
A Novel Splicing Variant of COL2A1 in a Fetus with Achondrogenesis Type II: Interpretation of Pathogenicity of In-Frame Deletions

 , , , and
, , , and 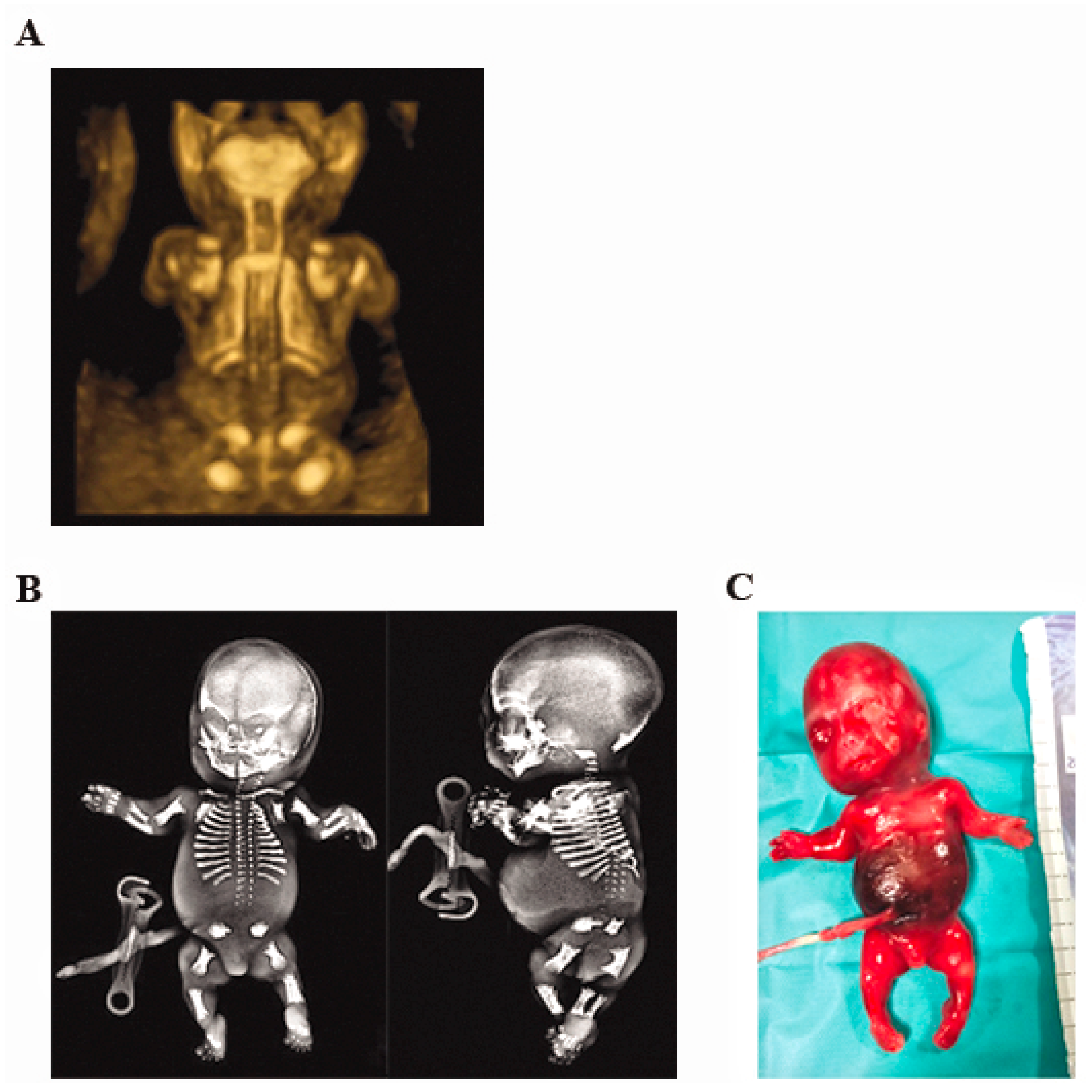{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Case Report
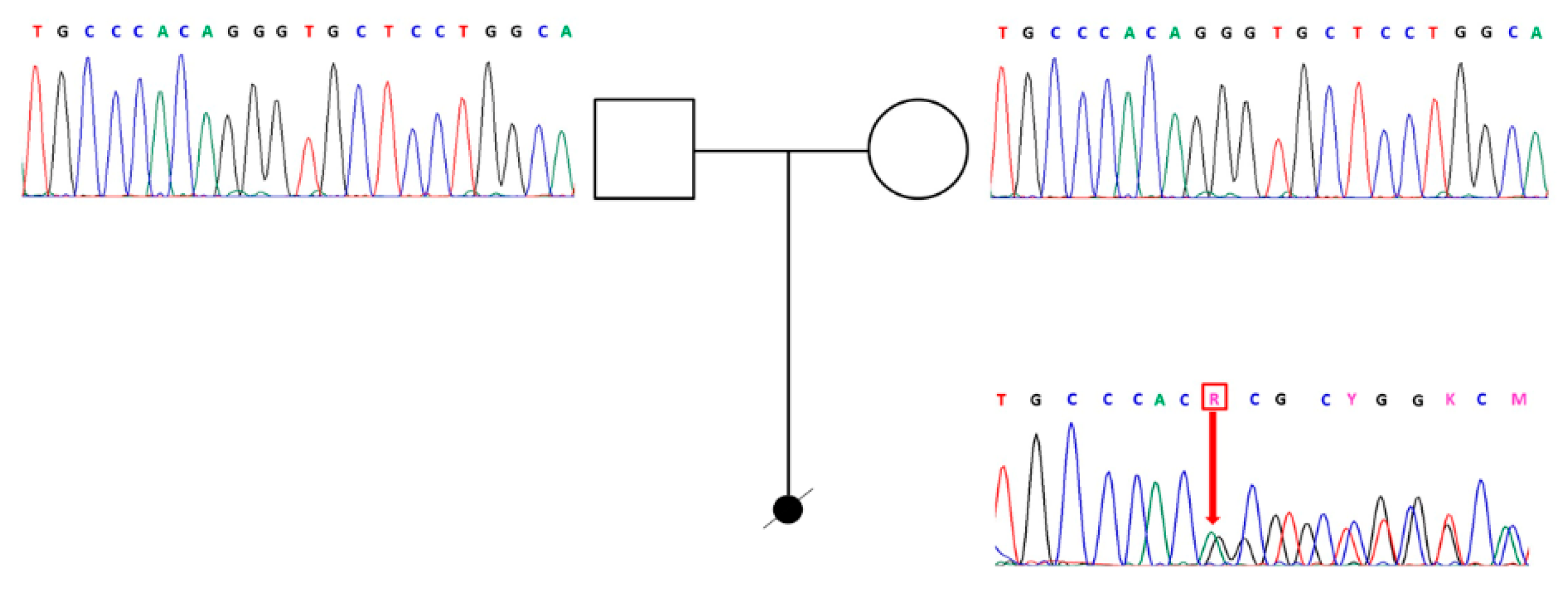
2.2. Gene Sequencing
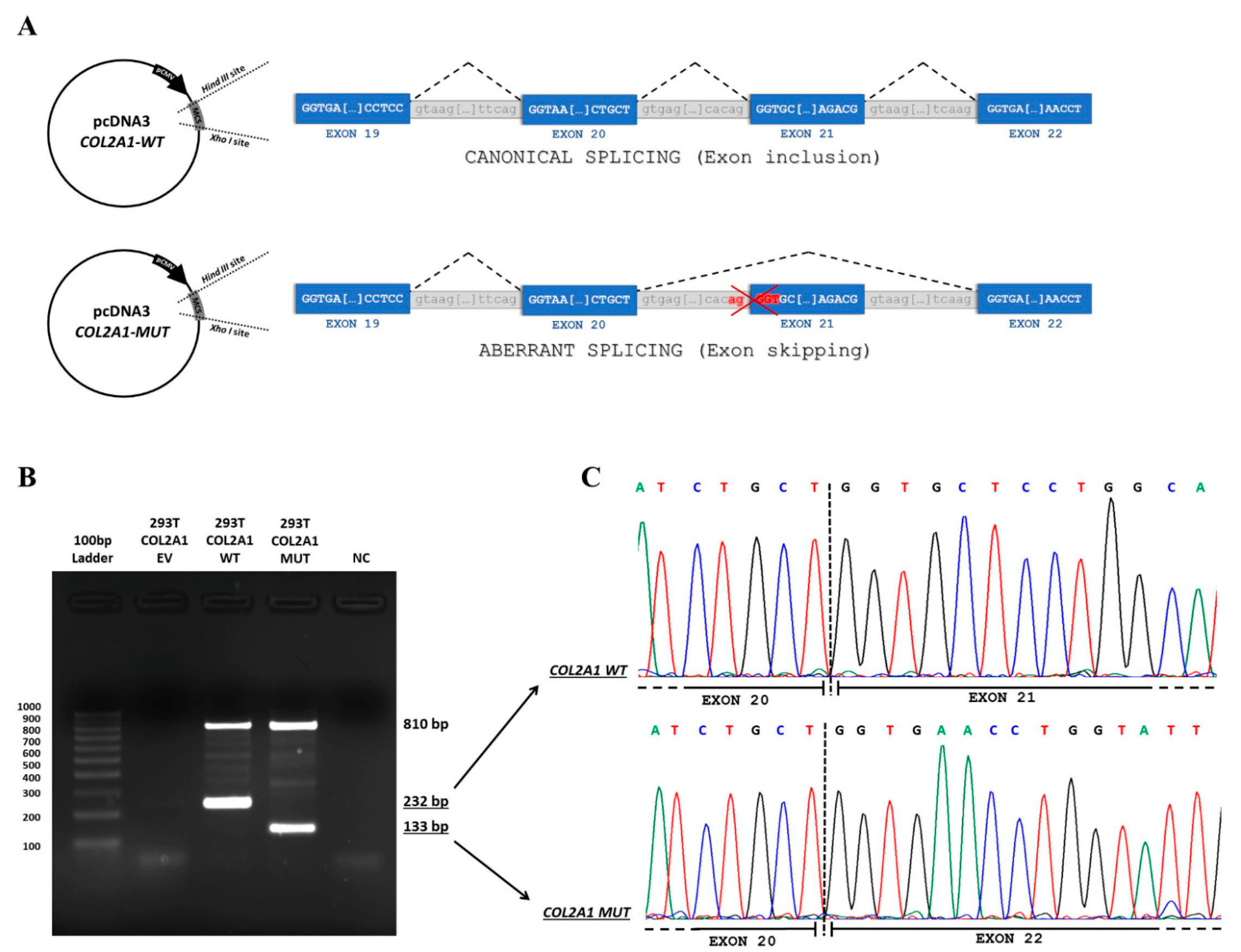
2.3. Minigene Assay
3. Results
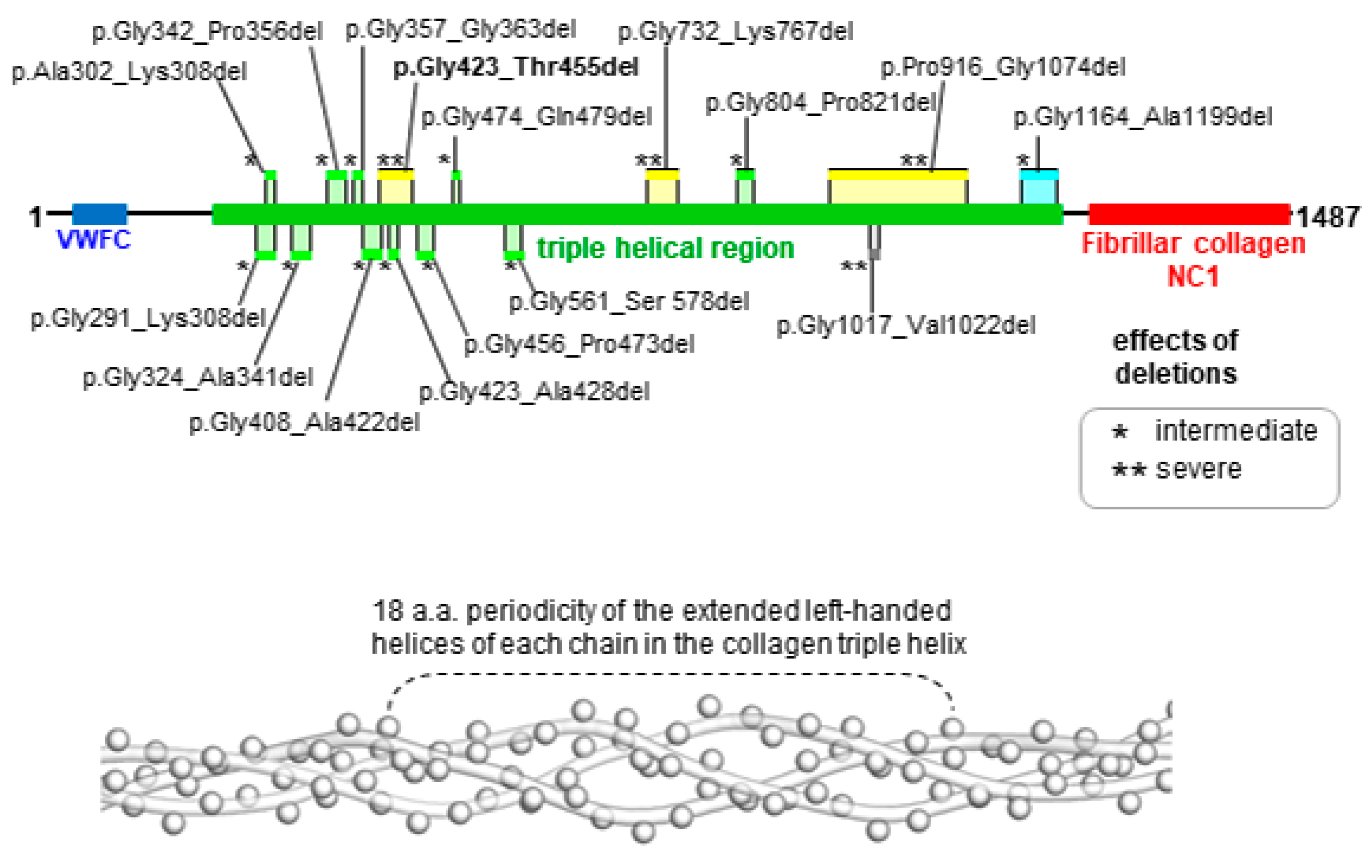
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Chen, H.; Liu, C.T.; Yang, S.S.; Opitz, J.M. Achondrogenesis: A review with special consideration of achondrogenesis type II (Langer-Saldino). Am. J. Med. Genet. 1981, 10, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Langer, L.O., Jr.; Spranger, J.W.; Greinacher, I.; Herdman, R.C. Thanatophoric dwarfism: A condition confused with achondroplasia in the neonate, with brief comments on achondrogenesis and homozygous achondroplasia. Radiology 1969, 92, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Saldino, M.D.; Ronald, M. Lethal short-limbed dwarfism: Achondrogenesis and thanatophoric dwarfism. Am. J. Roentgen. Radium Ther. Nucl. Med. 1971, 112, 185–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warman, M.L.; Cormier-Daire, V.; Hall, C.; Krakow, D.; Lachman, R.; LeMerrer, M.; Mortier, G.; Mundlos, S.; Nishimura, G.; Rimoin, D.L.; et al. Nosology and classification of genetic skeletal disorders: 2010 revision. Am. J. Med. Genet. Part A 2011, 155, 943–968. [Google Scholar] [CrossRef] [PubMed]
- Terhal, P.A.; Nievelstein, R.J.; Verver, E.J.; Topsakal, V.; van Dommelen, P.; Hoornaert, K.; Le Merrer, M.; Zankl, A.; Simon, M.E.; Smithson, S.F.; et al. A study of the clinical and radiological features in a cohort of 93 patients with a COL2A1 mutation causing spondyloepiphyseal dysplasia congenita or a related phenotype. Am. J. Med. Genet. Part A 2015, 167, 461–475. [Google Scholar] [CrossRef] [PubMed]
- Francomano, C.A.; McIntosh, I.; Wilkin, D.J. Bone dysplasias in man: Molecular insights. Curr. Opin. Genet. Dev. 1996, 6, 301–308. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, Y.; Wu, N.; Li, J.; Liu, H.; Wang, J. Integrated analysis of COL2A1 variant data and classification of type II collagenopathies. Clin. Genet. 2020, 97, 383–395. [Google Scholar] [CrossRef] [Green Version]
- Lovell-Badge, R.H.; Bygrave, A.; Bradley, A.; Robertson, E.; Tilly, R.; Cheah, K.S.E. Tissue-specific expression of the human type II collagen gene in mice. Proc. Nat. Acad. Sci. USA 1987, 84, 2803–2807. [Google Scholar] [CrossRef] [Green Version]
- Barat-Houari, M.; Sarrabay, G.; Gatinois, V.; Fabre, A.; Dumont, B.; Genevieve, D.; Touitou, I. Mutation Update for COL2A1 Gene Variants Associated with Type II Collagenopathies. Hum. Mutat. 2016, 37, 7–15. [Google Scholar] [CrossRef]
- Barat-Houari, M.; Dumont, B.; Fabre, A.; Them, F.T.; Alembik, Y.; Alessandri, J.L.; Amiel, J.; Audebert, S.; Baumann-Morel, C.; Blanchet, P.; et al. The expanding spectrum of COL2A1 gene variants IN 136 patients with a skeletal dysplasia phenotype. Eur. J. Hum. Genet. 2016, 24, 992–1000. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Chen, C.; Wang, Z.; Sun, L.; Li, S.; Zhang, T.; Luo, X.; Ding, X. Mutation Spectrum and De Novo Mutation Analysis in Stickler Syndrome Patients with High Myopia or Retinal Detachment. Genes 2020, 11, 882. [Google Scholar] [CrossRef]
- Wilkin, D.J.; Artz, A.S.; South, S.; Lachman, R.S.; Rimoin, D.L.; Wilcox, W.R.; McKusick, V.A.; Stratakis, C.A.; Francomano, C.A.; Cohn, D.H. Small deletions in the type II collagen triple helix produce kniest dysplasia. Am. J. Med. Genet. 1999, 85, 105–112. [Google Scholar] [CrossRef]
- Weis, M.A.; Wilkin, D.J.; Kim, H.J.; Wilcox, W.R.; Lachman, R.S.; Rimoin, D.L.; Cohn, D.H.; Eyre, D.R. Structurally abnormal type II collagen in a severe form of Kniest dysplasia caused by an exon 24 skipping mutation. J. Biol. Chem. 1998, 273, 4761–4768. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [Green Version]
- Richards, A.J.; Snead, M.P. The influence of pre-mRNA splicing on phenotypic modification in Stickler’s syndrome and other type II collagenopathies. Eye 2008, 22, 1243–1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelse, K.; Pöschl, E.; Aigner, T. Collagens-structure, function, and biosynthesis. Adv. Drug Deliv. Rev. 2003, 55, 1531–1546. [Google Scholar] [CrossRef] [Green Version]
- Körkkö, J.; Cohn, D.H.; Ala-Kokko, L.; Krakow, D.; Prockop, D.J. Widely Distributed Mutations in the COL2A1 Gene Produce Achondrogenesis Type II/Hypochondrogenesis. Am. J. Med. Genet. 2000, 92, 95–100. [Google Scholar] [CrossRef]
- Winterpacht, A.; Hilbert, M.; Schwarze, U.; Mundlos, S.; Spranger, J.; Zabel, B.U. Kniest and Stickler dysplasia phenotypes caused by collagen type II gene (COL2A1) defect. Nat. Genet. 1993, 3, 323–326. [Google Scholar] [CrossRef] [PubMed]
- Bogaert, R.; Wilkin, D.; Wilcox, W.R.; Lachman, R.; Rimoin, D.; Cohn, D.H.; Eyre, D.R. Expression, in cartilage, of a 7-amino-acid deletion in type II collagen from two unrelated individuals with Kniest dysplasia. Am. J. Hum. Genet. 1994, 55, 1128–1136. [Google Scholar] [PubMed]
- Fernandes, R.J.; Wilkin, D.J.; Weis, M.A.; Wilcox, W.R.; Cohn, D.H.; Rimoin, D.L.; Eyre, D.R. Incorporation of structurally defective type II collagen into cartilage matrix in kniest chondrodysplasia. Arch. Biochem. Biophys. 1998, 355, 282–290. [Google Scholar] [CrossRef]
- Wada, R.; Sawai, H.; Nishimura, G.; Isono, K.; Minagawa, K.; Takenobu, T.; Harada, K.; Tanaka, H.; Ishikura, R.; Komori, S. Prenatal diagnosis of Kniest dysplasia with three-dimensional helical computed tomography. J. Matern. Fetal Neonatal Med. 2011, 24, 1181–1184. [Google Scholar] [CrossRef]
- Spranger, J.; Winterpacht, A.; Zabel, B. Kniest dysplasia: Dr. W. Kniest, his patient, the molecular defect. Am. J. Med. Genet. 1997, 69, 79–84. [Google Scholar] [CrossRef]
- Winterpacht, A.; Schwarze, U.; Mundlos, S.; Menger, H.; Spranger, J.; Zabel, B. Alternative splicing as the result of a type II procollagen gene (COL2A1) mutation in a patient with Kniest dysplasia. Hum. Mol. Genet. 1994, 3, 1891–1893. [Google Scholar] [CrossRef] [PubMed]
- Mortier, G.R.; Weis, M.; Nuytinck, L.; King, L.M.; Wilkin, D.J.; De Paepe, A.; Lachman, R.S.; Rimoin, D.L.; Eyre, D.R.; Cohn, D.H. Report of five novel and one recurrent COL2A1 mutations with analysis of genotype-phenotype correlation in patients with a lethal type II collagen disorder. J. Med. Genet. 2000, 37, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.; Vissing, H.; Ramirez, F.; Rogers, D.; Rimoin, D. (Identification of the molecular defect in a family with spondyloepiphyseal dysplasia. Science 1989, 244, 978–980. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, P.; Khillan, J.S.; Prockop, D.J.; Helminen, H.; Kontusaari, S.; Ala-Kokko, L. Expression of a partially deleted gene of human type II procollagen (COL2A1) in transgenic mice produces a chondrodysplasia. Proc. Natl. Acad. Sci. USA 1991, 88, 7640–7644. [Google Scholar] [CrossRef] [Green Version]
- Metsäranta, M.; Garofalo, S.; Decker, G.; Rintala, M.; de Crombrugghe, B.; Vuorio, E. Chondrodysplasia in transgenic mice harboring a 15-amino acid deletion in the triple helical domain of pro α 1(II) collagen chain. J. Cell Biol. 1992, 118, 203–212. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bruni, V.; Spoleti, C.B.; La Barbera, A.; Dattilo, V.; Colao, E.; Votino, C.; Bellacchio, E.; Perrotti, N.; Giglio, S.; Iuliano, R. A Novel Splicing Variant of COL2A1 in a Fetus with Achondrogenesis Type II: Interpretation of Pathogenicity of In-Frame Deletions. Genes 2021, 12, 1395. https://doi.org/10.3390/genes12091395
Bruni V, Spoleti CB, La Barbera A, Dattilo V, Colao E, Votino C, Bellacchio E, Perrotti N, Giglio S, Iuliano R. A Novel Splicing Variant of COL2A1 in a Fetus with Achondrogenesis Type II: Interpretation of Pathogenicity of In-Frame Deletions. Genes. 2021; 12(9):1395. https://doi.org/10.3390/genes12091395
Chicago/Turabian StyleBruni, Valentina, Cristina Barbara Spoleti, Andrea La Barbera, Vincenzo Dattilo, Emma Colao, Carmela Votino, Emanuele Bellacchio, Nicola Perrotti, Sabrina Giglio, and Rodolfo Iuliano. 2021. "A Novel Splicing Variant of COL2A1 in a Fetus with Achondrogenesis Type II: Interpretation of Pathogenicity of In-Frame Deletions" Genes 12, no. 9: 1395. https://doi.org/10.3390/genes12091395