

The Role of the Plasminogen/Plasmin System in Inflammation of the Oral Cavity
 , , ,
, , , {kind=link}
{kind=link}
Abstract
:1. Introduction
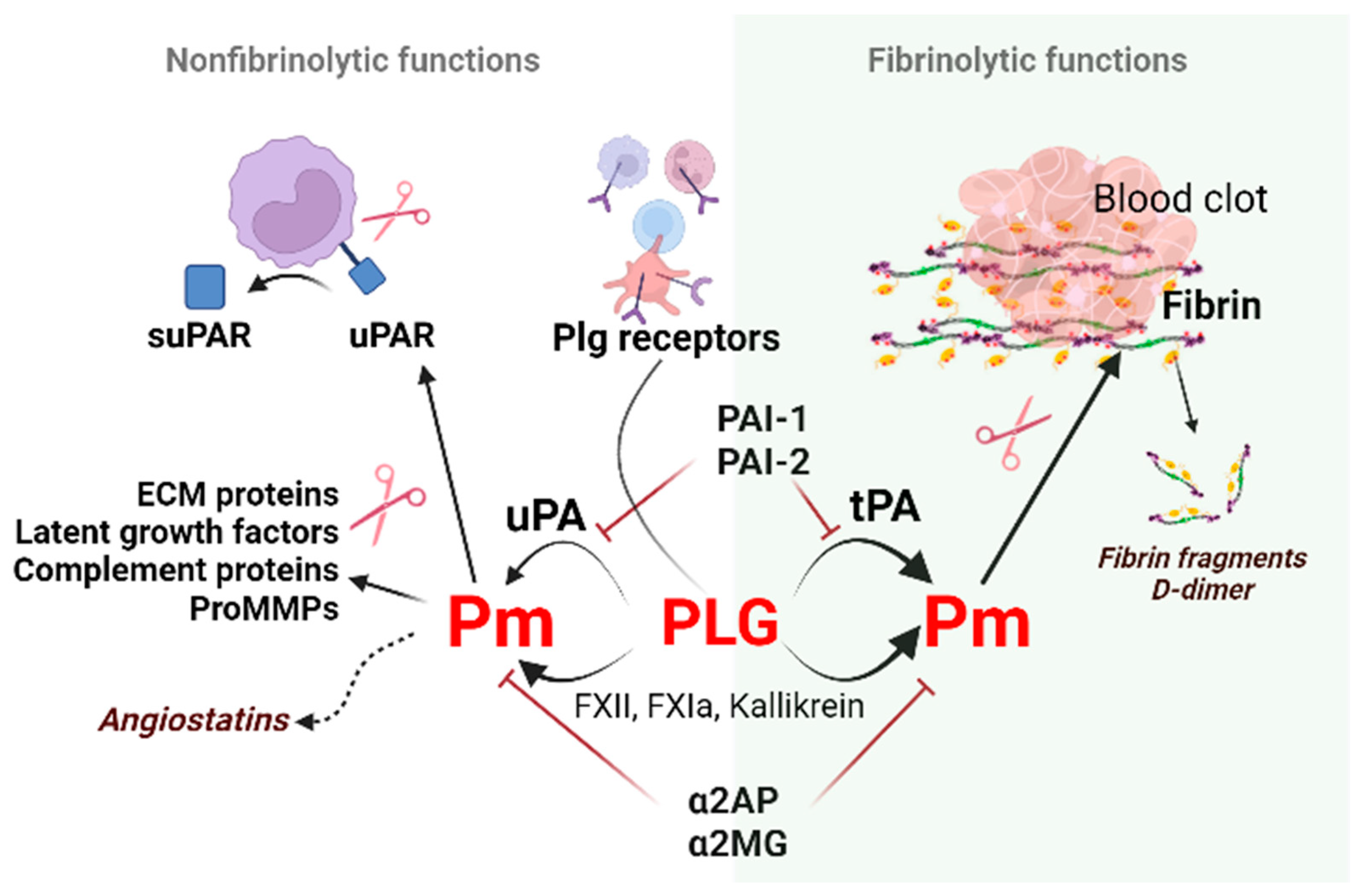
2. Regulators of Fibrinolysis
2.1. Plasminogen (Plg)
2.2. Plasminogen Activators (PAs)
2.3. Plasminogen Activator Inhibitors
3. Soluble uPAR, a New Biomarker of Inflammation
4. Fibrinolytic Factors during Inflammation in the Oral Cavity
Plg Deficiency and the Oral Cavity
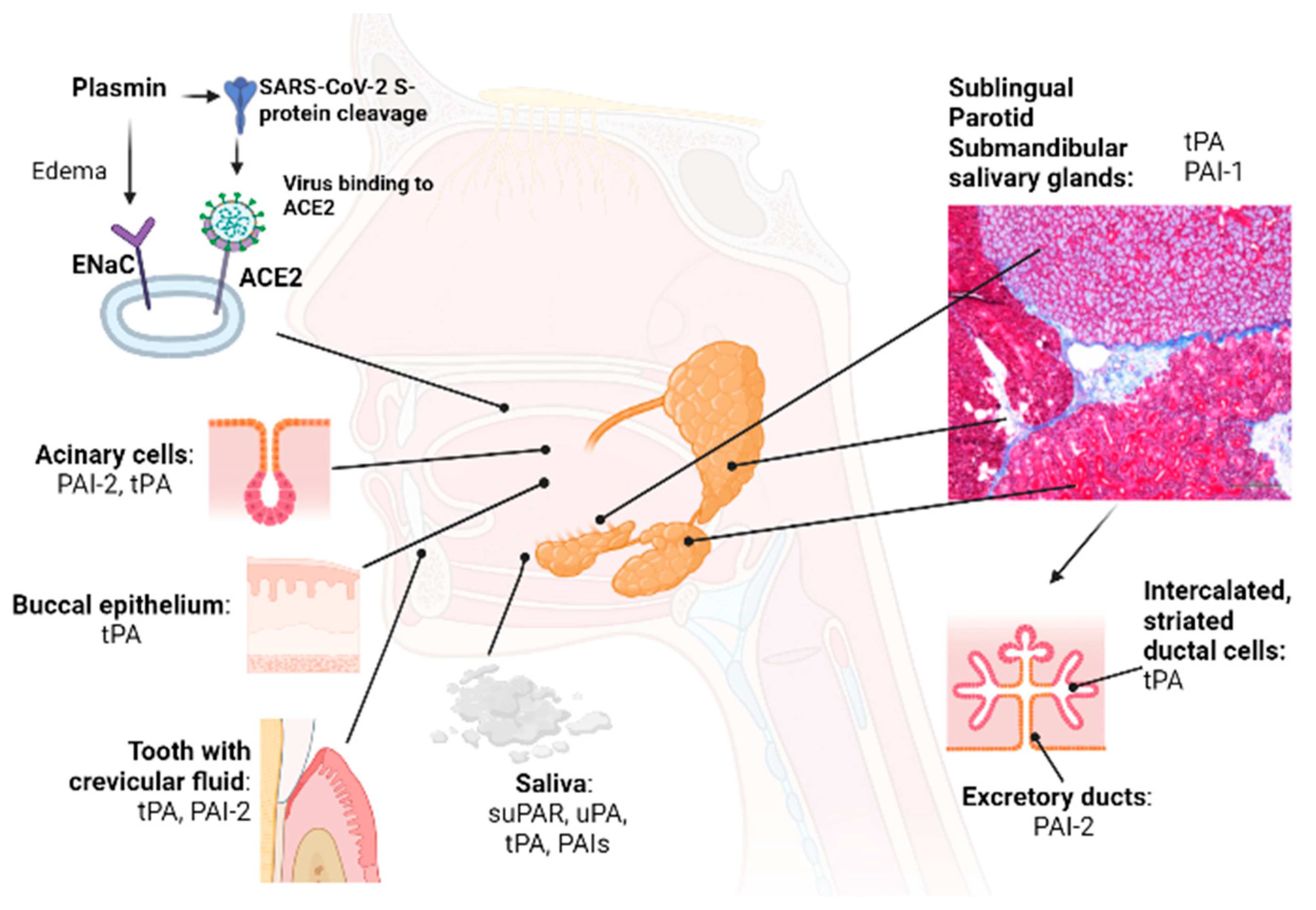
5. Fibrinolytic Factors during Bacterial and Viral Infection
5.1. Viral Entry of SARS-CoV-2 in the Oral Cavity
5.2. Coagulopathy with Hyper-/Hypocoagulation during COVID-19
6. Concluding Remarks and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Bharadwaj, A.G.; Holloway, R.W.; Miller, V.A.; Waisman, D.M. Plasmin and Plasminogen System in the Tumor Microenvironment: Implications for Cancer Diagnosis, Prognosis, and Therapy. Cancers 2021, 13, 1838. [Google Scholar] [CrossRef]
- Heissig, B.; Salama, Y.; Osada, T.; Okumura, K.; Hattori, K. The Multifaceted Role of Plasminogen in Cancer. Int. J. Mol. Sci. 2021, 22, 2304. [Google Scholar] [CrossRef]
- Heissig, B.; Ohki-Koizumi, M.; Tashiro, Y.; Gritli, I.; Sato-Kusubata, K.; Hattori, K. New functions of the fibrinolytic system in bone marrow cell-derived angiogenesis. Int. J. Hematol. 2012, 95, 131–137. [Google Scholar] [CrossRef]
- Medcalf, R.L.; Keragala, C.B. The Fibrinolytic System: Mysteries and Opportunities. HemaSphere 2021, 5, e570. [Google Scholar] [CrossRef]
- Myöhänen, H.; Vaheri, A. Regulation and interactions in the activation of cell-associated plasminogen. Cell. Mol. Life Sci. CMLS 2004, 61, 2840–2858. [Google Scholar] [CrossRef]
- Rijken, D.C.; Lijnen, H.R. New insights into the molecular mechanisms of the fibrinolytic system. J. Thromb. Haemost. 2009, 7, 4–13. [Google Scholar] [CrossRef]
- Urano, T.; Castellino, F.J.; Suzuki, Y. Regulation of plasminogen activation on cell surfaces and fibrin. J. Thromb. Haemost. 2018, 16, 1487–1497. [Google Scholar] [CrossRef] [Green Version]
- Heissig, B.; Salama, Y.; Takahashi, S.; Osada, T.; Hattori, K. The multifaceted role of plasminogen in inflammation. Cell. Signal. 2020, 75, 109761. [Google Scholar] [CrossRef]
- Baker, S.K.; Strickland, S. A critical role for plasminogen in inflammation. J. Exp. Med. 2020, 217, e20191865. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Guo, Y.; Mikus, P.; Sulniute, R.; Wilczynska, M.; Ny, T.; Li, J. Plasminogen is a key proinflammatory regulator that accelerates the healing of acute and diabetic wounds. Blood 2012, 119, 5879–5887. [Google Scholar] [CrossRef]
- Kessler, A.T.; Bhatt, A.A. Review of the Major and Minor Salivary Glands, Part 1: Anatomy, Infectious, and Inflammatory Processes. J. Clin. Imaging Sci. 2018, 8, 47. [Google Scholar] [CrossRef] [PubMed]
- Rijken, D.C.; Sakharov, D.V. Basic Principles in Thrombolysis: Regulatory Role of Plasminogen. Thromb. Res. 2001, 103, S41–S49. [Google Scholar] [CrossRef] [PubMed]
- Pryzdial, E.L.G.; Leatherdale, A.; Conway, E.M. Coagulation and complement: Key innate defense participants in a seamless web. Front. Immunol. 2022, 13, 918775. [Google Scholar] [CrossRef]
- Twining, S.S.; Wilson, P.M.; Ngamkitidechakul, C. Extrahepatic synthesis of plasminogen in the human cornea is up-regulated by interleukins-1alpha and -1beta. Biochem. J. 1999, 339 Pt 3, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Castellino, F.J.; McCance, S.G. The Kringle Domains of Human Plasminogen. In Ciba Foundation Symposium 212—Plasminogen-Related Growth Factors; Novartis Foundation Symposia; John Wiley & Sons: Hoboken, NJ, USA, 2007; pp. 46–65. [Google Scholar]
- Rudd, P.M.; Woods, R.J.; Wormald, M.R.; Opdenakker, G.; Downing, A.K.; Campbell, I.D.; Dwek, R.A. The effects of variable glycosylation on the functional activities of ribonuclease, plasminogen and tissue plasminogen activator. Biochim. Biophys. Acta (BBA)—Protein Struct. Mol. Enzymol. 1995, 1248, 1–10. [Google Scholar] [CrossRef]
- Zhang, L.; Gong, Y.; Grella, D.K.; Castellino, F.J.; Miles, L.A. Endogenous plasmin converts Glu-plasminogen to Lys-plasminogen on the monocytoid cell surface. J. Thromb. Haemost. 2003, 1, 1264–1270. [Google Scholar] [CrossRef]
- Silverstein, R.L.; Friedlander, R.J., Jr.; Nicholas, R.L.; Nachman, R.L. Binding of Lys-plasminogen to monocytes/macrophages. J. Clin. Investig. 1988, 82, 1948–1955. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Xue, L. Angiostatin. Semin. Thromb. Hemost. 2004, 30, 83–93. [Google Scholar] [CrossRef]
- O’Reilly, M.S.; Holmgren, L.; Shing, Y.; Chen, C.; Rosenthal, R.A.; Moses, M.; Lane, W.S.; Cao, Y.; Sage, E.H.; Folkman, J. Angiostatin: A novel angiogenesis inhibitor that mediates the suppression of metastases by a lewis lung carcinoma. Cell 1994, 79, 315–328. [Google Scholar] [CrossRef]
- Syed, S.P.; Martin, A.-M.; Haupt, H.M.; Arenas-Elliot, C.P.; Brooks, J.J. Angiostatin receptor annexin II in vascular tumors including angiosarcoma. Hum. Pathol. 2007, 38, 508–513. [Google Scholar] [CrossRef]
- Yatsenko, T.A.; Rybachuk, V.M.; Yusova, O.I.; Kharchenko, S.M.; Grinenko, T.V. Effect of fibrin degradation products on fibrinolytic process. Ukr. Biochem. J. 2016, 88, 16–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, M.; Nesheim, M. A Study of the Protection of Plasmin from Antiplasmin Inhibition within an Intact Fibrin Clot during the Course of Clot Lysis. J. Biol. Chem. 2004, 279, 13333–13339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamarre, J.; Vasudevan, J.; Gonias, S.L. Plasmin cleaves betaglycan and releases a 60 kDa transforming growth factor-β complex from the cell surface. Biochem. J. 1994, 302, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Sahni, A.; Francis, C.W. Plasmic degradation modulates activity of fibrinogen-bound fibroblast growth factor-2. J. Thromb. Haemost. 2003, 1, 1271–1277. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, H.; Sisson, T.H.; Nishiuma, T.; Simon, R.H. Plasminogen-Mediated Activation and Release of Hepatocyte Growth Factor from Extracellular Matrix. Am. J. Respir. Cell Mol. Biol. 2006, 35, 705–713. [Google Scholar] [CrossRef] [Green Version]
- Friedrich, C.; Neugebauer, L.; Zamora, M.; Robles, J.P.; Martínez de la Escalera, G.; Clapp, C.; Bertsch, T.; Triebel, J. Plasmin generates vasoinhibin-like peptides by cleaving prolactin and placental lactogen. Mol. Cell. Endocrinol. 2021, 538, 111471. [Google Scholar] [CrossRef]
- Novak, J.F.; Hayes, J.D.; Nishimoto, S.K. Plasmin-Mediated Proteolysis of Osteocalcin. J. Bone Miner. Res. 1997, 12, 1035–1042. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zhang, L.; Miles, L.; Hoover-Plow, J. Plasminogen regulates pro-opiomelanocortin processing. J. Thromb. Haemost. 2004, 2, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Fukano, K.; Shimada, K.; Kamikawa, A.; Okamatsu-Ogura, Y.; Terao, A.; Yoshida, T.; Saito, M.; Kimura, K. Proinsulin C-peptide activates α-enolase: Implications for C-peptide–cell membrane interaction. J. Biochem. 2012, 152, 53–62. [Google Scholar] [CrossRef] [Green Version]
- Okaji, Y.; Tashiro, Y.; Gritli, I.; Nishida, C.; Sato, A.; Ueno, Y.; Del Canto Gonzalez, S.; Ohki-Koizumi, M.; Akiyama, H.; Nakauchi, H.; et al. Plasminogen deficiency attenuates postnatal erythropoiesis in male C57BL/6 mice through decreased activity of the LH-testosterone axis. Exp. Hematol. 2012, 40, 143–154. [Google Scholar] [CrossRef]
- Magnussen, S.N.; Hadler-Olsen, E.; Costea, D.E.; Berg, E.; Jacobsen, C.C.; Mortensen, B.; Salo, T.; Martinez-Zubiaurre, I.; Winberg, J.O.; Uhlin-Hansen, L.; et al. Cleavage of the urokinase receptor (uPAR) on oral cancer cells: Regulation by transforming growth factor—beta1 (TGF-beta1) and potential effects on migration and invasion. BMC Cancer 2017, 17, 350. [Google Scholar] [CrossRef] [Green Version]
- Monea, S.; Lehti, K.; Keski-Oja, J.; Mignatti, P. Plasmin activates pro-matrix metalloproteinase-2 with a membrane-type 1 matrix metalloproteinase-dependent mechanism. J. Cell. Physiol. 2002, 192, 160–170. [Google Scholar] [CrossRef]
- Heissig, B.; Lund, L.R.; Akiyama, H.; Ohki, M.; Morita, Y.; Rømer, J.; Nakauchi, H.; Okumura, K.; Ogawa, H.; Werb, Z.; et al. The Plasminogen Fibrinolytic Pathway Is Required for Hematopoietic Regeneration. Cell Stem Cell 2008, 3, 120. [Google Scholar] [CrossRef] [Green Version]
- Plow, E.F.; Doeuvre, L.; Das, R. So Many Plasminogen Receptors: Why? J. Biomed. Biotechnol. 2012, 2012, 141806. [Google Scholar] [CrossRef] [Green Version]
- Miles, L.A.; Vago, J.P.; Sousa, L.P.; Parmer, R.J. Functions of the plasminogen receptor Plg-RKT. J. Thromb. Haemost. 2020, 18, 2468–2481. [Google Scholar] [CrossRef]
- Bharadwaj, A.G.; Kempster, E.; Waisman, D.M. The ANXA2/S100A10 Complex—Regulation of the Oncogenic Plasminogen Receptor. Biomolecules 2021, 11, 1772. [Google Scholar] [CrossRef]
- Bharadwaj, A.; Kempster, E.; Waisman, D.M. The Annexin A2/S100A10 Complex: The Mutualistic Symbiosis of Two Distinct Proteins. Biomolecules 2021, 11, 1849. [Google Scholar] [CrossRef]
- Salama, Y.; Lin, S.Y.; Dhahri, D.; Hattori, K.; Heissig, B. The fibrinolytic factor tPA drives LRP1-mediated melanoma growth and metastasis. FASEB J. 2019, 33, 3465–3480. [Google Scholar] [CrossRef]
- Ichinose, A.; Kisiel, W.; Fujikawa, K. Proteolytic activation of tissue plasminogen activator by plasma and tissue enzymes. FEBS Lett. 1984, 175, 412–418. [Google Scholar] [CrossRef] [Green Version]
- Sappino, A.P.; Huarte, J.; Vassalli, J.D.; Belin, D. Sites of synthesis of urokinase and tissue-type plasminogen activators in the murine kidney. J. Clin. Investig. 1991, 87, 962–970. [Google Scholar] [CrossRef]
- Padró, T.; van den Hoogen, C.M.; Emeis, J.J. Distribution of tissue-type plasminogen activator (activity and antigen) in rat tissues. Blood Coagul. Fibrinolysis 1990, 1, 601–608. [Google Scholar] [PubMed]
- Kristensen, P.; Larsson, L.-I.; Nielsen, L.S.; Grøndahl-Hansen, J.; Andreasen, P.A.; Danø, K. Human endothelial cells contain one type of plasminogen activator. FEBS Lett. 1984, 168, 33–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, M.M.C.G.; Thelwell, C.; Williams, S.C.; Longstaff, C. Regulation of fibrinolysis by C-terminal lysines operates through plasminogen and plasmin but not tissue-type plasminogen activator. J. Thromb. Haemost. 2012, 10, 2354–2360. [Google Scholar] [CrossRef] [PubMed]
- Collen, D.; Lijnen, H.R. The Tissue-Type Plasminogen Activator Story. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1151–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, X.-F.; Brohlin, M.; Pohl, G.; Bäck, O.; Wallén, P. Binding of tissue plasminogen activator to endothelial cells: The effect on functional properties. Localization of a ligand in the B-chain of tPA. Thromb. Res. 1995, 77, 149–164. [Google Scholar] [CrossRef] [PubMed]
- Sashindranath, M.; Sales, E.; Daglas, M.; Freeman, R.; Samson, A.L.; Cops, E.J.; Beckham, S.; Galle, A.; McLean, C.; Morganti-Kossmann, C.; et al. The tissue-type plasminogen activator–plasminogen activator inhibitor 1 complex promotes neurovascular injury in brain trauma: Evidence from mice and humans. Brain 2012, 135, 3251–3264. [Google Scholar] [CrossRef] [Green Version]
- Heissig, B.; Eiamboonsert, S.; Salama, Y.; Shimazu, H.; Dhahri, D.; Munakata, S.; Tashiro, Y.; Hattori, K. Cancer therapy targeting the fibrinolytic system. Adv. Drug Deliv. Rev. 2016, 99, 172–179. [Google Scholar] [CrossRef]
- Leu, S.; Day, Y.-J.; Sun, C.-K.; Yip, H.-K. tPA-MMP-9 Axis Plays a Pivotal Role in Mobilization of Endothelial Progenitor Cells from Bone Marrow to Circulation and Ischemic Region for Angiogenesis. Stem Cells Int. 2016, 2016, 5417565. [Google Scholar] [CrossRef] [Green Version]
- Seillier, C.; Hélie, P.; Petit, G.; Vivien, D.; Clemente, D.; Le Mauff, B.; Docagne, F.; Toutirais, O. Roles of the tissue-type plasminogen activator in immune response. Cell. Immunol. 2022, 371, 104451. [Google Scholar] [CrossRef]
- Hu, K.; Lin, L.; Tan, X.; Yang, J.; Bu, G.; Mars, W.M.; Liu, Y. tPA Protects Renal Interstitial Fibroblasts and Myofibroblasts from Apoptosis. J. Am. Soc. Nephrol. 2008, 19, 503. [Google Scholar] [CrossRef]
- Das, L.; Azmoon, P.; Banki, M.A.; Mantuano, E.; Gonias, S.L. Tissue-type plasminogen activator selectively inhibits multiple toll-like receptors in CSF-1-differentiated macrophages. PLoS ONE 2019, 14, e0224738. [Google Scholar] [CrossRef] [PubMed]
- Husain, S.S. Single-chain urokinase-type plasminogen activator does not possess measurable intrinsic amidolytic or plasminogen activator activities. Biochem.-Us 1991, 30, 5797–5805. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, N.; Mihalcioiu, C.; Rabbani, S.A. Multifaceted Role of the Urokinase-Type Plasminogen Activator (uPA) and Its Receptor (uPAR): Diagnostic, Prognostic, and Therapeutic Applications. Front. Oncol. 2018, 8, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bansal, V.; Roychoudhury, P.K. Production and purification of urokinase: A comprehensive review. Protein Expr. Purif. 2006, 45, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Sui, Y.; Wang, J.; Li, Y.; Li, H.; Cao, Y.; Chen, L.; Jiang, L.; Yuan, C.; Huang, M. Crystal structure and cellular functions of uPAR dimer. Nat. Commun. 2022, 13, 1665. [Google Scholar] [CrossRef] [PubMed]
- Sillen, M.; Declerck, P.J. Thrombin Activatable Fibrinolysis Inhibitor (TAFI): An Updated Narrative Review. Int. J. Mol. Sci. 2021, 22, 3670. [Google Scholar] [CrossRef]
- Tjärnlund-Wolf, A.; Brogren, H.; Lo, E.H.; Wang, X. Plasminogen Activator Inhibitor-1 and Thrombotic Cerebrovascular Diseases. Stroke 2012, 43, 2833–2839. [Google Scholar] [CrossRef]
- Kubala, M.H.; DeClerck, Y.A. The plasminogen activator inhibitor-1 paradox in cancer: A mechanistic understanding. Cancer Metastasis Rev. 2019, 38, 483–492. [Google Scholar] [CrossRef]
- Yasar Yildiz, S.; Kuru, P.; Toksoy Oner, E.; Agirbasli, M. Functional Stability of Plasminogen Activator Inhibitor-1. Sci. World J. 2014, 2014, 858293. [Google Scholar] [CrossRef] [Green Version]
- Sillen, M.; Miyata, T.; Vaughan, D.E.; Strelkov, S.V.; Declerck, P.J. Structural Insight into the Two-Step Mechanism of PAI-1 Inhibition by Small Molecule TM5484. Int. J. Mol. Sci. 2021, 22, 1482. [Google Scholar] [CrossRef]
- Smith, H.W.; Marshall, C.J. Regulation of cell signalling by uPAR. Nat. Rev. Mol. Cell Biol. 2010, 11, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, L.J.H.; Petersen, J.E.V.; Eugen-Olsen, J. Soluble Urokinase Plasminogen Activator Receptor (suPAR) as a Biomarker of Systemic Chronic Inflammation. Front. Immunol. 2021, 12, 780641. [Google Scholar] [CrossRef]
- Yuan, C.; Guo, Z.; Yu, S.; Jiang, L.; Huang, M. Development of inhibitors for uPAR: Blocking the interaction of uPAR with its partners. Drug Discov. Today 2021, 26, 1076–1085. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Liu, G.; Zhang, Y.; Cui, Z.; Wang, F.; Liu, X.; Chu, R.; Zhao, M. Urinary soluble urokinase receptor levels are elevated and pathogenic in patients with primary focal segmental glomerulosclerosis. BMC Med. 2014, 12, 81. [Google Scholar] [CrossRef] [Green Version]
- Montuori, N.; Ragno, P. Multiple activities of a multifaceted receptor: Roles of cleaved and soluble uPAR. Front. Biosci. (Landmark Ed.) 2009, 14, 2494–2503. [Google Scholar] [CrossRef] [Green Version]
- Huai, Q.; Mazar, A.P.; Kuo, A.; Parry, G.C.; Shaw, D.E.; Callahan, J.; Li, Y.; Yuan, C.; Bian, C.; Chen, L.; et al. Structure of human urokinase plasminogen activator in complex with its receptor. Science 2006, 311, 656–659. [Google Scholar] [CrossRef]
- Montuori, N.; Visconte, V.; Rossi, G.; Ragno, P. Soluble and cleaved forms of the urokinase-receptor: Degradation products or active molecules? Thromb. Haemost. 2005, 93, 192–198. [Google Scholar] [CrossRef]
- Fietz, T.; Hattori, K.; Thiel, E.; Heissig, B. Increased soluble urokinase plasminogen activator receptor (suPAR) serum levels after granulocyte colony-stimulating factor treatment do not predict successful progenitor cell mobilization in vivo. Blood 2006, 107, 3408–3409. [Google Scholar] [CrossRef] [Green Version]
- Jo, M.; Thomas, K.S.; Wu, L.; Gonias, S.L. Soluble urokinase-type plasminogen activator receptor inhibits cancer cell growth and invasion by direct urokinase-independent effects on cell signaling. J. Biol. Chem. 2003, 278, 46692–46698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chew-Harris, J.; Appleby, S.; Richards, A.M.; Troughton, R.W.; Pemberton, C.J. Analytical, biochemical and clearance considerations of soluble urokinase plasminogen activator receptor (suPAR) in healthy individuals. Clin. Biochem. 2019, 69, 36–44. [Google Scholar] [CrossRef]
- Wei, C.; El Hindi, S.; Li, J.; Fornoni, A.; Goes, N.; Sageshima, J.; Maiguel, D.; Karumanchi, S.A.; Yap, H.K.; Saleem, M.; et al. Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis. Nat. Med. 2011, 17, 952–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoenigl, M.; Raggam, R.B.; Wagner, J.; Valentin, T.; Leitner, E.; Seeber, K.; Zollner-Schwetz, I.; Krammer, W.; Prüller, F.; Grisold, A.J.; et al. Diagnostic accuracy of soluble urokinase plasminogen activator receptor (suPAR) for prediction of bacteremia in patients with systemic inflammatory response syndrome. Clin. Biochem. 2013, 46, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Skrypnyk, M.; Petrushanko, T.; Neporada, K.; Vynnyk, N.; Petrushanko, V.; Skrypnyk, R. Colonization resistance of oral mucosa in individuals with diverse body mass index. J. Stomatol. 2022, 75, 171–175. [Google Scholar] [CrossRef]
- Virtanen, O.J.; Sirén, V.; Multanen, J.; Färkkilä, M.; Leivo, I.; Vaheri, A.; Koskiniemi, M. Plasminogen activators and their inhibitors in human saliva and salivary gland tissue. Eur. J. Oral Sci. 2006, 114, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Botran, R.; Szabo, Y.Z.; Lyle, K.B.; Newton, T.L. The levels of soluble urokinase plasminogen activator receptor (suPAR) in saliva are influenced by acute stress. Biol. Psychol. 2021, 165, 108147. [Google Scholar] [CrossRef]
- El-Patal, M.A.E.; Khalil, M.A.; Shipl, W.; Barakat, I.; Youssef, E.M.I.; El Attar, S.; Fathi, A.; Abdallah, A.A. Detection of soluble urokinase type plasminogen activator receptors in children with gingivitis and normal subjects. BMC Oral Health 2022, 22, 436. [Google Scholar] [CrossRef]
- Skottrup, P.D.; Dahlén, G.; Baelum, V.; Lopez, R. Soluble urokinase-type plasminogen activator receptor is associated with signs of periodontitis in adolescents. Eur. J. Oral Sci. 2018, 126, 292–299. [Google Scholar] [CrossRef]
- Taşdemir, İ.; Erbak Yılmaz, H.; Narin, F.; Sağlam, M. Assessment of saliva and gingival crevicular fluid soluble urokinase plasminogen activator receptor (suPAR), galectin-1, and TNF-α levels in periodontal health and disease. J. Periodontal Res. 2020, 55, 622–630. [Google Scholar] [CrossRef]
- Gustafsson, A.; Ajeti, V.; Ljunggren, L. Detection of suPAR in the Saliva of Healthy Young Adults: Comparison with Plasma Levels. Biomark. Insights 2011, 6, BMI–S8326. [Google Scholar] [CrossRef]
- Wen, J.; Nikitakis, N.G.; Chaisuparat, R.; Greenwell-Wild, T.; Gliozzi, M.; Jin, W.; Adli, A.; Moutsopoulos, N.; Wu, T.; Warburton, G.; et al. Secretory leukocyte protease inhibitor (SLPI) expression and tumor invasion in oral squamous cell carcinoma. Am. J. Pathol. 2011, 178, 2866–2878. [Google Scholar] [CrossRef]
- Sejima, T.; Holtappels, G.; Bachert, C. The Expression of Fibrinolytic Components in Chronic Paranasal Sinus Disease. Am. J. Rhinol. Allergy 2011, 25, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Firinu, D.; Arba, M.; Vincenzoni, F.; Iavarone, F.; Costanzo, G.; Cabras, T.; Castagnola, M.; Messana, I.; Del Giacco, S.R.; Sanna, M.T. Proteomic Analysis of the Acid-Insoluble Fraction of Whole Saliva from Patients Affected by Different Forms of Non-histaminergic Angioedema. J. Clin. Immunol. 2020, 40, 840–850. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, Y.; Pan, L.; Li, J.; Yu, Y.; Liu, B.; Zubair, M.; Wei, Y.; Pillay, B.; Olaniran, A.O.; et al. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) moonlights as an adhesin in Mycoplasma hyorhinis adhesion to epithelial cells as well as a plasminogen receptor mediating extracellular matrix degradation. Vet. Res. 2021, 52, 80. [Google Scholar] [CrossRef] [PubMed]
- Sindet-Pedersen, S.; Gram, J.; Jespersen, J. The Possible Role of Oral Epithelial Cells in Tissue-type Plasminogen Activator-related Fibrinolysis in Human Saliva. J. Dent. Res. 1990, 69, 1283–1286. [Google Scholar] [CrossRef] [PubMed]
- Kjaeldgaard, A.; Kjaeldgaard, M. Immunological characterization of plasminogen activators in human mixed saliva. Acta Physiol. Scand. 1986, 126, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Dimeski, G.; Punyadeera, C. Validation of an immunoassay to measure plasminogen-activator inhibitor-1 concentrations in human saliva. Biochem. Med. 2014, 24, 258–265. [Google Scholar] [CrossRef]
- Guru, S.R.; Aghanashini, S. Impact of scaling and root planing on salivary and serum plasminogen activator inhibitor-1 expression in patients with periodontitis with and without type 2 diabetes mellitus. J. Periodontol. 2023, 94, 20–30. [Google Scholar] [CrossRef]
- Joshipura, K.J.; Wand, H.C.; Merchant, A.T.; Rimm, E.B. Periodontal disease and biomarkers related to cardiovascular disease. J. Dent. Res. 2004, 83, 151–155. [Google Scholar] [CrossRef]
- Silva, L.M.; Doyle, A.D.; Greenwell-Wild, T.; Dutzan, N.; Tran, C.L.; Abusleme, L.; Juang, L.J.; Leung, J.; Chun, E.M.; Lum, A.G.; et al. Fibrin is a critical regulator of neutrophil effector function at the oral mucosal barrier. Science 2021, 374, eabl5450. [Google Scholar] [CrossRef]
- Schuster, V.; Hugle, B.; Tefs, K. Plasminogen deficiency. J. Thromb. Haemost. 2007, 5, 2315–2322. [Google Scholar] [CrossRef]
- Recke, A.; Massalme, E.G.; Jappe, U.; Steinmüller-Magin, L.; Schmidt, J.; Hellenbroich, Y.; Hüning, I.; Gillessen-Kaesbach, G.; Zillikens, D.; Hartmann, K. Identification of the recently described plasminogen gene mutation p.Lys330Glu in a family from Northern Germany with hereditary angioedema. Clin. Transl. Allergy 2019, 9, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivolella, S.; De Biagi, M.; Sartori, M.T.; Berengo, M.; Bressan, E. Destructive Membranous Periodontal Disease (Ligneous Gingivitis): A Literature Review. J. Periodontol. 2012, 83, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Sulniute, R.; Lindh, T.; Wilczynska, M.; Li, J.; Ny, T. Plasmin is essential in preventing periodontitis in mice. Am. J. Pathol. 2011, 179, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Kurtulus Waschulewski, I.; Gökbuget, A.Y.; Christiansen, N.M.; Ziegler, M.; Schuster, V.; Wahl, G.; Götz, W. Immunohistochemical analysis of the gingiva with periodontitis of type I plasminogen deficiency compared to gingiva with gingivitis and periodontitis and healthy gingiva. Arch. Oral Biol. 2016, 72, 75–86. [Google Scholar] [CrossRef]
- Sadasivan, A.; Ramesh, R.; Mathew, D.G. Ligneous Periodontitis in a Patient with Type 1 Plasminogen Deficiency: A Case Report and Review of the Literature. Case Rep. Dent. 2020, 2020, 5680535. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, A.D.; Nakar, C.; Parker, J.M.; Albert, G.R.; Moran, J.E.; Thibaudeau, K.; Thukral, N.; Hardesty, B.M.; Laurin, P.; Sandset, P.M. Plasminogen replacement therapy for the treatment of children and adults with congenital plasminogen deficiency. Blood 2018, 131, 1301–1310. [Google Scholar] [CrossRef] [Green Version]
- Castellino, F.J.; Ploplis, V.A. Structure and function of the plasminogen/plasmin system. Thromb. Haemost. 2005, 93, 647–654. [Google Scholar] [CrossRef]
- Urban, C.F.; Ermert, D.; Schmid, M.; Abu-Abed, U.; Goosmann, C.; Nacken, W.; Brinkmann, V.; Jungblut, P.R.; Zychlinsky, A. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 2009, 5, e1000639. [Google Scholar] [CrossRef] [Green Version]
- Thammavongsa, V.; Kim, H.K.; Missiakas, D.; Schneewind, O. Staphylococcal manipulation of host immune responses. Nat. Rev. Microbiol. 2015, 13, 529–543. [Google Scholar] [CrossRef] [Green Version]
- Munz, M.; Chen, H.; Jockel-Schneider, Y.; Adam, K.; Hoffman, P.; Berger, K.; Kocher, T.; Meyle, J.; Eickholz, P.; Doerfer, C.; et al. A haplotype block downstream of plasminogen is associated with chronic and aggressive periodontitis. J. Clin. Periodontol. 2017, 44, 962–970. [Google Scholar] [CrossRef]
- Sochaj-Gregorczyk, A.; Ksiazek, M.; Waligorska, I.; Straczek, A.; Benedyk, M.; Mizgalska, D.; Thøgersen, I.B.; Enghild, J.J.; Potempa, J. Plasmin inhibition by bacterial serpin: Implications in gum disease. FASEB J. 2020, 34, 619–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avilan, L.; Calcagno, M.; Figuera, M.; Lemus, L.; Puig, J.; Rodriguez, A.M. Interaction of Leishmania mexicana promastigotes with the plasminogen-plasmin system. Mol. Biochem. Parasitol. 2000, 110, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Ayón-Núñez, D.A.; Fragoso, G.; Bobes, R.J.; Laclette, J.P. Plasminogen-binding proteins as an evasion mechanism of the host’s innate immunity in infectious diseases. Biosci. Rep. 2018, 38, BSR20180705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, S.; Ploplis, V.A.; Castellino, F.J. Bacterial plasminogen receptors utilize host plasminogen system for effective invasion and dissemination. J. Biomed. Biotechnol. 2012, 2012, 482096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tapper, H.; Herwald, H. Modulation of hemostatic mechanisms in bacterial infectious diseases. Blood 2000, 96, 2329–2337. [Google Scholar] [CrossRef] [PubMed]
- Degen, J.L.; Bugge, T.H.; Goguen, J.D. Fibrin and fibrinolysis in infection and host defense. J. Thromb. Haemost. 2007, 5, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Loof, T.G.; Deicke, C.; Medina, E. The role of coagulation/fibrinolysis during Streptococcus pyogenes infection. Front. Cell. Infect. Microbiol. 2014, 4, 128. [Google Scholar] [CrossRef] [Green Version]
- Marcos, C.M.; de Fátima da Silva, J.; de Oliveira, H.C.; Moraes da Silva, R.A.; Mendes-Giannini, M.J.S.; Fusco-Almeida, A.M. Surface-expressed enolase contributes to the adhesion of Paracoccidioides brasiliensis to host cells. FEMS Yeast Res. 2012, 12, 557–570. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, V.; Talens, S.; Grandits, A.M.; Blom, A.M. A Novel Interaction between Complement Inhibitor C4b-binding Protein and Plasminogen That Enhances Plasminogen Activation. J. Biol. Chem. 2015, 290, 18333–18342. [Google Scholar] [CrossRef] [Green Version]
- Barthel, D.; Schindler, S.; Zipfel, P.F. Plasminogen is a complement inhibitor. J. Biol. Chem. 2012, 287, 18831–18842. [Google Scholar] [CrossRef]
- Sato, A.; Nishida, C.; Sato-Kusubata, K.; Ishihara, M.; Tashiro, Y.; Gritli, I.; Shimazu, H.; Munakata, S.; Yagita, H.; Okumura, K.; et al. Inhibition of plasmin attenuates murine acute graft-versus-host disease mortality by suppressing the matrix metalloproteinase-9-dependent inflammatory cytokine storm and effector cell trafficking. Leukemia 2015, 29, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Heissig, B.; Salama, Y.; Tateno, M.; Takahashi, S.; Hattori, K. siRNA against CD40 delivered via a fungal recognition receptor ameliorates murine acute graft-versus-host disease. EJHaem 2022, 3, 849–861. [Google Scholar] [CrossRef] [PubMed]
- Shimazu, H.; Munakata, S.; Tashiro, Y.; Salama, Y.; Dhahri, D.; Eiamboonsert, S.; Ota, Y.; Onoda, H.; Tsuda, Y.; Okada, Y.; et al. Pharmacological targeting of plasmin prevents lethality in a murine model of macrophage activation syndrome. Blood 2017, 130, 59–72. [Google Scholar] [CrossRef] [Green Version]
- Kwaan, H.C.; Lindholm, P.F. The Central Role of Fibrinolytic Response in COVID-19—A Hematologist’s Perspective. Int. J. Mol. Sci. 2021, 22, 1283. [Google Scholar] [CrossRef] [PubMed]
- Berri, F.; Rimmelzwaan, G.F.; Hanss, M.; Albina, E.; Foucault-Grunenwald, M.-L.; Lê, V.B.; Vogelzang-van Trierum, S.E.; Gil, P.; Camerer, E.; Martinez, D.; et al. Plasminogen Controls Inflammation and Pathogenesis of Influenza Virus Infections via Fibrinolysis. PLoS Pathog. 2013, 9, e1003229. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Wang, Y.; Ji, M.; Pei, F.; Zhao, Q.; Zhou, Y.; Hong, Y.; Han, S.; Wang, J.; Wang, Q.; et al. Transmission Routes Analysis of SARS-CoV-2: A Systematic Review and Case Report. Front. Cell Dev. Biol. 2020, 8, 618. [Google Scholar] [CrossRef]
- Elisetti, N. Periodontal pocket and COVID-19: Could there be a possible link? Med. Hypotheses 2021, 146, 110355. [Google Scholar] [CrossRef]
- Belouzard, S.; Millet, J.K.; Licitra, B.N.; Whittaker, G.R. Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses 2012, 4, 1011–1033. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.; Cho, J.G.; Chae, S.W.; Lee, H.M.; Hwang, S.J.; Woo, J.S. Epithelial Na+ channel (ENaC) expression in obstructive sialadenitis of the submandibular gland. Arch. Oral Biol. 2011, 56, 121–126. [Google Scholar] [CrossRef]
- Zhu, F.; Zhong, Y.; Ji, H.; Ge, R.; Guo, L.; Song, H.; Wu, H.; Jiao, P.; Li, S.; Wang, C.; et al. ACE2 and TMPRSS2 in human saliva can adsorb to the oral mucosal epithelium. J. Anat. 2022, 240, 398–409. [Google Scholar] [CrossRef]
- Matuck, B.F.; Dolhnikoff, M.; Duarte-Neto, A.N.; Maia, G.; Gomes, S.C.; Sendyk, D.I.; Zarpellon, A.; de Andrade, N.P.; Monteiro, R.A.; Pinho, J.R.R.; et al. Salivary glands are a target for SARS-CoV-2: A source for saliva contamination. J. Pathol. 2021, 254, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e286. [Google Scholar] [CrossRef] [PubMed]
- Takeda, M. Proteolytic activation of SARS-CoV-2 spike protein. Microbiol. Immunol. 2022, 66, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Meng, B.; Abdullahi, A.; Ferreira, I.A.T.M.; Goonawardane, N.; Saito, A.; Kimura, I.; Yamasoba, D.; Gerber, P.P.; Fatihi, S.; Rathore, S.; et al. Altered TMPRSS2 usage by SARS-CoV-2 Omicron impacts infectivity and fusogenicity. Nature 2022, 603, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Iwata-Yoshikawa, N.; Okamura, T.; Shimizu, Y.; Hasegawa, H.; Takeda, M.; Nagata, N. TMPRSS2 Contributes to Virus Spread and Immunopathology in the Airways of Murine Models after Coronavirus Infection. J. Virol. 2019, 93, e01815-18. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.W.; Mao, H.J.; Wu, Y.L.; Tanaka, Y.; Zhang, W. TMPRSS2: A potential target for treatment of influenza virus and coronavirus infections. Biochimie 2017, 142, 1–10. [Google Scholar] [CrossRef]
- Dittmann, M.; Hoffmann, H.-H.; Scull, M.A.; Gilmore, R.H.; Bell, K.L.; Ciancanelli, M.; Wilson, S.J.; Crotta, S.; Yu, Y.; Flatley, B.; et al. A Serpin Shapes the Extracellular Environment to Prevent Influenza A Virus Maturation. Cell 2015, 160, 631–643. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Yu, T.; Wang, T.; Ding, Y.; Cui, Y.; Nie, H. Competitive cleavage of SARS-CoV-2 spike protein and epithelial sodium channel by plasmin as a potential mechanism for COVID-19 infection. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2022, 323, L569–L577. [Google Scholar] [CrossRef]
- Passero, C.J.; Mueller, G.M.; Rondon-Berrios, H.; Tofovic, S.P.; Hughey, R.P.; Kleyman, T.R. Plasmin activates epithelial Na+ channels by cleaving the gamma subunit. J. Biol. Chem. 2008, 283, 36586–36591. [Google Scholar] [CrossRef]
- Matsuyama, A.; Okura, H.; Hashimoto, S.; Tanaka, T. A prospective, randomized, open-label trial of early versus late povidone-iodine gargling in patients with COVID-19. Sci. Rep. 2022, 12, 20449. [Google Scholar] [CrossRef] [PubMed]
- Marouf, N.; Cai, W.; Said, K.N.; Daas, H.; Diab, H.; Chinta, V.R.; Hssain, A.A.; Nicolau, B.; Sanz, M.; Tamimi, F. Association between periodontitis and severity of COVID-19 infection: A case–control study. J. Clin. Periodontol. 2021, 48, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.R.; Scully, M. Clinical features of thrombosis and bleeding in COVID-19. Blood 2022, 140, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Flaumenhaft, R.; Enjyoji, K.; Schmaier, A.A. Vasculopathy in COVID-19. Blood 2022, 140, 222–235. [Google Scholar] [CrossRef]
- Semiz, S. COVID-19 biomarkers: What did we learn from systematic reviews? Front. Cell. Infect. Microbiol. 2022, 12, 1038908. [Google Scholar] [CrossRef] [PubMed]
- Rovina, N.; Akinosoglou, K.; Eugen-Olsen, J.; Hayek, S.; Reiser, J.; Giamarellos-Bourboulis, E.J. Soluble urokinase plasminogen activator receptor (suPAR) as an early predictor of severe respiratory failure in patients with COVID-19 pneumonia. Crit. Care 2020, 24, 187. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Warnock, M.; Harbaugh, A.; Yalavarthi, S.; Gockman, K.; Zuo, M.; Madison, J.A.; Knight, J.S.; Kanthi, Y.; Lawrence, D.A. Plasma tissue plasminogen activator and plasminogen activator inhibitor-1 in hospitalized COVID-19 patients. Sci. Rep. 2021, 11, 1580. [Google Scholar] [CrossRef] [PubMed]
- Seheult, J.N.; Seshadri, A.; Neal, M.D. Fibrinolysis Shutdown and Thrombosis in Severe COVID-19. J. Am. Coll. Surg. 2020, 231, 203–204. [Google Scholar] [CrossRef]
- Della-Morte, D.; Pacifici, F.; Ricordi, C.; Massoud, R.; Rovella, V.; Proietti, S.; Iozzo, M.; Lauro, D.; Bernardini, S.; Bonassi, S.; et al. Low level of plasminogen increases risk for mortality in COVID-19 patients. Cell Death Dis. 2021, 12, 773. [Google Scholar] [CrossRef]
- Heissig, B.; Salama, Y.; Iakoubov, R.; Vehreschild, J.J.; Rios, R.; Nogueira, T.; Vehreschild, M.J.G.T.; Stecher, M.; Mori, H.; Lanznaster, J.; et al. COVID-19 Severity and Thrombo-Inflammatory Response Linked to Ethnicity. Biomedicines 2022, 10, 2549. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yatsenko, T.; Skrypnyk, M.; Troyanovska, O.; Tobita, M.; Osada, T.; Takahashi, S.; Hattori, K.; Heissig, B. The Role of the Plasminogen/Plasmin System in Inflammation of the Oral Cavity. Cells 2023, 12, 445. https://doi.org/10.3390/cells12030445
Yatsenko T, Skrypnyk M, Troyanovska O, Tobita M, Osada T, Takahashi S, Hattori K, Heissig B. The Role of the Plasminogen/Plasmin System in Inflammation of the Oral Cavity. Cells. 2023; 12(3):445. https://doi.org/10.3390/cells12030445
Chicago/Turabian StyleYatsenko, Tetiana, Maksym Skrypnyk, Olga Troyanovska, Morikuni Tobita, Taro Osada, Satoshi Takahashi, Koichi Hattori, and Beate Heissig. 2023. "The Role of the Plasminogen/Plasmin System in Inflammation of the Oral Cavity" Cells 12, no. 3: 445. https://doi.org/10.3390/cells12030445