

A Comprehensive Analysis of Triplophysa labiata (Kessler, 1874) Mitogenome and Its Phylogenetic Implications within the Triplophysa Genus

Abstract
:1. Introduction
2. Materials and Methods
2.1. Research Ethics Clearance
2.2. Experimental Fish and Sampling
2.3. DNA Isolation, Library Preparation, and Sequencing
2.4. T. labiata Mitogenome Assembly and Annotation
2.5. Sequence Analyses
2.6. Phylogenetic Analyses

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genus | Species | Size (bp) | Accession No | Resource |
|---|---|---|---|---|
| Triplophysa | T. labiata | 16,573 | OQ559481 | this study |
| Triplophysa bombifrons | 16,568 | OP499856 | [23] | |
| Triplophysa bombifrons | 16,569 | NC_027189 | [20] | |
| Triplophysa tenuis | 16,571 | NC_030511 | ||
| Triplophysa dorsalis | 16,572 | NC_029423 | [16] | |
| Triplophysa strauchii | 16,590 | NC_026714 | [20] | |
| Triplophysa stoliczkai | 16,571 | NC_017890 | ||
| Triplophysa xichangensis | 16,570 | NC_030513 | ||
| Triplophysa dalaica | 16,569 | NC_037925 | ||
| Triplophysa wuweiensis | 16,681 | NC_030512 | ||
| Triplophysa venusta | 16,574 | NC_029330 | [19] | |
| Triplophysa angeli | 16,569 | NC_065113 | ||
| Triplophysa bleekeri | 16,568 | NC_018774 | [12] | |
| Triplophysa anterodorsalis | 16,567 | NC_024597 | [11] | |
| Triplophysa orientalis | 16,562 | NC_030505 | ||
| Triplophysa stenura | 16,569 | NC_032692 | [13] | |
| Triplophysa stewarti | 16,567 | NC_030506 | ||
| Triplophysa lixianensis | 16,570 | NC_030521 | [8] | |
| Triplophysa tibetana | 16,574 | NC_030483 | [10] | |
| Triplophysa pappenheimi | 16,571 | NC_037924 | [7] | |
| Triplophysa pappenheimi | 16,572 | NC_033972 | ||
| Triplophysa robusta | 16,570 | NC_025632 | [22] | |
| Triplophysa siluroides | 16,574 | NC_024611 | [6] | |
| Triplophysa wangmoensis | 16,569 | NC_037704 | [9] | |
| Triplophysa xiangxiensis | 16,598 | NC_029492 | [14] | |
| Triplophysa rosa | 16,585 | NC_019587 | [15] | |
| Triplophysa nasobarbatula | 16,605 | NC_058005 | [21] | |
| Triplophysa baotianensis | 16,576 | NC_056365 | [5] | |
| Triplophysa zhenfengensis | 16,564 | NC_063617 | [39] | |
| Triplophysa yarkandensis | 16,574 | NC_027517 | [4] | |
| Aborichthys | Aborichthys elongatus | 16,544 | NC_031582 |
3. Results and Discussion
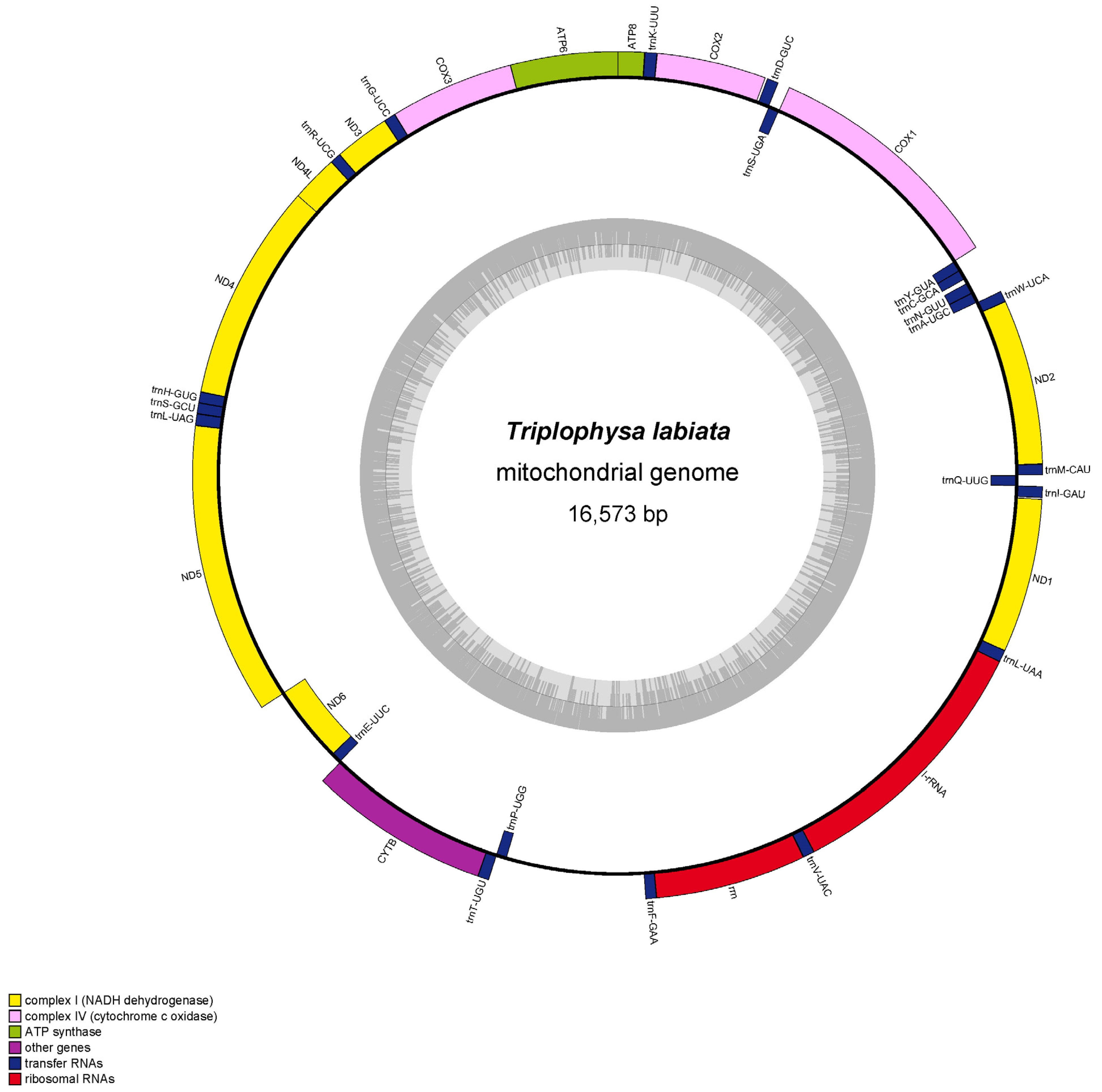
3.1. Genome Structure and Base Composition
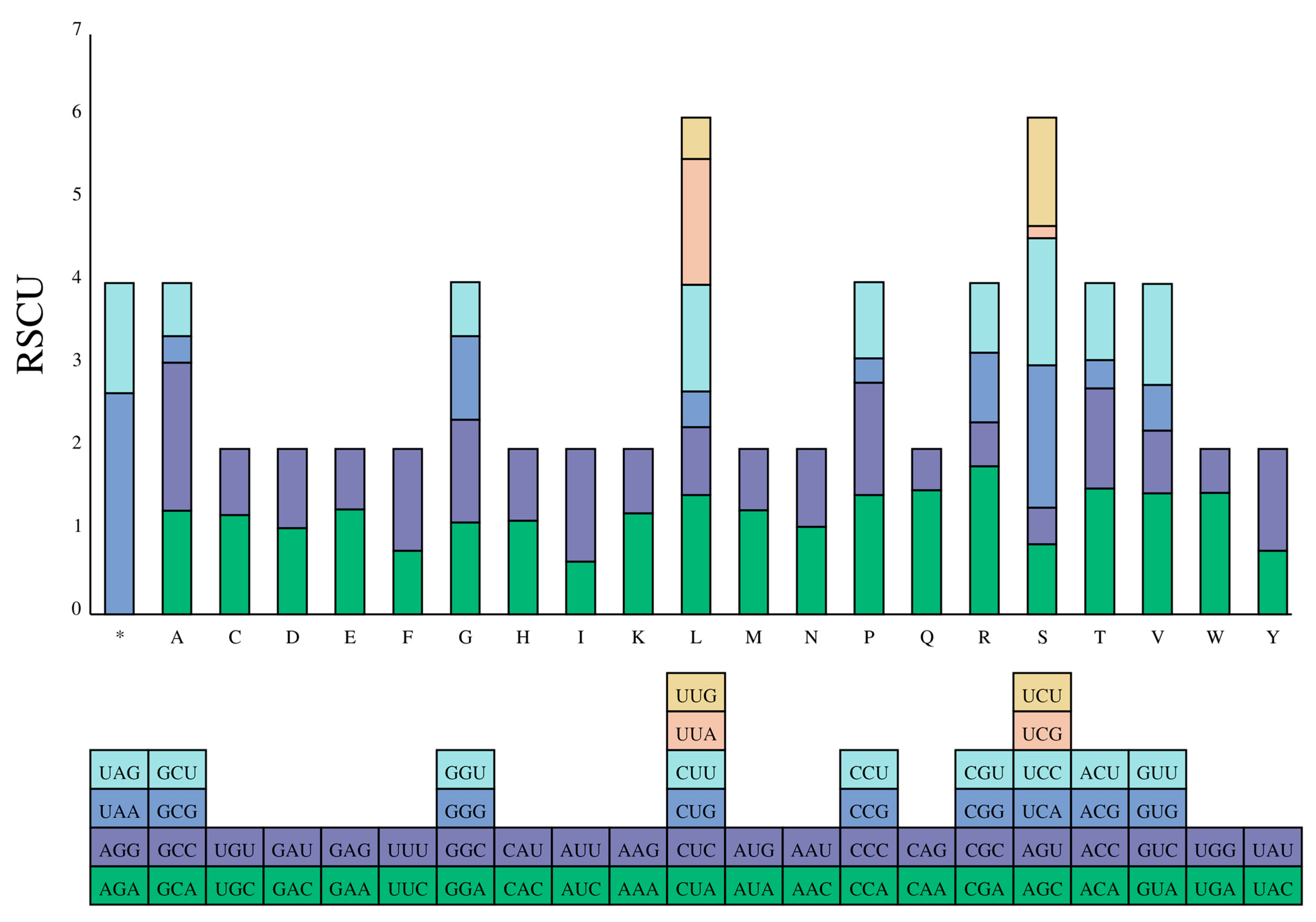
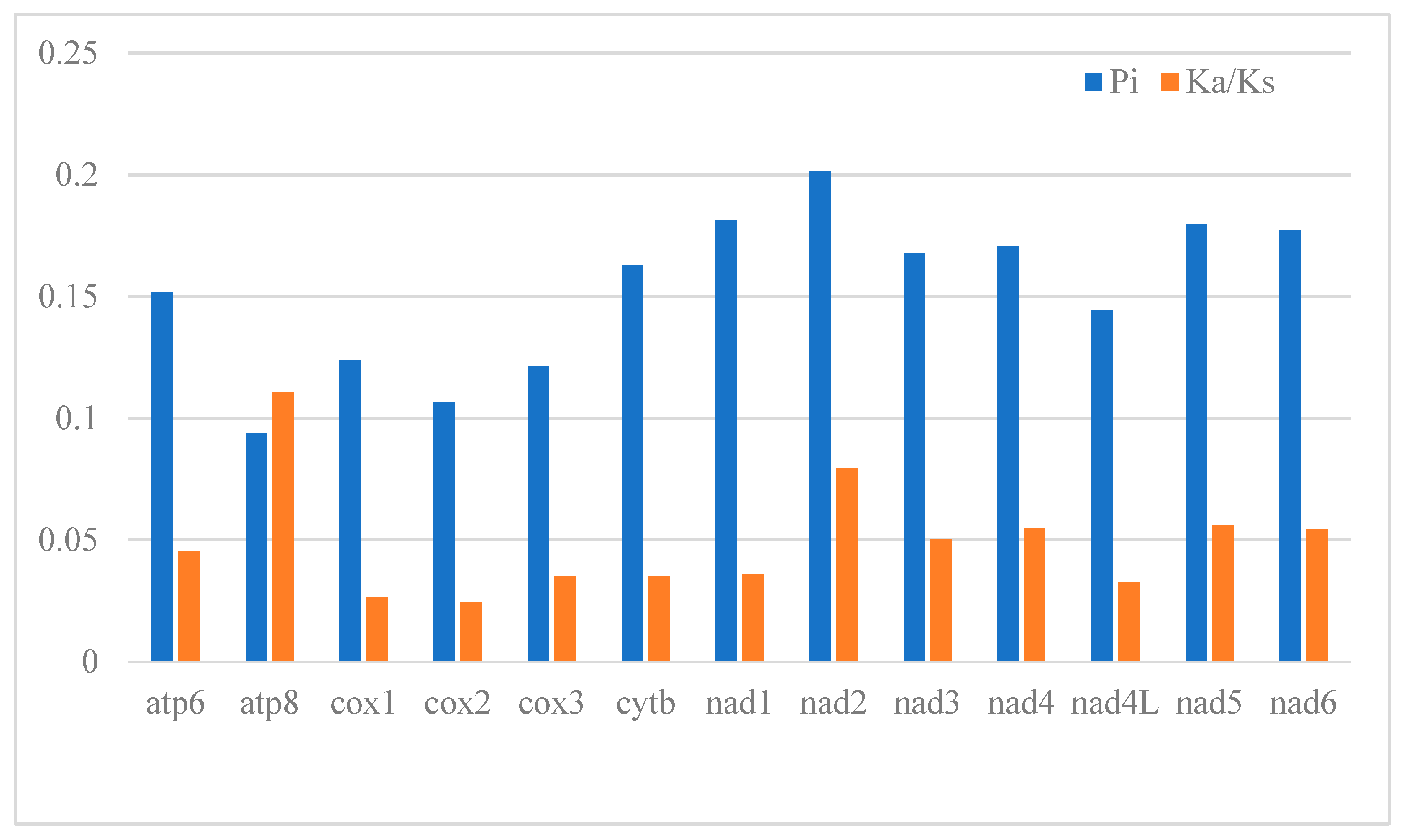
3.2. Description of Protein-Coding Genes (PCGs)
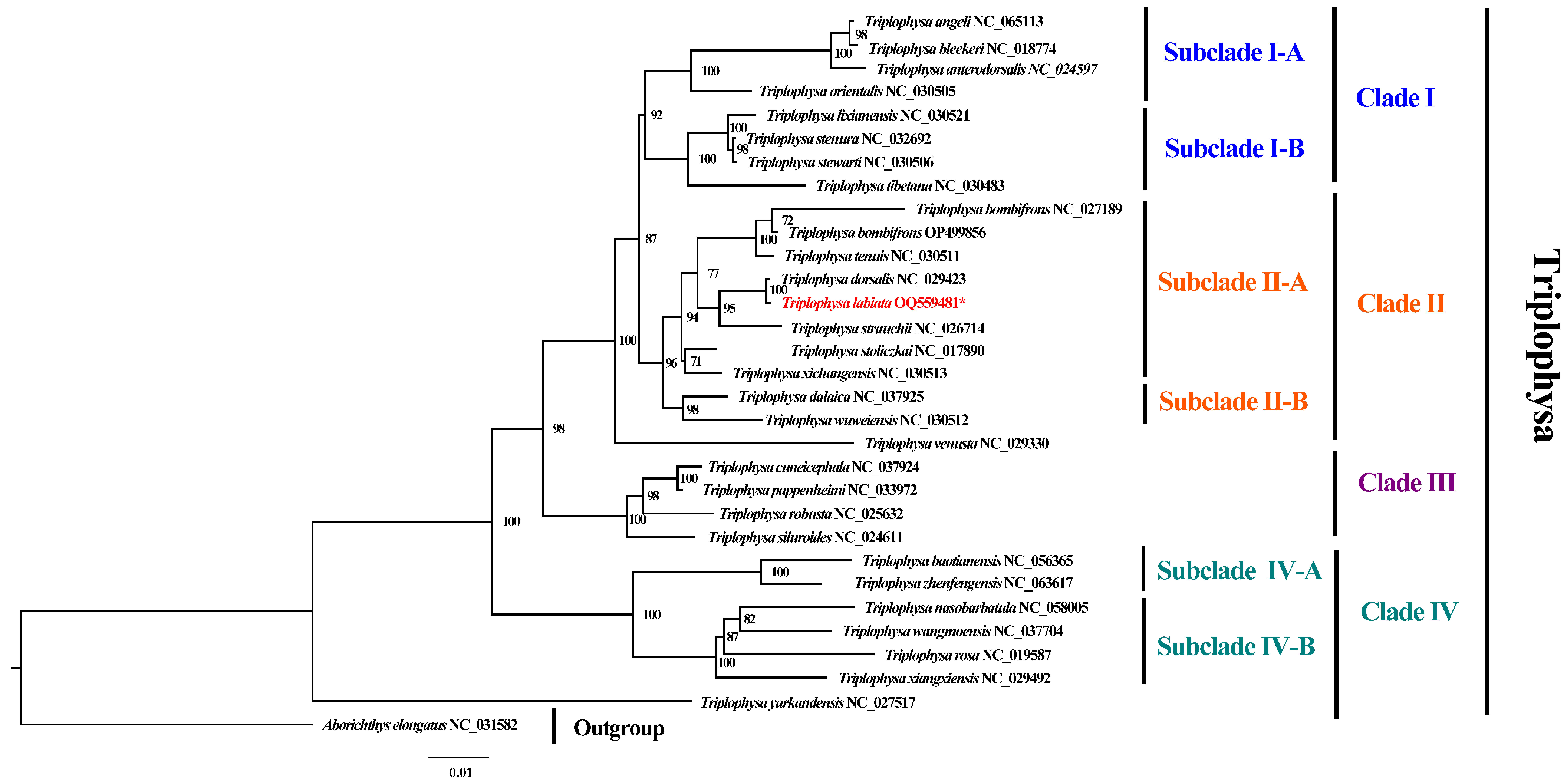
3.3. Phylogenetic Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nelson, J.S.; Grande, T.C.; Wilson, M.V. Fishes of the World; John Wiley & Sons: Hoboken, NJ, USA, 2016. [Google Scholar]
- Wen, J.; Yu, Y.; Xie, D.-F.; Peng, C.; Liu, Q.; Zhou, S.-D.; He, X.-J. A transcriptome-based study on the phylogeny and evolution of the taxonomically controversial subfamily Apioideae (Apiaceae). Ann. Bot. 2020, 125, 937–953. [Google Scholar] [CrossRef]
- He, B.; Zhao, Y.; Su, C.; Lin, G.; Wang, Y.; Li, L.; Ma, J.; Yang, Q.; Hao, J. Phylogenomics reveal extensive phylogenetic discordance due to incomplete lineage sorting following the rapid radiation of alpine butterflies (Papilionidae: Parnassius). Syst. Entomol. 2023, 2, 1–15. [Google Scholar] [CrossRef]
- Ning, X.; Zhang, Y.-Z.; Sui, Z.-H.; Quan, X.-Q.; Zhang, H.-G.; Liu, L.-X.; Han, Q.-D.; Liu, Y.-G. The complete mitochondrial DNA sequence of Kashgarian loach (Triplophysa yarkandensis) from Bosten Lake. Mitochondrial DNA Part B 2020, 5, 821–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Xiao, N.; Wang, S.; Luo, T.; Yang, X.; Liu, T.; Zhou, J. The complete mitochondrial genome of a cave-dwelling loach Triplophysa baotianensis (Teleostei: Nemacheilidae). Mitochondrial DNA Part B 2021, 6, 1209–1211. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.-S.; Liu, G.-D.; Prokofiev, A.M. The complete mitochondrial genome of giant stone loach Triplophysa siluroides (Cypriniformes: Balitoridae). Mitochondrial DNA Part A 2016, 27, 998–1000. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Chen, Y.; Sui, X.; Chen, Y. The complete mitochondrial genome of Triplophysa cuneicephala (Cypriniformes: Balitoridae) with phylogenetic consideration. Mitochondrial DNA Part B 2019, 4, 1239–1240. [Google Scholar] [CrossRef] [Green Version]
- Jing, H.; Yan, P.; Li, W.; Li, X.; Song, Z. The complete mitochondrial genome of Triplophysa lixianensis (Teleostei: Cypriniformes: Balitoridae) with phylogenetic consideration. Biochem. Syst. Ecol. 2016, 66, 254–264. [Google Scholar] [CrossRef]
- Liu, T.; You, P. The complete mitochondrial genome of Triplophysa sp. (Teleostei: Cypriniformes: Balitoridae). Mitochondrial DNA Part A 2016, 27, 4557–4558. [Google Scholar] [CrossRef]
- Wang, J.; Li, L.; Jin, X.; Wang, P.; Du, Y.; Ma, B. The complete mitochondrial genome of Triplophysa tibetana. Mitochondrial DNA Part B. 2019, 4, 1411–1412. [Google Scholar] [CrossRef] [Green Version]
- Que, Y.; Liao, X.; Xu, D.; Yang, Z.; Tang, H.; Zhu, B. The complete mitochondrial genome sequence of Triplophysa anterodorsalis (Teleostei, Balitoridae, Nemacheilinae). Mitochondrial DNA Part A 2016, 27, 937–938. [Google Scholar] [CrossRef]
- Tang, Q.; Huang, Y.; Wang, J.; Huang, J.; Wang, Z.; Peng, Z. The complete mitochondrial genome sequence of Triplophysa bleekeri (Teleostei, Balitoridae, Nemacheilinae). Mitochondrial DNA 2013, 24, 25–27. [Google Scholar] [CrossRef]
- Yan, Y.; Luo, D. The complete mitochondrial genome sequence of Triplophysa stenura (Teleostei, Cypriniformes): Genome characterization and phylogenetic analysis. Mitochondrial DNA Part B 2016, 1, 607–608. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Cao, L.; Zhang, E. The complete mitochondrial genome sequence of Triplophysa xiangxiensis (Teleostei: Nemacheilidae). Mitochondrial DNA Part A 2017, 28, 171–172. [Google Scholar] [CrossRef]
- Wang, J.; Tang, Q.; Wang, Z.; Zhang, Y.; Wu, Q.; Peng, Z. The complete mitogenome sequence of a cave loach Triplophysa rosa (Teleostei, Balitoridae, Nemacheilinae). Mitochondrial DNA 2012, 23, 366–368. [Google Scholar] [CrossRef]
- Lei, D.; Conteh Kanu, U.; Zhao, G.; Xie, P.; Yuan, H.; Li, Y.; Niu, J.; Ma, X. The complete mtDNA genome of Triplophysa dorsalis (Cypriniformes, Balitoridae, Cobitoidea): Genome characterization and phylogenetic analysis. Mitochondrial DNA Part A 2016, 27, 3745–3746. [Google Scholar] [CrossRef]
- Sun, X.; Cheng, J. Comparative mitogenomic analyses and new insights into the phylogeny of thamnocephalidae (branchiopoda: Anostraca). Genes 2022, 13, 1765. [Google Scholar] [CrossRef]
- Greaves, L.C.; Elson, J.L.; Nooteboom, M.; Grady, J.P.; Taylor, G.A.; Taylor, R.W.; Mathers, J.C.; Kirkwood, T.B.L.; Turnbull, D.M. Comparison of Mitochondrial Mutation Spectra in Ageing Human Colonic Epithelium and Disease: Absence of Evidence for Purifying Selection in Somatic Mitochondrial DNA Point Mutations. PLoS Genet. 2012, 8, e1003082. [Google Scholar] [CrossRef]
- Wang, C.; Liang, Y.-Q.; Li, M.; Zhang, Y.; Shen, Z.-J.; Jiang, Z.-W. Complete mitochondrial DNA genome of Triplophysa venusta (cypriniformes: Cobitida). Mitochondrial DNA Part A 2016, 27, 4617–4619. [Google Scholar] [CrossRef]
- Ming Han, M.; Lu, J.; Wang, L.; Mahboob, S.; Al-Ghanim, K.A.; Sun, X.-W. Complete mitochondrial genome of the Triplophysa bombifrons and Triplophysa strauchii. Mitochondrial DNA Part A 2016, 27, 4710–4711. [Google Scholar] [CrossRef]
- Yang, X.; Wen, H.; Luo, T.; Zhou, J. Complete mitochondrial genome of Triplophysa nasobarbatula. Mitochondrial DNA Part B 2020, 5, 3771–3772. [Google Scholar] [CrossRef]
- Yan, P.; Li, J.; Ma, Q.; Deng, Y.; Song, Z. Complete mitochondrial genome of Triplophysa robusta (teleostei: Cypriniformes: Balitoridae). Mitochondrial DNA Part A 2016, 27, 1715–1716. [Google Scholar]
- Wang, X.; Song, Y.; Xie, H.; Zi, F.; Chen, S.; Luo, S. Complete mitogenome of the Triplophysa bombifrons: Comparative analysis and phylogenetic relationships among the members of Triplophysa. Genes 2023, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.Y.; Ahn, S.J.; Seo, J.S.; Jeon, E.J.; Cho, M.Y.; Choi, H.S. Characterization of the complete mitochondrial genome of Miamiensis avidus causing flatfish scuticociliatosis. Genetica 2022, 150, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [Green Version]
- Cao, L.; Causse, R.; Zhang, E. Revision of the loach species Barbatula nuda (Bleeker 1865) (Pisces: Balitoridae) from North China, with a description of a new species from Inner Mongolia. Zootaxa 2012, 3586, 236–248. [Google Scholar] [CrossRef] [Green Version]
- Jeon, S.A.; Park, J.L.; Park, S.-J.; Kim, J.H.; Goh, S.-H.; Han, J.-Y.; Kim, S.-Y. Comparison between MGI and Illumina sequencing platforms for whole genome sequencing. Genes Genom. 2021, 43, 713–724. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Jin, J.-J.; Yu, W.-B.; Yang, J.-B.; Song, Y.; Yi, T.-S.; Li, D.-Z. GetOrganelle: A simple and fast pipeline for de novo assembly of a complete circular chloroplast genome using genome skimming data. BioRxiv 2018, 4, 256479. [Google Scholar]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997v2. [Google Scholar]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef] [Green Version]
- Iriarte, A.; Lamolle, G.; Musto, H. Codon usage bias: An endless tale. J. Mol. Evol. 2021, 89, 589–593. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Horiike, T. An introduction to molecular phylogenetic analysis. Rev. Agric. Sci. 2016, 4, 36–45. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Carraretto, D.; Aketarawong, N.; Di Cosimo, A.; Manni, M.; Scolari, F.; Valerio, F.; Malacrida, A.R.; Gomulski, L.M.; Gasperi, G. Transcribed sex-specific markers on the Y chromosome of the oriental fruit fly, Bactrocera dorsalis. BMC Genet. 2020, 21, 125. [Google Scholar] [CrossRef]
- Zhou, L.; Huang, S.; Wang, Q.; Li, Z.; Li, Z.; He, A.; Chen, J.; Liu, L.; Zou, K. Novel evolutionary insights into nemacheilid cavefish: Evidence from comparative analysis of mitochondrial genomes. J. Oceanol. Limnol. 2022, 40, 1640–1653. [Google Scholar] [CrossRef]





| Locus | Start | Stop | Size (bp) | Start Coding | Stop Coding | Strand |
|---|---|---|---|---|---|---|
| tRNAMet | 1 | 69 | 69 | H | ||
| nad2 | 70 | 1116 | 1047 | ATG | T | H |
| tRNATrp | 1115 | 1184 | 70 | H | ||
| tRNAAla | 1187 | 1255 | 69 | L | ||
| tRNAAsn | 1257 | 1329 | 73 | L | ||
| tRNACys | 1361 | 1426 | 66 | L | ||
| tRNATyr | 1427 | 1495 | 69 | L | ||
| cox1 | 1497 | 3047 | 1551 | GTG | TAA | H |
| tRNASer | 3048 | 3118 | 71 | L | ||
| tRNAAsp | 3121 | 3193 | 73 | H | ||
| cox2 | 3207 | 3911 | 705 | ATG | T | H |
| tRNALys | 3898 | 3973 | 76 | H | ||
| atp8 | 3975 | 4142 | 168 | ATG | TAA | H |
| atp6 | 4133 | 4816 | 684 | ATG | TAA | H |
| cox3 | 4816 | 5616 | 801 | ATG | T | H |
| tRNAGly | 5600 | 5672 | 73 | H | ||
| nad3 | 5673 | 6023 | 351 | ATG | TAG | H |
| tRNAArg | 6022 | 6091 | 70 | H | ||
| nad4l | 6092 | 6388 | 297 | ATG | TAA | H |
| nad4 | 6382 | 7764 | 1383 | ATG | TAG | H |
| tRNAHis | 7764 | 7833 | 70 | H | ||
| tRNASer | 7834 | 7901 | 68 | H | ||
| tRNALeu | 7903 | 7975 | 73 | H | ||
| nad5 | 7976 | 9814 | 1839 | ATG | TAG | H |
| nad6 | 9811 | 10,332 | 522 | ATG | TAA | L |
| tRNAGlu | 10,333 | 10,401 | 69 | L | ||
| cob | 10,406 | 11,566 | 1161 | ATG | T | H |
| tRNAThr | 11,547 | 11,617 | 71 | H | ||
| tRNAPro | 11,616 | 11,685 | 70 | L | ||
| tRNAPhe | 12,604 | 12,672 | 69 | H | ||
| 12S rRNA | 12,673 | 13,622 | 950 | H | ||
| tRNAVal | 13,625 | 13,696 | 72 | H | ||
| 16S rRNA | 13,697 | 15,374 | 1678 | H | ||
| tRNALeu | 15,375 | 15,449 | 75 | H | ||
| nad1 | 15,450 | 16,424 | 975 | ATG | TAA | H |
| tRNAIle | 16,432 | 16,503 | 72 | H | ||
| tRNAGln | 16,502 | 16,572 | 71 | L |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.; Luo, S.; Yao, N.; Wang, X.; Song, Y.; Chen, S. A Comprehensive Analysis of Triplophysa labiata (Kessler, 1874) Mitogenome and Its Phylogenetic Implications within the Triplophysa Genus. Genes 2023, 14, 1356. https://doi.org/10.3390/genes14071356
Wang C, Luo S, Yao N, Wang X, Song Y, Chen S. A Comprehensive Analysis of Triplophysa labiata (Kessler, 1874) Mitogenome and Its Phylogenetic Implications within the Triplophysa Genus. Genes. 2023; 14(7):1356. https://doi.org/10.3390/genes14071356
Chicago/Turabian StyleWang, Chengxin, Site Luo, Na Yao, Xinyue Wang, Yong Song, and Shengao Chen. 2023. "A Comprehensive Analysis of Triplophysa labiata (Kessler, 1874) Mitogenome and Its Phylogenetic Implications within the Triplophysa Genus" Genes 14, no. 7: 1356. https://doi.org/10.3390/genes14071356