. Lancet Oncol. 2009, 10, 258–266. [Google Scholar] [CrossRef] [Green Version]
- Baliga, S.; Gandola, L.; Timmermann, B.; Gail, H.; Padovani, L.; Janssens, G.O.; Yock, T.I. Brain tumors: Medulloblastoma, ATRT, ependymoma. Pediatr. Blood Cancer 2021, 68, e28395. [Google Scholar] [CrossRef] [PubMed]
- Merchant, T.E.; Fouladi, M. Ependymoma: New Therapeutic Approaches Including Radiation and Chemotherapy. J. Neuro Oncol. 2005, 75, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, S.M.; Sethi, R.; Lavally, B.; Yeap, B.Y.; Marcus, K.J.; Caruso, P.; Pulsifer, M.; Huang, M.; Ebb, D.; Tarbell, N.J.; et al. Proton radiotherapy for pediatric central nervous system ependymoma: Clinical outcomes for 70 patients. Neuro Oncol. 2013, 15, 1552–1559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacDonald, S.M.; Safai, S.; Trofimov, A.; Wolfgang, J.; Fullerton, B.; Yeap, B.Y.; Bortfeld, T.; Tarbell, N.J.; Yock, T. Proton Radiotherapy for Childhood Ependymoma: Initial Clinical Outcomes and Dose Comparisons. Int. J. Radiat. Oncol. 2008, 71, 979–986. [Google Scholar] [CrossRef]
- DeNunzio, N.J.; Yock, T.I. Modern Radiotherapy for Pediatric Brain Tumors. Cancers 2020, 12, 1533. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Chung, S.Y.; Han, J.W.; Kim, D.-S.; Kim, J.; Moon, J.Y.; Yoon, H.I.; Suh, C.-O. Treatment outcome of anaplastic ependymoma under the age of 3 treated by intensity-modulated radiotherapy. Radiat. Oncol. J. 2020, 38, 26–34. [Google Scholar] [CrossRef] [Green Version]
- Gornet, M.; Buckner, J.; Marks, R.; Scheithauer, B.; Erickson, B. Chemotherapy for advanced CNS ependymoma. J. Neuro Oncol. 1999, 45, 61–67. [Google Scholar] [CrossRef]
- Rojas-Marcos, I.; Calvet, D.; Janoray, P.; Delattre, J.Y. Response of recurrent anaplastic ependymoma to a combination of tamoxifen and isotretinoin. Neurology 2003, 61, 1019. [Google Scholar] [CrossRef]
- Miller, J.P.; Stoodley, R.J. Studies directed towards anthracyclinone syntheses: The use of d-glucose as a chiral auxiliary in asymmetric Diels–Alder reactions. J. Saudi Chem. Soc. 2013, 17, 29–42. [Google Scholar] [CrossRef] [Green Version]
- Edwardson, D.; Narendrula, R.; Chewchuk, S.; Mispel-Beyer, K.; Mapletoft, J.; Parissenti, A. Role of Drug Metabolism in the Cytotoxicity and Clinical Efficacy of Anthracyclines. Curr. Drug Metab. 2015, 16, 412–426. [Google Scholar] [CrossRef] [Green Version]
- Boogerd, W.; Tjahja, I.; Van De Sandt, M.; Beijnen, J. Penetration of idarubicin into malignant brain tumor tissue. J. Neuro Oncol. 1999, 44, 65–69. [Google Scholar] [CrossRef]
- Dreyer, Z.E.; Kadota, R.P.; Stewart, C.F.; Friedman, H.S.; Mahoney, D.H.; Kun, L.E.; McCluggage, C.W.; Burger, P.C.; Kepner, J.; Heideman, R.L. Phase 2 study of idarubicin in pediatric brain tumors: Pediatric Oncology Group study POG 9237. Neuro Oncol. 2003, 5, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Engstrom, P.; Benson, A., 3rd; Cohen, A.; Doroshow, J.; Kiel, K.; Niederhuber, J.; Roh, M.; Tempero, M. NCCN Colorectal Cancer Practice Guidelines. The National Comprehensive Cancer Network. Oncology 1996, 10, 140–175. [Google Scholar]
- Guérin, E.; Raffelsberger, W.; Pencreach, E.; Maier, A.; Neuville, A.; Schneider, A.; Bachellier, P.; Rohr, S.; Petitprez, A.; Poch, O.; et al. In Vivo Topoisomerase I Inhibition Attenuates the Expression of Hypoxia-Inducible Factor 1α Target Genes and Decreases Tumor Angiogenesis. Mol. Med. 2011, 18, 83–94. [Google Scholar] [CrossRef]
- Jannier, S.; Kemmel, V.; Sancho, C.S.; Chammas, A.; Sabo, A.-N.; Pencreach, E.; Farace, F.; Chenard, M.P.; Lhermitte, B.; Geoerger, B.; et al. SFCE-RAPIRI Phase I Study of Rapamycin Plus Irinotecan: A New Way to Target Intra-Tumor Hypoxia in Pediatric Refractory Cancers. Cancers 2020, 12, 3051. [Google Scholar] [CrossRef] [PubMed]
- Vredenburgh, J.J.; Desjardins, A.; Ii, J.E.H.; Marcello, J.; Reardon, D.A.; Quinn, J.A.; Rich, J.N.; Sathornsumetee, S.; Gururangan, S.; Sampson, J.; et al. Bevacizumab Plus Irinotecan in Recurrent Glioblastoma Multiforme. J. Clin. Oncol. 2007, 25, 4722–4729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vredenburgh, J.J.; Desjardins, A.; Herndon, J.E.; Dowell, J.M.; Reardon, D.A.; Quinn, J.A.; Rich, J.N.; Sathornsumetee, S.; Gururangan, S.; Wagner, M.; et al. Phase II Trial of Bevacizumab and Irinotecan in Recurrent Malignant Glioma. Clin. Cancer Res. 2007, 13, 1253–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gururangan, S.; Chi, S.N.; Poussaint, T.Y.; Onar-Thomas, A.; Gilbertson, R.J.; Vajapeyam, S.; Friedman, H.S.; Packer, R.J.; Rood, B.N.; Boyett, J.M. Lack of efficacy of bevacizumab plus irinotecan in children with recurrent malignant glioma and diffuse brainstem glioma: A Pediatric Brain Tumor Consortium study. J. Clin. Oncol. 2010, 28, 3069. [Google Scholar] [CrossRef]
- Gordaliza, M.; García, P.; del Corral, J.M.; Castro, M.; Gómez-Zurita, M. Podophyllotoxin: Distribution, sources, applications and new cytotoxic derivatives. Toxicon 2004, 44, 441–459. [Google Scholar] [CrossRef]
- Chamberlain, M.C. Recurrent intracranial ependymoma in children: Salvage therapy with oral etoposide. Pediatr. Neurol. 2001, 24, 117–121. [Google Scholar] [CrossRef]
- Jakacki, R.I.; Foley, M.A.; Horan, J.; Wang, J.; Kieran, M.W.; Bowers, D.; Bouffet, E.; Zacharoulis, S.; Gill, S.C. Single-agent erlotinib versus oral etoposide in patients with recurrent or refractory pediatric ependymoma: A randomized open-label study. J. Neuro Oncol. 2016, 129, 131–138. [Google Scholar] [CrossRef]
- Chastagner, P.; Sommelet-Olive, D.; Kalifa, C.; Brunat-Mentigny, M.; Zucker, J.M.; Demeocq, F.; Baranzelli, M.C.; Tron, P.; Bergeron, C.; Pein, F. Phase II study of ifosfamide in childhood brain tumors: A report by the French Society of Pediatric Oncology (SFOP). Med. Pediatr. Oncol. 1993, 21, 49–53. [Google Scholar] [CrossRef]
- Leeper, H.; Felicella, M.M.; Walbert, T. Recent Advances in the Classification and Treatment of Ependymomas. Curr. Treat. Options Oncol. 2017, 18, 55. [Google Scholar] [CrossRef] [PubMed]
- Garvin, J.H., Jr.; Selch, M.T.; Holmes, E.; Berger, M.S.; Finlay, J.L.; Flannery, A.; Goldwein, J.W.; Packer, R.J.; Rorke-Adams, L.B.; Shiminski-Maher, T. Phase II study of pre-irradiation chemotherapy for childhood intracranial ependymoma. Children’s Cancer Group protocol 9942: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2012, 59, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Venkatramani, R.; Ji, L.; Lasky, J.; Haley, K.; Judkins, A.; Zhou, S.; Sposto, R.; Olshefski, R.; Garvin, J.; Tekautz, T.; et al. Outcome of infants and young children with newly diagnosed ependymoma treated on the “Head Start” III prospective clinical trial. J. Neuro Oncol. 2013, 113, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Massimino, M.; Miceli, R.; Giangaspero, F.; Boschetti, L.; Modena, P.; Antonelli, M.; Ferroli, P.; Bertin, D.; Pecori, E.; Valentini, L.; et al. Final results of the second prospective AIEOP protocol for pediatric intracranial ependymoma. Neuro Oncol. 2016, 18, 1451–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.; Onar-Thomas, A.; Ellison, D.; Owens-Pickle, E.; Wu, S.; Leary, S.E.S.; Fouladi, M.; Merchant, T.; Gajjar, A.; Foreman, N. Epen-acns0831, phase iii randomized trial of post-radiation chemotherapy in patients with newly diagnosed ependymoma ages 1 to 21 years. Neuro Oncol. 2020, 22, iii318–iii319. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Manabe, H.; Izumi, B.; Shima, T.; Adachi, N. Remarkable efficacy of temozolomide for relapsed spinal myxopapillary ependymoma with multiple recurrence and cerebrospinal dissemination: A case report and literature review. Eur. Spine J. 2017, 27, 421–425. [Google Scholar] [CrossRef]
- Schreck, K.C.; Grossman, S.A. Role of Temozolomide in the Treatment of Cancers Involving the Central Nervous System. Oncology 2018, 32, 555. [Google Scholar]
- Buccoliero, A.; Castiglione, F.; Degl’Innocenti, D.R.; Paglierani, M.; Maio, V.; Gheri, C.; Garbini, F.; Moncini, D.; Taddei, A.; Sardi, I.; et al. O6- Methylguanine-DNA-Methyltransferase in Recurring Anaplastic Ependymomas: PCR and Immunohistochemistry. J. Chemother. 2008, 20, 263–268. [Google Scholar] [CrossRef]
- Gilbert, M.R.; Yuan, Y.; Wu, J.; Mendoza, T.; Vera, E.; Omuro, A.; Lieberman, F.; Robins, H.I.; Gerstner, E.R.; Wu, J.; et al. A phase II study of dose-dense temozolomide and lapatinib for recurrent low-grade and anaplastic supratentorial, infratentorial, and spinal cord ependymoma. Neuro Oncol. 2021, 23, 468–477. [Google Scholar] [CrossRef]
- Pajtler, K.W.; Witt, H.; Sill, M.; Jones, D.; Hovestadt, V.; Johann, P.; Reimand, J.; Lichter, P.; Taylor, M.D.; Gilbertson, R.; et al. EP-03 Molecular classification of ependymal tumors across all cns compartments, histopathological grades and age groups. Neuro Oncol. 2015, 17, iii6. [Google Scholar] [CrossRef] [Green Version]
- Wright, K.D.; Daryani, V.M.; Turner, D.C.; Onar-Thomas, A.; Boulos, N.; Orr, B.A.; Gilbertson, R.J.; Stewart, C.F.; Gajjar, A. Phase I study of 5-fluorouracil in children and young adults with recurrent ependymoma. Neuro Oncol. 2015, 17, 1620–1627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeWire, M.; Fouladi, M.; Turner, D.C.; Wetmore, C.; Hawkins, C.; Jacobs, C.; Yuan, Y.; Liu, D.; Goldman, S.; Fisher, P.; et al. An open-label, two-stage, phase II study of bevacizumab and lapatinib in children with recurrent or refractory ependymoma: A collaborative ependymoma research network study (CERN). J. Neuro Oncol. 2015, 123, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Fouladi, M.; Park, J.R.; Stewart, C.F.; Gilbertson, R.J.; Schaiquevich, P.; Sun, J.; Reid, J.M.; Ames, M.M.; Speights, R.; Ingle, A.M. Pediatric phase I trial and pharmacokinetic study of vorinostat: A Children’s Oncology Group phase I consortium report. J. Clin. Oncol. 2010, 28, 3623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lester, A.; McDonald, K.L. Intracranial ependymomas: Molecular insights and translation to treatment. Brain Pathol. 2020, 30, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Temozolomide: Uses, Interactions, Mechanism of Action DrugBank Online. Available online: https://go.drugbank.com/drugs/DB00853 (accessed on 3 June 2021).
- Marinoff, A.E.; Matija, S.; Guo, D.; Snuderl, M.; Wright, K.D.; Manley, P.E.; Al-Sayegh, H.; Sinai, C.E.; Ullrich, N.J.; Marcus, K.; et al. Rethinking childhood ependymoma: A retrospective, multi-center analysis reveals poor long-term overall survival. J. Neuro Oncol. 2017, 135, 201–211. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WHO Classifications | Tumor Names | Characteristics |
|---|---|---|
| WHO grade I | Myxopapillary ependymoma |
|
| Subependymoma |
| |
| WHO grade II | Ependymoma |
|
| WHO grade II or III | RELA fusion-positive ependymoma |
|
| WHO grade III | Anaplastic ependymoma |
|
| Age | Incidence of Primary Brain or CNS Tumors | Percentage of Ependymoma |
|---|---|---|
| 0–14 years (children) | 16,366 | 5.7% |
| 15–19 years | 6747 | 4% |
| >19 years (Adults) | 356,858 | 1.9% |
| Age | Percentage of Survival |
|---|---|
| 75+ years | 57.8% |
| 20–44 years | 91% |
| 0–19 years | 75.2% |
| Anatomical/Molecular Classification | WHO Grade | Age Group | |
|---|---|---|---|
| Supratentorial ependymoma (ST-) | |||
| ST-SE | Subependymoma loss of chromosome 19 | WHO-grade I | Adults |
| ST-EPN-YAP1 | (Anaplastic) Ependymoma YAP1-fusion | WHO-grade II/III | Infants, Children |
| ST-EPN-RELA | (Anaplastic) Ependymoma Chromothripsis; RELA-fusion | WHO-grade II/III | Infants, Children, Adults |
| Posterior fossa ependymoma (PF-) | |||
| PF-SE | Subependymoma loss of chromosome 19, 6p TERT-mutation | WHO-grade I | Adults |
| PF-EPN-A | (Anaplastic) Ependymoma Balanced genome | WHO-grade II/III | Infants, Children |
| PF-EPN-B | (Anaplastic) Ependymoma Chromosomal instability | WHO-grade II/III | Children, Adults |
| Spinal ependymoma (SP-) | |||
| SP-SE | Subependymoma loss of chromosome 19, 6p | WHO-grade I | Adults |
| SP-MPE | Myxopapillary Ependymoma Chromosomal instability | WHO-grade I | Adults |
| SP-EPN | (Anaplastic) Ependymoma Chromosomal instability, NF2 mutation | WHO-grade II/III | Adults |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seo, S.-H.; Paul, S.K.; Shikder, M.; Khanam, M.; Ghosh, P.; Hasib, T.A.; Ahmed, K.A.; Sikdar, S.; Uddin, M.J.; Kwon, Y. An Insight into Pathophysiological Features and Therapeutic Advances on Ependymoma. Cancers 2021, 13, 3221. https://doi.org/10.3390/cancers13133221
Seo S-H, Paul SK, Shikder M, Khanam M, Ghosh P, Hasib TA, Ahmed KA, Sikdar S, Uddin MJ, Kwon Y. An Insight into Pathophysiological Features and Therapeutic Advances on Ependymoma. Cancers. 2021; 13(13):3221. https://doi.org/10.3390/cancers13133221
Chicago/Turabian StyleSeo, Seung-Hee, Shamrat Kumar Paul, Mita Shikder, Mushira Khanam, Popy Ghosh, Tasnin Al Hasib, Kazi Ahsan Ahmed, Suranjana Sikdar, Md Jamal Uddin, and Youngjoo Kwon. 2021. "An Insight into Pathophysiological Features and Therapeutic Advances on Ependymoma" Cancers 13, no. 13: 3221. https://doi.org/10.3390/cancers13133221