
Pulmonary Vasculitides: A Radiological Review Emphasizing Parenchymal HRCT Features

 ,
,  , , , , and
, , , , and
Abstract
:1. Introduction
2. Pulmonary Vasculitis
2.1. Overview
2.2. Radiological Features
2.3. Microscopic Polyangiitis (MPA)
2.4. Granulomatosis with Polyangiitis (GPA-Wegener’s Disease)
2.5. Eosinophilic Granulomatosis with Polyangiitis (EGPA-Churg-Strauss)
2.6. Takayasu Arteritis (TA)
2.7. Giant-Cell Arteritis (GCA-Temporal Arteritis)
2.8. Variable Vessel Vasculitis (VVV)
2.9. Vasculitis Associated with Probable Aetiology
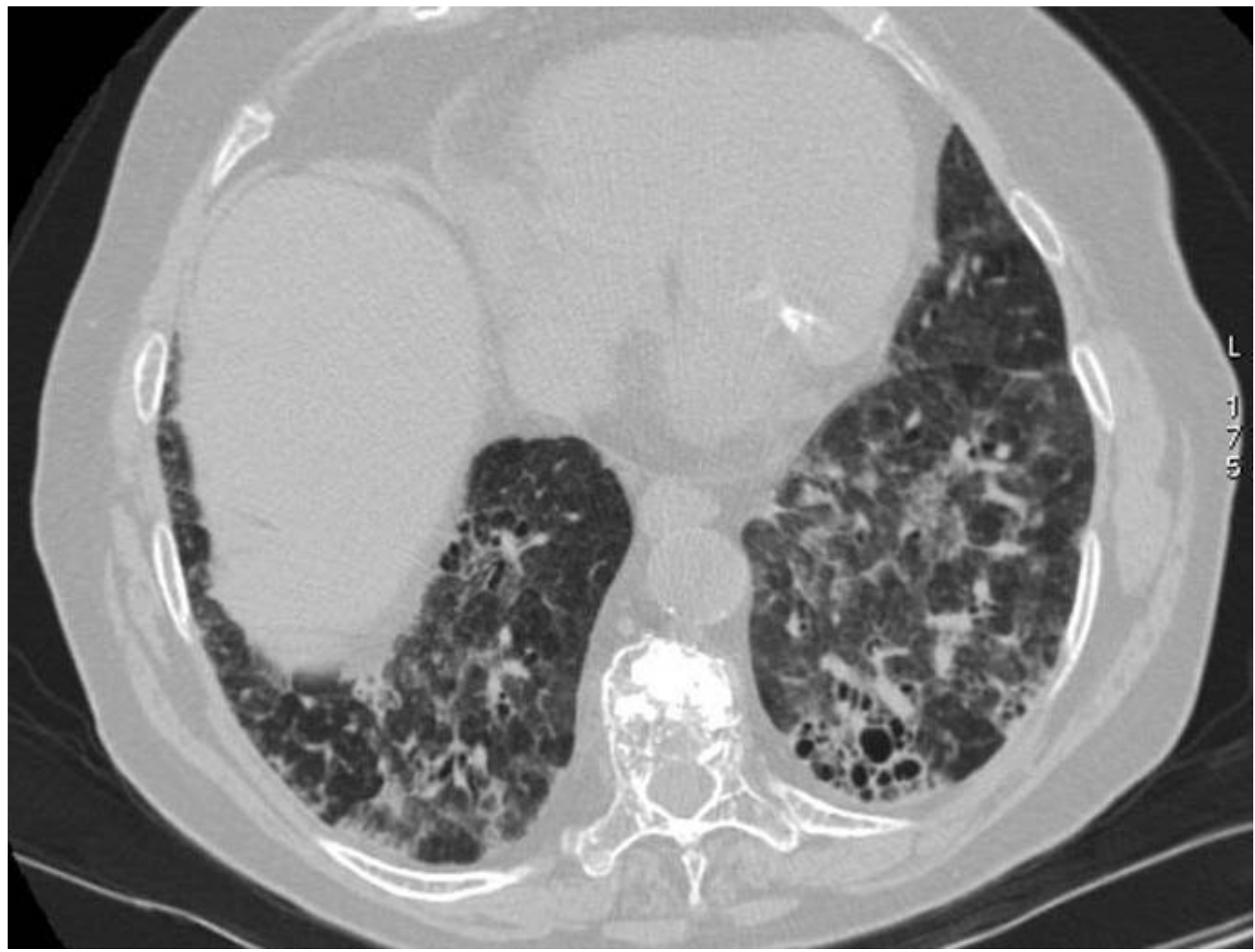
2.10. ILD in ANCA-Associated Vasculitides
2.10.1. Epidemiology
2.10.2. Morphological Patterns and Imaging
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Jennette, J.C.; Falk, R.J.; Andrassy, K.; Bacon, A.P.; Churg, J.; Wolfgang, L.G.; Christiaan Hagen, E.; Hoffman, G.S.; Hunder, G.G.; Kallenberg, C.G.M.; et al. Nomenclature of systemic vasculitides: The proposal of an international consensus conference. Arthritis Rheum. 1994, 37, 187–192. [Google Scholar] [CrossRef]
- Jennette, J.C.; Falk, R.J.; Bacon, A.P.; Basu, N.; Cid, M.C.; Ferrario, F.; Flores-Suarez, L.F.; Gross, W.L.; Guillevin, L.; Hagen, E.C.; et al. 2012 revised International Chapel Hill Consensus Conference nomenclature of vasculitides. Arthritis Rheum. 2013, 65, 1–11. [Google Scholar] [CrossRef]
- Brown, K.K. Pulmonary vasculitis. Proc. Am. Thorac. Soc. 2006, 3, 48–57. [Google Scholar] [CrossRef]
- Maffessanti, M.; Dalpiaz, G. Diffuse Lung Disease—Clinical Features, Pathology, HRCT; Springer: Berlin/Heidelberg, Germany, 2004; ISBN 8847004292. [Google Scholar]
- Alba, M.A.; Flores-Suárez, L.F.; Henderson, A.G.; Xiao, H.; Hu, P.; Nachman, P.H.; Falk, R.J.; Jennetteet, J.C. Interstitial lung disease in ANCA vasculitis. Autoimmun. Rev. 2017, 16, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Castañer, E.; Alguersuari, A.; Andreu, M.; Gallardo, X.; Spinu, C.; Mata, J.M. Imaging findings in pulmonary vasculitis. Semin. Ultrasound CT MR 2012, 33, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Nasser, M.; Cottin, V. Alveolar hemorrhage in vasculitis (primary and secondary). Semin. Respir. Crit. Care Med. 2018, 39, 482–493. [Google Scholar] [CrossRef] [PubMed]
- Ceylan, N.; Bayraktaroglu, S.; Erturk, S.M.; Savas, R.; Alper, H. Pulmonary and vascular manifestations of Behcet disease: Imaging findings. AJR Am. J. Roentgenol. 2010, 194, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, M.; Ikeda, K.; Tsushima, K.; Iesato, K.; Abe, M.; Ito, T.; Kashiwakuma, D.; Kagami, S.; Iwamoto, I.; Nakagomi, D.; et al. Prevalence and responsiveness to treatment of lung abnormalities on chest computed tomography in patients with microscopic polyangiitis: A multicenter, longitudinal, retrospective study of one hundred fifty consecutive hospital-based Japanese patients. Arthritis Rheumatol. 2016, 68, 713–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casal, A.; Pereiro, T.; Valdés, L. Pulmonary vasculitis: An update. Arch. Bronconeumol. 2018, 54, 407–408. [Google Scholar] [CrossRef] [PubMed]
- Jennet, J.C.; Falk, R.J. Small-vessel vasculitis. N. Engl. J. Med. 1997, 337, 1512–1523. [Google Scholar] [CrossRef]
- Schnabel, A.; Reuter, M.; Csernok, E.; Richter, C.; Gross, W.L. Subclinical alveolar bleeding in pulmonary vasculitides: Correlation with indices of disease activity. Eur. Respir. J. 1999, 14, 118–124. [Google Scholar] [CrossRef] [Green Version]
- Chung, M.P.; Yi, C.A.; Lee, H.Y.; Han, J.; Lee, K.S. Imaging of pulmonary vasculitis. Radiology 2010, 255, 322–341. [Google Scholar] [CrossRef] [PubMed]
- Gaudin, P.B.; Askin, F.B.; Falk, R.J.; Jennette, J.C. The pathologic spectrum of pulmonary lesions in patients with anti-neutrophil cytoplasmic autoantibodies specific for antiproteinase 3 and anti-myeloperoxidase. Am. J. Clin. Pathol. 1995, 104, 7–16. [Google Scholar] [CrossRef]
- Mahmoud, S.; Ghosh, S.; Farver, C.; Lempel, J.; Azok, J.; Renapurkar, R.D. Pulmonary vasculitis: Spectrum of imaging appearances. Radiol. Clin. N. Am. 2016, 54, 1097–1118. [Google Scholar] [CrossRef] [PubMed]
- Alba, M.A.; Jennette, J.C.; Falk, R.J. Pathogenesis of ANCA-associated pulmonary vasculitis. Semin. Respir. Crit. Care Med. 2018, 39, 413–424. [Google Scholar] [CrossRef]
- Katzenstein, A.L. Diagnostic features and differential diagnosis of Churg-Strauss syndrome in the lung: A review. Am. J. Clin. Pathol. 2000, 114, 767–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Vietinghoff, S. Pulmonary manifestations of vasculitis. Pneumologie 2016, 70, 747–757. [Google Scholar] [CrossRef]
- Guillevin, L. Infections in vasculitis. Best Pract. Res. Clin. Rheumatol. 2013, 27, 19–31. [Google Scholar] [CrossRef]
- Matsunaga, N.; Hayashi, K.; Sakamoto, I.; Ogawa, Y.; Matsumoto, T. Takayasu arteritis: Protean radiologic manifestations and diagnosis. Radiographics 1997, 17, 579–594. [Google Scholar] [CrossRef]
- Travis, W.; Colby, T.; Lombard, C.; Carpenter, H. A clinicopathologic study of 34 cases of diffuse pulmonary hemorrhage with lung biopsy confirmation. Am. J. Surg. Pathol. 1990, 14, 1112–1125. [Google Scholar] [CrossRef]
- Anaev, E.K.; Baranova, I.A.; Belevsky, A.S. Pulmonary vasculitis: Diagnosis and treatment. Ter. Arkhiv 2018, 90, 99–106. [Google Scholar] [CrossRef]
- Adams, T.N.; Zhang, D.; Batra, K.; Fitzgerald, J.E. Pulmonary manifestations of large, medium, and variable vessel vasculitis. Respir. Med. 2018, 145, 182–191. [Google Scholar] [CrossRef]
- Erkan, F.; Gül, A.; Tasali, E. Pulmonary manifestations of Behçet’s disease. Thorax 2001, 56, 572–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feragalli, B.; Mantini, C.; Sperandeo, M.; Galluzzo, M.; Belcaro, G.; Tartaro, A.; Cotroneo, A.R. The lung in systemic vasculitis: Radiological patterns and differential diagnosis. Br. J. Radiol. 2016, 89, 20150992. [Google Scholar] [CrossRef] [Green Version]
- Comarmond, C.; Crestani, B.; Tazi, A.; Hervier, B.; Adam-Marchand, S.; Nunes, H.; Cohen-Aubart, F.; Wislez, M.; Cadranel, J.; Housset, B.; et al. Pulmonary fibrosis in antineutrophil cytoplasmic antibodies (ANCA)-associated vasculitis: A series of 49 patients and review of the literature. Medicine 2014, 93, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Foulon, G.; Delaval, P.; Valeyre, D.; Wallaert, B.; Debray, M.P.; Brauner, M.; Nicaise, P.; Cadranel, J.; Cottin, V.; Tazi, A.; et al. ANCA-associated lung fibrosis: Analysis of 17 patients. Respir. Med. 2008, 102, 1392–1398. [Google Scholar] [CrossRef] [PubMed]
- Arulkumaran, N.; Periselneris, N.; Gaskin, G.; Strickland, N.; Ind, P.W.; Pusey, C.D.; Salamae, A.D. Interstitial lung disease and ANCA-associated vasculitis: A retrospective observational cohort study. Rheumatology 2011, 50, 2035–2043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzelepis, G.E.; Kokosi, M.; Tzioufas, A.; Toya, S.P.; Boki, K.A.; Zormpala, A.; Moutsopoulos, H.M. Prevalence and outcome of pulmonary fibrosis in microscopic polyangiitis. Eur. Respir. J. 2010, 36, 116–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casares, F.M.; Gonzalez, A.; Fielli, M.; Caputo, F.; Bottinelli, Y.; Zamboni, M. Microscopic polyangiitis associated with pulmonary fibrosis. Clin. Rheumatol. 2015, 34, 1273–1277. [Google Scholar] [CrossRef]
- Huang, H.; Wang, Y.X.; Jiang, C.G.; Liu, J.; Li, J.; Xu, K.; Xu, Z.J. A retrospective study of microscopic polyangiitis patients presenting with pulmonary fibrosis in China. BMC Pulm. Med. 2014, 14, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kagiyama, N.; Takayanagi, N.; Kanauchi, T.; Ishiguro, T.; Yanagisawa, T.; Sugita, Y. Antineutrophil cytoplasmic antibody-positive conversion and microscopic paolyangiitis development in patients with idiopathic pulmonary fibrosis. BMJ Open Respir. Res. 2015, 2, e000058. [Google Scholar] [CrossRef] [PubMed]
- Hervier, B.; Pagnoux, C.; Agard, C.; Haroche, J.; Amoura, Z.; Guillevin, L.; Hamidou, M.A.; French Vasculitis Study Group. Pulmonary fibrosis associated with ANCA-positive vasculitides. Retrospective study of 12 cases and review of the literature. Ann. Rheum. Dis. 2009, 68, 404–407. [Google Scholar] [CrossRef]
- Katsumata, Y.; Kawaguchi, Y.; Yamanaka, H. Interstitial lung disease with ANCA-associated Vasculitis. Clin. Med. Insights Circ. Respir. Pulm. Med. 2015, 9, 51–56. [Google Scholar] [CrossRef]
- Homma, S.; Matsushita, H.; Nakata, K. Pulmonary fibrosis in myeloperoxidase antineutrophil cytoplasmic antibody-associated vasculitides. Respirology 2004, 9, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Ando, Y.; Okada, F.; Matsumoto, S.; Mori, H. Thoracic manifestation of myeloperoxidase-antineutrophil cytoplasmic antibody (MPO-ANCA)-related disease. CT findings in 51 patients. J. Comput. Assist. Tomogr. 2004, 28, 710–716. [Google Scholar] [CrossRef] [Green Version]
- Bargagli, E.; Conticini, E.; Mazzei, M.A.; Cameli, P.; Guerrini, S.; D’Alessandro, M.; Frediani, B.; Pleuroparenchymal Fibroelastosis (PPFE) Siena Unit, Italy. Pleuroparenchymal fibroelastosis in interstitial lung disease with antineutrophil cytoplasmic antibody-associated vasculitis. Clin. Exp. Rheumatol. 2021, 39, 190. [Google Scholar]
- Shumar, J.N.; Chandel, A.; King, C.S. Antifibrotic therapies and progressive fibrosing interstitial lung disease (PF-ILD): Building on INBUILD. J. Clin. Med. 2021, 10, 2285. [Google Scholar] [CrossRef] [PubMed]
- Skolnik, K.; Ryerson, C.J. Unclassifiable interstitial lung disease: A review. Respirology 2016, 21, 51–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
















{kind=link}
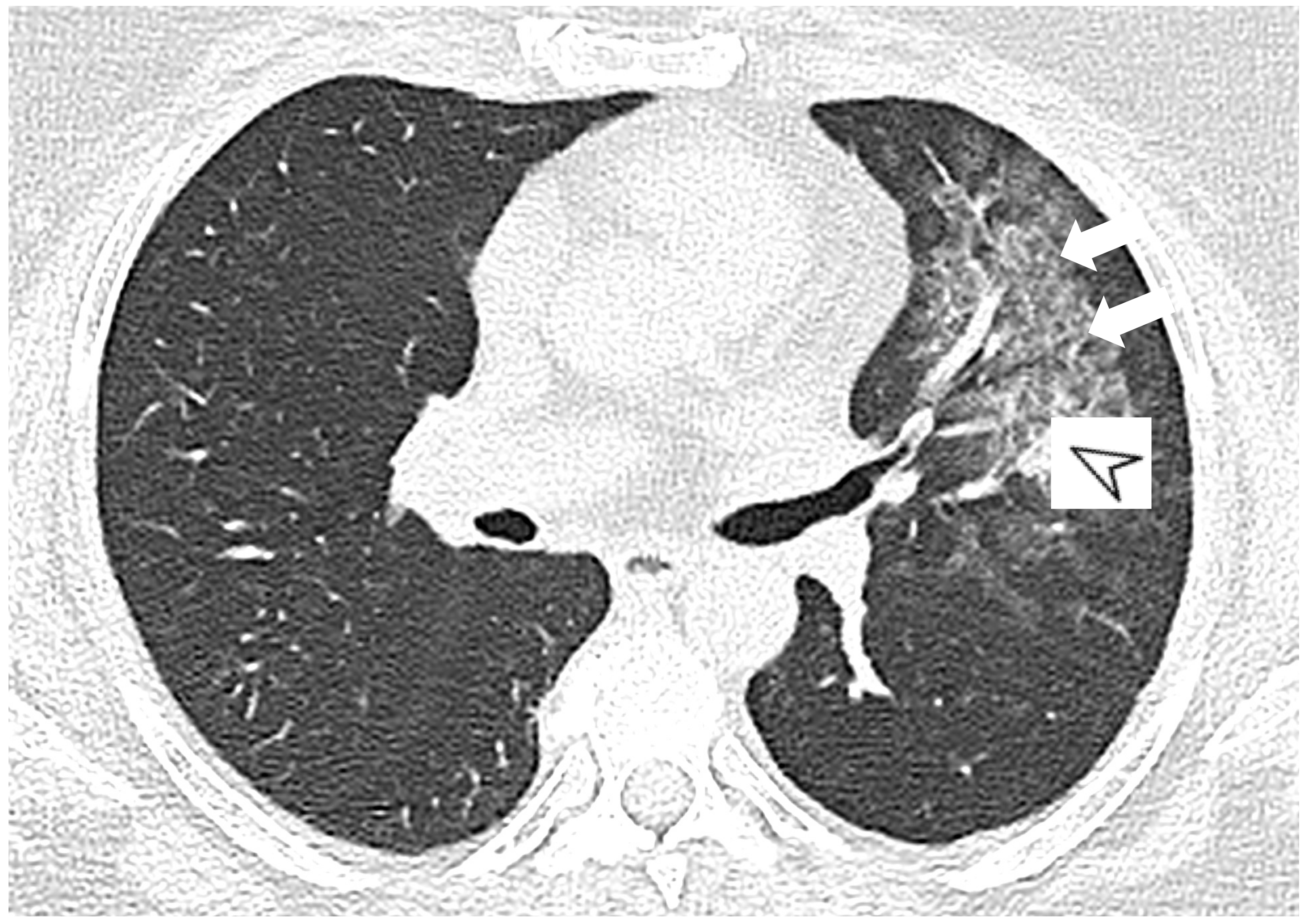{kind=link}
{kind=link}
{kind=link}
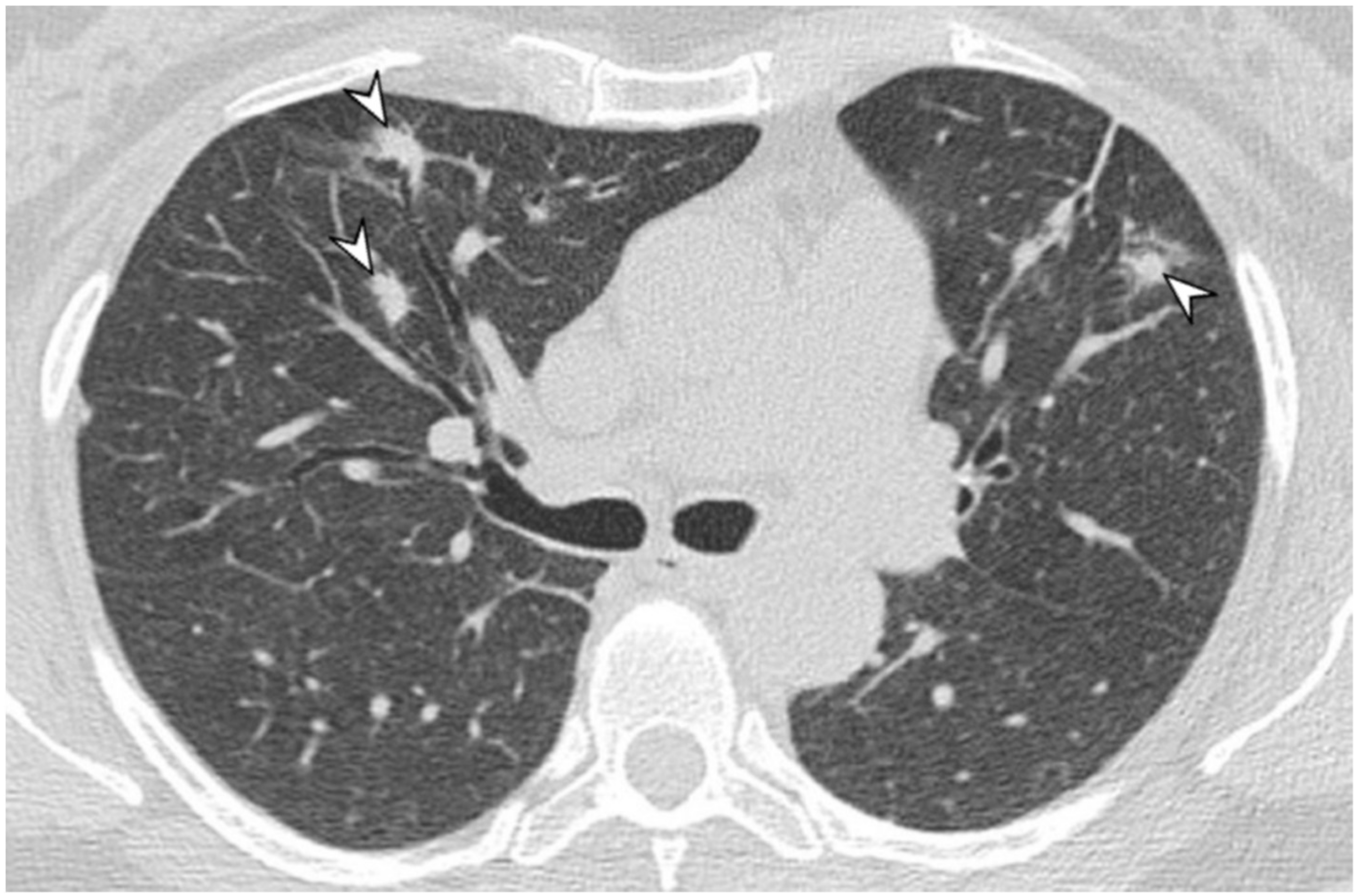{kind=link}
{kind=link}
{kind=link}
{kind=link}
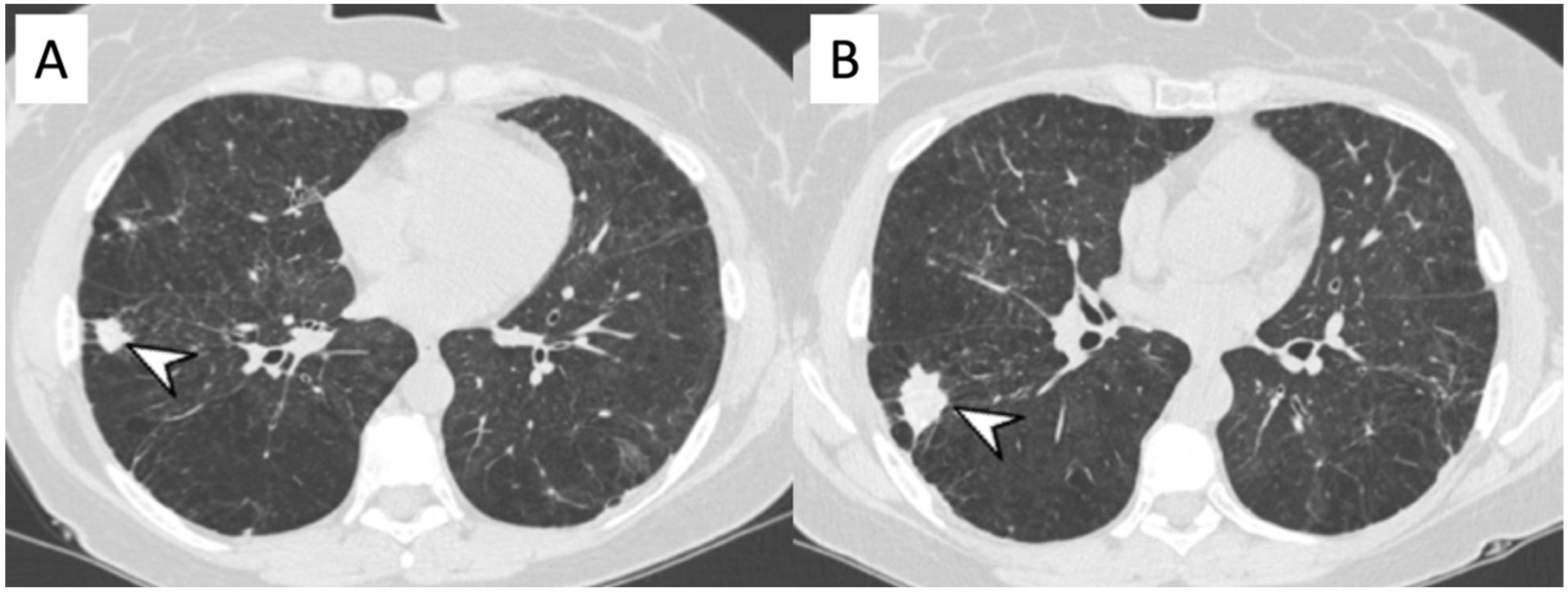{kind=link}
{kind=link}
{kind=link}
{kind=link}
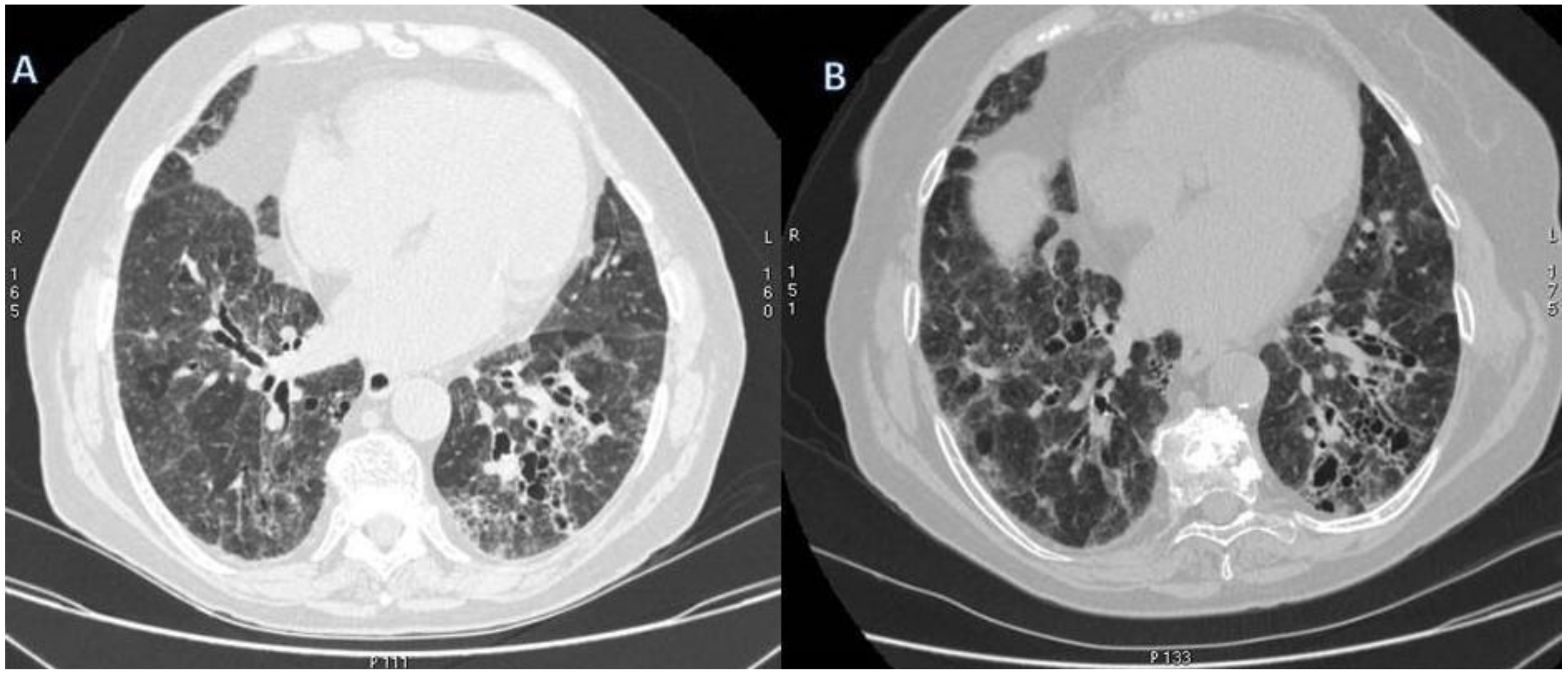{kind=link}
{kind=link}
{kind=link}
| Vasculitis | HRCT Features |
|---|---|
| MPA | GGOs due to hemorrhagic alveolitis (common); consolidation, nodules with centrilobular distribution (less common) |
| GPA | Solid nodules, GGOs due to hemorrhagic alveolitis (common); halo sign, crazy paving (less common) |
| EGPA | Migrant GGOs, transient consolidation, irregular bronchial wall thickening, small nodules with peribronchial and centrilobular distribution, pleural effusion. |
| Takayasu arteritis | Stenosis and/or occlusion of segmental arteries; stenosis and/or occlusion of lobular or main pulmonary arteries (less common); C.E. of vessel wall may be evident |
| Giant-cell arteritis | Aneurism or dissection of the thoracic aorta (common); nodules, GGOs, monolateral or bilateral pleural effusion (less common) |
| Bechet’s disease | Subpleural alveolar infiltrates and wedge-shaped or ill-defined rounded areas with increased opacity, pulmonary artery aneurism |
| Author | Ref | Patients | Onset of ILD |
|---|---|---|---|
| Comarmond et al., 2014 | [26] | 49 ANCA and PF: Typical UIP 43% Atypical UIP 14% Fibrotic NSIP 7% NSIP 9,5% | Preceding 45% Concomitant 43% Posterior 12% |
| Foulon et al., 2008 | [27] | 17 ANCA and ILD: Probable PF 70% UIP 17% | Preceding 76% Concomitant 24% |
| Arulkumaran et al., 2011 | [28] | 194 MPA-14 MPA and ILD: Idiopathic pulmonary fibrosis: 57% DIP: 14% NSIP: 7% | Preceding 14% Concomitant 64% Posterior 21% |
| Tzelepis et al., 2010 | [29] | 33 MPA-13 MPA with PF: UIP 54% NSIP 31% | Preceding 53% |
| Casares et al., 2015 | [30] | 28 MPA-9 MPA with PF: UIP 66% UIP probable 22% | Preceding 55% Concomitant 45% |
| Huang et al., 2014 | [31] | 67 MPA-19 MPA with PF: UIP: 28% | Preceding 68% Concomitant 32% |
| Kagiyama et al., 2015 | [32] | 504 ILD-36 ANCA with ILD | Preceding 100% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palmucci, S.; Inì, C.; Cosentino, S.; Fanzone, L.; Di Pietro, S.; Di Mari, A.; Galioto, F.; Tiralongo, F.; Vignigni, G.; Toscano, S.; et al. Pulmonary Vasculitides: A Radiological Review Emphasizing Parenchymal HRCT Features. Diagnostics 2021, 11, 2318. https://doi.org/10.3390/diagnostics11122318
Palmucci S, Inì C, Cosentino S, Fanzone L, Di Pietro S, Di Mari A, Galioto F, Tiralongo F, Vignigni G, Toscano S, et al. Pulmonary Vasculitides: A Radiological Review Emphasizing Parenchymal HRCT Features. Diagnostics. 2021; 11(12):2318. https://doi.org/10.3390/diagnostics11122318
Chicago/Turabian StylePalmucci, Stefano, Corrado Inì, Salvatore Cosentino, Luigi Fanzone, Stefano Di Pietro, Alessia Di Mari, Federica Galioto, Francesco Tiralongo, Giovanna Vignigni, Stefano Toscano, and et al. 2021. "Pulmonary Vasculitides: A Radiological Review Emphasizing Parenchymal HRCT Features" Diagnostics 11, no. 12: 2318. https://doi.org/10.3390/diagnostics11122318