
Mast Cell Leukemia: An Update with a Practical Review
 , , , , , , ,
, , , , , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Overview on Mastocytosis, SM Diagnostic Criteria and Different Subtypes of SM
- cutaneous mastocytosis (CM) with skin-limited disease, which is more common in children and which presents good biological behavior;
- SM with involvement of one or more extra-cutaneous organs, which is more frequent in adults and has a less favorable outcome;
- The major criterion consists of multifocal, dense aggregates, each of at least 15 MCs, in BM biopsy and/or another extra-cutaneous organ.
- The following features are recognized as B-findings: >30% of BM cellularity consisting of MCs and serum total tryptase >200 ng/mL; dysplasia or myeloproliferative features in non-MC lineages that are not enough for the diagnosis of other myeloid neoplasms with normal or only slightly abnormal blood counts; hepatomegaly without impairment of liver function; splenomegaly without hypersplenism and/or abnormal size of lymph nodes.
- The following features qualify as C-findings: one or more cytopenia due to diffuse BM infiltration; hepatomegaly with damaged liver function; splenomegaly with hypersplenism; malabsorption and weight loss due to gastrointestinal (GI) MC infiltration; and osteolytic bone lesions.
3. MCL: Defining Criteria, Leukemic and Aleukemic Variants, Primary and Secondary Variants
4. MCL: Epidemiology and Clinical Manifestations
5. MCL: Acute Variant and Chronic Variant
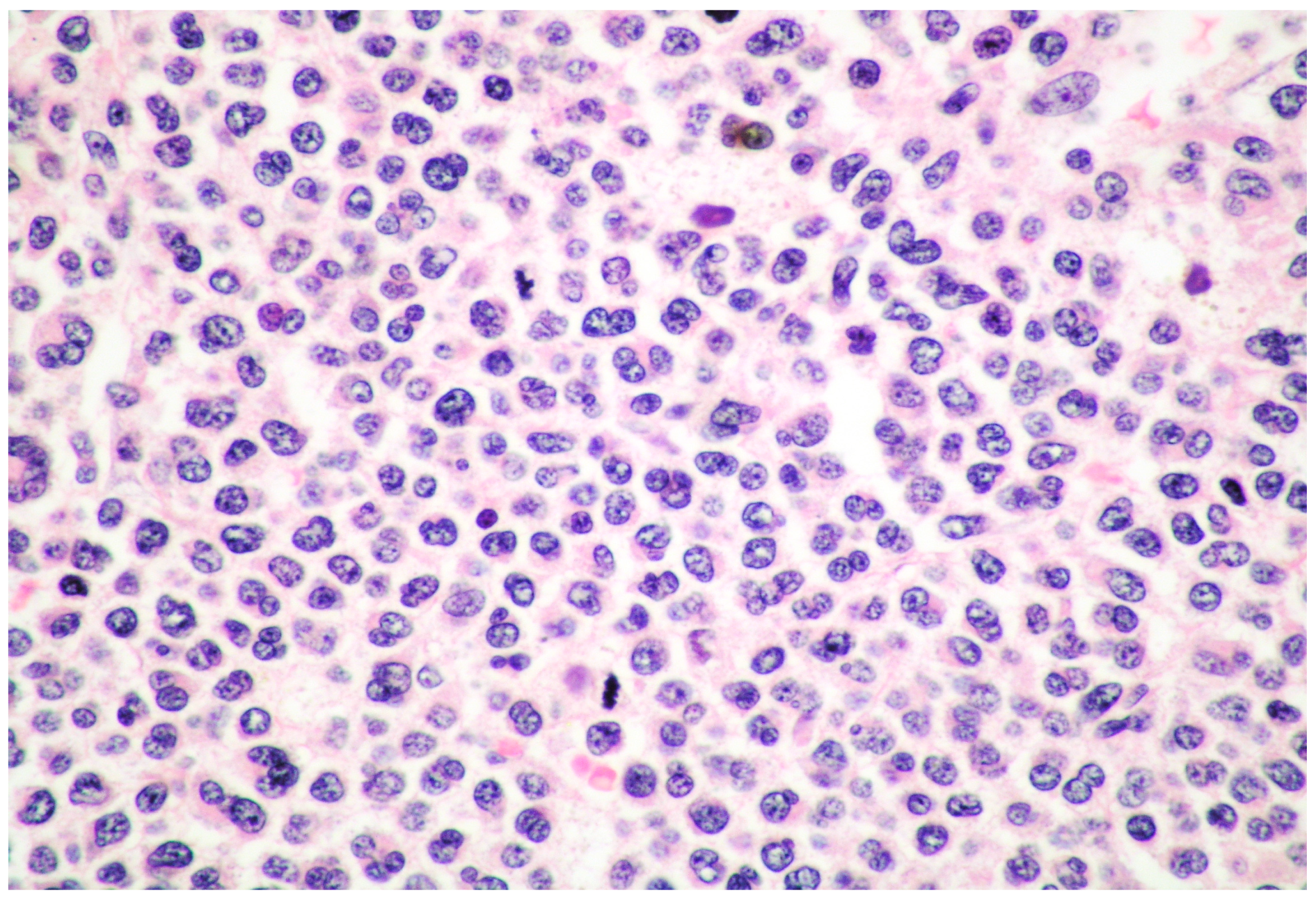
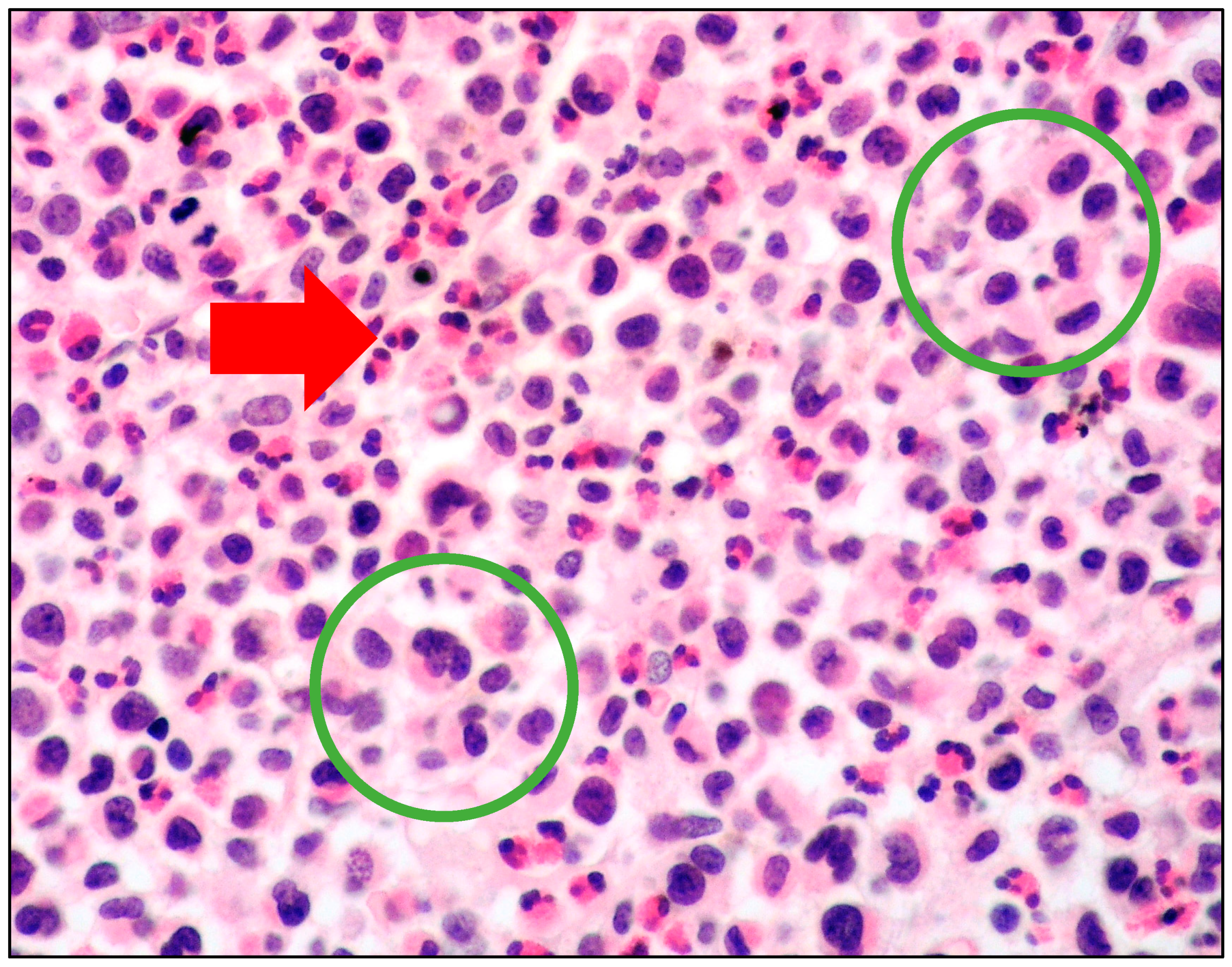
6. MCL: Morphology of MCs
- spindle-shaped MCs characterized by an oval nucleus, elongated cytoplasmic processes and hypo-granulated cytoplasm, often with focal granule accumulation (atypical MC type I);
- well-differentiated MCs characterized by a round-shaped morphology, with a round nucleus and granulated cytoplasm.
- promastocytes which have bi-lobated or multi-lobated nuclei (Figure 2);
- metachromatic, granulated blasts;
- pleomorphic, multinucleated MCs.
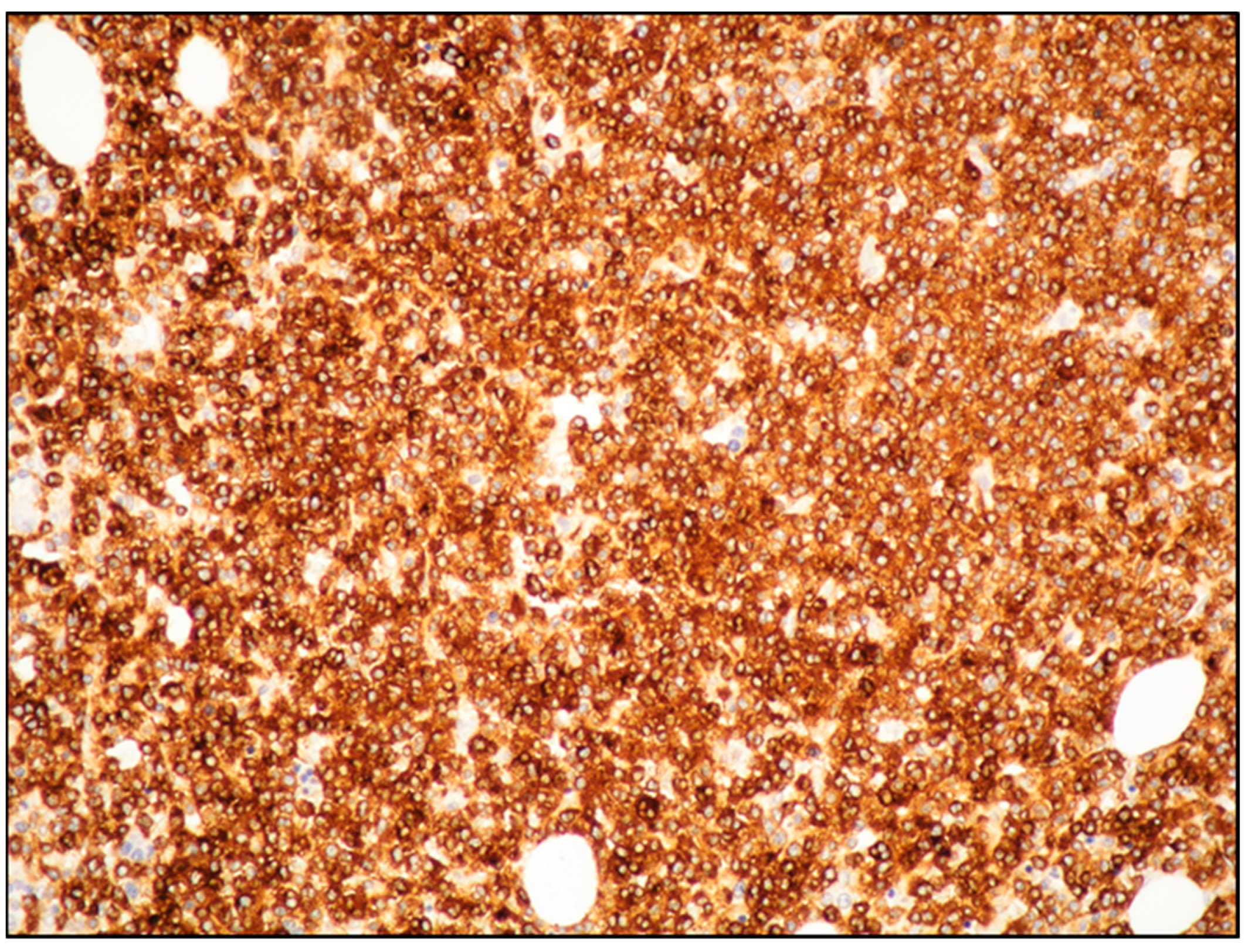
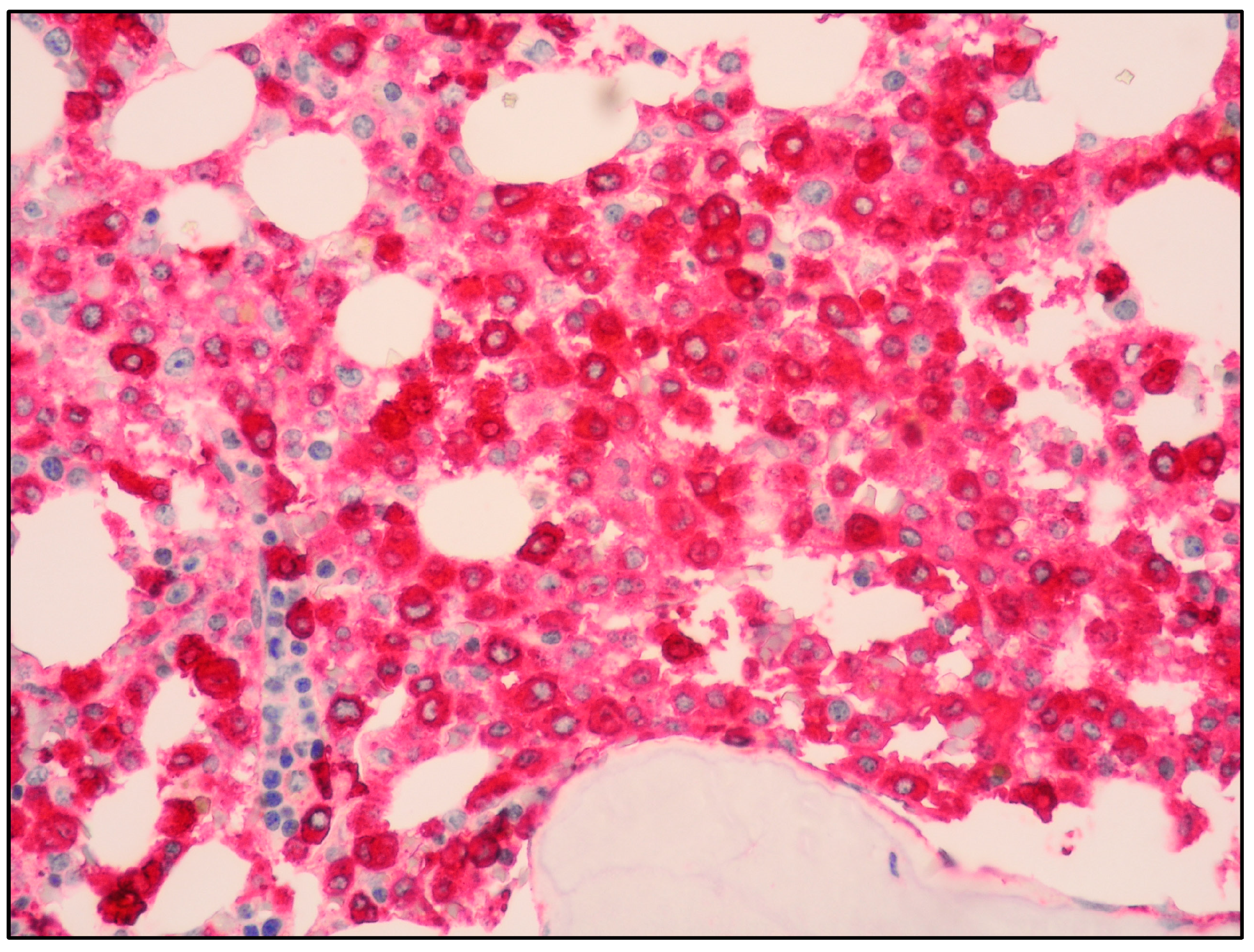
7. MCL: Immunophenotyping
8. MCL: Molecular Features
9. SM-AHN (or SM/AMN) and MCL-AHN (or MCL-AMN)
10. Differential Diagnosis between MCL and Myelomastocytic leukemia (MML) and Other Rare Entities
- at least 10% immature MCs/metachromatic blasts in BM smear in the context of an advanced myeloid neoplasm;
- >5% myeloblasts in BM smear or PB (sign of advanced myeloid neoplasm);
- at least 10% of BM cells should be MCs (positivity for tryptase and CD117);
- absence of SM diagnostic criteria (in particular, MML is CD25-negative and lacks KIT mutations).
11. MCL: Outcome and Treatment
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviation
References
- Joachim, G. Uber Mastzellenleukamien. Dtsch. Arch. Fur Klin. Medizin. 1906, 87, 437. [Google Scholar]
- WHO Classification of Tumours Editorial Board (Ed.) WHO Classification of Tumours Haematopoietic and Lymphoid Tissues, 4th ed.; IARC: Lyon, France, 2017.
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef] [PubMed]
- Georgin-Lavialle, S.; Lhermitte, L.; Dubreuil, P.; Chandesris, M.O.; Hermine, O.; Damaj, G. Mast cell leukemia. Blood 2013, 121, 1285–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jawhar, M.; Schwaab, J.; Meggendorfer, M.; Naumann, N.; Horny, H.P.; Sotlar, K.; Haferlach, T.; Schmitt, K.; Fabarius, A.; Valent, P.; et al. The clinical and molecular diversity of mast cell leukemia with or without associated hematologic neoplasm. Haematologica 2017, 102, 1035–1043. [Google Scholar] [CrossRef] [Green Version]
- Jain, P.; Wang, S.; Patel, K.P.; Sarwari, N.; Cortes, J.; Kantarjian, H.; Verstovsek, S. Mast cell leukemia (MCL): Clinico-pathologic and molecular features and survival outcome. Leuk. Res. 2017, 59, 105–109. [Google Scholar] [CrossRef]
- Galura, G.M.; Cherukuri, S.V.; Hakim, N.; Gaur, S.; Orazi, A. Acute aleukemic mast cell leukemia: Report of a case and review of the literature. Leuk. Res. Rep. 2020, 14, 100230. [Google Scholar] [CrossRef]
- Leguit, R.; Hebeda, K.; Kremer, M.; van der Walt, J.; Gianelli, U.; Tzankiv, A.; Orazi, A. The spectrum of aggressive mastocytosis: A workshop report and literature review. Pathobiology 2020, 87, 2–19. [Google Scholar] [CrossRef]
- Kennedy, V.E.; Perkins, C.; Reiter, A.; Jawhar, M.; Lubke, J.; Kluin-Nelemans, H.C.; Shomali, W.; Langford, C.; Abuel, J.; Hermine, O.; et al. Mast cell leukemia: Clinical and molecular features and survival outcomes of patients in the ECNM Registry. Blood Adv. 2022. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.-M.; Wang, S.A.; Bagg, A.; Tiziano Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef]
- Valent, P.; Sotlar, K.; Sperr, W.R.; Escribano, L.; Yavuz, S.; Reiter, A.; George, T.I.; Kluin-Nelemans, H.C.; Hermine, O.; Butterfield, J.H.; et al. Refined diagnostic classification of mast cell leukemia (MCL) and myelomastocytic leukemia (MML): A consensus proposal. Ann. Oncol. 2014, 25, 1691–1700. [Google Scholar] [CrossRef]
- Metcalfe, D.D. Mast cells and mastocytosis. Blood 2008, 112, 946–956. [Google Scholar] [CrossRef] [Green Version]
- Valent, P.; Akin, C.; Arock, M.; Brockow, K.; Butterfield, J.H.; Carter, M.C.; Castelss, M.; Escribano, L.; Hartmann, K.; Lieberman, P.; et al. Definitions, criteria and global classification of mast cell disorders with special reference to mast cell activation syndromes: A consensus proposal. Int. Arch. Allergy. Immunol. 2012, 157, 215–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valent, P. Mastocytosis: A paradigmatic example of a rare disease with complex biology and pathology. Am. J. Cancer Res. 2013, 3, 159–172. [Google Scholar]
- Valent, P.; Akin, C.; Metcalfe, D.D. Mastocytosis: 2016 updated WHO classification and novel emerging treatment concepts. Blood 2017, 129, 1420–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanelli, M.; Pai, R.K.; Zorzi, M.G.; Zizzo, M.; Martino, G.; Morelli, L.; Parisi, A.; De Marco, L.; Annessi, V.; Ascani, S. Gastrointestinal mastocytosis: A potential diagnostic pitfall to be aware. Int. J. Surg. Pathol. 2019, 27, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Zanelli, M.; Pizzi, M.; Sanguedolce, F.; Zizzo, M.; Palicelli, A.; Soriano, A.; Bisagni, A.; Martino, G.; Caprera, C.; Moretti, M.; et al. Gastrointestinal manifestations in systemic mastocytosis: The need of a multidisciplinary approach. Cancers 2021, 13, 3316. [Google Scholar] [CrossRef]
- Scherber, R.M.; Borate, U. How we diagnose and treat systemic mastocytosis in adults. Br. J. Haematol. 2018, 180, 11–23. [Google Scholar] [CrossRef]
- Pardani, A. Systemic mastocytosis in adults: 2021 update on diagnosis, risk stratification and management. Am. J. Hematol. 2021, 96, 508–525. [Google Scholar]
- El Hussein, S.; Chifotides, H.T.; Khoury, J.D.; Verstovsek, S.; Thakral, B. Systemic mastocytosis and other entities involving mast cells: A practical review and update. Cancers 2022, 14, 3474. [Google Scholar] [CrossRef]
- Lim, K.H.; Tefferi, A.; Lasho, T.L.; Finke, C.; Patnaik, M.; Butterfield, J.H.; McClure, R.F.; Li, C.-Y.; Pardani, A. Systemic mastocytosis in 342 consecutive adults: Survival studies and prognostic factors. Blood 2009, 113, 5727–5736. [Google Scholar] [CrossRef] [Green Version]
- Monnier, J.; Georgin-Lavialle, S.; Canioni, D.; Lhermitte, L.; Soussan, M.; Arock, M.; Bruneau, J.; Dubreuil, P.; Bodemer, C.; Chandesris, M.-O.; et al. Mast cell sarcoma: New cases and literature review. Oncotarget 2016, 7, 66299–66309. [Google Scholar] [CrossRef] [Green Version]
- Horny, H.P.; Sotlar, K.; Sperr, W.R.; Valent, P. Systemic mastocytosis with associated clonal haematological non-mast cell lineage diseases: A histopathological challenge. J. Clin. Pathol. 2004, 57, 604–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanelli, M.; Ricci, S.; Zizzo, M.; Sanguedolce, F.; De Giorgi, F.; Palicelli, A.; Martino, G.; Ascani, S. Systemic mastocytosis associated with “smoldering” multiple myeloma. Diagnostics 2021, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.C.; Ward, D.; Dunn, P.; Chang, C.-C. Acute mast cell leukemia associated with t(4;5)(q21;q33). Hum. Pathol. 2017, 67, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Suh, M.C.; Ham, J.Y.; Park, T.-I.; Moon, J.H.; Soo Suh, J.S. Highly Aggressive de novo Aleukemic Variant of Mast Cell Leukemia without KIT D816V Mutation. Ann. Lab. Med. 2017, 37, 547–549. [Google Scholar] [CrossRef]
- Costopoulos, M.; Uzunov, M.; Bories, D.; Charlotte, F.; Maloum, K.; Arock, M. Acute mast cell leukemia: A rare but highly aggressive hematopoietic neoplasm. Diagn. Cytopathol. 2018, 6, 639–641. [Google Scholar] [CrossRef]
- Valent, P.; Sotlar, K.; Sperr, W.R.; Reiter, A.; Arock, M.; Horny, H.-P. Chronic mast cell leukemia: A novel leukemia-variant with distinct morphological and clinical features. Leuk. Res. 2015, 39, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Valent, P.; Berger, J.; Cerny-Reiterer, S.; Peter, B.; Eisenwort, G.; Hoermann, G.; Mullauer, L.; Mannhalter, C.; Steurer, M.; Bettelheim, P.; et al. Chronic mast cell leukemia (MCL) with KIT S476I: A rare entity defined by leukemic expansion of mature mast cells and absence of organ damage. Ann. Hematol. 2015, 94, 223–231. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Twose, I.; Jara-Acevedo, M.; Morgado, J.M.; Garcia-Montero, A.; Sanchez-Munoz, L.; Teodosio, C.; Matito, A.; Mayado, A.; Caldas, C.; Mollejo, M.; et al. Clinical, immunophenotypic and molecular characteristics of well-differentiated systemic mastocytosis. J. Allergy Clin. Immunol. 2016, 137, 168–178. [Google Scholar] [CrossRef]
- Sperr, W.R.; Escribano, L.; Jordan, J.H.; Schernthaner, G.H.; Kundi, M.; Horny, H.-P.; Valent, P. Morphologic properties of neoplastic mast cells: Delineation of stages of maturation and implication for cytological grading of mastocytosis. Leuk. Res. 2001, 25, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Leguit, R.J.; Wang, S.A.; George, T.I.; Tzankov, A.; Orazi, A. The international consensus classification of mastocytosis sand related entities. Virchows Arch. 2022, 482, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Sotlar, K.; Horny, H.-P. Aberrant expression of CD30 in aggressive systemic mastocytosis and mast cell leukemia: A differential diagnosis to consider in aggressive hematopoietic CD30-positive neoplasms. Leuk. Lymphoma 2011, 52, 740–744. [Google Scholar] [CrossRef]
- Jawhar, M.; Schwaab, J.; Schnittger, S.; Meggerdorfer, M.; Pfirrmann, M.; Sotlar, K.; Horny, H.-P.; Metzgeroth, G.; Kluger, S.; Naumann, N.; et al. Additional mutations in SRSF2, ASXL1 and/or RUNX1 identify a high-risk group of patients with KIT D816V (+) advanced systemic mastocytosis. Leukemia 2016, 30, 136–143. [Google Scholar] [CrossRef]
- Sperr, W.R.; Horny, H.-P.; Valent, P. Spectrum of associated clonal hematologic non-mast cell lineage disorders occurring in patients with systemic mastocytosis. Int. Arch. Allergy Immunol. 2002, 127, 140–142. [Google Scholar] [CrossRef]
- Sperr, W.R.; Horny, H.-P.; Lechner, K.; Valent, P. Clinical and biologic diversity of leukemias occurring in patients with mastocytosis. Leuk. Lymphoma 2000, 37, 473–486. [Google Scholar] [CrossRef]
- Bernd, H.-W.; Sotlar, K.; Lorenzen, J.; Osieka, R.; Fabry, U.; Valent, P.; Horny, H.-P. Acute myeloid leukaemia (AML) with t (8;21) associated with “occult” mastocytosis (SM-AHNMD). Report of an unusual case and review of the literature. J. Clin. Pathol. 2004, 57, 324–328. [Google Scholar] [CrossRef] [Green Version]
- Pullarkat, V.A.; Bueso-Ramos, C.; Lai, R.; Kroft, S.; Wilson, C.S.; Pullarkat, S.T.; Bu, X.; Thein, M.; Lee, M.; Brynes, R.K. Systemic mastocytosis with associated clonal hematological non-mast-cell lineage disease: Analysis of clinicopathologic features and activating c-kit mutations. Am. J. Hematol. 2003, 73, 12–17. [Google Scholar] [CrossRef]
- Valent, P.; Samorapoompichi, P.; Sperr, W.R.; Horny, H.P.; Lechner, K. Myelomastocytic leukemia: Myeloid neoplasm characterized by partial differentiation of mast cell lineage cells. Hematol. J. 2002, 3, 90–94. [Google Scholar] [CrossRef]
- Horny, H.P.; Sotlar, K.; Reiter, A.; Valent, P. Myelomastocytic leukemia: Histopathological features, diagnostic criteria and differential diagnosis. Expert Rev. Hematol. 2014, 7, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Sperr, W.R.; Hauswirth, A.W.; Valent, P. Tryptase: A novel biochemical marker of acute myeloid leukemia. Leuk Lymphoma 2002, 43, 2257–2261. [Google Scholar] [CrossRef] [PubMed]
- Ustun, C.; Reiter, A.; Scott, B.L.; Nakamura, R.; Damaj, G.; Kreil, S.; Shanley, R.; Hogan, W.J.; Perales, M.-A.; Shore, T.; et al. Hematopoietic stem-cell transplantation for advanced systemic mastocytosis. J. Clin. Oncol. 2014, 32, 3264–3274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jawhar, M.; Schwaab, J.; Hausmann, D.; Clemens, J.; Naumann, N.; Henzler, T.; Horny, H.-P.; Sotlar, K.; Schoenberg, S.O.; Cross, N.C.P.; et al. Splenomegaly, elevated alkaline phosphatase and mutations in the SRSF2/ASXL1/RUNX1 gene panel are strong adverse prognostic markers in patients with systemic mastocytosis. Leukemia. 2016, 30, 2342–2350. [Google Scholar] [CrossRef] [PubMed]
- Gotlib, J.; Reiter, A.; Radia, D.H. Efficacy and safety of avapritinib in advanced systemic mastocytosis: Interim analysis of the phase 2 PATHFINDER trial. Nat Med. 2021, 27, 2192–2199. [Google Scholar] [CrossRef] [PubMed]
- DeAngelo, D.J.; Radia, D.H.; George, T.I.; Robinson, W.A.; Quiery, A.T.; Drummond, M.W.; Bose, P.; Hexner, E.O.; Winton, E.F.; Horny, H.-P.; et al. Safety and efficacy of avapritinib in advanced systemic mastocytosis: The phase 1 EXPLORER trial. Nat Med. 2021, 27, 2183–2191. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
|
|
|
|
|
| 2022 WHO: The diagnosis of SM can be performed when the major criterion plus one minor criterion or at least 3 minor criteria are fulfilled. 2022 ICC: The diagnosis of SM can be performed when the major criterion or, in its absence, at least 3 minor criteria are fulfilled. |
| Major criterion |
|
| Minor criteria |
|
| 2022 WHO: SM-AHN AHN includes neoplasms of myeloid, plasma-cell and lymphoid-cell origin |
| 2022 ICC: SM-AMN AMN includes only neoplasms of myeloid origin |
| Myelomastocytic Leukemia | Mast Cell Leukemia |
|---|---|
| At least 10% immature mast cells or metachromatic blasts in BM | =/>20% immature mast cells or metachromatic blasts in BM |
| >5% myeloblasts in BM or PB | Immature mast cells may be present or not in PB |
| CD25-negative | CD25-positive |
| Wild-type KIT | KIT mutations in most cases |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zanelli, M.; Quintini, M.; Magnasco, S.; Aprile, L.; Palicelli, A.; Zizzo, M.; Sanguedolce, F.; Ricci, S.; Pancetti, S.; Zuccalà, V.; et al. Mast Cell Leukemia: An Update with a Practical Review. Cancers 2023, 15, 1664. https://doi.org/10.3390/cancers15061664
Zanelli M, Quintini M, Magnasco S, Aprile L, Palicelli A, Zizzo M, Sanguedolce F, Ricci S, Pancetti S, Zuccalà V, et al. Mast Cell Leukemia: An Update with a Practical Review. Cancers. 2023; 15(6):1664. https://doi.org/10.3390/cancers15061664
Chicago/Turabian StyleZanelli, Magda, Martina Quintini, Salvatore Magnasco, Lara Aprile, Andrea Palicelli, Maurizio Zizzo, Francesca Sanguedolce, Stefano Ricci, Saverio Pancetti, Valeria Zuccalà, and et al. 2023. "Mast Cell Leukemia: An Update with a Practical Review" Cancers 15, no. 6: 1664. https://doi.org/10.3390/cancers15061664