
The Future of Regenerative Medicine: Cell Therapy Using Pluripotent Stem Cells and Acellular Therapies Based on Extracellular Vesicles

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Regenerative Medicine: State of the Art
2.1. Introduction
2.2. Cell Sources
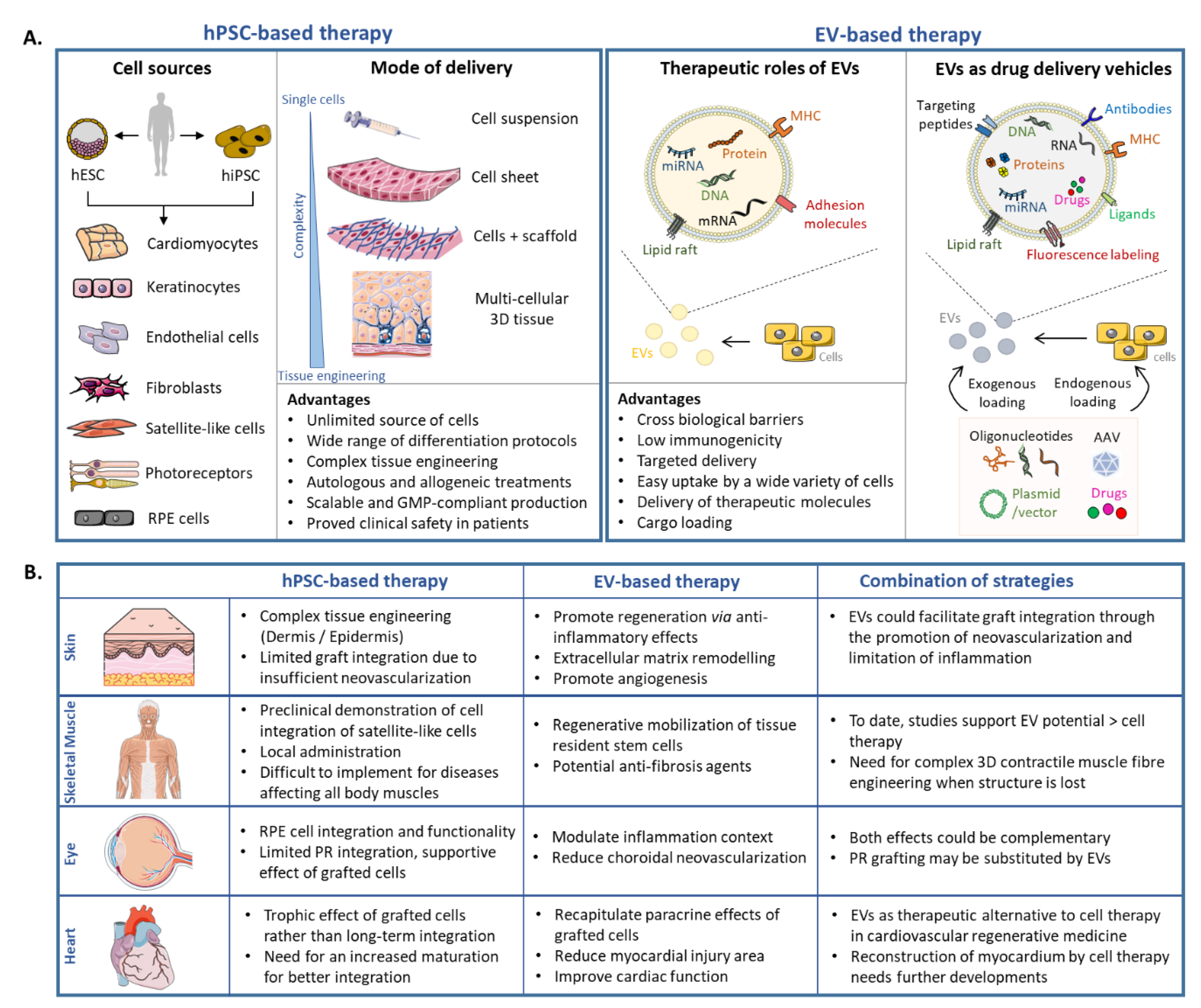
2.3. hPSC-Based Cell Therapy: Selected Examples
2.3.1. Retinal Degeneration
2.3.2. Cutaneous Wounds
2.3.3. Heart Infarction
2.3.4. Skeletal Muscle Regeneration
2.4. hPSC-Based Clinical Trials
2.5. Limitations/Challenges
3. Application of EVs in Regenerative Medicine
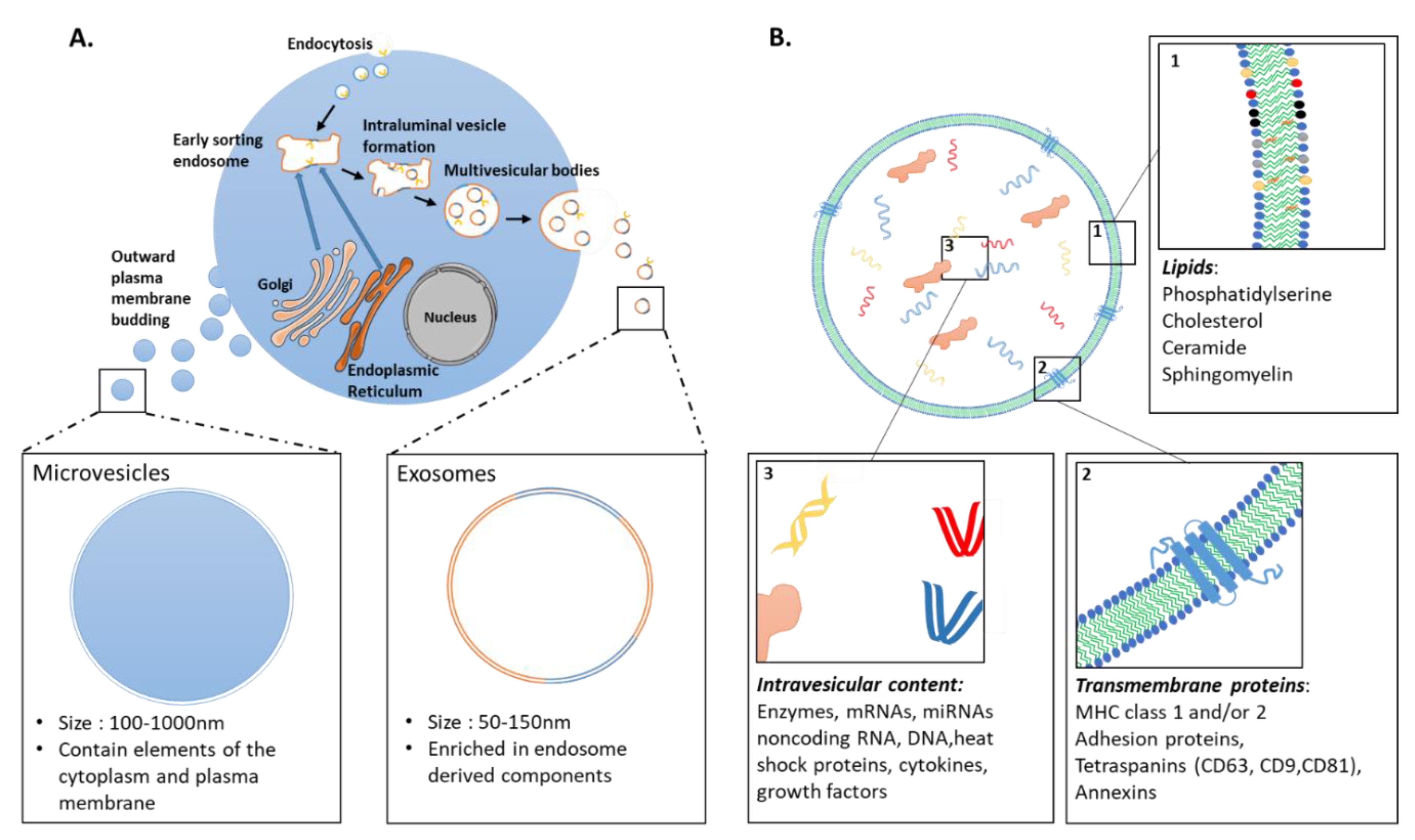
3.1. Extracellular Vesicles: Definition
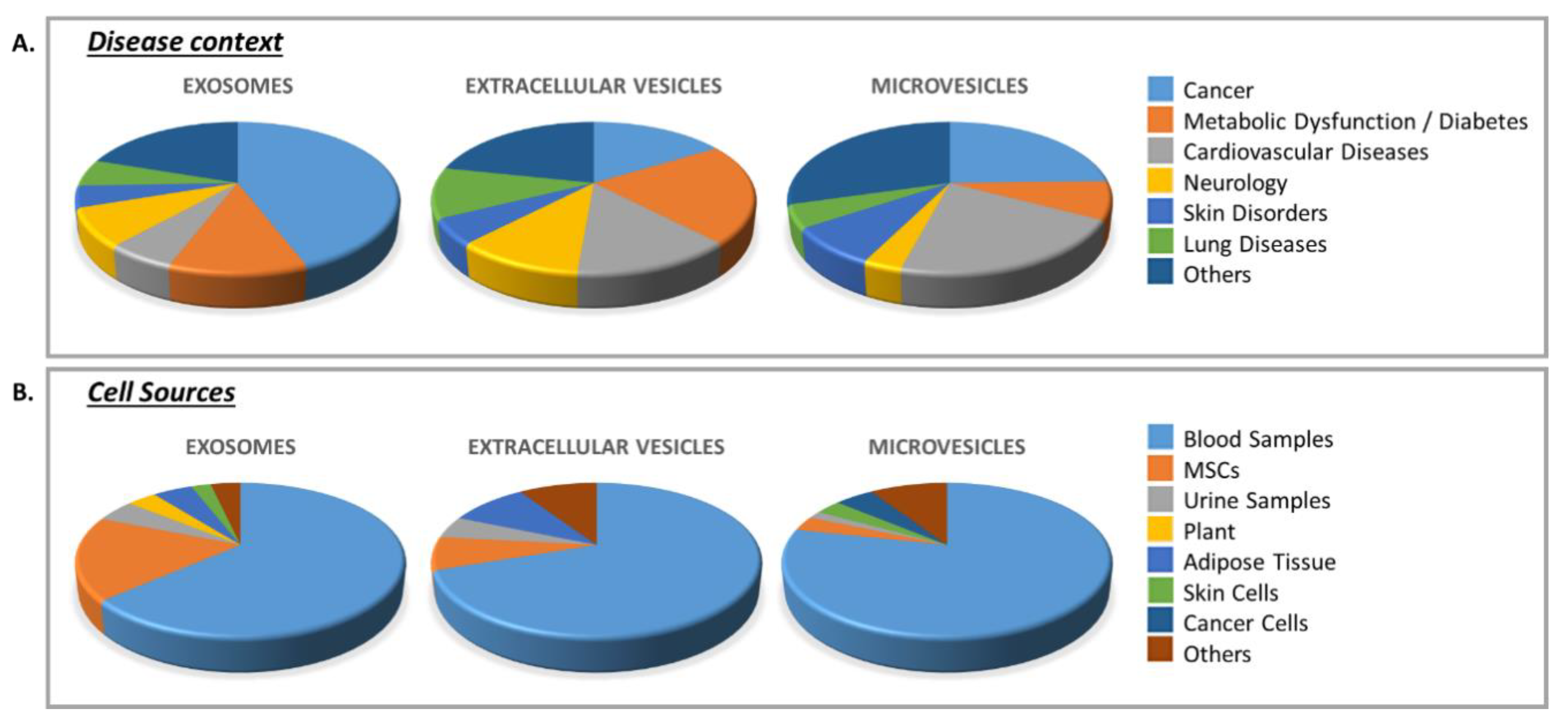
3.2. Clinical Trials Using EVs
3.3. EVs as Potential Therapeutic Tools: Selected Examples
3.3.1. Tissue Restoration in the Eye
3.3.2. Cutaneous Wound Healing
3.3.3. Heart
3.3.4. Skeletal Muscle
3.4. Challenges
3.4.1. Manufacturing EVs: Considerations
3.4.2. EV-Based Therapeutics: Regulatory Aspects
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kobold, S.; Guhr, A.; Mah, N.; Bultjer, N.; Seltmann, S.; Seiler Wulczyn, A.E.M.; Stacey, G.; Jie, H.; Liu, W.; Loser, P.; et al. A Manually Curated Database on Clinical Studies Involving Cell Products Derived from Human Pluripotent Stem Cells. Stem Cell Rep. 2020, 15, 546–555. [Google Scholar] [CrossRef]
- Schwartz, S.D.; Hubschman, J.P.; Heilwell, G.; Franco-Cardenas, V.; Pan, C.K.; Ostrick, R.M.; Mickunas, E.; Gay, R.; Klimanskaya, I.; Lanza, R. Embryonic stem cell trials for macular degeneration: A preliminary report. Lancet 2012, 379, 713–720. [Google Scholar] [CrossRef]
- Ben M’Barek, K.; Monville, C. Cell Therapy for Retinal Dystrophies: From Cell Suspension Formulation to Complex Retinal Tissue Bioengineering. Stem Cells Int. 2019, 2019, 4568979. [Google Scholar] [CrossRef]
- Ghareeb, A.E.; Lako, M.; Steel, D.H. Coculture techniques for modeling retinal development and disease, and enabling regenerative medicine. Stem Cells Transl. Med. 2020, 9, 1531–1548. [Google Scholar] [CrossRef]
- Sun, B.K.; Siprashvili, Z.; Khavari, P.A. Advances in skin grafting and treatment of cutaneous wounds. Science 2014, 346, 941–945. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Mandai, M.; Kamao, H.; Hashiguchi, T.; Shikamura, M.; Kawamata, S.; Sugita, S.; Takahashi, M. Protective Effects of Human iPS-Derived Retinal Pigmented Epithelial Cells in Comparison with Human Mesenchymal Stromal Cells and Human Neural Stem Cells on the Degenerating Retina in rd1 mice. Stem Cells 2015, 33, 1543–1553. [Google Scholar] [CrossRef] [PubMed]
- Morizur, L.; Herardot, E.; Monville, C.; Ben M’Barek, K. Human pluripotent stem cells: A toolbox to understand and treat retinal degeneration. Mol. Cell Neurosci. 2020, 107, 103523. [Google Scholar] [CrossRef] [PubMed]
- Hargett, L.A.; Bauer, N.N. On the origin of microparticles: From “platelet dust” to mediators of intercellular communication. Pulm. Circ. 2013, 3, 329–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiklander, O.P.B.; Brennan, M.A.; Lotvall, J.; Breakefield, X.O.; El Andaloussi, S. Advances in therapeutic applications of extracellular vesicles. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- S, E.L.A.; Mager, I.; Breakefield, X.O.; Wood, M.J. Extracellular vesicles: Biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 2013, 12, 347–357. [Google Scholar] [CrossRef]
- Bachoud-Levi, A.C.; Perrier, A.L. Regenerative medicine in Huntington’s disease: Current status on fetal grafts and prospects for the use of pluripotent stem cell. Rev. Neurol. (Paris) 2014, 170, 749–762. [Google Scholar] [CrossRef] [PubMed]
- Bachoud-Levi, A.C.; Massart, R.; Rosser, A. Cell therapy in Huntington’s disease: Taking stock of past studies to move the field forward. Stem Cells 2020. [Google Scholar] [CrossRef]
- Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Okita, K.; Mochiduki, Y.; Takizawa, N.; Yamanaka, S. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat. Biotechnol. 2008, 26, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [Green Version]
- Flaxman, S.R.; Bourne, R.R.A.; Resnikoff, S.; Ackland, P.; Braithwaite, T.; Cicinelli, M.V.; Das, A.; Jonas, J.B.; Keeffe, J.; Kempen, J.H.; et al. Global causes of blindness and distance vision impairment 1990-2020: A systematic review and meta-analysis. Lancet Glob. Health 2017, 5, e1221–e1234. [Google Scholar] [CrossRef] [Green Version]
- Gagliardi, G.; Ben M’Barek, K.; Goureau, O. Photoreceptor cell replacement in macular degeneration and retinitis pigmentosa: A pluripotent stem cell-based approach. Prog. Retin. Eye Res. 2019, 71, 1–25. [Google Scholar] [CrossRef]
- Ben M’Barek, K.; Habeler, W.; Plancheron, A.; Jarraya, M.; Regent, F.; Terray, A.; Yang, Y.; Chatrousse, L.; Domingues, S.; Masson, Y.; et al. Human ESC-derived retinal epithelial cell sheets potentiate rescue of photoreceptor cell loss in rats with retinal degeneration. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Klimanskaya, I.; Hipp, J.; Rezai, K.A.; West, M.; Atala, A.; Lanza, R. Derivation and comparative assessment of retinal pigment epithelium from human embryonic stem cells using transcriptomics. Cloning Stem Cells 2004, 6, 217–245. [Google Scholar] [CrossRef]
- Buchholz, D.E.; Hikita, S.T.; Rowland, T.J.; Friedrich, A.M.; Hinman, C.R.; Johnson, L.V.; Clegg, D.O. Derivation of functional retinal pigmented epithelium from induced pluripotent stem cells. Stem Cells 2009, 27, 2427–2434. [Google Scholar] [CrossRef]
- Buchholz, D.E.; Pennington, B.O.; Croze, R.H.; Hinman, C.R.; Coffey, P.J.; Clegg, D.O. Rapid and efficient directed differentiation of human pluripotent stem cells into retinal pigmented epithelium. Stem Cells Transl. Med. 2013, 2, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Leach, L.L.; Buchholz, D.E.; Nadar, V.P.; Lowenstein, S.E.; Clegg, D.O. Canonical/beta-catenin Wnt pathway activation improves retinal pigmented epithelium derivation from human embryonic stem cells. Investig. Ophthalmol. Vis. Sci. 2015, 56, 1002–1013. [Google Scholar] [CrossRef] [PubMed]
- Idelson, M.; Alper, R.; Obolensky, A.; Ben-Shushan, E.; Hemo, I.; Yachimovich-Cohen, N.; Khaner, H.; Smith, Y.; Wiser, O.; Gropp, M.; et al. Directed differentiation of human embryonic stem cells into functional retinal pigment epithelium cells. Cell Stem Cell 2009, 5, 396–408. [Google Scholar] [CrossRef] [Green Version]
- Maruotti, J.; Sripathi, S.R.; Bharti, K.; Fuller, J.; Wahlin, K.J.; Ranganathan, V.; Sluch, V.M.; Berlinicke, C.A.; Davis, J.; Kim, C.; et al. Small-molecule-directed, efficient generation of retinal pigment epithelium from human pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2015, 112, 10950–10955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crombie, D.E.; Daniszewski, M.; Liang, H.H.; Kulkarni, T.; Li, F.; Lidgerwood, G.E.; Conquest, A.; Hernandez, D.; Hung, S.S.; Gill, K.P.; et al. Development of a Modular Automated System for Maintenance and Differentiation of Adherent Human Pluripotent Stem Cells. SLAS Discov. 2017, 22, 1016–1025. [Google Scholar] [CrossRef] [Green Version]
- Regent, F.; Morizur, L.; Lesueur, L.; Habeler, W.; Plancheron, A.; Ben M’Barek, K.; Monville, C. Automation of human pluripotent stem cell differentiation toward retinal pigment epithelial cells for large-scale productions. Sci. Rep. 2019, 9, 10646. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, E.; Koide, N.; Hanzawa, H.; Kiyama, M.; Ohta, M.; Kuwabara, J.; Takeda, S.; Takahashi, M. Fabricating retinal pigment epithelial cell sheets derived from human induced pluripotent stem cells in an automated closed culture system for regenerative medicine. PLoS ONE 2019, 14, e0212369. [Google Scholar] [CrossRef] [Green Version]
- Carr, A.J.; Vugler, A.A.; Hikita, S.T.; Lawrence, J.M.; Gias, C.; Chen, L.L.; Buchholz, D.E.; Ahmado, A.; Semo, M.; Smart, M.J.; et al. Protective effects of human iPS-derived retinal pigment epithelium cell transplantation in the retinal dystrophic rat. PLoS ONE 2009, 4, e8152. [Google Scholar] [CrossRef]
- Lu, B.; Malcuit, C.; Wang, S.; Girman, S.; Francis, P.; Lemieux, L.; Lanza, R.; Lund, R. Long-term safety and function of RPE from human embryonic stem cells in preclinical models of macular degeneration. Stem Cells 2009, 27, 2126–2135. [Google Scholar] [CrossRef]
- Lund, R.D.; Wang, S.; Klimanskaya, I.; Holmes, T.; Ramos-Kelsey, R.; Lu, B.; Girman, S.; Bischoff, N.; Sauve, Y.; Lanza, R. Human embryonic stem cell-derived cells rescue visual function in dystrophic RCS rats. Cloning Stem Cells 2006, 8, 189–199. [Google Scholar] [CrossRef] [Green Version]
- Diniz, B.; Thomas, P.; Thomas, B.; Ribeiro, R.; Hu, Y.; Brant, R.; Ahuja, A.; Zhu, D.; Liu, L.; Koss, M.; et al. Subretinal implantation of retinal pigment epithelial cells derived from human embryonic stem cells: Improved survival when implanted as a monolayer. Investig. Ophthalmol. Vis. Sci. 2013, 54, 5087–5096. [Google Scholar] [CrossRef] [Green Version]
- Sharma, R.; Khristov, V.; Rising, A.; Jha, B.S.; Dejene, R.; Hotaling, N.; Li, Y.; Stoddard, J.; Stankewicz, C.; Wan, Q.; et al. Clinical-grade stem cell-derived retinal pigment epithelium patch rescues retinal degeneration in rodents and pigs. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Stanzel, B.V.; Liu, Z.; Somboonthanakij, S.; Wongsawad, W.; Brinken, R.; Eter, N.; Corneo, B.; Holz, F.G.; Temple, S.; Stern, J.H.; et al. Human RPE stem cells grown into polarized RPE monolayers on a polyester matrix are maintained after grafting into rabbit subretinal space. Stem Cell Rep. 2014, 2, 64–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Cruz, L.; Fynes, K.; Georgiadis, O.; Kerby, J.; Luo, Y.H.; Ahmado, A.; Vernon, A.; Daniels, J.T.; Nommiste, B.; Hasan, S.M.; et al. Phase 1 clinical study of an embryonic stem cell-derived retinal pigment epithelium patch in age-related macular degeneration. Nat. Biotechnol. 2018, 36, 328–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandes, R.A.B.; Stefanini, F.R.; Falabella, P.; Koss, M.J.; Wells, T.; Diniz, B.; Ribeiro, R.; Schor, P.; Maia, M.; Penha, F.M.; et al. Development of a new tissue injector for subretinal transplantation of human embryonic stem cell derived retinal pigmented epithelium. Int. J. Retina Vitr. 2017, 3, 41. [Google Scholar] [CrossRef] [Green Version]
- Ben M’Barek, K.; Bertin, S.; Brazhnikova, E.; Jaillard, C.; Habeler, W.; Plancheron, A.; Fovet, C.M.; Demilly, J.; Jarraya, M.; Bejanariu, A.; et al. Clinical-grade production and safe delivery of human ESC derived RPE sheets in primates and rodents. Biomaterials 2020, 230, 119603. [Google Scholar] [CrossRef]
- Nakano, T.; Ando, S.; Takata, N.; Kawada, M.; Muguruma, K.; Sekiguchi, K.; Saito, K.; Yonemura, S.; Eiraku, M.; Sasai, Y. Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell 2012, 10, 771–785. [Google Scholar] [CrossRef] [Green Version]
- Zhong, X.; Gutierrez, C.; Xue, T.; Hampton, C.; Vergara, M.N.; Cao, L.H.; Peters, A.; Park, T.S.; Zambidis, E.T.; Meyer, J.S.; et al. Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat. Commun. 2014, 5, 4047. [Google Scholar] [CrossRef] [Green Version]
- Capowski, E.E.; Samimi, K.; Mayerl, S.J.; Phillips, M.J.; Pinilla, I.; Howden, S.E.; Saha, J.; Jansen, A.D.; Edwards, K.L.; Jager, L.D.; et al. Reproducibility and staging of 3D human retinal organoids across multiple pluripotent stem cell lines. Development 2019, 146. [Google Scholar] [CrossRef] [Green Version]
- Reichman, S.; Slembrouck, A.; Gagliardi, G.; Chaffiol, A.; Terray, A.; Nanteau, C.; Potey, A.; Belle, M.; Rabesandratana, O.; Duebel, J.; et al. Generation of Storable Retinal Organoids and Retinal Pigmented Epithelium from Adherent Human iPS Cells in Xeno-Free and Feeder-Free Conditions. Stem Cells 2017, 35, 1176–1188. [Google Scholar] [CrossRef] [Green Version]
- Welby, E.; Lakowski, J.; Di Foggia, V.; Budinger, D.; Gonzalez-Cordero, A.; Lun, A.T.L.; Epstein, M.; Patel, A.; Cuevas, E.; Kruczek, K.; et al. Isolation and Comparative Transcriptome Analysis of Human Fetal and iPSC-Derived Cone Photoreceptor Cells. Stem Cell Rep. 2017, 9, 1898–1915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacLaren, R.E.; Pearson, R.A.; MacNeil, A.; Douglas, R.H.; Salt, T.E.; Akimoto, M.; Swaroop, A.; Sowden, J.C.; Ali, R.R. Retinal repair by transplantation of photoreceptor precursors. Nature 2006, 444, 203–207. [Google Scholar] [CrossRef]
- Singh, M.S.; Balmer, J.; Barnard, A.R.; Aslam, S.A.; Moralli, D.; Green, C.M.; Barnea-Cramer, A.; Duncan, I.; MacLaren, R.E. Transplanted photoreceptor precursors transfer proteins to host photoreceptors by a mechanism of cytoplasmic fusion. Nat. Commun. 2016, 7, 13537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos-Ferreira, T.; Llonch, S.; Borsch, O.; Postel, K.; Haas, J.; Ader, M. Retinal transplantation of photoreceptors results in donor-host cytoplasmic exchange. Nat. Commun. 2016, 7, 13028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decembrini, S.; Martin, C.; Sennlaub, F.; Chemtob, S.; Biel, M.; Samardzija, M.; Moulin, A.; Behar-Cohen, F.; Arsenijevic, Y. Cone Genesis Tracing by the Chrnb4-EGFP Mouse Line: Evidences of Cellular Material Fusion after Cone Precursor Transplantation. Mol. Ther. 2017, 25, 634–653. [Google Scholar] [CrossRef] [Green Version]
- Tu, H.Y.; Watanabe, T.; Shirai, H.; Yamasaki, S.; Kinoshita, M.; Matsushita, K.; Hashiguchi, T.; Onoe, H.; Matsuyama, T.; Kuwahara, A.; et al. Medium- to long-term survival and functional examination of human iPSC-derived retinas in rat and primate models of retinal degeneration. EBioMedicine 2019, 39, 562–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iraha, S.; Tu, H.Y.; Yamasaki, S.; Kagawa, T.; Goto, M.; Takahashi, R.; Watanabe, T.; Sugita, S.; Yonemura, S.; Sunagawa, G.A.; et al. Establishment of Immunodeficient Retinal Degeneration Model Mice and Functional Maturation of Human ESC-Derived Retinal Sheets after Transplantation. Stem Cell Rep. 2018, 10, 1059–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLelland, B.T.; Lin, B.; Mathur, A.; Aramant, R.B.; Thomas, B.B.; Nistor, G.; Keirstead, H.S.; Seiler, M.J. Transplanted hESC-Derived Retina Organoid Sheets Differentiate, Integrate, and Improve Visual Function in Retinal Degenerate Rats. Investig. Ophthalmol. Vis. Sci. 2018, 59, 2586–2603. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.H.; Phillips, M.J.; Lee, J.; Xie, R.; Ludwig, A.L.; Chen, G.; Zheng, Q.; Kim, T.J.; Zhang, H.; Barney, P.; et al. 3D Microstructured Scaffolds to Support Photoreceptor Polarization and Maturation. Adv. Mater. 2018, 30, e1803550. [Google Scholar] [CrossRef]
- Worthington, K.S.; Wiley, L.A.; Kaalberg, E.E.; Collins, M.M.; Mullins, R.F.; Stone, E.M.; Tucker, B.A. Two-photon polymerization for production of human iPSC-derived retinal cell grafts. Acta Biomater. 2017, 55, 385–395. [Google Scholar] [CrossRef]
- Thompson, J.R.; Worthington, K.S.; Green, B.J.; Mullin, N.K.; Jiao, C.; Kaalberg, E.E.; Wiley, L.A.; Han, I.C.; Russell, S.R.; Sohn, E.H.; et al. Two-photon polymerized poly(caprolactone) retinal cell delivery scaffolds and their systemic and retinal biocompatibility. Acta Biomater. 2019, 94, 204–218. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Wang, S.B.; Xia, T.; Tainsh, L.; Ghiassi-Nejad, M.; Xu, T.; Peng, S.; Adelman, R.A.; Rizzolo, L.J. A biodegradable scaffold enhances differentiation of embryonic stem cells into a thick sheet of retinal cells. Biomaterials 2018, 154, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Garita-Hernandez, M.; Lampic, M.; Chaffiol, A.; Guibbal, L.; Routet, F.; Santos-Ferreira, T.; Gasparini, S.; Borsch, O.; Gagliardi, G.; Reichman, S.; et al. Restoration of visual function by transplantation of optogenetically engineered photoreceptors. Nat. Commun. 2019, 10, 4524. [Google Scholar] [CrossRef] [Green Version]
- Chu, G.Y.; Chen, Y.F.; Chen, H.Y.; Chan, M.H.; Gau, C.S.; Weng, S.M. Stem cell therapy on skin: Mechanisms, recent advances and drug reviewing issues. J. Food Drug Anal. 2018, 26, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Gurtner, G.C.; Werner, S.; Barrandon, Y.; Longaker, M.T. Wound repair and regeneration. Nature 2008, 453, 314–321. [Google Scholar] [CrossRef]
- Przekora, A. A Concise Review on Tissue Engineered Artificial Skin Grafts for Chronic Wound Treatment: Can We Reconstruct Functional Skin Tissue In Vitro? Cells 2020, 9, 1622. [Google Scholar] [CrossRef]
- Dai, C.; Shih, S.; Khachemoune, A. Skin substitutes for acute and chronic wound healing: An updated review. J. Dermatolog. Treat. 2020, 31, 639–648. [Google Scholar] [CrossRef]
- Nourian Dehkordi, A.; Mirahmadi Babaheydari, F.; Chehelgerdi, M.; Raeisi Dehkordi, S. Skin tissue engineering: Wound healing based on stem-cell-based therapeutic strategies. Stem Cell Res. Ther. 2019, 10, 111. [Google Scholar] [CrossRef] [Green Version]
- Vig, K.; Chaudhari, A.; Tripathi, S.; Dixit, S.; Sahu, R.; Pillai, S.; Dennis, V.A.; Singh, S.R. Advances in Skin Regeneration Using Tissue Engineering. Int. J. Mol. Sci. 2017, 18, 789. [Google Scholar] [CrossRef]
- Guenou, H.; Nissan, X.; Larcher, F.; Feteira, J.; Lemaitre, G.; Saidani, M.; Del Rio, M.; Barrault, C.C.; Bernard, F.X.; Peschanski, M.; et al. Human embryonic stem-cell derivatives for full reconstruction of the pluristratified epidermis: A preclinical study. Lancet 2009, 374, 1745–1753. [Google Scholar] [CrossRef]
- Umegaki-Arao, N.; Pasmooij, A.M.; Itoh, M.; Cerise, J.E.; Guo, Z.; Levy, B.; Gostynski, A.; Rothman, L.R.; Jonkman, M.F.; Christiano, A.M. Induced pluripotent stem cells from human revertant keratinocytes for the treatment of epidermolysis bullosa. Sci. Transl. Med. 2014, 6, 264ra164. [Google Scholar] [CrossRef] [PubMed]
- Sebastiano, V.; Zhen, H.H.; Haddad, B.; Bashkirova, E.; Melo, S.P.; Wang, P.; Leung, T.L.; Siprashvili, Z.; Tichy, A.; Li, J.; et al. Human COL7A1-corrected induced pluripotent stem cells for the treatment of recessive dystrophic epidermolysis bullosa. Sci. Transl. Med. 2014, 6, 264ra163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazaki, H.; Tsunoi, Y.; Akagi, T.; Sato, S.; Akashi, M.; Saitoh, D. A novel strategy to engineer pre-vascularized 3-dimensional skin substitutes to achieve efficient, functional engraftment. Sci. Rep. 2019, 9, 7797. [Google Scholar] [CrossRef] [PubMed]
- Itoh, M.; Umegaki-Arao, N.; Guo, Z.; Liu, L.; Higgins, C.A.; Christiano, A.M. Generation of 3D skin equivalents fully reconstituted from human induced pluripotent stem cells (iPSCs). PLoS ONE 2013, 8, e77673. [Google Scholar] [CrossRef] [Green Version]
- Abaci, H.E.; Guo, Z.; Coffman, A.; Gillette, B.; Lee, W.H.; Sia, S.K.; Christiano, A.M. Human Skin Constructs with Spatially Controlled Vasculature Using Primary and iPSC-Derived Endothelial Cells. Adv. Healthc. Mater. 2016, 5, 1800–1807. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.S.; Kwon, Y.W.; Kong, J.S.; Park, G.T.; Gao, G.; Han, W.; Kim, M.B.; Lee, H.; Kim, J.H.; Cho, D.W. 3D cell printing of in vitro stabilized skin model and in vivo pre-vascularized skin patch using tissue-specific extracellular matrix bioink: A step towards advanced skin tissue engineering. Biomaterials 2018, 168, 38–53. [Google Scholar] [CrossRef]
- Hosaka, C.; Kunisada, M.; Koyanagi-Aoi, M.; Masaki, T.; Takemori, C.; Taniguchi-Ikeda, M.; Aoi, T.; Nishigori, C. Induced pluripotent stem cell-derived melanocyte precursor cells undergoing differentiation into melanocytes. Pigment Cell Melanoma Res. 2019, 32, 623–633. [Google Scholar] [CrossRef]
- Ohta, S.; Imaizumi, Y.; Okada, Y.; Akamatsu, W.; Kuwahara, R.; Ohyama, M.; Amagai, M.; Matsuzaki, Y.; Yamanaka, S.; Okano, H.; et al. Generation of human melanocytes from induced pluripotent stem cells. PLoS ONE 2011, 6, e16182. [Google Scholar] [CrossRef]
- Nissan, X.; Larribere, L.; Saidani, M.; Hurbain, I.; Delevoye, C.; Feteira, J.; Lemaitre, G.; Peschanski, M.; Baldeschi, C. Functional melanocytes derived from human pluripotent stem cells engraft into pluristratified epidermis. Proc. Natl. Acad. Sci. USA 2011, 108, 14861–14866. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Rabbani, C.C.; Gao, H.; Steinhart, M.R.; Woodruff, B.M.; Pflum, Z.E.; Kim, A.; Heller, S.; Liu, Y.; Shipchandler, T.Z.; et al. Hair-bearing human skin generated entirely from pluripotent stem cells. Nature 2020, 582, 399–404. [Google Scholar] [CrossRef]
- Yang, R.; Zheng, Y.; Burrows, M.; Liu, S.; Wei, Z.; Nace, A.; Guo, W.; Kumar, S.; Cotsarelis, G.; Xu, X. Generation of folliculogenic human epithelial stem cells from induced pluripotent stem cells. Nat. Commun. 2014, 5, 3071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GBD 2017 Causes of Death Collaborators. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1736–1788. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Murry, C.E. Function Follows Form- A Review of Cardiac Cell Therapy. Circ. J. 2019, 83, 2399–2412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, P.; Lemcke, H.; David, R. Stem Cell Therapy in Heart Diseases—Cell Types, Mechanisms and Improvement Strategies. Cell Physiol. Biochem. 2018, 48, 2607–2655. [Google Scholar] [CrossRef]
- Kadota, S.; Tanaka, Y.; Shiba, Y. Heart regeneration using pluripotent stem cells. J. Cardiol. 2020, 76, 459–463. [Google Scholar] [CrossRef]
- Yang, L.; Soonpaa, M.H.; Adler, E.D.; Roepke, T.K.; Kattman, S.J.; Kennedy, M.; Henckaerts, E.; Bonham, K.; Abbott, G.W.; Linden, R.M.; et al. Human cardiovascular progenitor cells develop from a KDR+ embryonic-stem-cell-derived population. Nature 2008, 453, 524–528. [Google Scholar] [CrossRef]
- Laflamme, M.A.; Chen, K.Y.; Naumova, A.V.; Muskheli, V.; Fugate, J.A.; Dupras, S.K.; Reinecke, H.; Xu, C.; Hassanipour, M.; Police, S.; et al. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat. Biotechnol. 2007, 25, 1015–1024. [Google Scholar] [CrossRef]
- Karakikes, I.; Ameen, M.; Termglinchan, V.; Wu, J.C. Human induced pluripotent stem cell-derived cardiomyocytes: Insights into molecular, cellular, and functional phenotypes. Circ. Res. 2015, 117, 80–88. [Google Scholar] [CrossRef] [Green Version]
- Machiraju, P.; Greenway, S.C. Current methods for the maturation of induced pluripotent stem cell-derived cardiomyocytes. World J. Stem Cells 2019, 11, 33–43. [Google Scholar] [CrossRef]
- Burridge, P.W.; Matsa, E.; Shukla, P.; Lin, Z.C.; Churko, J.M.; Ebert, A.D.; Lan, F.; Diecke, S.; Huber, B.; Mordwinkin, N.M.; et al. Chemically defined generation of human cardiomyocytes. Nat. Methods 2014, 11, 855–860. [Google Scholar] [CrossRef] [Green Version]
- Kempf, H.; Kropp, C.; Olmer, R.; Martin, U.; Zweigerdt, R. Cardiac differentiation of human pluripotent stem cells in scalable suspension culture. Nat. Protoc. 2015, 10, 1345–1361. [Google Scholar] [CrossRef] [PubMed]
- Verma, V.; Purnamawati, K.; Manasi; Shim, W. Steering signal transduction pathway towards cardiac lineage from human pluripotent stem cells: A review. Cell Signal 2013, 25, 1096–1107. [Google Scholar] [CrossRef] [PubMed]
- Ellis, P.; Fagan, B.M.; Magness, S.T.; Hutton, S.; Taranova, O.; Hayashi, S.; McMahon, A.; Rao, M.; Pevny, L. SOX2, a persistent marker for multipotential neural stem cells derived from embryonic stem cells, the embryo or the adult. Dev. Neurosci. 2004, 26, 148–165. [Google Scholar] [CrossRef] [PubMed]
- Lian, X.; Hsiao, C.; Wilson, G.; Zhu, K.; Hazeltine, L.B.; Azarin, S.M.; Raval, K.K.; Zhang, J.; Kamp, T.J.; Palecek, S.P. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc. Natl. Acad. Sci. USA 2012, 109, E1848–E1857. [Google Scholar] [CrossRef] [Green Version]
- Kadota, S.; Minami, I.; Morone, N.; Heuser, J.E.; Agladze, K.; Nakatsuji, N. Development of a reentrant arrhythmia model in human pluripotent stem cell-derived cardiac cell sheets. Eur. Heart J. 2013, 34, 1147–1156. [Google Scholar] [CrossRef] [Green Version]
- Menasche, P.; Vanneaux, V.; Hagege, A.; Bel, A.; Cholley, B.; Parouchev, A.; Cacciapuoti, I.; Al-Daccak, R.; Benhamouda, N.; Blons, H.; et al. Transplantation of Human Embryonic Stem Cell-Derived Cardiovascular Progenitors for Severe Ischemic Left Ventricular Dysfunction. J. Am. Coll. Cardiol. 2018, 71, 429–438. [Google Scholar] [CrossRef]
- Guan, X.; Xu, W.; Zhang, H.; Wang, Q.; Yu, J.; Zhang, R.; Chen, Y.; Xia, Y.; Wang, J.; Wang, D. Transplantation of human induced pluripotent stem cell-derived cardiomyocytes improves myocardial function and reverses ventricular remodeling in infarcted rat hearts. Stem Cell Res. Ther. 2020, 11, 73. [Google Scholar] [CrossRef] [Green Version]
- Shiba, Y.; Gomibuchi, T.; Seto, T.; Wada, Y.; Ichimura, H.; Tanaka, Y.; Ogasawara, T.; Okada, K.; Shiba, N.; Sakamoto, K.; et al. Allogeneic transplantation of iPS cell-derived cardiomyocytes regenerates primate hearts. Nature 2016, 538, 388–391. [Google Scholar] [CrossRef]
- Liu, Y.W.; Chen, B.; Yang, X.; Fugate, J.A.; Kalucki, F.A.; Futakuchi-Tsuchida, A.; Couture, L.; Vogel, K.W.; Astley, C.A.; Baldessari, A.; et al. Erratum: Human embryonic stem cell-derived cardiomyocytes restore function in infarcted hearts of non-human primates. Nat. Biotechnol. 2018, 36, 899. [Google Scholar] [CrossRef]
- Chong, J.J.; Yang, X.; Don, C.W.; Minami, E.; Liu, Y.W.; Weyers, J.J.; Mahoney, W.M.; Van Biber, B.; Cook, S.M.; Palpant, N.J.; et al. Human embryonic-stem-cell-derived cardiomyocytes regenerate non-human primate hearts. Nature 2014, 510, 273–277. [Google Scholar] [CrossRef]
- Ishida, M.; Miyagawa, S.; Saito, A.; Fukushima, S.; Harada, A.; Ito, E.; Ohashi, F.; Watabe, T.; Hatazawa, J.; Matsuura, K.; et al. Transplantation of Human-induced Pluripotent Stem Cell-derived Cardiomyocytes Is Superior to Somatic Stem Cell Therapy for Restoring Cardiac Function and Oxygen Consumption in a Porcine Model of Myocardial Infarction. Transplantation 2019, 103, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Romagnuolo, R.; Masoudpour, H.; Porta-Sanchez, A.; Qiang, B.; Barry, J.; Laskary, A.; Qi, X.; Masse, S.; Magtibay, K.; Kawajiri, H.; et al. Human Embryonic Stem Cell-Derived Cardiomyocytes Regenerate the Infarcted Pig Heart but Induce Ventricular Tachyarrhythmias. Stem Cell Rep. 2019, 12, 967–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kehat, I.; Khimovich, L.; Caspi, O.; Gepstein, A.; Shofti, R.; Arbel, G.; Huber, I.; Satin, J.; Itskovitz-Eldor, J.; Gepstein, L. Electromechanical integration of cardiomyocytes derived from human embryonic stem cells. Nat. Biotechnol. 2004, 22, 1282–1289. [Google Scholar] [CrossRef]
- Zhang, J.Z.; Termglinchan, V.; Shao, N.Y.; Itzhaki, I.; Liu, C.; Ma, N.; Tian, L.; Wang, V.Y.; Chang, A.C.Y.; Guo, H.; et al. A Human iPSC Double-Reporter System Enables Purification of Cardiac Lineage Subpopulations with Distinct Function and Drug Response Profiles. Cell Stem Cell 2019, 24, 802–811. [Google Scholar] [CrossRef]
- Gerbin, K.A.; Yang, X.; Murry, C.E.; Coulombe, K.L. Enhanced Electrical Integration of Engineered Human Myocardium via Intramyocardial versus Epicardial Delivery in Infarcted Rat Hearts. PLoS ONE 2015, 10, e0131446. [Google Scholar] [CrossRef] [Green Version]
- Bargehr, J.; Ong, L.P.; Colzani, M.; Davaapil, H.; Hofsteen, P.; Bhandari, S.; Gambardella, L.; Le Novere, N.; Iyer, D.; Sampaziotis, F.; et al. Epicardial cells derived from human embryonic stem cells augment cardiomyocyte-driven heart regeneration. Nat. Biotechnol. 2019, 37, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Kim, R.Y.; Park, B.W.; Lee, S.; Choi, S.W.; Park, J.H.; Choi, J.J.; Kim, S.W.; Jang, J.; Cho, D.W.; et al. Dual stem cell therapy synergistically improves cardiac function and vascular regeneration following myocardial infarction. Nat. Commun. 2019, 10, 3123. [Google Scholar] [CrossRef] [Green Version]
- Judson, R.N.; Rossi, F.M.V. Towards stem cell therapies for skeletal muscle repair. NPJ Regen. Med. 2020, 5, 10. [Google Scholar] [CrossRef]
- Chal, J.; Pourquie, O. Making muscle: Skeletal myogenesis in vivo and in vitro. Development 2017, 144, 2104–2122. [Google Scholar] [CrossRef] [Green Version]
- Qazi, T.H.; Duda, G.N.; Ort, M.J.; Perka, C.; Geissler, S.; Winkler, T. Cell therapy to improve regeneration of skeletal muscle injuries. J. Cachexia Sarcopenia Muscle 2019, 10, 501–516. [Google Scholar] [CrossRef] [Green Version]
- Sacco, A.; Doyonnas, R.; Kraft, P.; Vitorovic, S.; Blau, H.M. Self-renewal and expansion of single transplanted muscle stem cells. Nature 2008, 456, 502–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerletti, M.; Jurga, S.; Witczak, C.A.; Hirshman, M.F.; Shadrach, J.L.; Goodyear, L.J.; Wagers, A.J. Highly efficient, functional engraftment of skeletal muscle stem cells in dystrophic muscles. Cell 2008, 134, 37–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montarras, D.; Morgan, J.; Collins, C.; Relaix, F.; Zaffran, S.; Cumano, A.; Partridge, T.; Buckingham, M. Direct isolation of satellite cells for skeletal muscle regeneration. Science 2005, 309, 2064–2067. [Google Scholar] [CrossRef] [PubMed]
- Arpke, R.W.; Darabi, R.; Mader, T.L.; Zhang, Y.; Toyama, A.; Lonetree, C.L.; Nash, N.; Lowe, D.A.; Perlingeiro, R.C.; Kyba, M. A new immuno-, dystrophin-deficient model, the NSG-mdx(4Cv) mouse, provides evidence for functional improvement following allogeneic satellite cell transplantation. Stem Cells 2013, 31, 1611–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chal, J.; Oginuma, M.; Al Tanoury, Z.; Gobert, B.; Sumara, O.; Hick, A.; Bousson, F.; Zidouni, Y.; Mursch, C.; Moncuquet, P.; et al. Differentiation of pluripotent stem cells to muscle fiber to model Duchenne muscular dystrophy. Nat. Biotechnol. 2015, 33, 962–969. [Google Scholar] [CrossRef] [Green Version]
- Chal, J.; Al Tanoury, Z.; Hestin, M.; Gobert, B.; Aivio, S.; Hick, A.; Cherrier, T.; Nesmith, A.P.; Parker, K.K.; Pourquie, O. Generation of human muscle fibers and satellite-like cells from human pluripotent stem cells in vitro. Nat. Protoc. 2016, 11, 1833–1850. [Google Scholar] [CrossRef] [Green Version]
- Hicks, M.R.; Hiserodt, J.; Paras, K.; Fujiwara, W.; Eskin, A.; Jan, M.; Xi, H.; Young, C.S.; Evseenko, D.; Nelson, S.F.; et al. ERBB3 and NGFR mark a distinct skeletal muscle progenitor cell in human development and hPSCs. Nat. Cell. Biol. 2018, 20, 46–57. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Matthias, N.; Lo, J.; Ortiz-Vitali, J.L.; Shieh, A.W.; Wang, S.H.; Darabi, R. A Myogenic Double-Reporter Human Pluripotent Stem Cell Line Allows Prospective Isolation of Skeletal Muscle Progenitors. Cell Rep. 2018, 25, 1966–1981.e4. [Google Scholar] [CrossRef] [Green Version]
- van der Wal, E.; Herrero-Hernandez, P.; Wan, R.; Broeders, M.; In ‘t Groen, S.L.M.; van Gestel, T.J.M.; van, I.W.F.J.; Cheung, T.H.; van der Ploeg, A.T.; Schaaf, G.J.; et al. Large-Scale Expansion of Human iPSC-Derived Skeletal Muscle Cells for Disease Modeling and Cell-Based Therapeutic Strategies. Stem Cell Rep. 2018, 10, 1975–1990. [Google Scholar] [CrossRef]
- Quarta, M.; Cromie, M.; Chacon, R.; Blonigan, J.; Garcia, V.; Akimenko, I.; Hamer, M.; Paine, P.; Stok, M.; Shrager, J.B.; et al. Bioengineered constructs combined with exercise enhance stem cell-mediated treatment of volumetric muscle loss. Nat. Commun. 2017, 8, 15613. [Google Scholar] [CrossRef]
- Wolf, M.T.; Dearth, C.L.; Sonnenberg, S.B.; Loboa, E.G.; Badylak, S.F. Naturally derived and synthetic scaffolds for skeletal muscle reconstruction. Adv. Drug Deliv. Rev. 2015, 84, 208–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alper, J. Geron gets green light for human trial of ES cell-derived product. Nat. Biotechnol. 2009, 27, 213–214. [Google Scholar] [CrossRef]
- Scott, C.T.; Magnus, D. Wrongful termination: Lessons from the Geron clinical trial. Stem Cells Transl. Med. 2014, 3, 1398–1401. [Google Scholar] [CrossRef] [PubMed]
- Desgres, M.; Menasche, P. Clinical Translation of Pluripotent Stem Cell Therapies: Challenges and Considerations. Cell Stem Cell 2019, 25, 594–606. [Google Scholar] [CrossRef]
- Qiu, T.G. Transplantation of human embryonic stem cell-derived retinal pigment epithelial cells (MA09-hRPE) in macular degeneration. NPJ Regen. Med. 2019, 4, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashani, A.H.; Lebkowski, J.S.; Rahhal, F.M.; Avery, R.L.; Salehi-Had, H.; Dang, W.; Lin, C.M.; Mitra, D.; Zhu, D.; Thomas, B.B.; et al. A bioengineered retinal pigment epithelial monolayer for advanced, dry age-related macular degeneration. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Mandai, M.; Watanabe, A.; Kurimoto, Y.; Hirami, Y.; Morinaga, C.; Daimon, T.; Fujihara, M.; Akimaru, H.; Sakai, N.; Shibata, Y.; et al. Autologous Induced Stem-Cell-Derived Retinal Cells for Macular Degeneration. N. Engl. J. Med. 2017, 376, 1038–1046. [Google Scholar] [CrossRef]
- Sneddon, J.B.; Tang, Q.; Stock, P.; Bluestone, J.A.; Roy, S.; Desai, T.; Hebrok, M. Stem Cell Therapies for Treating Diabetes: Progress and Remaining Challenges. Cell Stem Cell 2018, 22, 810–823. [Google Scholar] [CrossRef] [Green Version]
- Bravery, C.A. Do human leukocyte antigen-typed cellular therapeutics based on induced pluripotent stem cells make commercial sense? Stem Cells Dev. 2015, 24, 1–10. [Google Scholar] [CrossRef]
- Malat, G.; Culkin, C. The ABCs of Immunosuppression: A Primer for Primary Care Physicians. Med. Clin. N. Am. 2016, 100, 505–518. [Google Scholar] [CrossRef]
- Schwartz, S.D.; Regillo, C.D.; Lam, B.L.; Eliott, D.; Rosenfeld, P.J.; Gregori, N.Z.; Hubschman, J.P.; Davis, J.L.; Heilwell, G.; Spirn, M.; et al. Human embryonic stem cell-derived retinal pigment epithelium in patients with age-related macular degeneration and Stargardt’s macular dystrophy: Follow-up of two open-label phase 1/2 studies. Lancet 2015, 385, 509–516. [Google Scholar] [CrossRef]
- Taylor, C.J.; Bolton, E.M.; Pocock, S.; Sharples, L.D.; Pedersen, R.A.; Bradley, J.A. Banking on human embryonic stem cells: Estimating the number of donor cell lines needed for HLA matching. Lancet 2005, 366, 2019–2025. [Google Scholar] [CrossRef]
- Wilmut, I.; Leslie, S.; Martin, N.G.; Peschanski, M.; Rao, M.; Trounson, A.; Turner, D.; Turner, M.L.; Yamanaka, S.; Taylor, C.J. Development of a global network of induced pluripotent stem cell haplobanks. Regen. Med. 2015, 10, 235–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, C.J.; Peacock, S.; Chaudhry, A.N.; Bradley, J.A.; Bolton, E.M. Generating an iPSC bank for HLA-matched tissue transplantation based on known donor and recipient HLA types. Cell Stem Cell 2012, 11, 147–152. [Google Scholar] [CrossRef] [Green Version]
- Sugita, S.; Mandai, M.; Hirami, Y.; Takagi, S.; Maeda, T.; Fujihara, M.; Matsuzaki, M.; Yamamoto, M.; Iseki, K.; Hayashi, N.; et al. HLA-Matched Allogeneic iPS Cells-Derived RPE Transplantation for Macular Degeneration. J. Clin. Med. 2020, 9, 2217. [Google Scholar] [CrossRef]
- Gornalusse, G.G.; Hirata, R.K.; Funk, S.E.; Riolobos, L.; Lopes, V.S.; Manske, G.; Prunkard, D.; Colunga, A.G.; Hanafi, L.A.; Clegg, D.O.; et al. HLA-E-expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nat. Biotechnol. 2017, 35, 765–772. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Wang, B.; Ono, M.; Kagita, A.; Fujii, K.; Sasakawa, N.; Ueda, T.; Gee, P.; Nishikawa, M.; Nomura, M.; et al. Targeted Disruption of HLA Genes via CRISPR-Cas9 Generates iPSCs with Enhanced Immune Compatibility. Cell Stem Cell 2019, 24, 566–578.e7. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Wang, M.; Duan, S.; Franco, P.J.; Kenty, J.H.; Hedrick, P.; Xia, Y.; Allen, A.; Ferreira, L.M.R.; Strominger, J.L.; et al. Generation of hypoimmunogenic human pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2019, 116, 10441–10446. [Google Scholar] [CrossRef] [Green Version]
- Deuse, T.; Hu, X.; Gravina, A.; Wang, D.; Tediashvili, G.; De, C.; Thayer, W.O.; Wahl, A.; Garcia, J.V.; Reichenspurner, H.; et al. Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nat. Biotechnol. 2019, 37, 252–258. [Google Scholar] [CrossRef]
- Shi, Z.D.; Tchao, J.; Wu, L.; Carman, A.J. Precision installation of a highly efficient suicide gene safety switch in human induced pluripotent stem cells. Stem Cells Transl. Med. 2020, 9, 1378–1388. [Google Scholar] [CrossRef]
- Jing, H.; He, X.; Zheng, J. Exosomes and regenerative medicine: State of the art and perspectives. Transl. Res. 2018, 196, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Brini, A.T.; Amodeo, G.; Ferreira, L.M.; Milani, A.; Niada, S.; Moschetti, G.; Franchi, S.; Borsani, E.; Rodella, L.F.; Panerai, A.E.; et al. Therapeutic effect of human adipose-derived stem cells and their secretome in experimental diabetic pain. Sci. Rep. 2017, 7, 9904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maumus, M.; Rozier, P.; Boulestreau, J.; Jorgensen, C.; Noel, D. Mesenchymal Stem Cell-Derived Extracellular Vesicles: Opportunities and Challenges for Clinical Translation. Front. Bioeng. Biotechnol. 2020, 8, 997. [Google Scholar] [CrossRef]
- Cai, Y.; Liu, W.; Lian, L.; Xu, Y.; Bai, X.; Xu, S.; Zhang, J. Stroke treatment: Is exosome therapy superior to stem cell therapy? Biochimie 2020, 179, 190–204. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, T.L.; Russell, A.J.; Riley, P. Experimental limitations of extracellular vesicle-based therapies for the treatment of myocardial infarction. Trends Cardiovasc. Med. 2020. [Google Scholar] [CrossRef]
- Fuster-Matanzo, A.; Gessler, F.; Leonardi, T.; Iraci, N.; Pluchino, S. Acellular approaches for regenerative medicine: On the verge of clinical trials with extracellular membrane vesicles? Stem Cell Res. Ther. 2015, 6, 227. [Google Scholar] [CrossRef] [Green Version]
- Thery, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Skog, J.; Wurdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Sena-Esteves, M.; Curry, W.T., Jr.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3. [Google Scholar] [CrossRef] [Green Version]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Thery, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiner, A.T.; Witwer, K.W.; van Balkom, B.W.M.; de Beer, J.; Brodie, C.; Corteling, R.L.; Gabrielsson, S.; Gimona, M.; Ibrahim, A.G.; de Kleijn, D.; et al. Concise Review: Developing Best-Practice Models for the Therapeutic Use of Extracellular Vesicles. Stem Cells Transl. Med. 2017, 6, 1730–1739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandham, S.; Su, X.; Wood, J.; Nocera, A.L.; Alli, S.C.; Milane, L.; Zimmerman, A.; Amiji, M.; Ivanov, A.R. Technologies and Standardization in Research on Extracellular Vesicles. Trends Biotechnol. 2020, 38, 1066–1098. [Google Scholar] [CrossRef] [PubMed]
- Markov, O.; Oshchepkova, A.; Mironova, N. Immunotherapy Based on Dendritic Cell-Targeted/-Derived Extracellular Vesicles-A Novel Strategy for Enhancement of the Anti-tumor Immune Response. Front. Pharmacol. 2019, 10, 1152. [Google Scholar] [CrossRef] [Green Version]
- Besse, B.; Charrier, M.; Lapierre, V.; Dansin, E.; Lantz, O.; Planchard, D.; Le Chevalier, T.; Livartoski, A.; Barlesi, F.; Laplanche, A.; et al. Dendritic cell-derived exosomes as maintenance immunotherapy after first line chemotherapy in NSCLC. Oncoimmunology 2016, 5, e1071008. [Google Scholar] [CrossRef] [Green Version]
- Mead, B.; Tomarev, S. Extracellular vesicle therapy for retinal diseases. Prog. Retin. Eye Res. 2020, 100849. [Google Scholar] [CrossRef]
- Wang, S.; Girman, S.; Lu, B.; Bischoff, N.; Holmes, T.; Shearer, R.; Wright, L.S.; Svendsen, C.N.; Gamm, D.M.; Lund, R.D. Long-term vision rescue by human neural progenitors in a rat model of photoreceptor degeneration. Investig. Ophthalmol. Vis. Sci. 2008, 49, 3201–3206. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, S.J.; Li, S.Y.; Qu, L.H.; Meng, X.H.; Wang, Y.; Xu, H.W.; Liang, Z.Q.; Yin, Z.Q. Long-term safety of human retinal progenitor cell transplantation in retinitis pigmentosa patients. Stem Cell Res. Ther. 2017, 8, 209. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Baranov, P.; Patel, S.; Ouyang, H.; Quach, J.; Wu, F.; Qiu, A.; Luo, H.; Hicks, C.; Zeng, J.; et al. Human retinal progenitor cell transplantation preserves vision. J. Biol. Chem. 2014, 289, 6362–6371. [Google Scholar] [CrossRef] [Green Version]
- Bian, B.; Zhao, C.; He, X.; Gong, Y.; Ren, C.; Ge, L.; Zeng, Y.; Li, Q.; Chen, M.; Weng, C.; et al. Exosomes derived from neural progenitor cells preserve photoreceptors during retinal degeneration by inactivating microglia. J. Extracell. Vesicles 2020, 9, 1748931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, B.; Xiao, J.; Wang, K.; So, K.F.; Tipoe, G.L.; Lin, B. Suppression of microglial activation is neuroprotective in a mouse model of human retinitis pigmentosa. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 8139–8150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, G.H.; Zhang, W.; Ma, Y.X.; Yang, J.; Chen, L.; Song, J.; Chen, S. Mesenchymal stem cells-derived exosomes ameliorate blue light stimulation in retinal pigment epithelium cells and retinal laser injury by VEGF-dependent mechanism. Int. J. Ophthalmol. 2018, 11, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Shao, H.; Su, C.; Jiang, Y.; Chen, X.; Bai, L.; Zhang, Y.; Li, Q.; Zhang, X.; Li, X. Exosomes derived from MSCs ameliorate retinal laser injury partially by inhibition of MCP-1. Sci. Rep. 2016, 6, 34562. [Google Scholar] [CrossRef] [Green Version]
- Hajrasouliha, A.R.; Jiang, G.; Lu, Q.; Lu, H.; Kaplan, H.J.; Zhang, H.G.; Shao, H. Exosomes from retinal astrocytes contain antiangiogenic components that inhibit laser-induced choroidal neovascularization. J. Biol. Chem. 2013, 288, 28058–28067. [Google Scholar] [CrossRef] [Green Version]
- Moisseiev, E.; Anderson, J.D.; Oltjen, S.; Goswami, M.; Zawadzki, R.J.; Nolta, J.A.; Park, S.S. Protective Effect of Intravitreal Administration of Exosomes Derived from Mesenchymal Stem Cells on Retinal Ischemia. Curr. Eye Res. 2017, 42, 1358–1367. [Google Scholar] [CrossRef]
- Bartsch, U.; Oriyakhel, W.; Kenna, P.F.; Linke, S.; Richard, G.; Petrowitz, B.; Humphries, P.; Farrar, G.J.; Ader, M. Retinal cells integrate into the outer nuclear layer and differentiate into mature photoreceptors after subretinal transplantation into adult mice. Exp. Eye Res. 2008, 86, 691–700. [Google Scholar] [CrossRef]
- Pearson, R.A.; Barber, A.C.; Rizzi, M.; Hippert, C.; Xue, T.; West, E.L.; Duran, Y.; Smith, A.J.; Chuang, J.Z.; Azam, S.A.; et al. Restoration of vision after transplantation of photoreceptors. Nature 2012, 485, 99–103. [Google Scholar] [CrossRef] [Green Version]
- Barber, A.C.; Hippert, C.; Duran, Y.; West, E.L.; Bainbridge, J.W.; Warre-Cornish, K.; Luhmann, U.F.; Lakowski, J.; Sowden, J.C.; Ali, R.R.; et al. Repair of the degenerate retina by photoreceptor transplantation. Proc. Natl. Acad. Sci. USA 2013, 110, 354–359. [Google Scholar] [CrossRef] [Green Version]
- Pearson, R.A.; Gonzalez-Cordero, A.; West, E.L.; Ribeiro, J.R.; Aghaizu, N.; Goh, D.; Sampson, R.D.; Georgiadis, A.; Waldron, P.V.; Duran, Y.; et al. Donor and host photoreceptors engage in material transfer following transplantation of post-mitotic photoreceptor precursors. Nat. Commun. 2016, 7, 13029. [Google Scholar] [CrossRef] [Green Version]
- Ortin-Martinez, A.; Tsai, E.L.; Nickerson, P.E.; Bergeret, M.; Lu, Y.; Smiley, S.; Comanita, L.; Wallace, V.A. A Reinterpretation of Cell Transplantation: GFP Transfer From Donor to Host Photoreceptors. Stem Cells 2017, 35, 932–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, M.S.; Park, S.S.; Albini, T.A.; Canto-Soler, M.V.; Klassen, H.; MacLaren, R.E.; Takahashi, M.; Nagiel, A.; Schwartz, S.D.; Bharti, K. Retinal stem cell transplantation: Balancing safety and potential. Prog. Retin. Eye Res. 2020, 75, 100779. [Google Scholar] [CrossRef] [PubMed]
- Wassmer, S.J.; Carvalho, L.S.; Gyorgy, B.; Vandenberghe, L.H.; Maguire, C.A. Exosome-associated AAV2 vector mediates robust gene delivery into the murine retina upon intravitreal injection. Sci. Rep. 2017, 7, 45329. [Google Scholar] [CrossRef] [PubMed]
- van der Merwe, Y.; Steketee, M.B. Extracellular Vesicles: Biomarkers, Therapeutics, and Vehicles in the Visual System. Curr. Ophthalmol. Rep. 2017, 5, 276–282. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, A.C.; Costa, T.F.; Andrade, Z.A.; Medrado, A.R. Wound healing—A literature review. An. Bras. Dermatol. 2016, 91, 614–620. [Google Scholar] [CrossRef] [Green Version]
- Ha, D.H.; Kim, H.K.; Lee, J.; Kwon, H.H.; Park, G.H.; Yang, S.H.; Jung, J.Y.; Choi, H.; Lee, J.H.; Sung, S.; et al. Mesenchymal Stem/Stromal Cell-Derived Exosomes for Immunomodulatory Therapeutics and Skin Regeneration. Cells 2020, 9, 1157. [Google Scholar] [CrossRef]
- Ferreira, A.D.F.; Gomes, D.A. Stem Cell Extracellular Vesicles in Skin Repair. Bioengineering 2018, 6, 4. [Google Scholar] [CrossRef] [Green Version]
- Hesketh, M.; Sahin, K.B.; West, Z.E.; Murray, R.Z. Macrophage Phenotypes Regulate Scar Formation and Chronic Wound Healing. Int. J. Mol. Sci. 2017, 18, 1545. [Google Scholar] [CrossRef] [Green Version]
- Ti, D.; Hao, H.; Tong, C.; Liu, J.; Dong, L.; Zheng, J.; Zhao, Y.; Liu, H.; Fu, X.; Han, W. LPS-preconditioned mesenchymal stromal cells modify macrophage polarization for resolution of chronic inflammation via exosome-shuttled let-7b. J. Transl. Med. 2015, 13, 308. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Dong, Z.; Cao, Y.; Wang, H.; Liu, S.; Liao, L.; Jin, Y.; Yuan, L.; Li, B. MSC-Derived Exosome Promotes M2 Polarization and Enhances Cutaneous Wound Healing. Stem Cells Int. 2019, 2019, 7132708. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Yu, Y.; Hou, Y.; Chai, J.; Duan, H.; Chu, W.; Zhang, H.; Hu, Q.; Du, J. Human umbilical cord mesenchymal stem cells transplantation promotes cutaneous wound healing of severe burned rats. PLoS ONE 2014, 9, e88348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Liu, L.; Yang, J.; Yu, Y.; Chai, J.; Wang, L.; Ma, L.; Yin, H. Exosome Derived From Human Umbilical Cord Mesenchymal Stem Cell Mediates MiR-181c Attenuating Burn-induced Excessive Inflammation. EBioMedicine 2016, 8, 72–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werner, S.; Krieg, T.; Smola, H. Keratinocyte-fibroblast interactions in wound healing. J. Investig. Dermatol. 2007, 127, 998–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Wang, M.; Gong, A.; Zhang, X.; Wu, X.; Zhu, Y.; Shi, H.; Wu, L.; Zhu, W.; Qian, H.; et al. HucMSC-Exosome Mediated-Wnt4 Signaling Is Required for Cutaneous Wound Healing. Stem Cells 2015, 33, 2158–2168. [Google Scholar] [CrossRef] [PubMed]
- Shabbir, A.; Cox, A.; Rodriguez-Menocal, L.; Salgado, M.; Van Badiavas, E. Mesenchymal Stem Cell Exosomes Induce Proliferation and Migration of Normal and Chronic Wound Fibroblasts, and Enhance Angiogenesis In Vitro. Stem Cells Dev. 2015, 24, 1635–1647. [Google Scholar] [CrossRef]
- Hu, L.; Wang, J.; Zhou, X.; Xiong, Z.; Zhao, J.; Yu, R.; Huang, F.; Zhang, H.; Chen, L. Exosomes derived from human adipose mensenchymal stem cells accelerates cutaneous wound healing via optimizing the characteristics of fibroblasts. Sci. Rep. 2016, 6, 32993. [Google Scholar] [CrossRef]
- Ferreira, A.D.F.; Cunha, P.D.S.; Carregal, V.M.; da Silva, P.C.; de Miranda, M.C.; Kunrath-Lima, M.; de Melo, M.I.A.; Faraco, C.C.F.; Barbosa, J.L.; Frezard, F.; et al. Extracellular Vesicles from Adipose-Derived Mesenchymal Stem/Stromal Cells Accelerate Migration and Activate AKT Pathway in Human Keratinocytes and Fibroblasts Independently of miR-205 Activity. Stem Cells Int. 2017, 2017, 9841035. [Google Scholar] [CrossRef]
- Chen, C.Y.; Rao, S.S.; Ren, L.; Hu, X.K.; Tan, Y.J.; Hu, Y.; Luo, J.; Liu, Y.W.; Yin, H.; Huang, J.; et al. Exosomal DMBT1 from human urine-derived stem cells facilitates diabetic wound repair by promoting angiogenesis. Theranostics 2018, 8, 1607–1623. [Google Scholar] [CrossRef]
- Cooper, D.R.; Wang, C.; Patel, R.; Trujillo, A.; Patel, N.A.; Prather, J.; Gould, L.J.; Wu, M.H. Human Adipose-Derived Stem Cell Conditioned Media and Exosomes Containing MALAT1 Promote Human Dermal Fibroblast Migration and Ischemic Wound Healing. Adv. Wound Care (New Rochelle) 2018, 7, 299–308. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Lee, S.K.; Kim, H.; Kim, T.M. Exosomes Secreted from Induced Pluripotent Stem Cell-Derived Mesenchymal Stem Cells Accelerate Skin Cell Proliferation. Int. J. Mol. Sci. 2018, 19, 3119. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, H.; Ebisawa, K.; Kambe, M.; Kasai, T.; Suga, H.; Nakamura, K.; Narita, Y.; Ogata, A.; Kamei, Y. Editors’ Choice Effects of exosomes derived from the induced pluripotent stem cells on skin wound healing. Nagoya J. Med. Sci. 2018, 80, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Jiao, Y.; Pan, Y.; Zhang, L.; Gong, H.; Qi, Y.; Wang, M.; Gong, H.; Shao, M.; Wang, X.; et al. Fetal Dermal Mesenchymal Stem Cell-Derived Exosomes Accelerate Cutaneous Wound Healing by Activating Notch Signaling. Stem Cells Int. 2019, 2019, 2402916. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Wu, R.; Bo, Y.; Zhang, M.; Chen, Y.; Wang, X.; Huang, M.; Liu, B.; Zhang, L. Induced pluripotent stem cells-derived microvesicles accelerate deep second-degree burn wound healing in mice through miR-16-5p-mediated promotion of keratinocytes migration. Theranostics 2020, 10, 9970–9983. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Rao, S.S.; Wang, Z.X.; Cao, J.; Tan, Y.J.; Luo, J.; Li, H.M.; Zhang, W.S.; Chen, C.Y.; Xie, H. Exosomes from human umbilical cord blood accelerate cutaneous wound healing through miR-21-3p-mediated promotion of angiogenesis and fibroblast function. Theranostics 2018, 8, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Guan, J.; Niu, X.; Hu, G.; Guo, S.; Li, Q.; Xie, Z.; Zhang, C.; Wang, Y. Exosomes released from human induced pluripotent stem cells-derived MSCs facilitate cutaneous wound healing by promoting collagen synthesis and angiogenesis. J. Transl. Med. 2015, 13, 49. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.J.; Yoo, S.M.; Park, H.H.; Lim, H.J.; Kim, Y.L.; Lee, S.; Seo, K.W.; Kang, K.S. Exosomes derived from human umbilical cord blood mesenchymal stem cells stimulates rejuvenation of human skin. Biochem. Biophys. Res. Commun. 2017, 493, 1102–1108. [Google Scholar] [CrossRef]
- Wang, L.; Hu, L.; Zhou, X.; Xiong, Z.; Zhang, C.; Shehada, H.M.A.; Hu, B.; Song, J.; Chen, L. Exosomes secreted by human adipose mesenchymal stem cells promote scarless cutaneous repair by regulating extracellular matrix remodelling. Sci. Rep. 2017, 7, 13321. [Google Scholar] [CrossRef]
- Marino, D.; Luginbuhl, J.; Scola, S.; Meuli, M.; Reichmann, E. Bioengineering dermo-epidermal skin grafts with blood and lymphatic capillaries. Sci. Transl. Med. 2014, 6, 221ra214. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Chen, C.; Hu, B.; Niu, X.; Liu, X.; Zhang, G.; Zhang, C.; Li, Q.; Wang, Y. Exosomes Derived from Human Endothelial Progenitor Cells Accelerate Cutaneous Wound Healing by Promoting Angiogenesis Through Erk1/2 Signaling. Int. J. Biol. Sci. 2016, 12, 1472–1487. [Google Scholar] [CrossRef]
- Chen, B.; Sun, Y.; Zhang, J.; Zhu, Q.; Yang, Y.; Niu, X.; Deng, Z.; Li, Q.; Wang, Y. Human embryonic stem cell-derived exosomes promote pressure ulcer healing in aged mice by rejuvenating senescent endothelial cells. Stem Cell Res. Ther. 2019, 10, 142. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Wu, X.; Zhang, X.; Sun, Y.; Yan, Y.; Shi, H.; Zhu, Y.; Wu, L.; Pan, Z.; Zhu, W.; et al. Human umbilical cord mesenchymal stem cell exosomes enhance angiogenesis through the Wnt4/beta-catenin pathway. Stem Cells Transl. Med. 2015, 4, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Qian, Z.; Liu, D.; Sun, J.; Wang, X.; Liu, H.; Xu, J.; Guo, X. GMSC-Derived Exosomes Combined with a Chitosan/Silk Hydrogel Sponge Accelerates Wound Healing in a Diabetic Rat Skin Defect Model. Front. Physiol. 2017, 8, 904. [Google Scholar] [CrossRef] [PubMed]
- Tao, S.C.; Guo, S.C.; Li, M.; Ke, Q.F.; Guo, Y.P.; Zhang, C.Q. Chitosan Wound Dressings Incorporating Exosomes Derived from MicroRNA-126-Overexpressing Synovium Mesenchymal Stem Cells Provide Sustained Release of Exosomes and Heal Full-Thickness Skin Defects in a Diabetic Rat Model. Stem Cells Transl. Med. 2017, 6, 736–747. [Google Scholar] [CrossRef] [PubMed]
- Sze, S.K.; de Kleijn, D.P.; Lai, R.C.; Khia Way Tan, E.; Zhao, H.; Yeo, K.S.; Low, T.Y.; Lian, Q.; Lee, C.N.; Mitchell, W.; et al. Elucidating the secretion proteome of human embryonic stem cell-derived mesenchymal stem cells. Mol. Cell Proteom. MCP 2007, 6, 1680–1689. [Google Scholar] [CrossRef] [Green Version]
- Timmers, L.; Lim, S.K.; Hoefer, I.E.; Arslan, F.; Lai, R.C.; van Oorschot, A.A.; Goumans, M.J.; Strijder, C.; Sze, S.K.; Choo, A.; et al. Human mesenchymal stem cell-conditioned medium improves cardiac function following myocardial infarction. Stem Cell Res. 2011, 6, 206–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timmers, L.; Lim, S.K.; Arslan, F.; Armstrong, J.S.; Hoefer, I.E.; Doevendans, P.A.; Piek, J.J.; El Oakley, R.M.; Choo, A.; Lee, C.N.; et al. Reduction of myocardial infarct size by human mesenchymal stem cell conditioned medium. Stem Cell Res. 2007, 1, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Lai, R.C.; Arslan, F.; Lee, M.M.; Sze, N.S.; Choo, A.; Chen, T.S.; Salto-Tellez, M.; Timmers, L.; Lee, C.N.; El Oakley, R.M.; et al. Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res. 2010, 4, 214–222. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.; Nickoloff, E.; Abramova, T.; Johnson, J.; Verma, S.K.; Krishnamurthy, P.; Mackie, A.R.; Vaughan, E.; Garikipati, V.N.; Benedict, C.; et al. Embryonic stem cell-derived exosomes promote endogenous repair mechanisms and enhance cardiac function following myocardial infarction. Circ. Res. 2015, 117, 52–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oskouei, B.N.; Lamirault, G.; Joseph, C.; Treuer, A.V.; Landa, S.; Da Silva, J.; Hatzistergos, K.; Dauer, M.; Balkan, W.; McNiece, I.; et al. Increased potency of cardiac stem cells compared with bone marrow mesenchymal stem cells in cardiac repair. Stem Cells Transl. Med. 2012, 1, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Li, T.S.; Cheng, K.; Malliaras, K.; Smith, R.R.; Zhang, Y.; Sun, B.; Matsushita, N.; Blusztajn, A.; Terrovitis, J.; Kusuoka, H.; et al. Direct comparison of different stem cell types and subpopulations reveals superior paracrine potency and myocardial repair efficacy with cardiosphere-derived cells. J. Am. Coll. Cardiol. 2012, 59, 942–953. [Google Scholar] [CrossRef] [Green Version]
- Kervadec, A.; Bellamy, V.; El Harane, N.; Arakelian, L.; Vanneaux, V.; Cacciapuoti, I.; Nemetalla, H.; Perier, M.C.; Toeg, H.D.; Richart, A.; et al. Cardiovascular progenitor-derived extracellular vesicles recapitulate the beneficial effects of their parent cells in the treatment of chronic heart failure. J. Heart Lung Transpl. 2016, 35, 795–807. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Lee, B.W.; Nakanishi, K.; Villasante, A.; Williamson, R.; Metz, J.; Kim, J.; Kanai, M.; Bi, L.; Brown, K.; et al. Cardiac recovery via extended cell-free delivery of extracellular vesicles secreted by cardiomyocytes derived from induced pluripotent stem cells. Nat. Biomed. Eng. 2018, 2, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Bittel, D.C.; Jaiswal, J.K. Contribution of Extracellular Vesicles in Rebuilding Injured Muscles. Front. Physiol. 2019, 10, 828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.S.; Yoon, H.I.; Lee, K.S.; Choi, Y.C.; Yang, S.H.; Kim, I.S.; Cho, Y.W. Exosomes from differentiating human skeletal muscle cells trigger myogenesis of stem cells and provide biochemical cues for skeletal muscle regeneration. J. Control. Release 2016, 222, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Kharraz, Y.; Guerra, J.; Pessina, P.; Serrano, A.L.; Munoz-Canoves, P. Understanding the process of fibrosis in Duchenne muscular dystrophy. Biomed. Res. Int. 2014, 2014, 965631. [Google Scholar] [CrossRef]
- Fry, C.S.; Kirby, T.J.; Kosmac, K.; McCarthy, J.J.; Peterson, C.A. Myogenic Progenitor Cells Control Extracellular Matrix Production by Fibroblasts during Skeletal Muscle Hypertrophy. Cell Stem Cell 2017, 20, 56–69. [Google Scholar] [CrossRef] [Green Version]
- Zanotti, S.; Gibertini, S.; Blasevich, F.; Bragato, C.; Ruggieri, A.; Saredi, S.; Fabbri, M.; Bernasconi, P.; Maggi, L.; Mantegazza, R.; et al. Exosomes and exosomal miRNAs from muscle-derived fibroblasts promote skeletal muscle fibrosis. Matrix Biol. 2018, 74, 77–100. [Google Scholar] [CrossRef]
- Wang, H.; Wang, B.; Zhang, A.; Hassounah, F.; Seow, Y.; Wood, M.; Ma, F.; Klein, J.D.; Price, S.R.; Wang, X.H. Exosome-Mediated miR-29 Transfer Reduces Muscle Atrophy and Kidney Fibrosis in Mice. Mol. Ther. 2019, 27, 571–583. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Zhang, A.; Wang, H.; Klein, J.D.; Tan, L.; Wang, Z.M.; Du, J.; Naqvi, N.; Liu, B.C.; Wang, X.H. miR-26a Limits Muscle Wasting and Cardiac Fibrosis through Exosome-Mediated microRNA Transfer in Chronic Kidney Disease. Theranostics 2019, 9, 1864–1877. [Google Scholar] [CrossRef]
- Aminzadeh, M.A.; Rogers, R.G.; Fournier, M.; Tobin, R.E.; Guan, X.; Childers, M.K.; Andres, A.M.; Taylor, D.J.; Ibrahim, A.; Ding, X.; et al. Exosome-Mediated Benefits of Cell Therapy in Mouse and Human Models of Duchenne Muscular Dystrophy. Stem Cell Rep. 2018, 10, 942–955. [Google Scholar] [CrossRef] [Green Version]
- Murphy, C.; Withrow, J.; Hunter, M.; Liu, Y.; Tang, Y.L.; Fulzele, S.; Hamrick, M.W. Emerging role of extracellular vesicles in musculoskeletal diseases. Mol. Asp. Med. 2018, 60, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Ran, N.; Dong, X.; Zuo, B.; Yang, R.; Zhou, Q.; Moulton, H.M.; Seow, Y.; Yin, H. Anchor peptide captures, targets, and loads exosomes of diverse origins for diagnostics and therapy. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Rome, S.; Forterre, A.; Mizgier, M.L.; Bouzakri, K. Skeletal Muscle-Released Extracellular Vesicles: State of the Art. Front. Physiol. 2019, 10, 929. [Google Scholar] [CrossRef]
- Cho, H.; Blatchley, M.R.; Duh, E.J.; Gerecht, S. Acellular and cellular approaches to improve diabetic wound healing. Adv. Drug Deliv. Rev. 2019, 146, 267–288. [Google Scholar] [CrossRef] [PubMed]
- Paganini, C.; Capasso Palmiero, U.; Pocsfalvi, G.; Touzet, N.; Bongiovanni, A.; Arosio, P. Scalable Production and Isolation of Extracellular Vesicles: Available Sources and Lessons from Current Industrial Bioprocesses. Biotechnol. J. 2019, 14, e1800528. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, N.; Whiteside, T.L.; Reichert, T.E. Challenges in Exosome Isolation and Analysis in Health and Disease. Int. J. Mol. Sci. 2019, 20, 4684. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Lee, Y.; Johansson, H.J.; Mager, I.; Vader, P.; Nordin, J.Z.; Wiklander, O.P.; Lehtio, J.; Wood, M.J.; Andaloussi, S.E. Serum-free culture alters the quantity and protein composition of neuroblastoma-derived extracellular vesicles. J. Extracell. Vesicles 2015, 4, 26883. [Google Scholar] [CrossRef] [PubMed]
- Whitford, W.; Guterstam, P. Exosome manufacturing status. Future Med. Chem. 2019, 11, 1225–1236. [Google Scholar] [CrossRef] [Green Version]
- Shelke, G.V.; Lasser, C.; Gho, Y.S.; Lotvall, J. Importance of exosome depletion protocols to eliminate functional and RNA-containing extracellular vesicles from fetal bovine serum. J. Extracell. Vesicles 2014, 3. [Google Scholar] [CrossRef] [Green Version]
- Eitan, E.; Zhang, S.; Witwer, K.W.; Mattson, M.P. Extracellular vesicle-depleted fetal bovine and human sera have reduced capacity to support cell growth. J. Extracell. Vesicles 2015, 4, 26373. [Google Scholar] [CrossRef]
- Mateescu, B.; Kowal, E.J.; van Balkom, B.W.; Bartel, S.; Bhattacharyya, S.N.; Buzas, E.I.; Buck, A.H.; de Candia, P.; Chow, F.W.; Das, S.; et al. Obstacles and opportunities in the functional analysis of extracellular vesicle RNA—An ISEV position paper. J. Extracell. Vesicles 2017, 6, 1286095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lener, T.; Gimona, M.; Aigner, L.; Borger, V.; Buzas, E.; Camussi, G.; Chaput, N.; Chatterjee, D.; Court, F.A.; Del Portillo, H.A.; et al. Applying extracellular vesicles based therapeutics in clinical trials—An ISEV position paper. J. Extracell. Vesicles 2015, 4, 30087. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Kaslan, M.; Lee, S.H.; Yao, J.; Gao, Z. Progress in Exosome Isolation Techniques. Theranostics 2017, 7, 789–804. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, C.; Di Vizio, D.; Sahoo, S.; Thery, C.; Witwer, K.W.; Wauben, M.; Hill, A.F. Techniques used for the isolation and characterization of extracellular vesicles: Results of a worldwide survey. J. Extracell. Vesicles 2016, 5, 32945. [Google Scholar] [CrossRef] [PubMed]
- Brennan, K.; Martin, K.; FitzGerald, S.P.; O’Sullivan, J.; Wu, Y.; Blanco, A.; Richardson, C.; Mc Gee, M.M. A comparison of methods for the isolation and separation of extracellular vesicles from protein and lipid particles in human serum. Sci. Rep. 2020, 10, 1039. [Google Scholar] [CrossRef] [Green Version]
- Lamparski, H.G.; Metha-Damani, A.; Yao, J.Y.; Patel, S.; Hsu, D.H.; Ruegg, C.; Le Pecq, J.B. Production and characterization of clinical grade exosomes derived from dendritic cells. J. Immunol. Methods 2002, 270, 211–226. [Google Scholar] [CrossRef]
- Lobb, R.J.; Becker, M.; Wen, S.W.; Wong, C.S.; Wiegmans, A.P.; Leimgruber, A.; Moller, A. Optimized exosome isolation protocol for cell culture supernatant and human plasma. J. Extracell. Vesicles 2015, 4, 27031. [Google Scholar] [CrossRef]
- Andriolo, G.; Provasi, E.; Lo Cicero, V.; Brambilla, A.; Soncin, S.; Torre, T.; Milano, G.; Biemmi, V.; Vassalli, G.; Turchetto, L.; et al. Exosomes From Human Cardiac Progenitor Cells for Therapeutic Applications: Development of a GMP-Grade Manufacturing Method. Front. Physiol. 2018, 9, 1169. [Google Scholar] [CrossRef] [Green Version]
- Watson, D.C.; Yung, B.C.; Bergamaschi, C.; Chowdhury, B.; Bear, J.; Stellas, D.; Morales-Kastresana, A.; Jones, J.C.; Felber, B.K.; Chen, X.; et al. Scalable, cGMP-compatible purification of extracellular vesicles carrying bioactive human heterodimeric IL-15/lactadherin complexes. J. Extracell. Vesicles 2018, 7, 1442088. [Google Scholar] [CrossRef]
- Nordin, J.Z.; Lee, Y.; Vader, P.; Mager, I.; Johansson, H.J.; Heusermann, W.; Wiklander, O.P.; Hallbrink, M.; Seow, Y.; Bultema, J.J.; et al. Ultrafiltration with size-exclusion liquid chromatography for high yield isolation of extracellular vesicles preserving intact biophysical and functional properties. Nanomedicine 2015, 11, 879–883. [Google Scholar] [CrossRef] [Green Version]
- Mol, E.A.; Goumans, M.J.; Doevendans, P.A.; Sluijter, J.P.G.; Vader, P. Higher functionality of extracellular vesicles isolated using size-exclusion chromatography compared to ultracentrifugation. Nanomedicine 2017, 13, 2061–2065. [Google Scholar] [CrossRef] [PubMed]
- Yanez-Mo, M.; Siljander, P.R.; Andreu, Z.; Zavec, A.B.; Borras, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiklander, O.P.B.; Bostancioglu, R.B.; Welsh, J.A.; Zickler, A.M.; Murke, F.; Corso, G.; Felldin, U.; Hagey, D.W.; Evertsson, B.; Liang, X.M.; et al. Systematic Methodological Evaluation of a Multiplex Bead-Based Flow Cytometry Assay for Detection of Extracellular Vesicle Surface Signatures. Front. Immunol. 2018, 9, 1326. [Google Scholar] [CrossRef] [Green Version]
- Gorgens, A.; Bremer, M.; Ferrer-Tur, R.; Murke, F.; Tertel, T.; Horn, P.A.; Thalmann, S.; Welsh, J.A.; Probst, C.; Guerin, C.; et al. Optimisation of imaging flow cytometry for the analysis of single extracellular vesicles by using fluorescence-tagged vesicles as biological reference material. J. Extracell. Vesicles 2019, 8, 1587567. [Google Scholar] [CrossRef] [Green Version]
- Jeyaram, A.; Jay, S.M. Preservation and Storage Stability of Extracellular Vesicles for Therapeutic Applications. AAPS J. 2017, 20, 1. [Google Scholar] [CrossRef] [PubMed]
- Kusuma, G.D.; Barabadi, M.; Tan, J.L.; Morton, D.A.V.; Frith, J.E.; Lim, R. To Protect and to Preserve: Novel Preservation Strategies for Extracellular Vesicles. Front. Pharmacol. 2018, 9, 1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jarrige, M.; Frank, E.; Herardot, E.; Martineau, S.; Darle, A.; Benabides, M.; Domingues, S.; Chose, O.; Habeler, W.; Lorant, J.; et al. The Future of Regenerative Medicine: Cell Therapy Using Pluripotent Stem Cells and Acellular Therapies Based on Extracellular Vesicles. Cells 2021, 10, 240. https://doi.org/10.3390/cells10020240
Jarrige M, Frank E, Herardot E, Martineau S, Darle A, Benabides M, Domingues S, Chose O, Habeler W, Lorant J, et al. The Future of Regenerative Medicine: Cell Therapy Using Pluripotent Stem Cells and Acellular Therapies Based on Extracellular Vesicles. Cells. 2021; 10(2):240. https://doi.org/10.3390/cells10020240
Chicago/Turabian StyleJarrige, Margot, Elie Frank, Elise Herardot, Sabrina Martineau, Annabelle Darle, Manon Benabides, Sophie Domingues, Olivier Chose, Walter Habeler, Judith Lorant, and et al. 2021. "The Future of Regenerative Medicine: Cell Therapy Using Pluripotent Stem Cells and Acellular Therapies Based on Extracellular Vesicles" Cells 10, no. 2: 240. https://doi.org/10.3390/cells10020240