
Mitochondrial Genome of Strophopteryx fasciata (Plecoptera: Taeniopterygidae), with a Phylogenetic Analysis of Nemouroidea

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection, DNA Extraction and Sequencing
2.2. Genome Annotation and Sequence Analysis
2.3. Phylogenetic Analysis
3. Results and Discussion
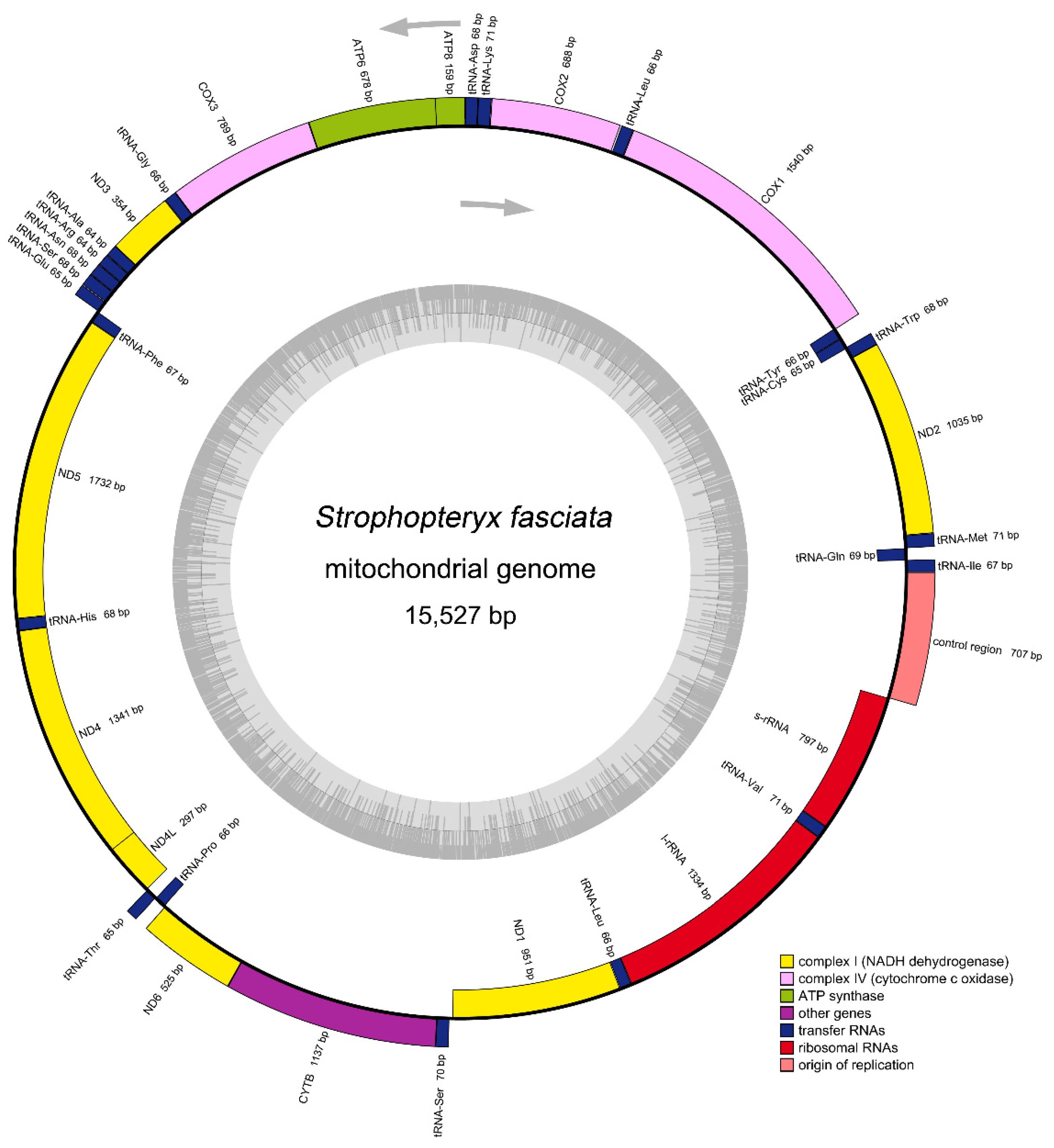
3.1. General Features of Mitogenome
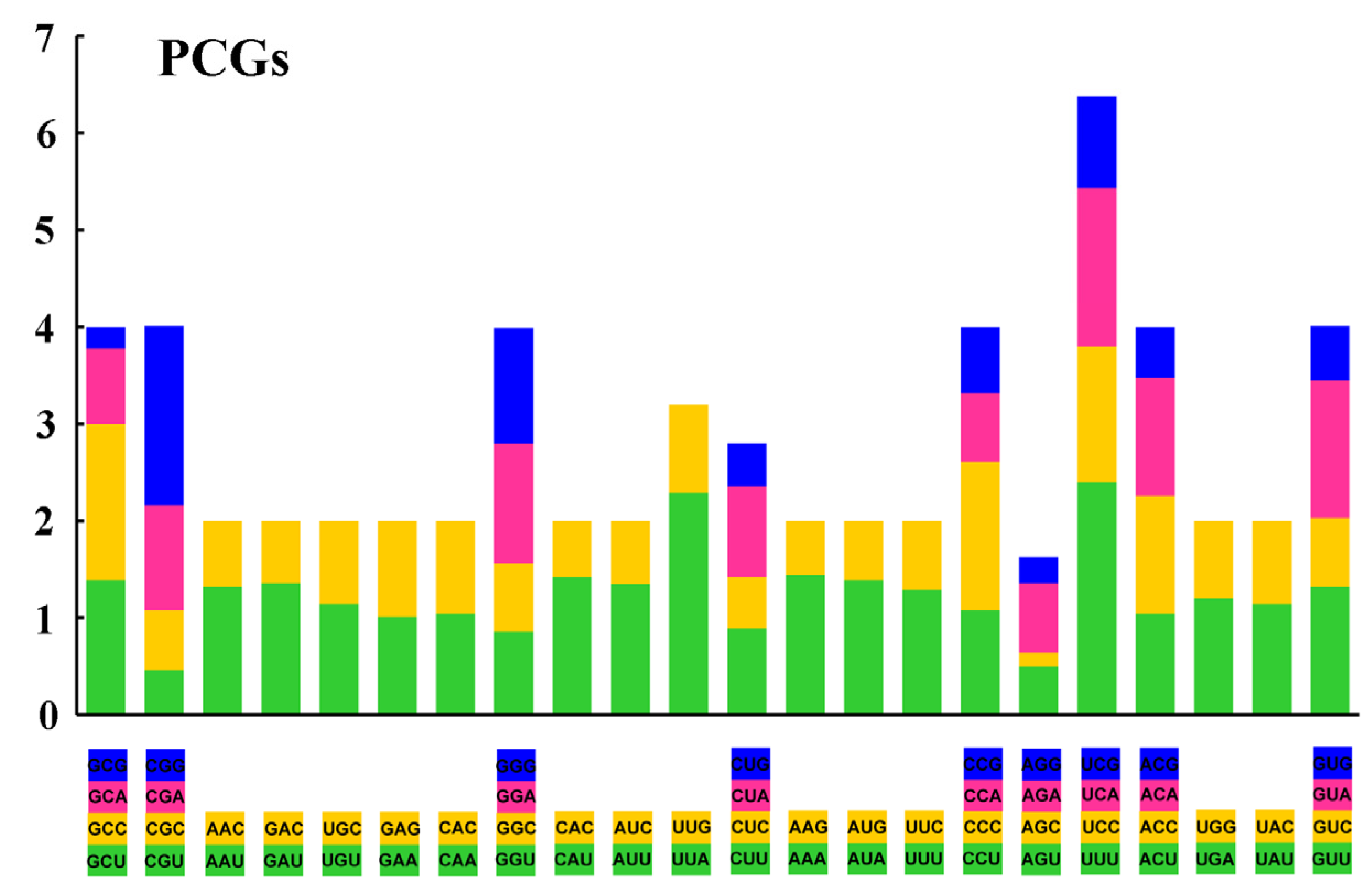
3.2. Nucleotide Composition and Codon Usage
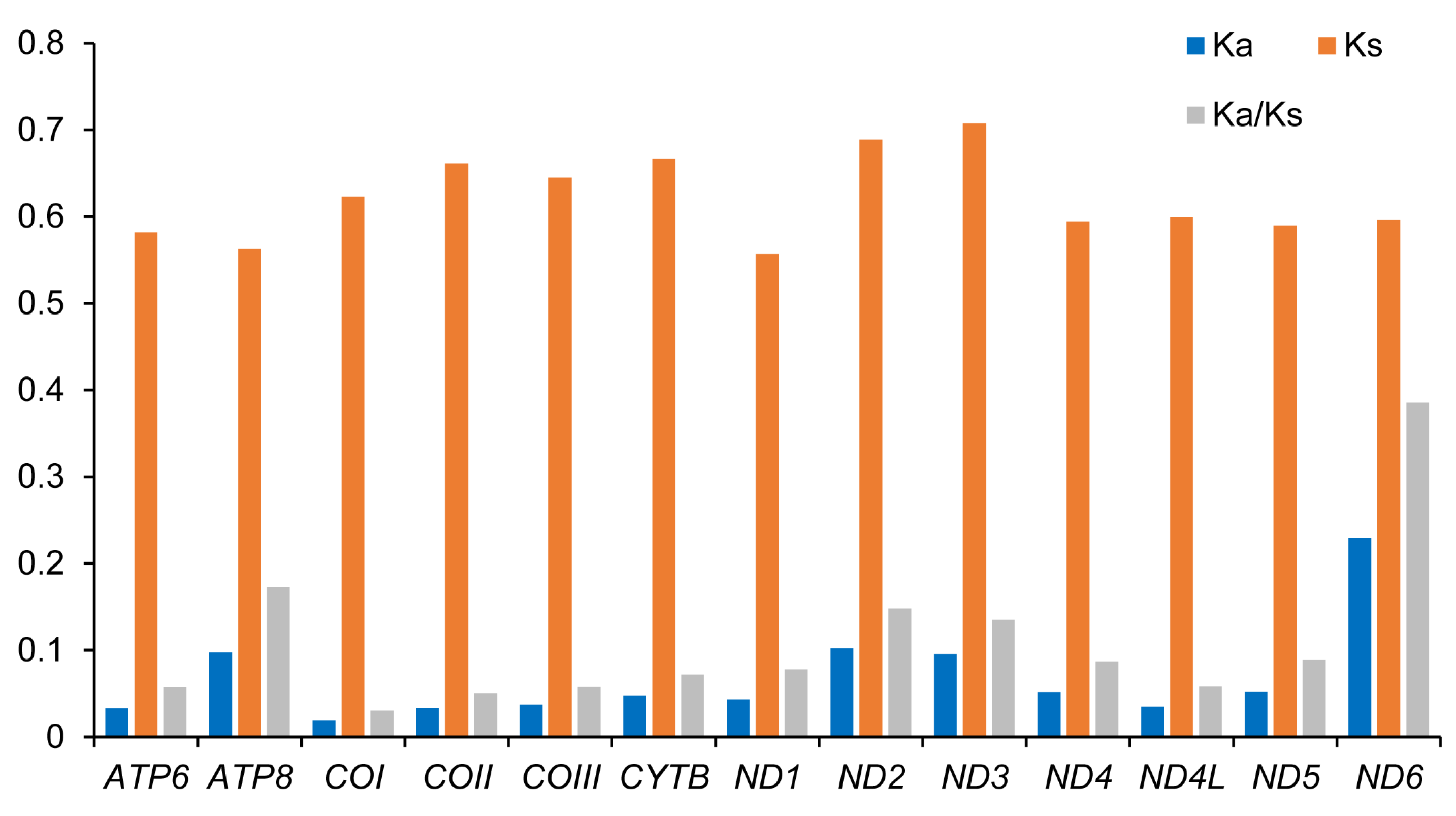
3.3. Protein-Coding Genes
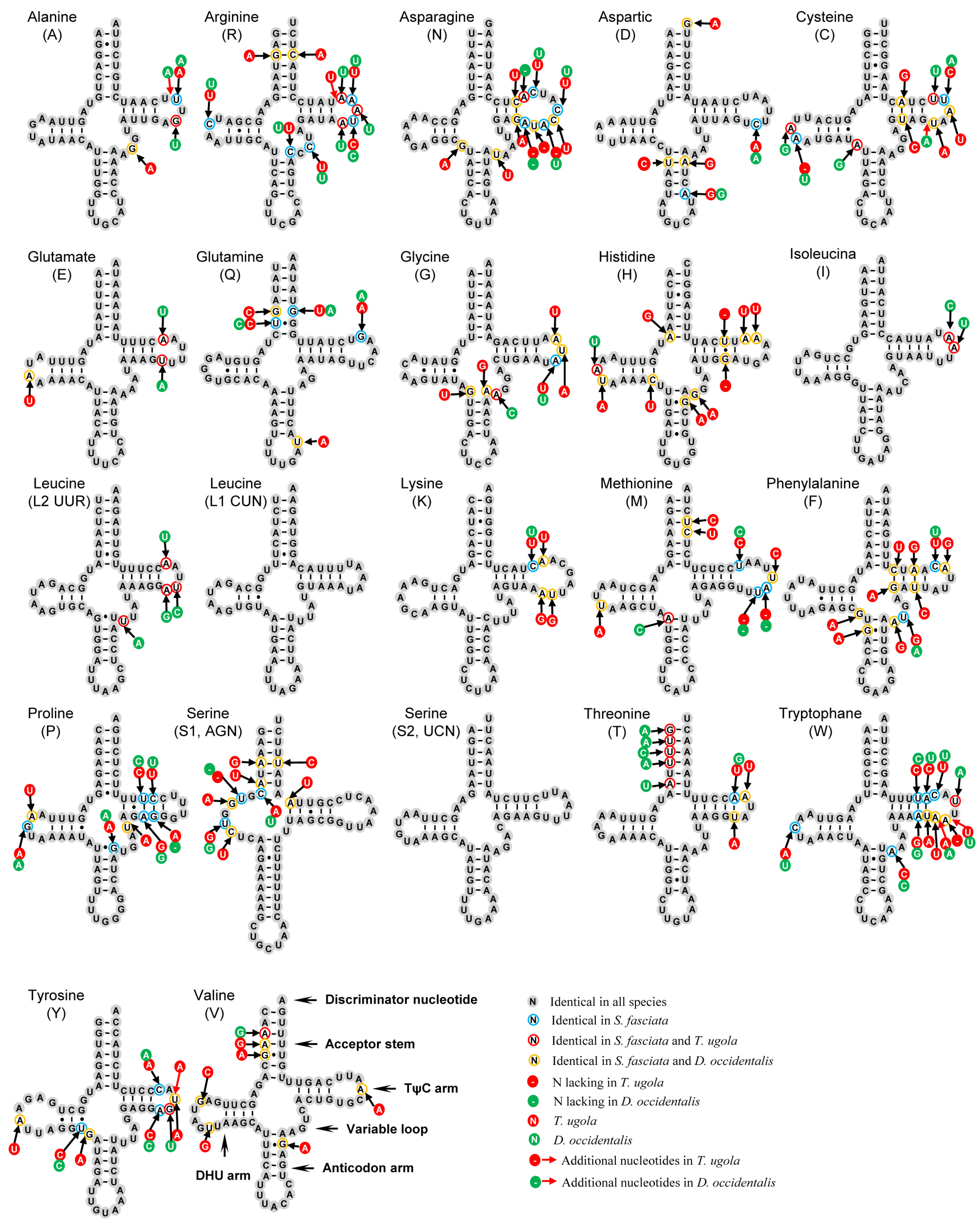
3.4. Transfer and Ribosomal RNA Genes
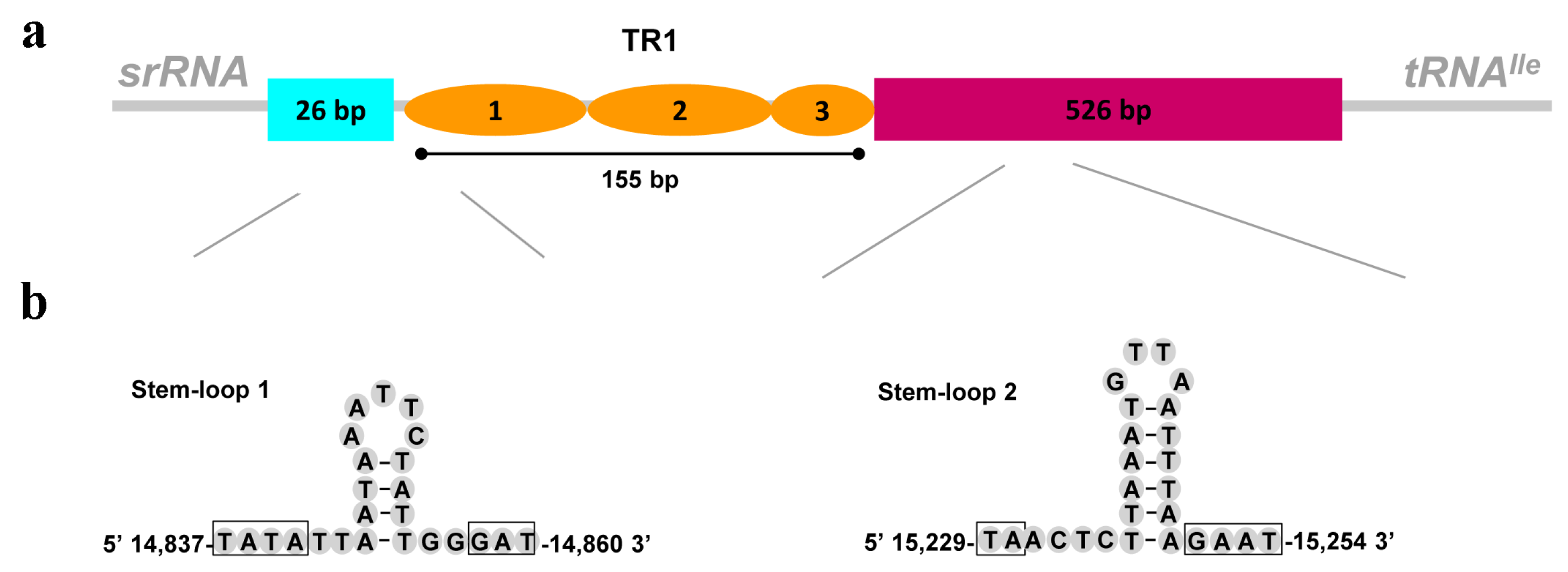
3.5. The Control Region
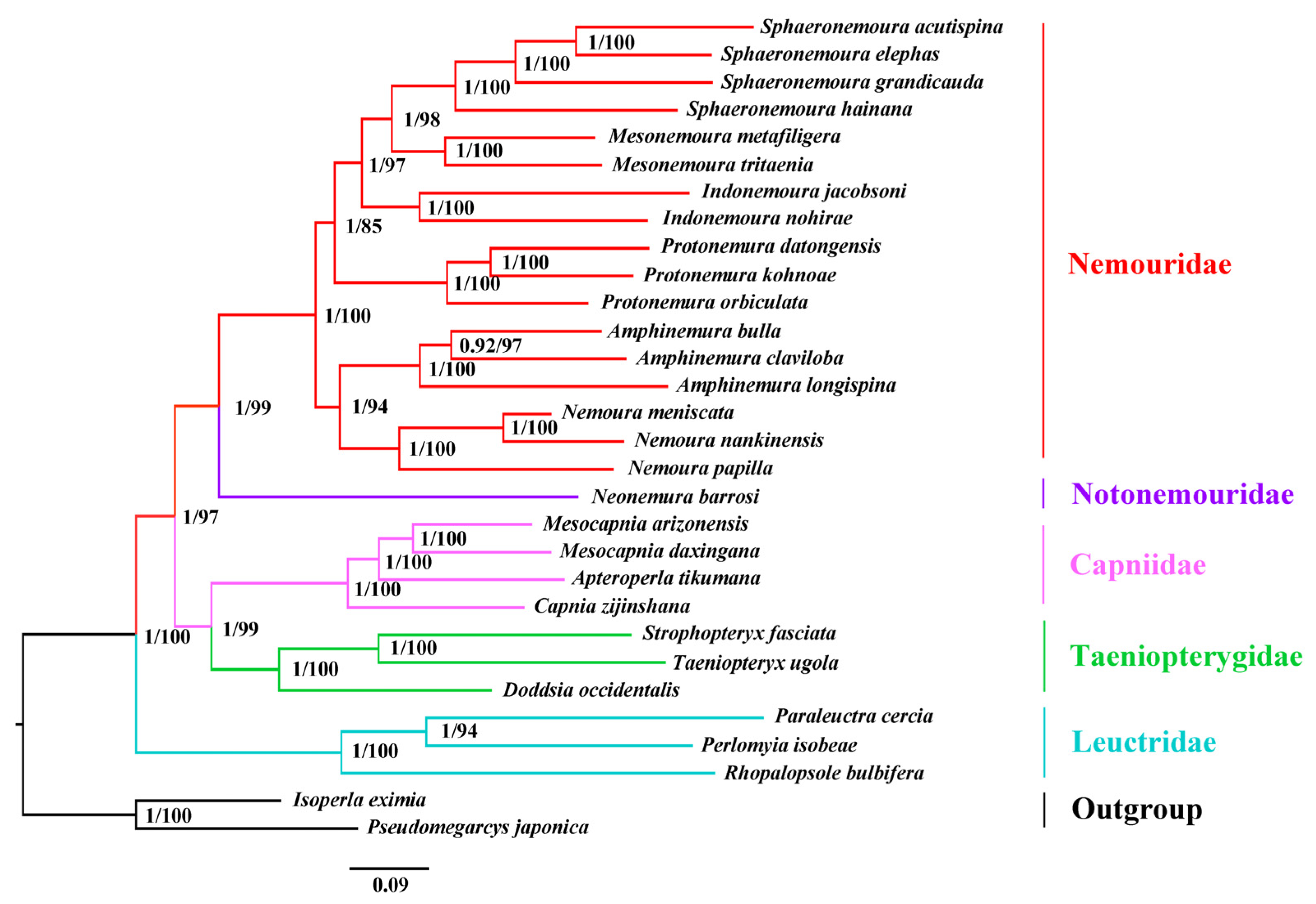
3.6. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- DeWalt, R.E.; Maehr, M.D.; Hopkins, H.; Neu-Becker, U.; Stueber, G. Plecoptera Species File Online. Available online: http://Plecoptera.SpeciesFile.org (accessed on 29 April 2022).
- Zwick, P. Phylogenetic System and Zoogeography of the Plecoptera. Annu. Rev. Entomol. 2000, 45, 709–746. [Google Scholar] [CrossRef] [PubMed]
- Hynes, H.B.N.; Hynes, M.E. The endemism of Tasmanian stoneflies (Plecoptera). Aquat. Insects 1980, 2, 81–89. [Google Scholar] [CrossRef]
- Thomas, M.A.; Walsh, K.A.; Wolf, M.R.; McPheron, B.A.; Marden, J.H. Molecular phylogenetic analysis of evolutionary trends in stonefly wing structure and locomotor behavior. Proc. Natl. Acad. Sci. USA 2000, 97, 13178–13183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terry, M.D. Phylogeny of the Polyneopterous Insects with Emphasis on Plecoptera: Molecular and Morpological Evidence. Ph.D. Thesis, Brigham Young University, Provo, UT, USA, 2003; pp. 1–118. [Google Scholar]
- Davis, N.G. Application of Next-Generation Transcriptomic Tools for Non-Model Organisms: Gene Discovery and Marker Development within Plecoptera (Insecta). Master’s Thesis, Brigham Young University, Provo, UT, USA, 2013; pp. 1–39. [Google Scholar]
- Chen, Z.T.; Du, Y.Z. The first two mitochondrial genomes from Taeniopterygidae (Insecta: Plecoptera): Structural features and phylogenetic implications. Int. J. Biol. Macromol. 2018, 111, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.J.; Wang, Y.; Li, W.H. Comparative mitogenomic analysis of species in the subfamily Amphinemurinae (Plecoptera: Nemouridae) reveal conserved mitochondrial genome organization. Int. J. Biol. Macromol. 2019, 138, 292–301. [Google Scholar] [CrossRef]
- Cao, J.J.; Wang, Y.; Guo, X.; Wang, G.Q.; Li, W.H.; Murányi, D. Two complete mitochondrial genomes from Leuctridae (Plecoptera: Nemouroidea): Implications for the phylogenetic relationships among stoneflies. J. Insect Sci. 2021, 21, 16. [Google Scholar] [CrossRef]
- Shen, Y.; Du, Y.Z. The mitochondrial genome of Leuctra sp. (Plecoptera: Leuctridae) and its performance in phylogenetic analyses. Zootaxa 2019, 4671, 571–580. [Google Scholar] [CrossRef]
- Zhao, M.Y.; Huo, Q.B.; Du, Y.Z. Molecular phylogeny inferred from the mitochondrial genomes of Plecoptera with Oyamia nigribasis (Plecoptera: Perlidae). Sci. Rep. 2020, 10, 20955. [Google Scholar] [CrossRef]
- Ding, S.M.; Li, W.H.; Wang, Y.; Cameron, S.L.; Murányi, D.; Yang, D. The phylogeny and evolutionary timescale of stoneflies (Insecta: Plecoptera) inferred from mitochondrial genomes. Mol. Phylogenet. Evol. 2019, 135, 123–135. [Google Scholar] [CrossRef]
- South, E.J.; Skinner, R.K.; DeWalt, R.E.; Kondratieff, B.C.; Johnson, K.P.; Davis, M.A.; Lee, J.J.; Durfee, R.S. Phylogenomics of the North American Plecoptera. Syst. Entomol. 2021, 46, 287–305. [Google Scholar] [CrossRef]
- Barr, C.M.; Neiman, M.; Taylor, D.R. Inheritance and recombination of mitochondrial genomes in plants, fungi and animals. New Phytol. 2005, 168, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curole, J.P.; Kocher, T.D. Mitogenomics: Digging deeper with complete mitochondrial genomes. Trends Ecol. Evol. 1999, 14, 394–398. [Google Scholar] [CrossRef]
- Cameron, S.L.; Lambkin, C.L.; Barker, S.C.; Whiting, M.F. Utility of mitochondrial genomes as phylogenetic markers for insect intraordinal relationships. A case study from flies (Diptera). Syst. Entomol. 2007, 32, 40–59. [Google Scholar] [CrossRef]
- Fenn, J.D.; Song, H.; Cameron, S.L.; Whiting, M.F. A preliminary mitochondrial genome phylogeny of Orthoptera (Insecta) and approaches to maximizing phylogenetic signal found within mitochondrial genome data. Mol. Phylogenet. Evol. 2008, 49, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Dowton, M.; Cameron, S.L.; Austin, A.D.; Whiting, M.F. Phylogenetic approaches for the analysis of mitochondrial genome sequence data in the Hymenoptera—A lineage with both rapidly and slowly evolving mitochondrial genomes. Mol. Phylogenet. Evol. 2009, 52, 512–519. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 29 April 2022).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA-UD: A de novo assembler for single cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Cao, J.J.; Li, W.H. The complete mitochondrial genome of the styloperlid stonefly species Styloperla spinicercia Wu (Insecta: Plecoptera) with family-level phylogenetic analyses of the Pteronarcyoidea. Zootaxa 2017, 4243, 125–138. [Google Scholar] [CrossRef]
- Wang, Y.; Cao, J.J.; Murányi, D.; Li, W.H. Comparison of two complete mitochondrial genomes from Perlodidae (Plecoptera: Perloidea) and the family-level phylogenetic implications of Perloidea. Gene 2018, 675, 254–264. [Google Scholar] [CrossRef]
- Wang, Y.; Cao, J.J.; Li, N.; Ma, G.Y.; Li, W.H. The first mitochondrial genome from Scopuridae (Insecta: Plecoptera) reveals structural features and phylogenetic implications. Int. J. Biol. Macromol. 2019, 122, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. Organellar genome DRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abascal, F.; Zardoya, R.; Telford, M.J. TranslatorX: Multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 2010, 38, W7–W13. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.V.D.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trifinopoulos, J.; Nguyen, L.T.; von Haeseler, A.; Minh, B.Q. W-IQTREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef] [Green Version]
- Boore, J.L. Complete mitochondrial genome sequence of the polychaete annelid Platynereis dumerilii. Mol. Biol. Evol. 2001, 18, 1413–1416. [Google Scholar] [CrossRef] [Green Version]
- Clary, D.O.; Wolstenholme, D.R. The ribosomal RNA genes of Drosophila mitochondrial DNA. Nucleic Acids Res. 1985, 13, 4029–4045. [Google Scholar] [CrossRef] [Green Version]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef]
- Boore, J.L. Complete mitochondrial genome sequence of Urechis caupo, a representative of the phylum Echiura. BMC Genom. 2004, 5, 67. [Google Scholar] [CrossRef] [Green Version]
- Roques, S.; Fox, C.J.; Villasana, M.I.; Rico, C. The complete mitochondrial genome of the whiting, Merlangius merlangus and the haddock, Melanogrammus aeglefinus: A detailed genomic comparison among closely related species of the Gadidae family. Gene 2006, 383, 12–23. [Google Scholar]
- Wolstenholme, D.R. Animal mitochondrial DNA: Structure and evolution. Int. Rev. Cytol. 1992, 141, 173–216. [Google Scholar]
- Li, H.; Liu, H.Y.; Song, F.; Shi, A.M.; Zhou, X.G.; Cai, W.Z. Comparative Mitogenomic analysis of damsel bugs representing three tribes in the family Nabidae (Insecta: Hemiptera). PLoS ONE 2012, 7, e45925. [Google Scholar]
- Clary, D.O.; Wolstenholme, D.R. Drosophila mitochondrial DNA: Conserved sequences in the A+T-rich region and supporting evidence for a secondary structure model of the small ribosomal RNA. J. Mol. Evol. 1987, 25, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.X.; Szymura, J.M.; Hewitt, G.M. Evolution and structural conservation of the control region of insect mitochondrial DNA. J. Mol. Evol. 1995, 40, 382–391. [Google Scholar] [CrossRef]
- Song, N.; Liang, A.P. Complete mitochondrial genome of the small brown planthopper, Laodelphax striatellus (Delphacidae: Hemiptera), with a novel gene order. Zool. Sci. 2009, 26, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.X.; Hewitt, F.M. Insect mitochondrial control region: A review of its structure, evolution and usefulness in evolutionary studies. Biochem. Syst. Ecol. 1997, 25, 99–120. [Google Scholar] [CrossRef]
- Cao, J.J.; Wang, Y.; Huang, Y.R.; Li, W.H. Mitochondrial genomes of the stoneflies Mesonemoura metafiligera and Mesonemoura tritaenia (Plecoptera, Nemouridae), with a phylogenetic analysis of Nemouroidea. ZooKeys 2019, 835, 43–63. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.T.; Du, Y.Z. Comparison of the complete mitochondrial genome of the stonefly Sweltsa longistyla (Plecoptera: Chloroperlidae) with mitogenomes of three other stoneflies. Gene 2015, 558, 82–87. [Google Scholar] [CrossRef]
- Chen, Z.T.; Du, Y.Z. First Mitochondrial genome from Nemouridae (Plecoptera) reveals novel features of the elongated control region and phylogenetic implications. Int. J. Mol. Sci. 2017, 18, 996. [Google Scholar] [CrossRef]
- Baumann, R.W. Revision of the stonefly family Nemouridae (Plecoptera): A study of the world fauna at the generic level. Smithson. Contrib. Zool. 1975, 211, 1–73. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Cao, J.J.; Murányi, D.; Chen, X.L.; Yan, F.M. The complete mitochondrial genome of Amphinemura bulla Shimizu, 1997 (Plecoptera: Nemouridae) from Japan. Mitochondrial DNA Part B 2021, 6, 846–847. [Google Scholar] [CrossRef]
- Shen, Y.; Du, Y.Z. The complete mitochondrial genome of Flavoperla biocellata Chu, 1929 (Plecoptera: Perlidae) and the phylogenetic analyses of Plecoptera. PeerJ 2020, 8, e8762. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Species | Number (bp) | Accession Number |
|---|---|---|---|
| Capniidae | Apteroperla tikumana | 15,564 | NC_027698 |
| Capnia zijinshana | 16,310 | KX094942 | |
| Mesocapnia arizonensis | 14,921 | KP642637 * | |
| Mesocapnia daxingana | 15,524 | KY568983 * | |
| Leuctridae | Rhopalopsole bulbifera | 15,599 | MK111419 * |
| Paraleuctra cercia | 15,625 | MK492251 | |
| Perlomyia isobeae | 15,795 | MK492252 | |
| Nemouridae | Nemoura meniscata | 15,895 | MN944386 |
| Nemoura nankinensis | 16,602 | KY940360 | |
| Nemoura papilla | 15,774 | MK290826 | |
| Amphinemura longispina | 15,709 | MH085446 | |
| Amphinemura bulla | 15,827 | MW339348 | |
| Amphinemura claviloba | 15,707 | MN720741 | |
| Indonemoura jacobsoni | 15,642 | MH085448 | |
| Indonemoura nohirae | 15,738 | MH085449 | |
| Mesonemoura metafiligera | 15,739 | MH085450 | |
| Mesonemoura tritaenia | 15,778 | MH085451 | |
| Protonemura kohnoae | 15,707 | MH085452 | |
| Protonemura orbiculata | 15,758 | MH085453 | |
| Protonemura datongensis | 15,756 | MT276842 | |
| Sphaeronemoura elephas | 15,846 | MN944385 | |
| Sphaeronemoura grandicauda | 15,661 | MH085454 | |
| Sphaeronemoura acutispina | 15,016 | MH085455 * | |
| Sphaeronemoura hainana | 15,260 | MK111420 * | |
| Notonemouridae | Neonemura barrosi | 14,852 | MK111418 * |
| Taeniopterygidae | Taeniopteryx ugola | 15,353 | MG589786 |
| Doddsia occidentalis | 16,020 | MG589787 | |
| Strophopteryx fasciata | 15,527 | ON500674 | |
| Perlodidae (Outgroup) | Isoperla eximia | 16,034 | MG910457 |
| Pseudomegarcys japonica | 16,067 | MG910458 |
| Region | Feature | S. fasciata | D. occidentalis | T. ugola |
|---|---|---|---|---|
| Whole mitgenome | Size (bp) | 15,527 | 16,020 | 15,353 |
| A + T% | 68.1 | 68.4 | 69.8 | |
| AT-skew | 0.03 | 0.02 | 0.02 | |
| GC-skew | −0.18 | −0.21 | −0.15 | |
| Protein-coding genes | Size (bp) | 11,193 | 11,148 | 11,223 |
| A + T% | 66.9 | 66.8 | 68.6 | |
| AT-skew | −0.19 | −0.20 | −0.20 | |
| GC-skew | −0.03 | −0.01 | 0.03 | |
| tRNAs | Size (bp) | 1479 | 1477 | 1471 |
| A + T% | 70.9 | 70.8 | 71.3 | |
| AT-skew | 0.03 | −0.01 | −0.02 | |
| GC-skew | −0.13 | 0.13 | 0.13 | |
| rRNAs | Size (bp) | 2131 | 2126 | 2119 |
| A + T% | 71.2 | 72.4 | 72.7 | |
| AT-skew | −0.05 | −0.06 | −0.05 | |
| GC–skew | 0.28 | 0.28 | 0.28 | |
| Control region | Size (bp) | 707 | 1248 | 537 |
| A + T% | 71.0 | 72.7 | 78.0 | |
| AT-skew | 0.07 | 0.01 | 0.02 | |
| GC-skew | −0.16 | −0.30 | −0.20 |
| Gene | Start Codon | Stop Codon | ||||
|---|---|---|---|---|---|---|
| S. fasciata | T. ugola | D. occidentalis | S. fasciata | T. ugola | D. occidentalis | |
| ND2 | ATG | GTG | ATG | TAA | TAA | TAA |
| COI | ATT | ATT | ATT | T- | T- | T- |
| COII | ATG | ATG | ATG | T- | T- | T- |
| ATP8 | ATT | ATC | ATT | TAA | TAA | TAA |
| ATP6 | ATG | ATG | ATG | TAA | TAA | TAA |
| COIII | ATG | ATG | ATG | TAA | TAA | TAA |
| ND3 | ATT | ATT | ATT | TAG | TAG | TAG |
| ND5 | ATG | GTG | ATG | T- | T- | T- |
| ND4 | ATG | ATG | ATG | TAA | TAA | TAA |
| ND4L | ATG | ATG | ATG | TAA | TAA | TAA |
| ND6 | ATG | ATT | ATT | TAA | TAA | TAA |
| CYTB | ATG | ATG | ATC | TAG | TAA | TAG |
| ND1 | TTG | TTG | TTG | TAG | TAG | TAG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, X.; Guo, C.; Dong, X.; Zhang, H.; Murányi, D.; Li, W.; Wang, Y. Mitochondrial Genome of Strophopteryx fasciata (Plecoptera: Taeniopterygidae), with a Phylogenetic Analysis of Nemouroidea. Genes 2022, 13, 1116. https://doi.org/10.3390/genes13071116
Guo X, Guo C, Dong X, Zhang H, Murányi D, Li W, Wang Y. Mitochondrial Genome of Strophopteryx fasciata (Plecoptera: Taeniopterygidae), with a Phylogenetic Analysis of Nemouroidea. Genes. 2022; 13(7):1116. https://doi.org/10.3390/genes13071116
Chicago/Turabian StyleGuo, Xuan, Caiyue Guo, Xiaojiao Dong, Heng Zhang, Dávid Murányi, Weihai Li, and Ying Wang. 2022. "Mitochondrial Genome of Strophopteryx fasciata (Plecoptera: Taeniopterygidae), with a Phylogenetic Analysis of Nemouroidea" Genes 13, no. 7: 1116. https://doi.org/10.3390/genes13071116