
scRNA-seq Reveals the Mechanism of Fatty Acid Desaturase 2 Mutation to Repress Leaf Growth in Peanut (Arachis hypogaea L.)
 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material, Growth Conditions, and Phenotype Assays
2.2. Protoplast Isolation and scRNA-seq Library Construction
2.3. Data Analysis of scRNA-seq of Peanut
2.4. Marker Genes for Specific Cell-Type Validation
2.5. Pseudo-Time Trajectory Analysis
2.6. RNA Velocity Analysis
2.7. Cell Cycle Analysis
2.8. Phytohormones Uptake and Detection Assay
2.9. Tissue Real-Time PCR Analysis
3. Results
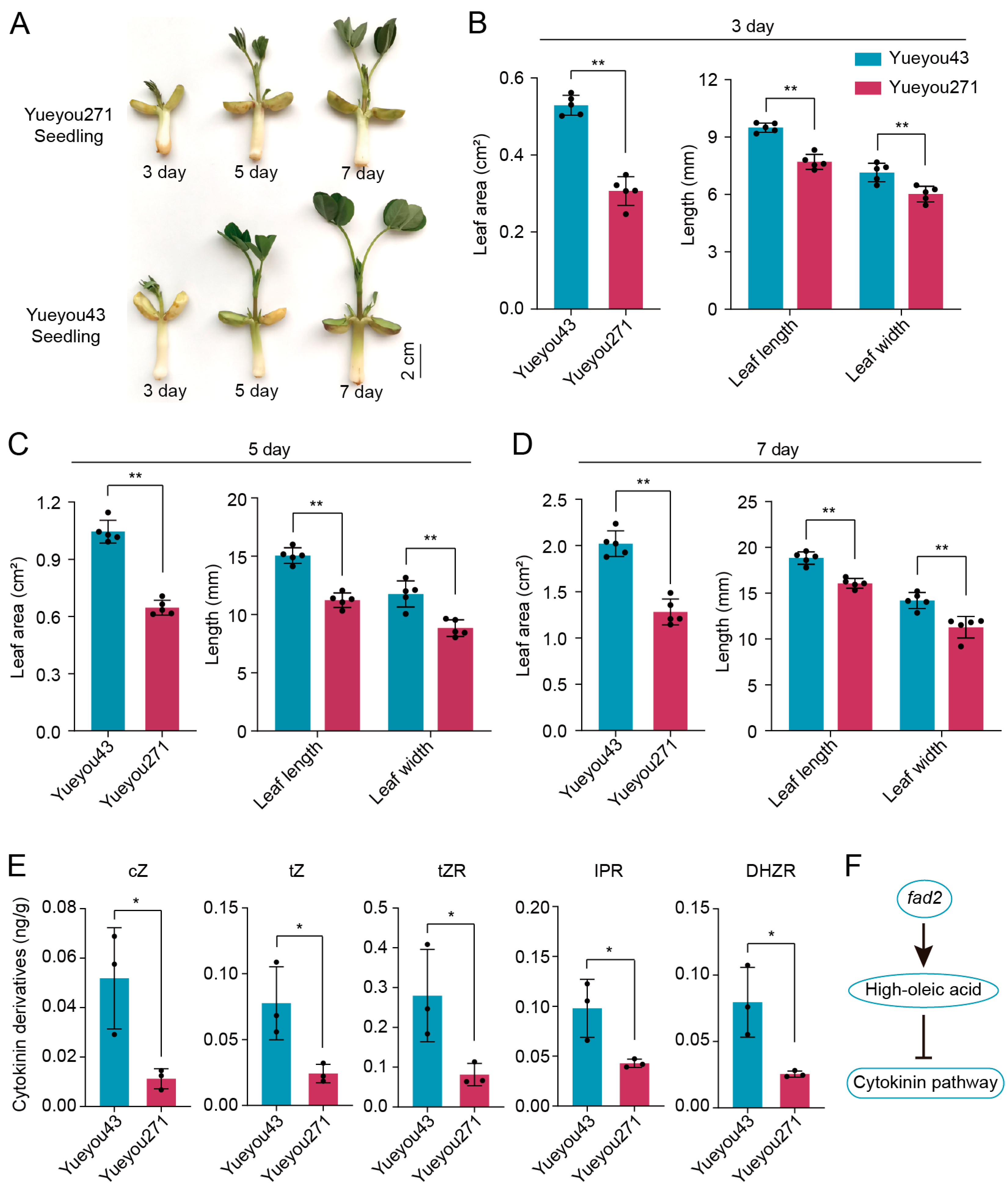
3.1. FAD2 Mutant Negatively Regulated the Growth Phenotypes of Peanut Seedling Leaf
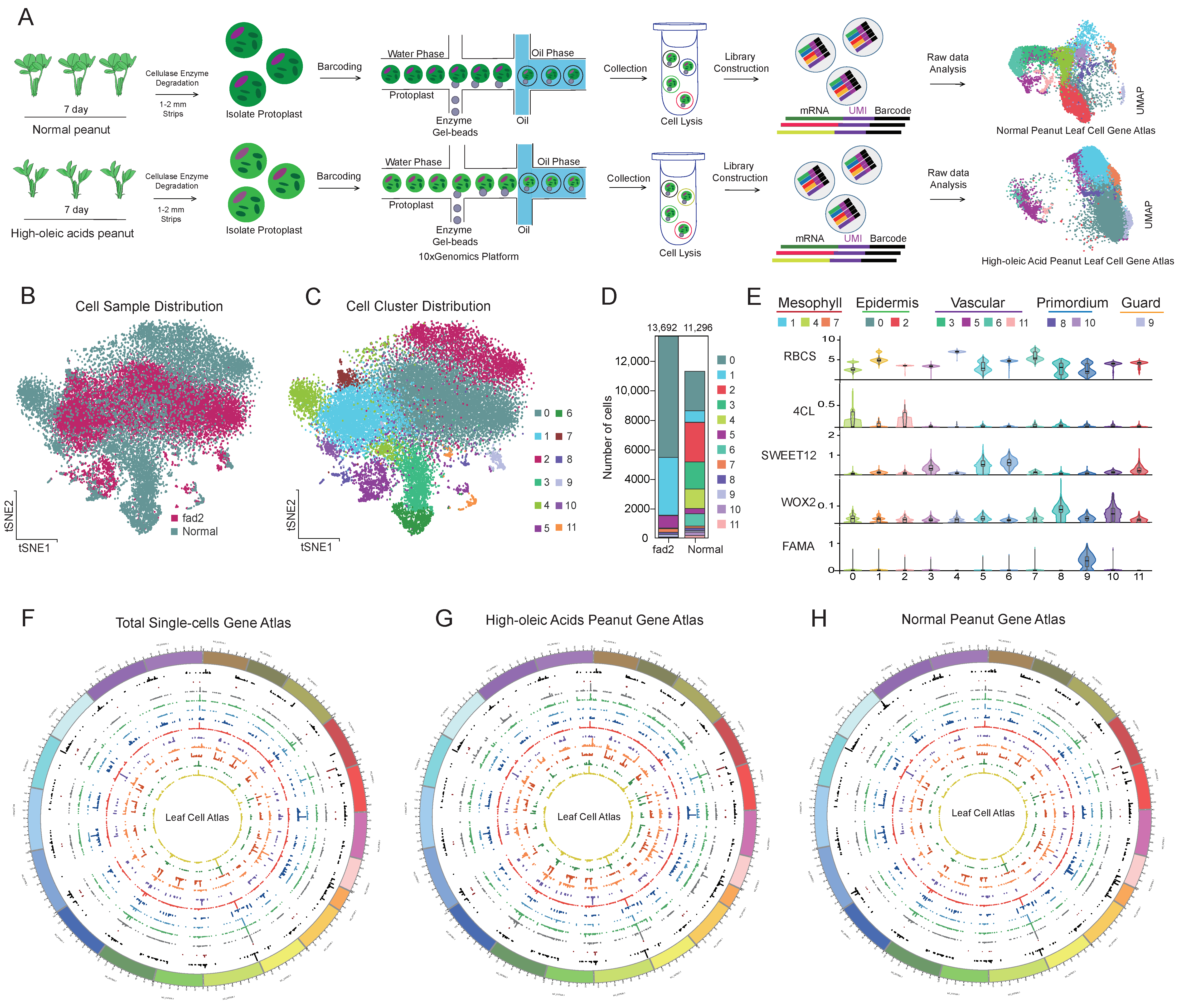
3.2. scRNA-seq Identified the Major Cell Clusters in High-OA and Normal Peanut Seedling Leaf
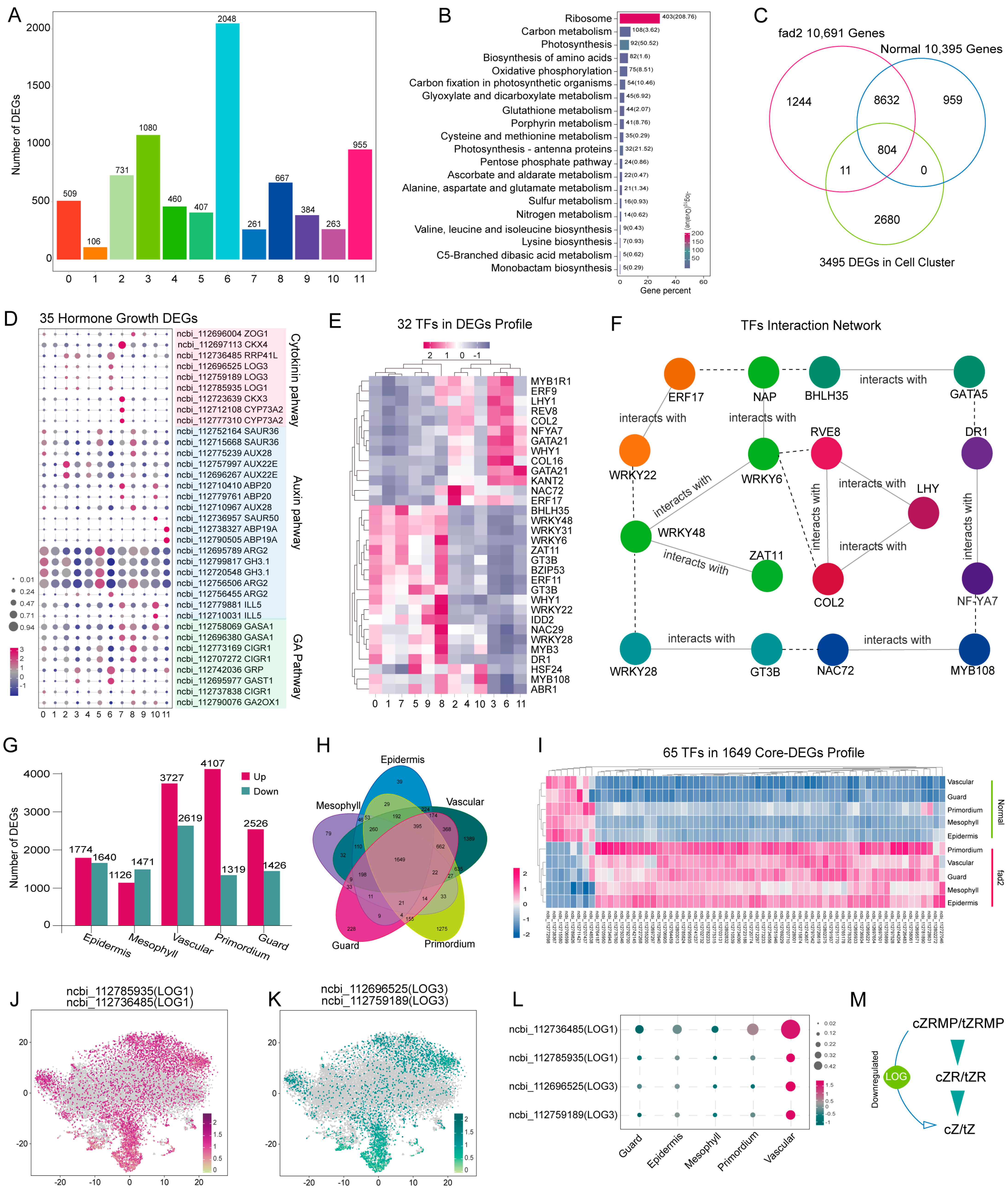
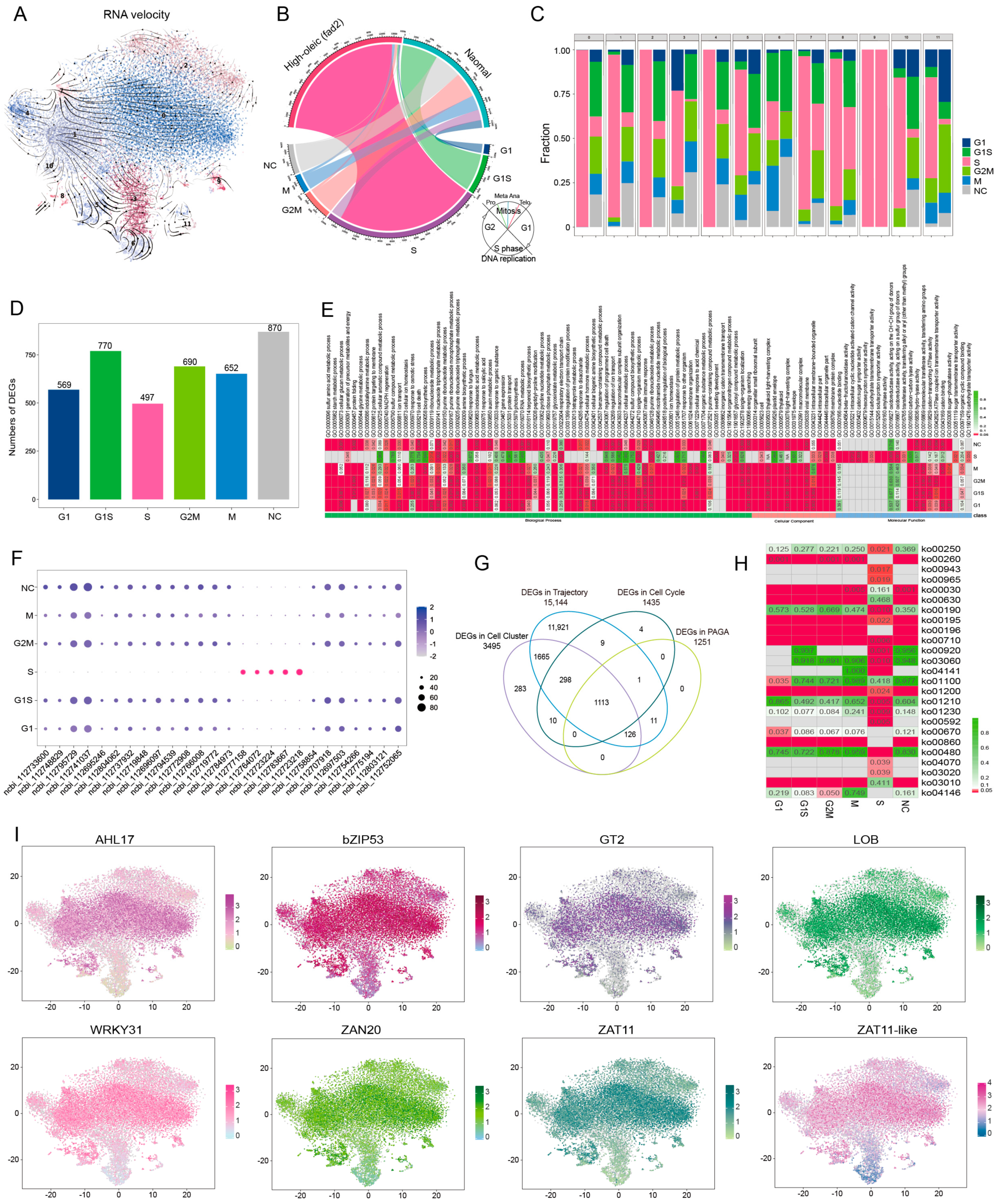
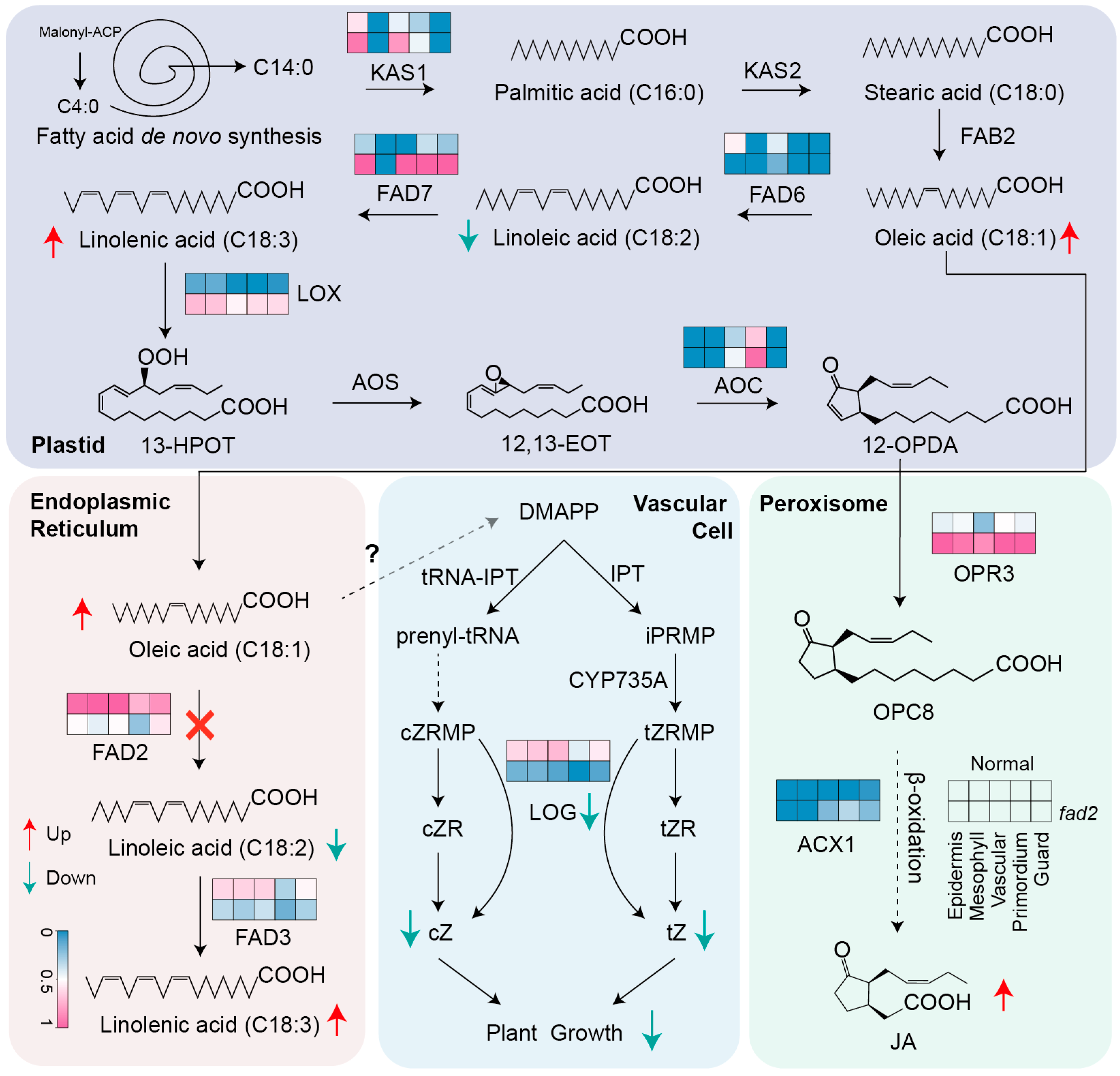
3.3. Identification of Important DEGs in Distinct Leaf Cell-Types Revealed fad2 Mutation Repressed the Cytokinin Pathway
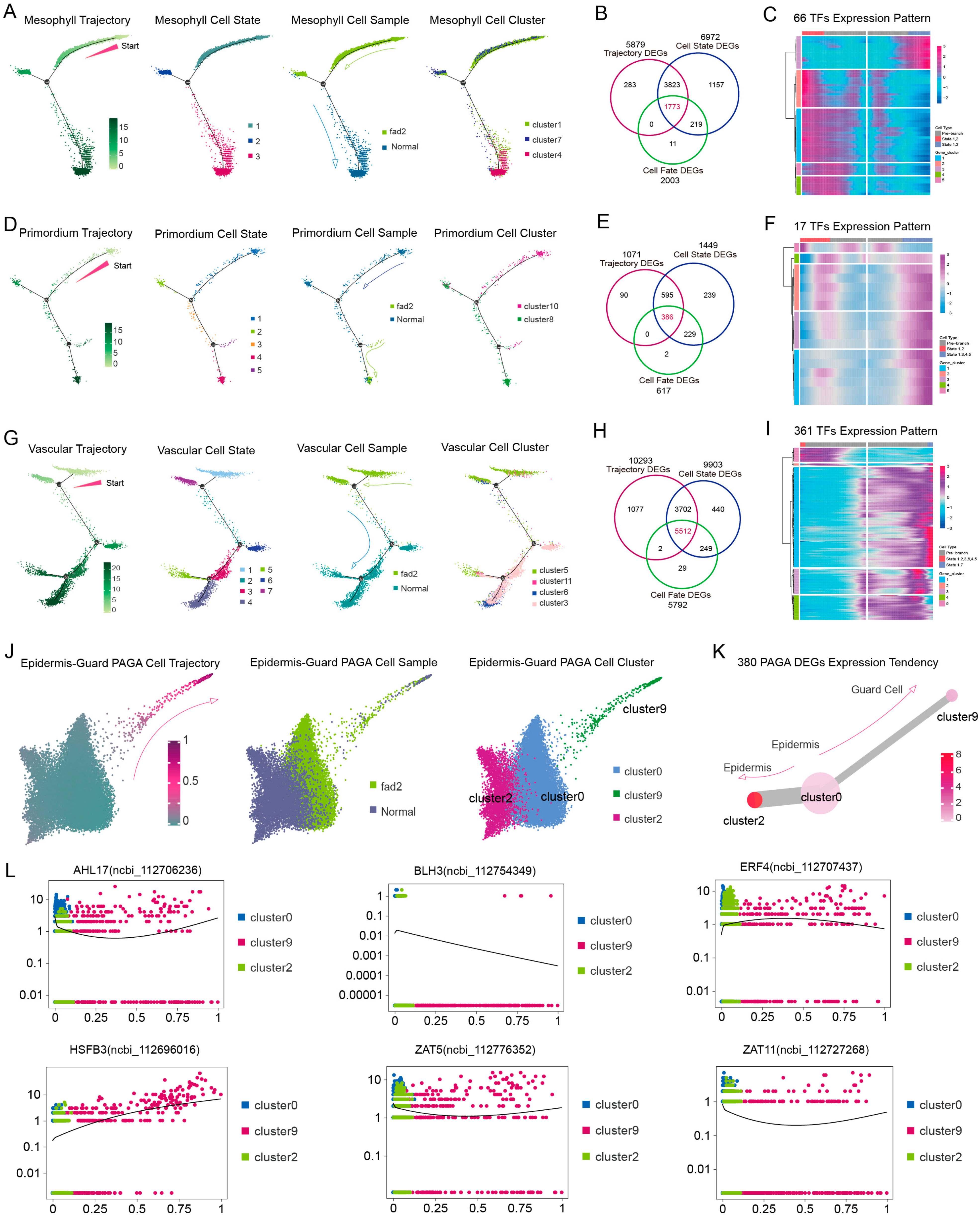
3.4. FAD2 Mutation Repressed the Cell Differentiation in Leaf Development Trajectory
3.5. High-OA Peanut Regulated Cell Development Features in Distinct Cell Type Trajectories
scRNA-seq Reveals That High-OA Accumulation Represses Leaf Cell Cycle Procedure
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Janila, P.; Pandey, M.K.; Shasidhar, Y.; Variath, M.T.; Sriswathi, M.; Khera, P.; Manohar, S.S.; Nagesh, P.; Vishwakarma, M.K.; Mishra, G.P.; et al. Molecular Breeding for Introgression of Fatty Acid Desaturase Mutant Alleles (AhFAD2A and AhFAD2B) Enhances Oil Quality in High and Low Oil Containing Peanut Genotypes. Plant Sci. 2016, 242, 203–213. [Google Scholar] [CrossRef]
- Liu, H.; Hong, Y.; Lu, Q.; Li, H.; Gu, J.; Ren, L.; Deng, L.; Zhou, B.; Chen, X.; Liang, X. Integrated Analysis of Comparative Lipidomics and Proteomics Reveals the Dynamic Changes of Lipid Molecular Species in High-Oleic Acid Peanut Seed. J. Agric. Food Chem. 2020, 68, 426–438. [Google Scholar] [CrossRef]
- Wang, D.D.; Hu, F.B. Dietary Fat and Risk of Cardiovascular Disease: Recent Controversies and Advances. Annu. Rev. Nutr. 2017, 37, 423–446. [Google Scholar] [CrossRef]
- Lim, G.H.; Singhal, R.; Kachroo, A.; Kachroo, P. Fatty Acid- and Lipid-Mediated Signaling in Plant Defense. Annu. Rev. Phytopathol. 2017, 55, 505–536. [Google Scholar] [CrossRef]
- Wen, S.; Liu, H.; Li, X.; Chen, X.; Hong, Y.; Li, H.; Lu, Q.; Liang, X. TALEN-Mediated Targeted Mutagenesis of Fatty Acid Desaturase 2 (FAD2) in Peanut (Arachis Hypogaea L.) Promotes the Accumulation of Oleic Acid. Plant Mol. Biol. 2018, 97, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Dhaliwal, L.K.; Angeles-Shim, R.B. Cell Membrane Features as Potential Breeding Targets to Improve Cold Germination Ability of Seeds. Plants 2022, 11, 3400. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.C.; Nakamura, Y.; Kanehara, K. Membrane Lipid Polyunsaturation Mediated by FATTY ACID DESATURASE 2 (FAD2) Is Involved in Endoplasmic Reticulum Stress Tolerance in Arabidopsis Thaliana. Plant J. 2019, 99, 478–493. [Google Scholar] [CrossRef] [PubMed]
- Sui, N.; Wang, Y.; Liu, S.; Yang, Z.; Wang, F.; Wan, S. Transcriptomic and Physiological Evidence for the Relationship between Unsaturated Fatty Acid and Salt Stress in Peanut. Front. Plant Sci. 2018, 9, 7. [Google Scholar] [CrossRef]
- Yang, W.; Dong, R.; Liu, L.; Hu, Z.; Li, J.; Wang, Y.; Ding, X.; Chu, Z. A novel mutant allele of SSI2 confers a better balance between disease resistance and plant growth inhibition on Arabidopsis thaliana. BMC Plant Biol. 2016, 16, 208. [Google Scholar] [CrossRef]
- Bawa, G.; Liu, Z.; Yu, X.; Qin, A.; Sun, X. Single-Cell RNA Sequencing for Plant Research: Insights and Possible Benefits. Int. J. Mol. Sci. 2022, 23, 4497. [Google Scholar] [CrossRef]
- Hedlund, E.; Deng, Q. Single-Cell RNA Sequencing: Technical Advancements and Biological Applications. Mol. Asp. Med. 2018, 59, 36–46. [Google Scholar] [CrossRef]
- Rodriguez-Villalon, A.; Brady, S.M. Single Cell RNA Sequencing and Its Promise in Reconstructing Plant Vascular Cell Lineages. Curr. Opin. Plant Biol. 2019, 48, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Ryu, K.H.; Huang, L.; Kang, H.M.; Schiefelbein, J. Single-Cell RNA Sequencing Resolves Molecular Relationships Among Individual Plant Cells. Plant Physiol. 2019, 179, 1444–1456. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.Q.; Xu, Z.G.; Shang, G.D.; Wang, J.W. A Single-Cell RNA Sequencing Profiles the Developmental Landscape of Arabidopsis Root. Mol. Plant 2019, 12, 648–660. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huan, Q.; Li, K.; Qian, W. Single-Cell Transcriptome Atlas of the Leaf and Root of Rice Seedlings. J. Genet. Genom. 2021, 48, 881–898. [Google Scholar] [CrossRef]
- Martín, M.P.; Grosso, A.L.; Nepote, V.; Grosso, N.R. Sensory and Chemical Stabilities of High-Oleic and Normal-Oleic Peanuts in Shell During Long-Term Storage. J. Food Sci. 2018, 83, 2362–2368. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Hu, D.; Du, P.; Wang, L.; Liang, X.; Li, H.; Lu, Q.; Li, S.; Liu, H.; Chen, X.; et al. Single-Cell RNA-Seq Describes the Transcriptome Landscape and Identifies Critical Transcription Factors in the Leaf Blade of the Allotetraploid Peanut (Arachis Hypogaea L.). Plant Biotechnol. J. 2021, 19, 2261–2276. [Google Scholar] [CrossRef]
- Zheng, G.X.Y.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively Parallel Digital Transcriptional Profiling of Single Cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M.; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated Analysis of Multimodal Single-Cell Data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING Database in 2021: Customizable Protein-Protein Networks, and Functional Characterization of User-Uploaded Gene/Measurement Sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wang, Q.; Zhu, X.; Wang, G.; Qin, Y.; Ding, F.; Tu, L.; Daniell, H.; Zhang, X.; Jin, S. Plant Single Cell Transcriptome Hub (PsctH): An Integrated Online Tool to Explore the Plant Single-Cell Transcriptome Landscape. Plant Biotechnol. J. 2022, 20, 10–12. [Google Scholar] [CrossRef]
- Jin, J.; Lu, P.; Xu, Y.; Tao, J.; Li, Z.; Wang, S.; Yu, S.; Wang, C.; Xie, X.; Gao, J.; et al. PCMDB: A Curated and Comprehensive Resource of Plant Cell Markers. Nucleic Acids Res. 2022, 50, D1448–D1455. [Google Scholar] [CrossRef]
- Chen, H.; Yin, X.; Guo, L.; Yao, J.; Ding, Y.; Xu, X.; Liu, L.; Zhu, Q.-H.; Chu, Q.; Fan, L. PlantscRNAdb: A Database for Plant Single-Cell RNA Analysis. Mol. Plant 2021, 14, 855–857. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wu, Y.; Peng, A.; Cui, J.; Zhao, M.; Pan, Y.; Zhang, M.; Tian, K.; Schwab, W.; Song, C. Single-Cell Transcriptome Atlas Reveals Developmental Trajectories and a Novel Metabolic Pathway of Catechin Esters in Tea Leaves. Plant Biotechnol. J. 2022, 20, 2089–2106. [Google Scholar] [CrossRef]
- Cao, J.; Spielmann, M.; Qiu, X.; Huang, X.; Ibrahim, D.M.; Hill, A.J.; Zhang, F.; Mundlos, S.; Christiansen, L.; Steemers, F.J.; et al. The Single-Cell Transcriptional Landscape of Mammalian Organogenesis. Nature 2019, 566, 496–502. [Google Scholar] [CrossRef]
- Plass, M.; Solana, J.; Wolf, F.A.; Ayoub, S.; Misios, A.; Glažar, P.; Obermayer, B.; Theis, F.J.; Kocks, C.; Rajewsky, N. Cell Type Atlas and Lineage Tree of a Whole Complex Animal by Single-Cell Transcriptomics. Science 2018, 360, eaaq1723. [Google Scholar] [CrossRef]
- La Manno, G.; Soldatov, R.; Zeisel, A.; Braun, E.; Hochgerner, H.; Petukhov, V.; Lidschreiber, K.; Kastriti, M.E.; Lönnerberg, P.; Furlan, A.; et al. RNA Velocity of Single Cells. Nature 2018, 560, 494–498. [Google Scholar] [CrossRef]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-Wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef]
- Liu, H.; Gu, J.; Lu, Q.; Li, H.; Hong, Y.; Chen, X.; Ren, L.; Deng, L.; Liang, X. Transcriptomic Analysis Reveals the High-Oleic Acid Feedback Regulating the Homologous Gene Expression of Stearoyl-ACP Desaturase 2 (SAD2) in Peanuts. Int. J. Mol. Sci. 2019, 20, 3091. [Google Scholar] [CrossRef]
- Pan, X.; Li, H.; Wei, H.; Su, W.; Jiang, X.; Lu, H. Analysis of the Spatial and Temporal Expression Pattern Directed by the Populus Tomentosa 4-Coumarate:CoA Ligase Pto4CL2 Promoter in Transgenic Tobacco. Mol. Biol. Rep. 2013, 40, 2309–2317. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.Q.; Chen, Y.; Wang, J.W. A Single-Cell Analysis of the Arabidopsis Vegetative Shoot Apex. Dev. Cell 2021, 56, 1056–1074.e8. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Symeonidi, E.; Pang, T.Y.; Denyer, T.; Weidauer, D.; Bezrutczyk, M.; Miras, M.; Zöllner, N.; Hartwig, T.; Wudick, M.M.; et al. Distinct Identities of Leaf Phloem Cells Revealed by Single Cell Transcriptomics. Plant Cell 2021, 33, 511–530. [Google Scholar] [CrossRef]
- Kawai, T.; Shibata, K.; Akahoshi, R.; Nishiuchi, S.; Takahashi, H.; Nakazono, M.; Kojima, T.; Nosaka-Takahashi, M.; Sato, Y.; Toyoda, A.; et al. WUSCHEL-Related Homeobox Family Genes in Rice Control Lateral Root Primordium Size. Proc. Natl. Acad. Sci. USA 2022, 119, e2101846119. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Anido, C.B.; Vatén, A.; Smoot, N.K.; Sharma, N.; Guo, V.; Gong, Y.; Anleu Gil, M.X.; Weimer, A.K.; Bergmann, D.C. Single-Cell Resolution of Lineage Trajectories in the Arabidopsis Stomatal Lineage and Developing Leaf. Dev. Cell 2021, 56, 1043–1055.e4. [Google Scholar] [CrossRef]
- Liu, Z.; Yu, X.; Qin, A.; Zhao, Z.; Liu, Y.; Sun, S.; Liu, H.; Guo, C.; Wu, R.; Yang, J.; et al. Research Strategies for Single-Cell Transcriptome Analysis in Plant Leaves. Plant J. 2022, 112, 27–37. [Google Scholar] [CrossRef]
- Wang, R.; Liu, L.; Kong, Z.; Li, S.; Lu, L.; Kabir, N.; Chen, G.; Zhang, J.; Qanmber, G.; Liu, Z. Identification of GhLOG Gene Family Revealed That GhLOG3 Is Involved in Regulating Salinity Tolerance in Cotton (Gossypium Hirsutum L.). Plant Physiol. Biochem. 2021, 166, 328–340. [Google Scholar] [CrossRef]
- Chen, L.; Jameson, G.B.; Guo, Y.; Song, J.; Jameson, P.E. The LONELY GUY Gene Family: From Mosses to Wheat, the Key to the Formation of Active Cytokinins in Plants. Plant Biotechnol. J. 2022, 20, 625–645. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Liu, H.; Du, P.; Liang, X.; Li, H.; Lu, Q.; Li, S.; Liu, H.; Hong, Y.; Varshney, R.K.; et al. Impact of Different Cooking Methods on the Chemical Profile of High-Oleic Acid Peanut Seeds. Food Chem. 2022, 379, 131970. [Google Scholar] [CrossRef]
- Neelakandan, A.K.; Wright, D.A.; Traore, S.M.; Chen, X.; Spalding, M.H.; He, G. CRISPR/Cas9 Based Site-Specific Modification of FAD2 Cis-Regulatory Motifs in Peanut (Arachis Hypogaea L). Front. Genet. 2022, 13, 849961. [Google Scholar] [CrossRef]
- Denyer, T.; Timmermans, M.C.P. Crafting a Blueprint for Single-Cell RNA Sequencing. Trends Plant Sci. 2022, 27, 92–103. [Google Scholar] [CrossRef]
- Song, G.; Li, X.; Munir, R.; Khan, A.R.; Azhar, W.; Yasin, M.U.; Jiang, Q.; Bancroft, I.; Gan, Y. The WRKY6 transcription factor affects seed oil accumulation and alters fatty acid compositions in Arabidopsis thaliana. Physiol. Plant 2020, 169, 612–624. [Google Scholar] [CrossRef]
- Cai, X.T.; Xu, P.; Zhao, P.X.; Liu, R.; Yu, L.H.; Xiang, C.B. Arabidopsis ERF109 mediates cross-talk between jasmonic acid and auxin biosynthesis during lateral root formation. Nat. Commun. 2014, 5, 5833. [Google Scholar] [CrossRef]
- Singh, D.; Debnath, P.; Sane, A.P.; Sane, V.A. Tomato (Solanum lycopersicum) WRKY23 enhances salt and osmotic stress tolerance by modulating the ethylene and auxin pathways in transgenic Arabidopsis. Plant Physiol. Biochem. 2023, 195, 330–340. [Google Scholar] [CrossRef]
- Dubois, M.; Skirycz, A.; Claeys, H.; Maleux, K.; Dhondt, S.; De Bodt, S.; Vanden Bossche, R.; De Milde, L.; Yoshizumi, T.; Matsui, M.; et al. Ethylene Response Factor6 acts as a central regulator of leaf growth under water-limiting conditions in Arabidopsis. Plant Physiol. 2013, 162, 319–332. [Google Scholar] [CrossRef]
- Piao, W.; Sakuraba, Y.; Paek, N.C. Transgenic expression of rice MYB102 (OsMYB102) delays leaf senescence and decreases abiotic stress tolerance in Arabidopsis thaliana. BMB Rep. 2019, 52, 653–658. [Google Scholar] [CrossRef]
- Besseau, S.; Li, J.; Palva, E.T. WRKY54 and WRKY70 co-operate as negative regulators of leaf senescence in Arabidopsis thaliana. J. Exp. Bot. 2012, 63, 2667–2679. [Google Scholar] [CrossRef]
- Dar, A.A.; Choudhury, A.R.; Kancharla, P.K.; Arumugam, N. The FAD2 Gene in Plants: Occurrence, Regulation, and Role. Front. Plant Sci. 2017, 8, 1789. [Google Scholar] [CrossRef]
- Xie, J.; Li, M.; Zeng, J.; Li, X.; Zhang, D. Single-Cell RNA Sequencing Profiles of Stem-Differentiating Xylem in Poplar. Plant Biotechnol. J. 2022, 20, 417–419. [Google Scholar] [CrossRef]
- Kang, M.; Choi, Y.; Kim, H.; Kim, S.G. Single-Cell RNA-Sequencing of Nicotiana Attenuata Corolla Cells Reveals the Biosynthetic Pathway of a Floral Scent. New Phytol. 2022, 234, 527–544. [Google Scholar] [CrossRef]
- Shaw, R.; Tian, X.; Xu, J. Single-Cell Transcriptome Analysis in Plants: Advances and Challenges. Mol. Plant 2021, 14, 115–126. [Google Scholar] [CrossRef]
- Farmer, A.; Thibivilliers, S.; Ryu, K.H.; Schiefelbein, J.; Libault, M. Single-Nucleus RNA and ATAC Sequencing Reveals the Impact of Chromatin Accessibility on Gene Expression in Arabidopsis Roots at the Single-Cell Level. Mol. Plant 2021, 14, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Xia, M.; Li, J.; Ma, W.; Li, Q.; Xie, J.; Bai, S.; Fang, S.; Sun, T.; Feng, X.; et al. The Maize Single-Nucleus Transcriptome Comprehensively Describes Signaling Networks Governing Movement and Development of Grass Stomata. Plant Cell 2022, 34, 1890–1911. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, P.; Deng, Q.; Wang, W.; Garg, V.; Lu, Q.; Huang, L.; Wang, R.; Li, H.; Huai, D.; Chen, X.; et al. scRNA-seq Reveals the Mechanism of Fatty Acid Desaturase 2 Mutation to Repress Leaf Growth in Peanut (Arachis hypogaea L.). Cells 2023, 12, 2305. https://doi.org/10.3390/cells12182305
Du P, Deng Q, Wang W, Garg V, Lu Q, Huang L, Wang R, Li H, Huai D, Chen X, et al. scRNA-seq Reveals the Mechanism of Fatty Acid Desaturase 2 Mutation to Repress Leaf Growth in Peanut (Arachis hypogaea L.). Cells. 2023; 12(18):2305. https://doi.org/10.3390/cells12182305
Chicago/Turabian StyleDu, Puxuan, Quanqing Deng, Wenyi Wang, Vanika Garg, Qing Lu, Lu Huang, Runfeng Wang, Haifen Li, Dongxin Huai, Xiaoping Chen, and et al. 2023. "scRNA-seq Reveals the Mechanism of Fatty Acid Desaturase 2 Mutation to Repress Leaf Growth in Peanut (Arachis hypogaea L.)" Cells 12, no. 18: 2305. https://doi.org/10.3390/cells12182305