
Intradural Pediatric Spinal Tumors: An Overview from Imaging to Novel Molecular Findings

 , , , and
, , , and
Abstract
:1. Introduction
2. Gliomas and Mixed Neuronal–Glial Tumors
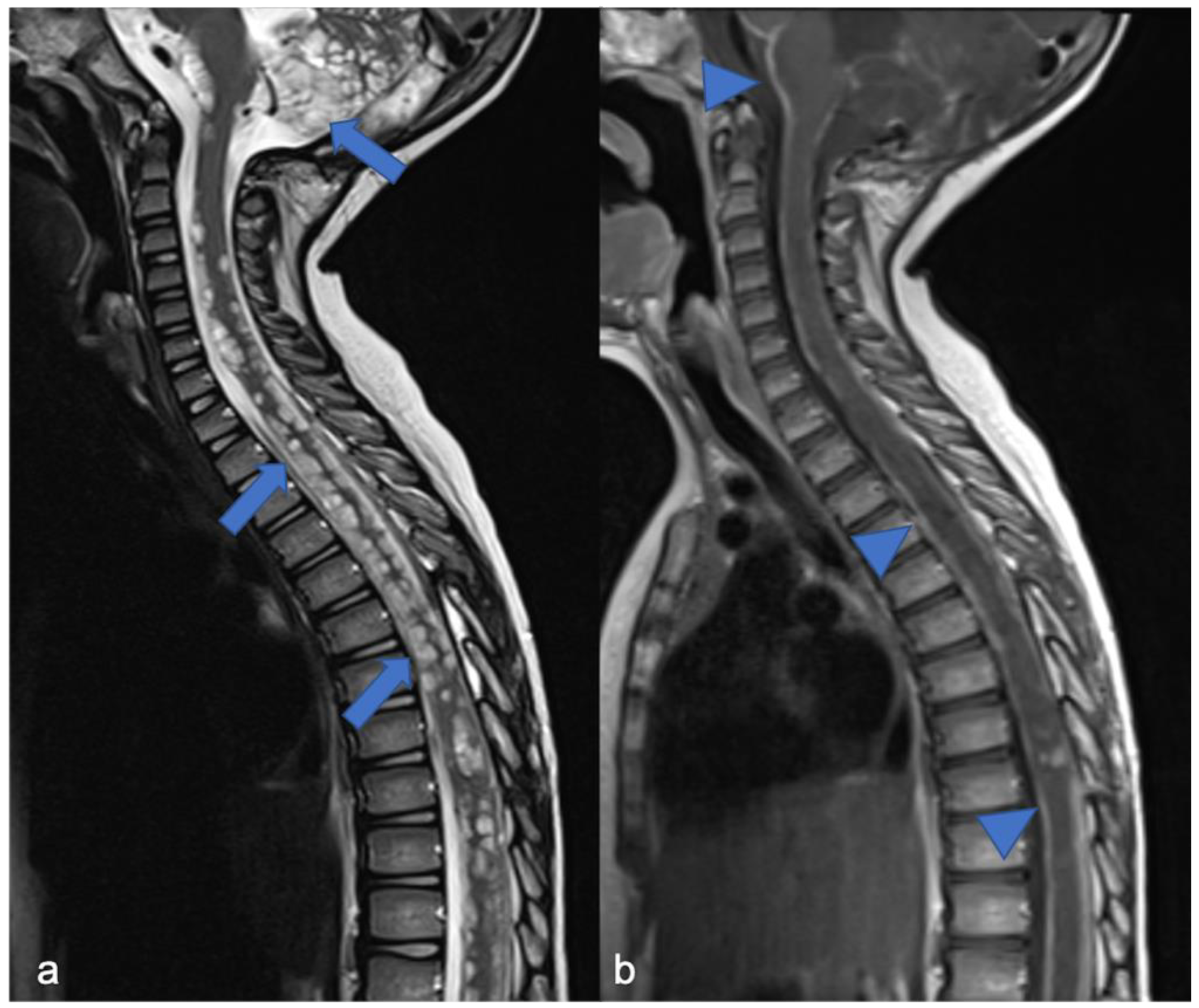
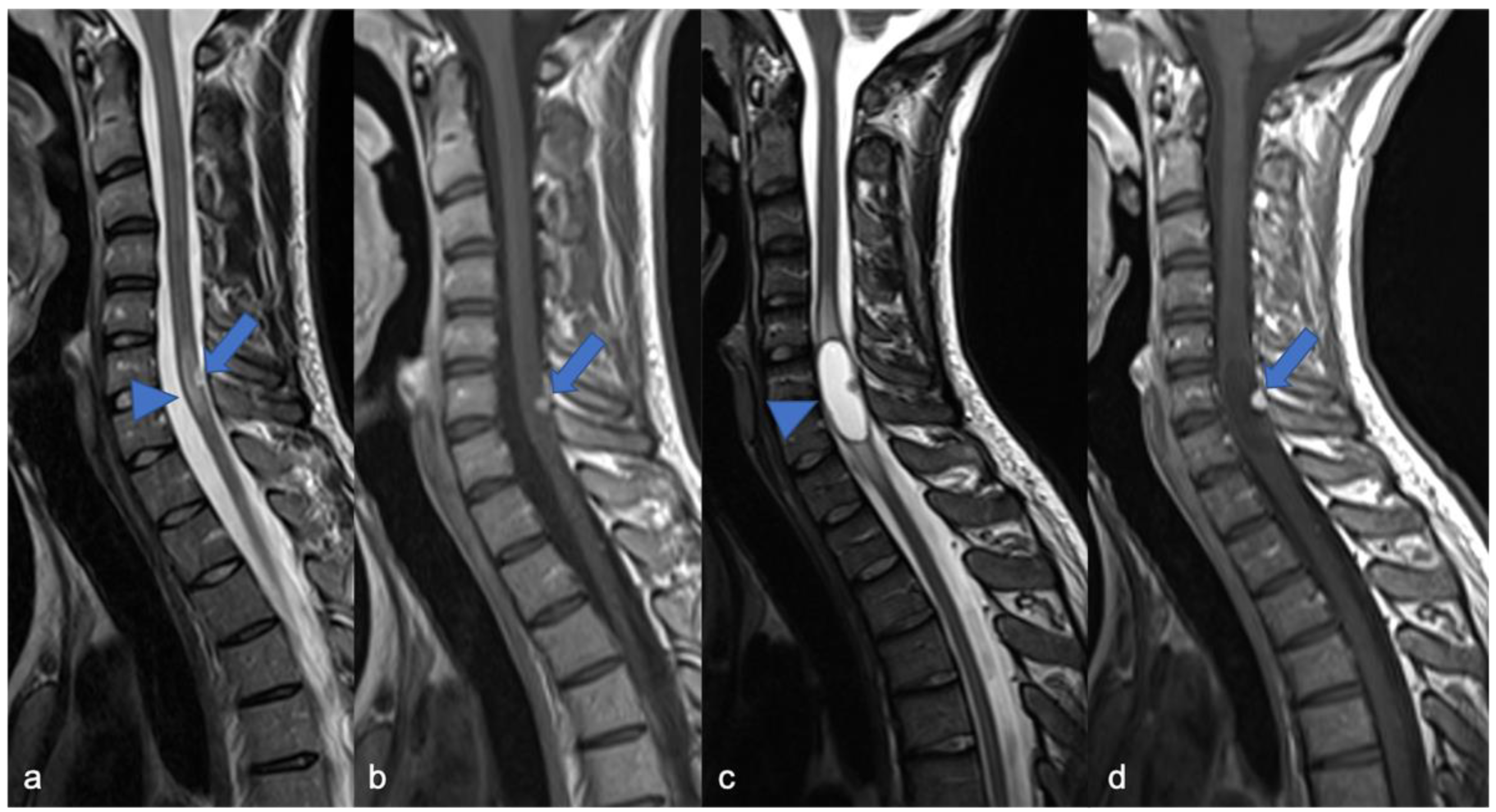
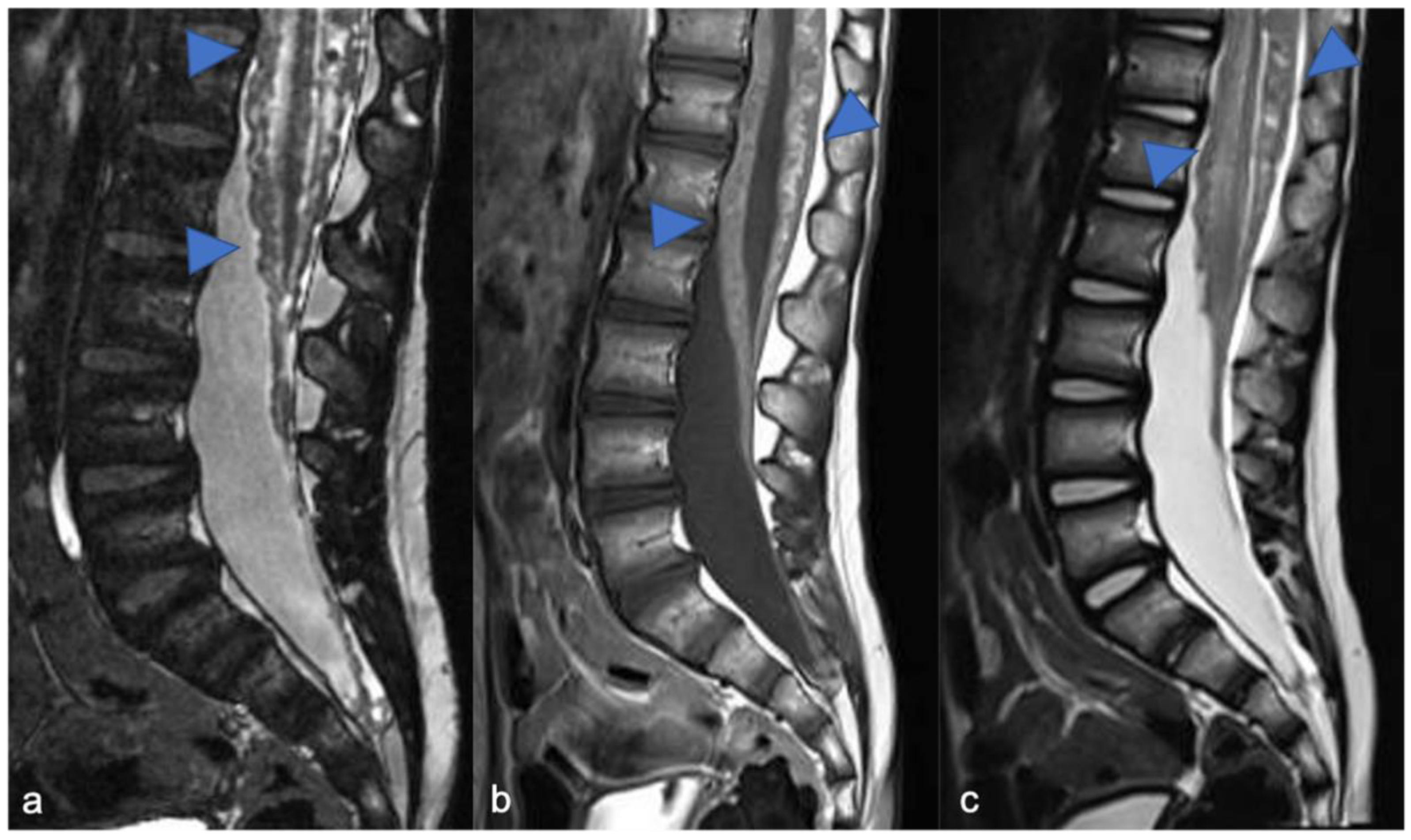
2.1. Gliomas
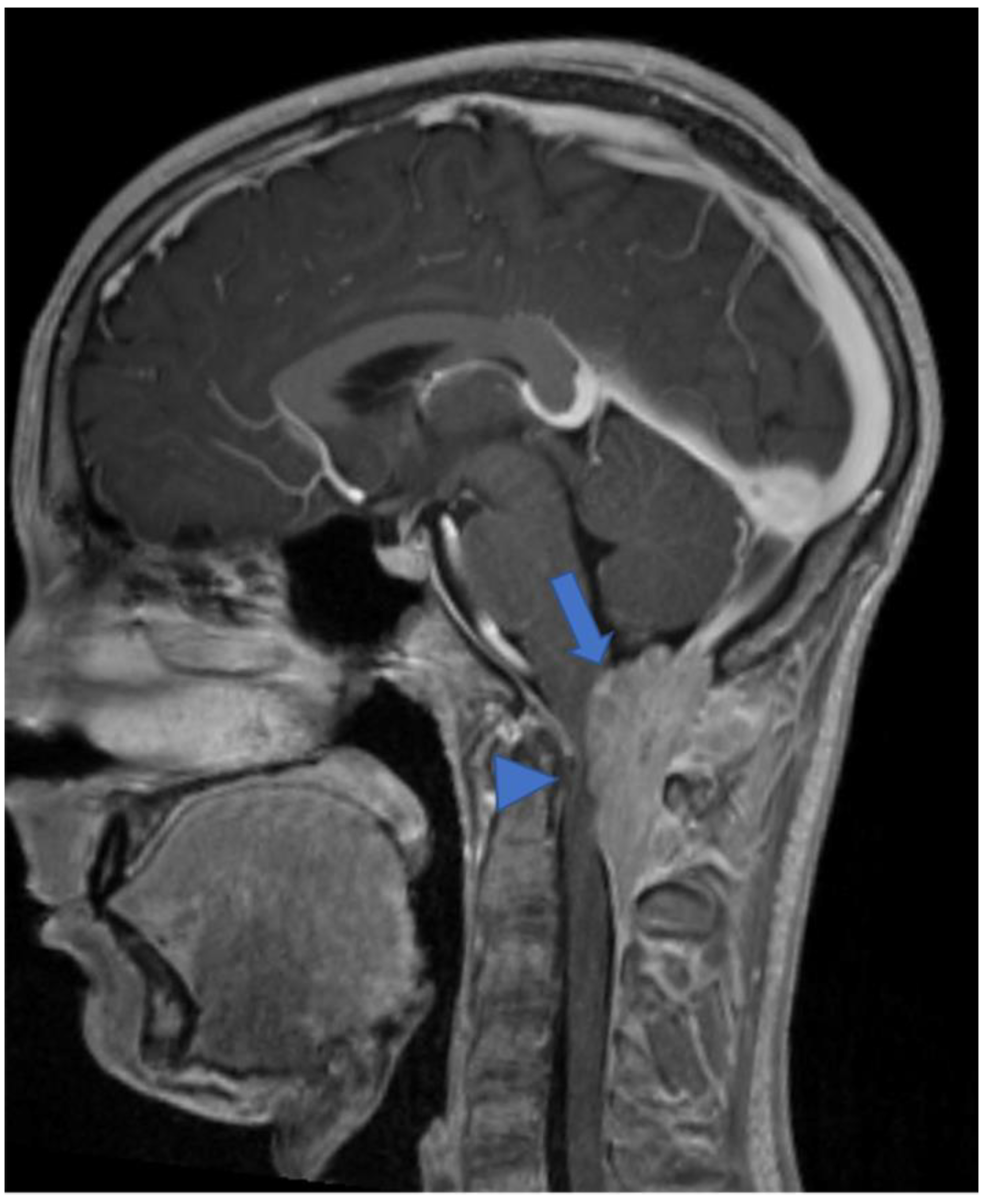
2.2. Diffuse Leptomeningeal Glioneuronal Tumor
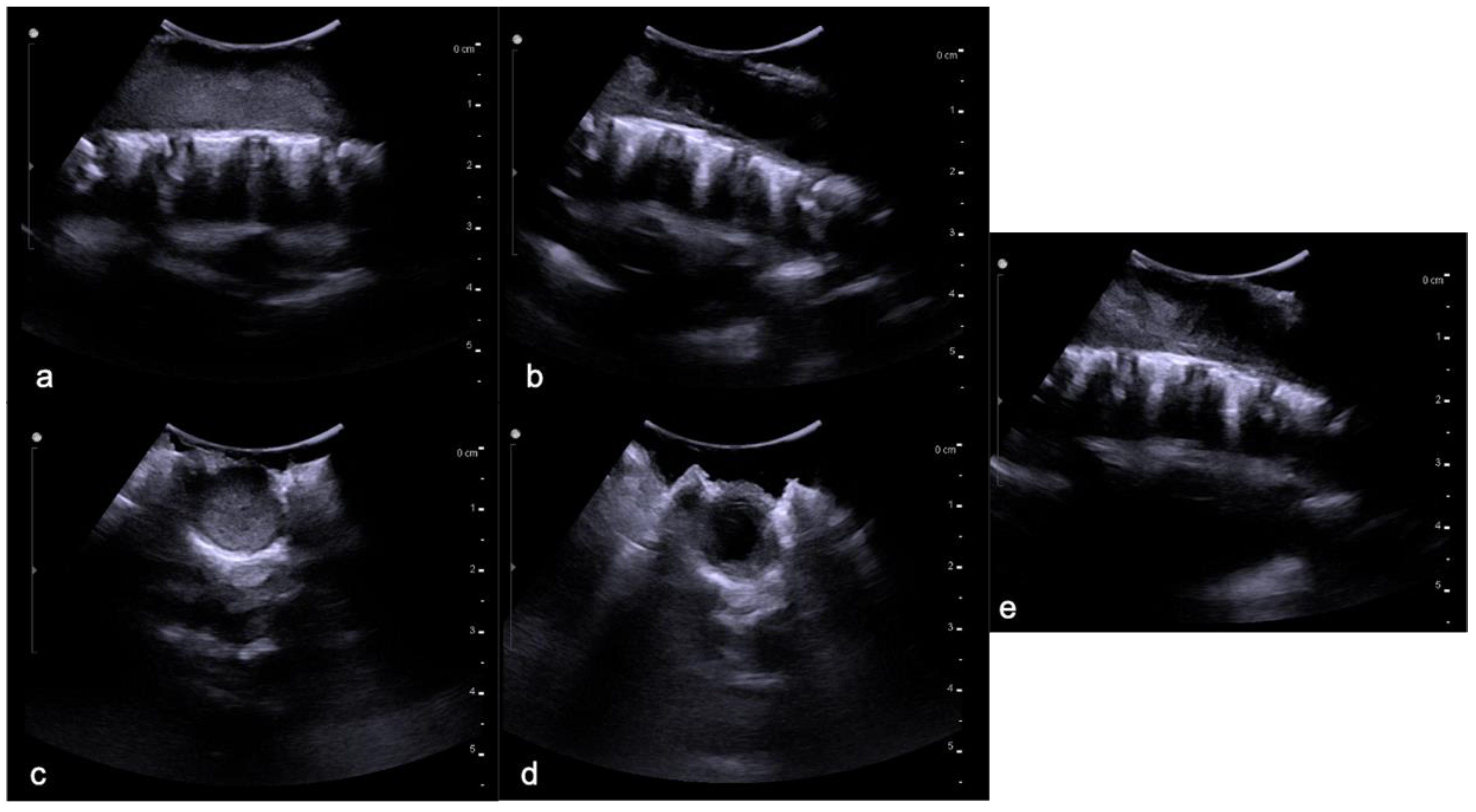
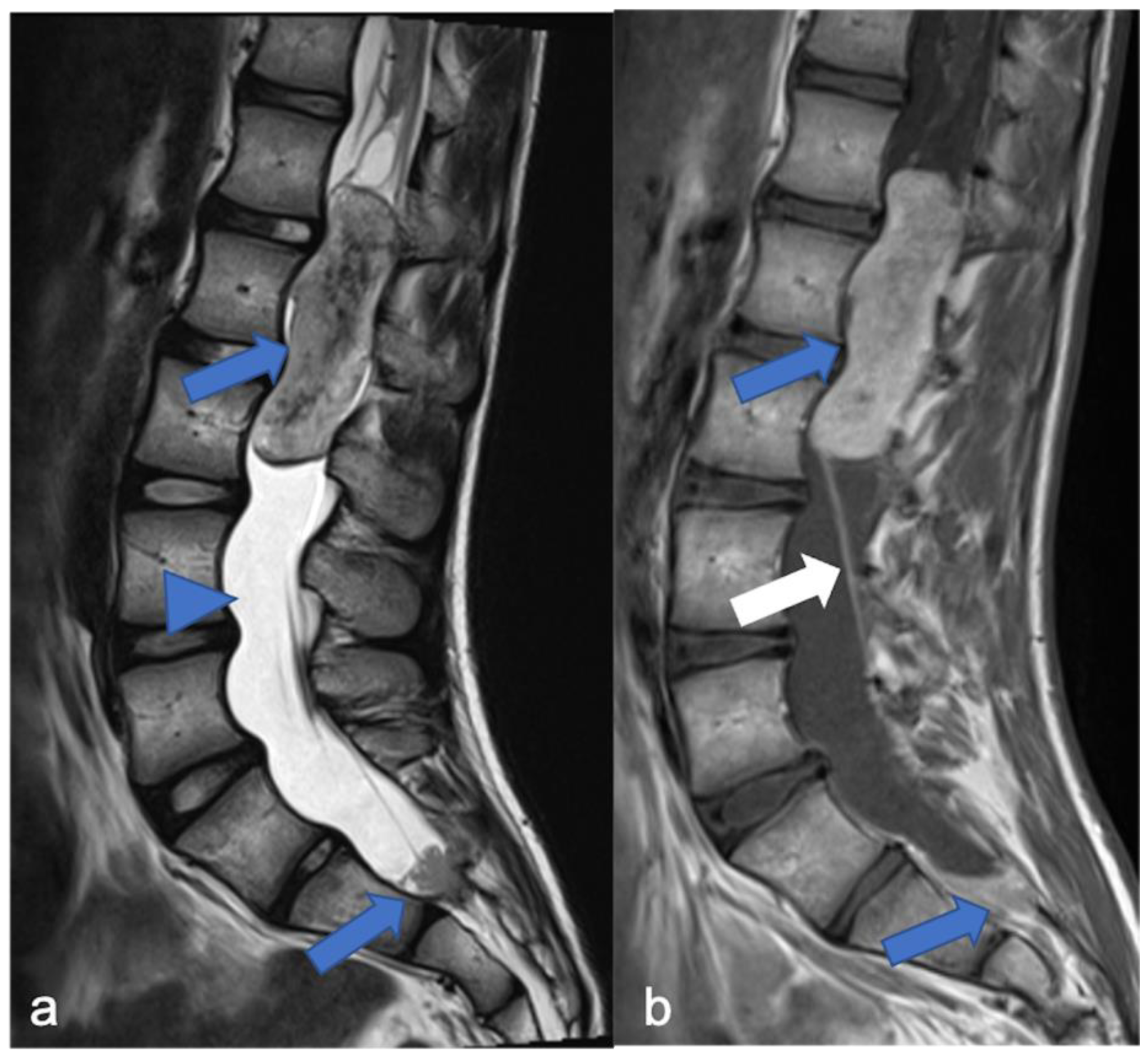
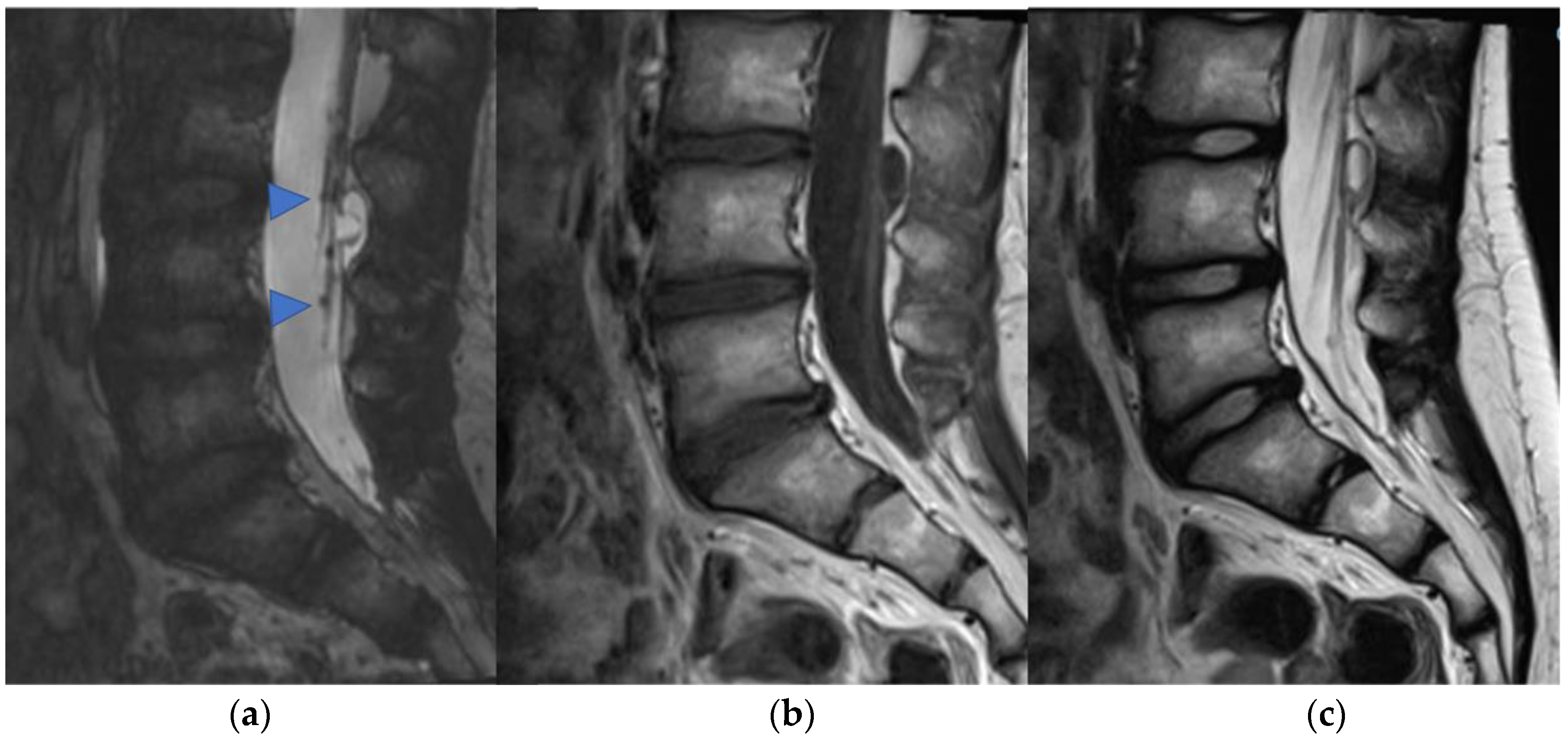
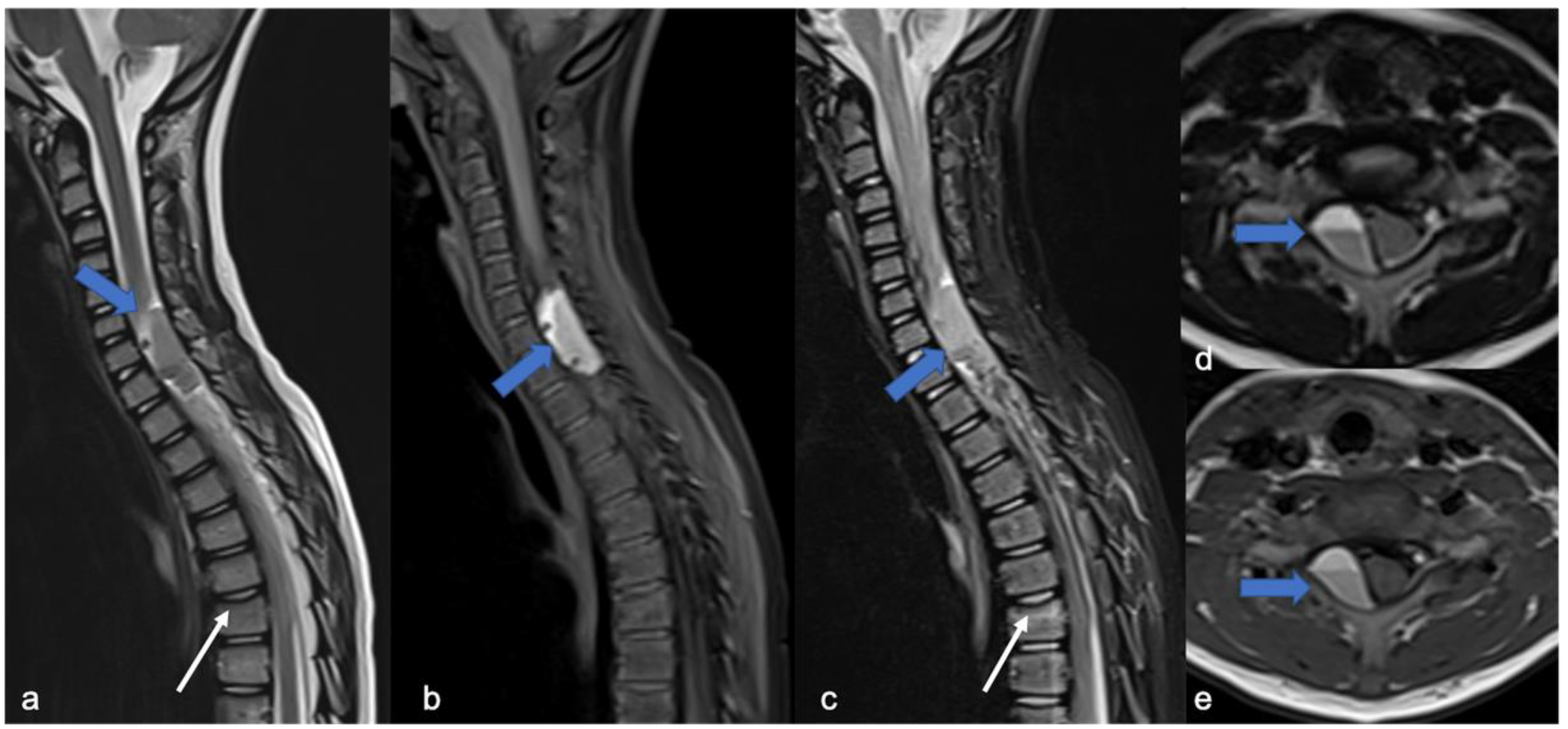
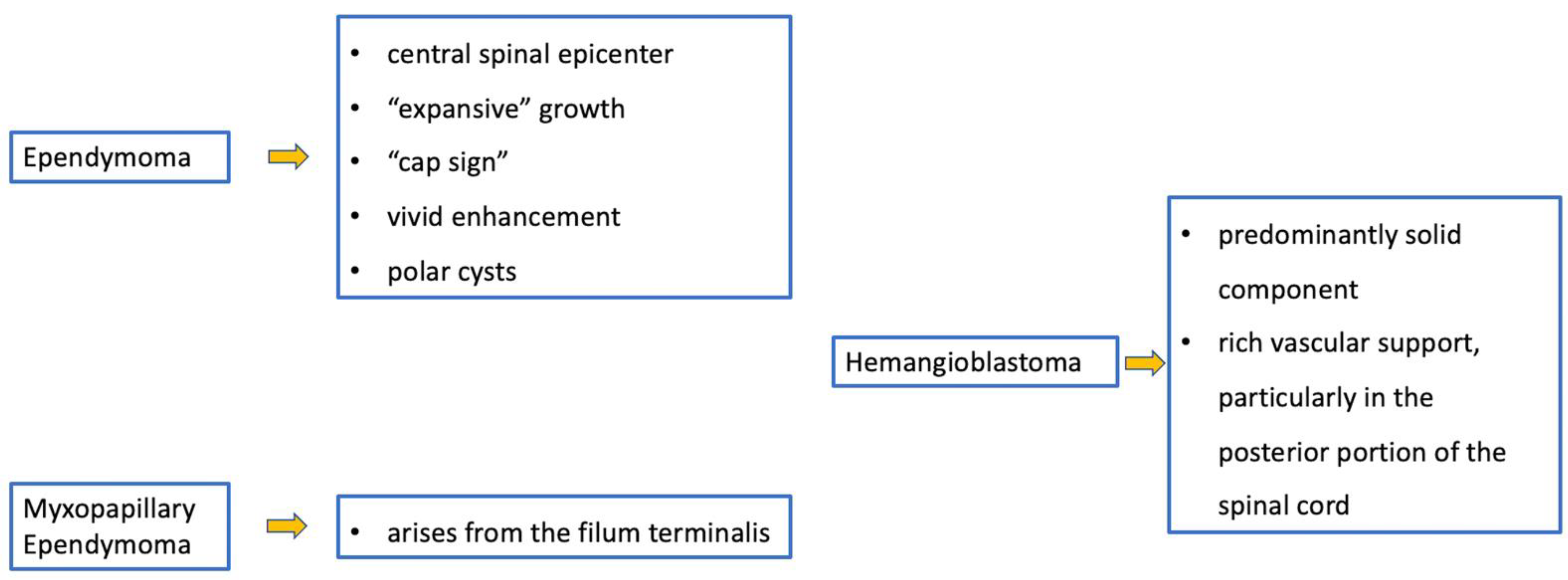
3. Ependymomas
4. Mesenchymal, Non-Meningothelial Tumors
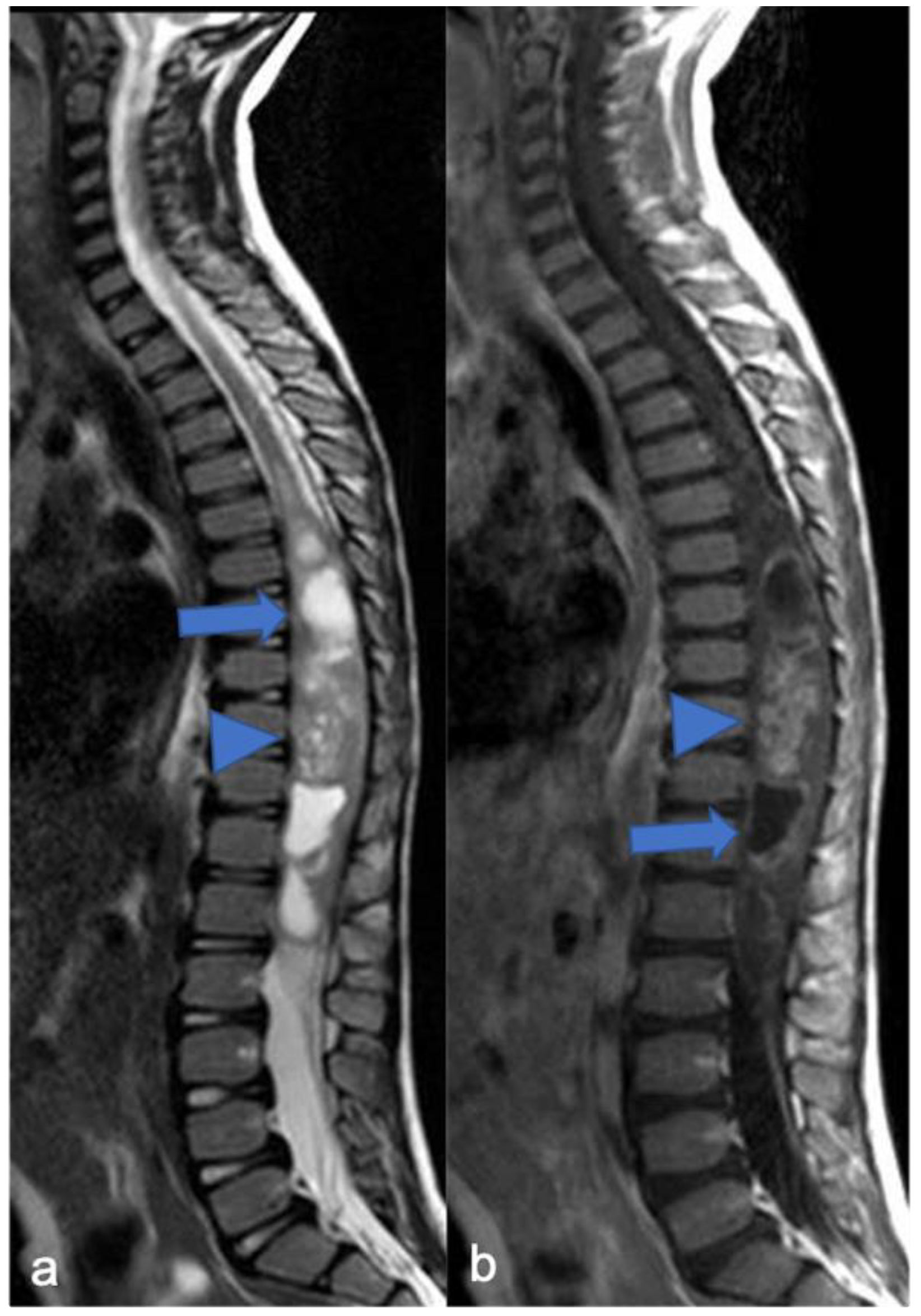
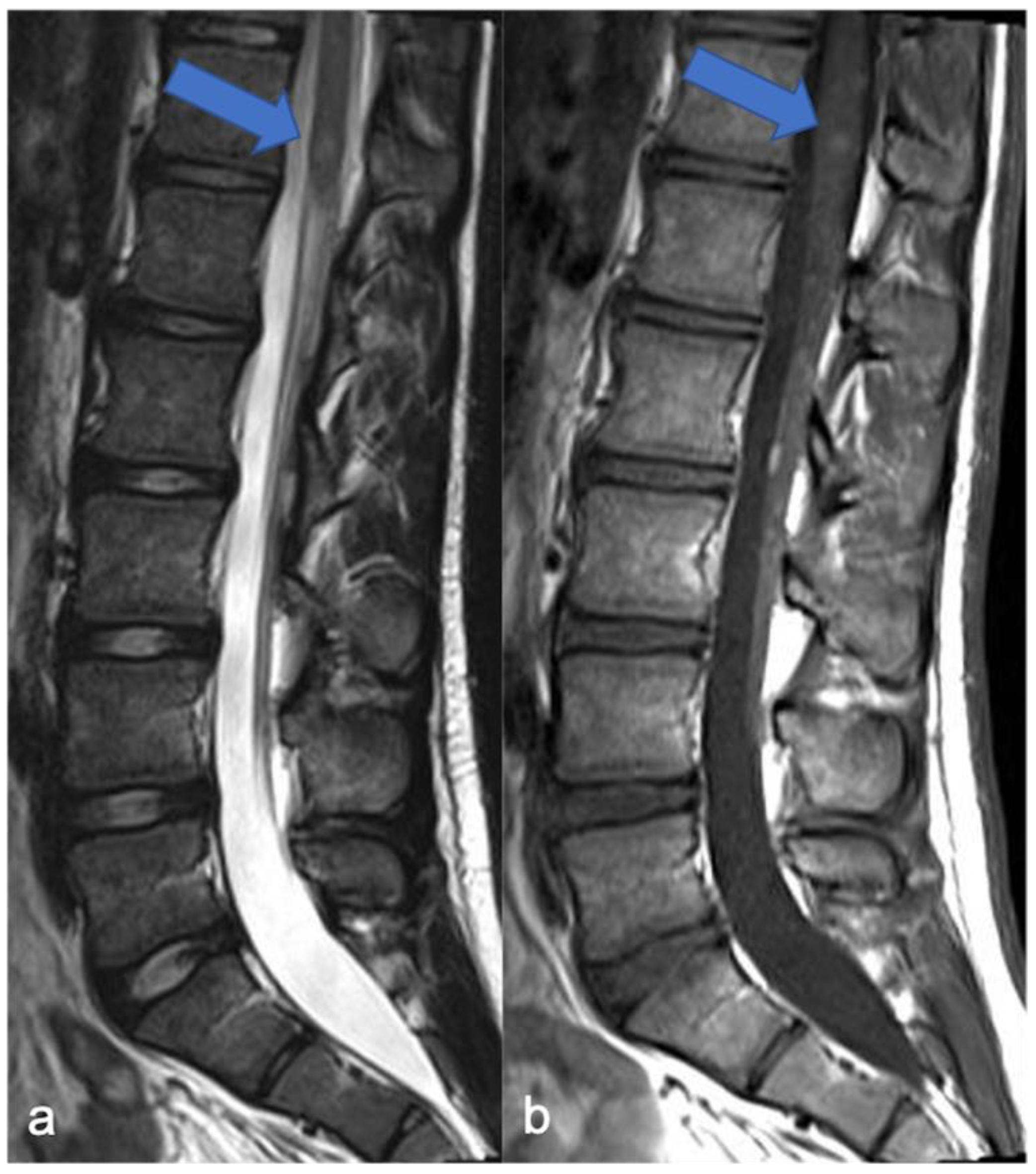
4.1. Hemangioblastomas
4.2. Mesenchymal Chondrosarcomas
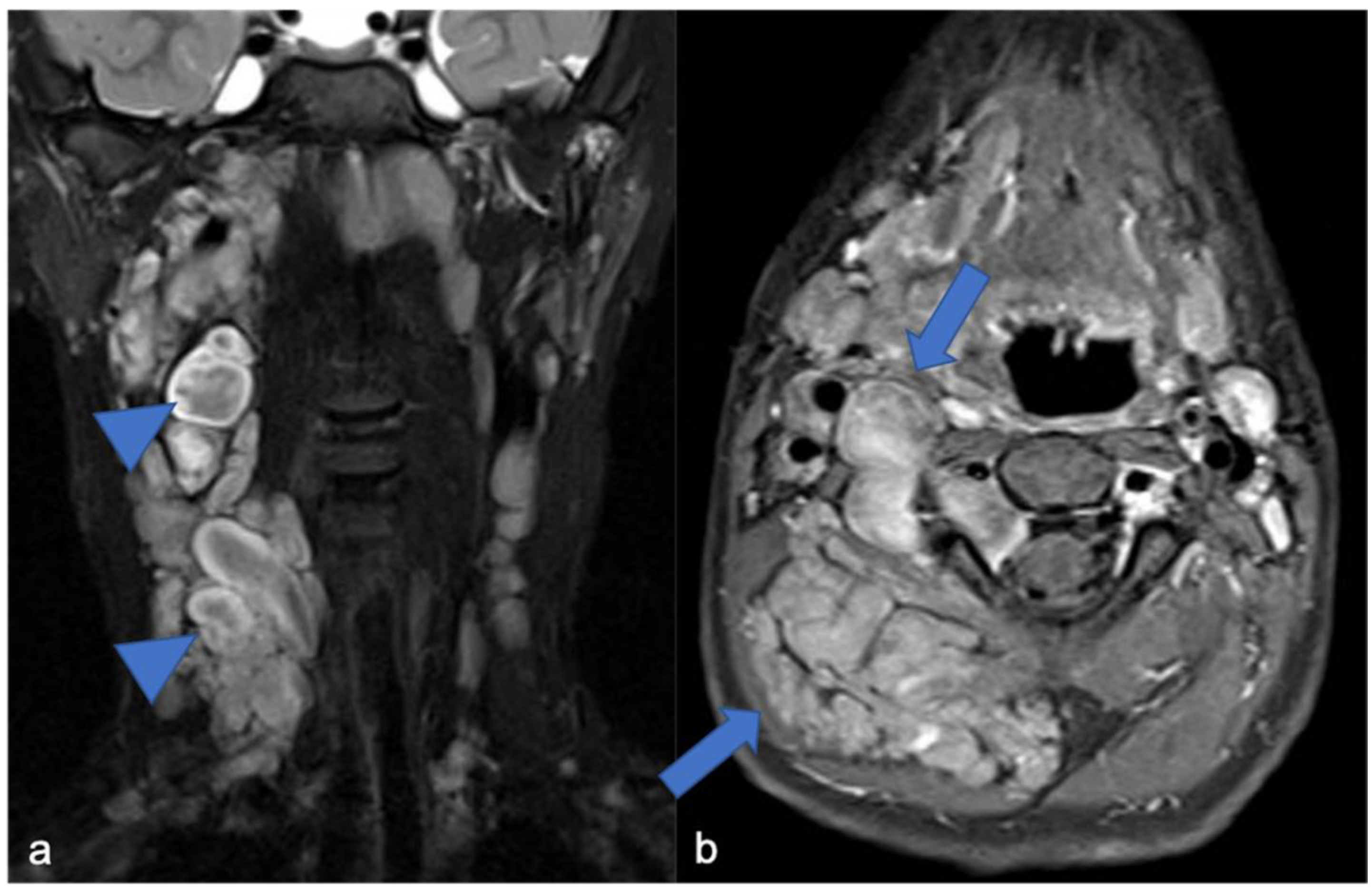
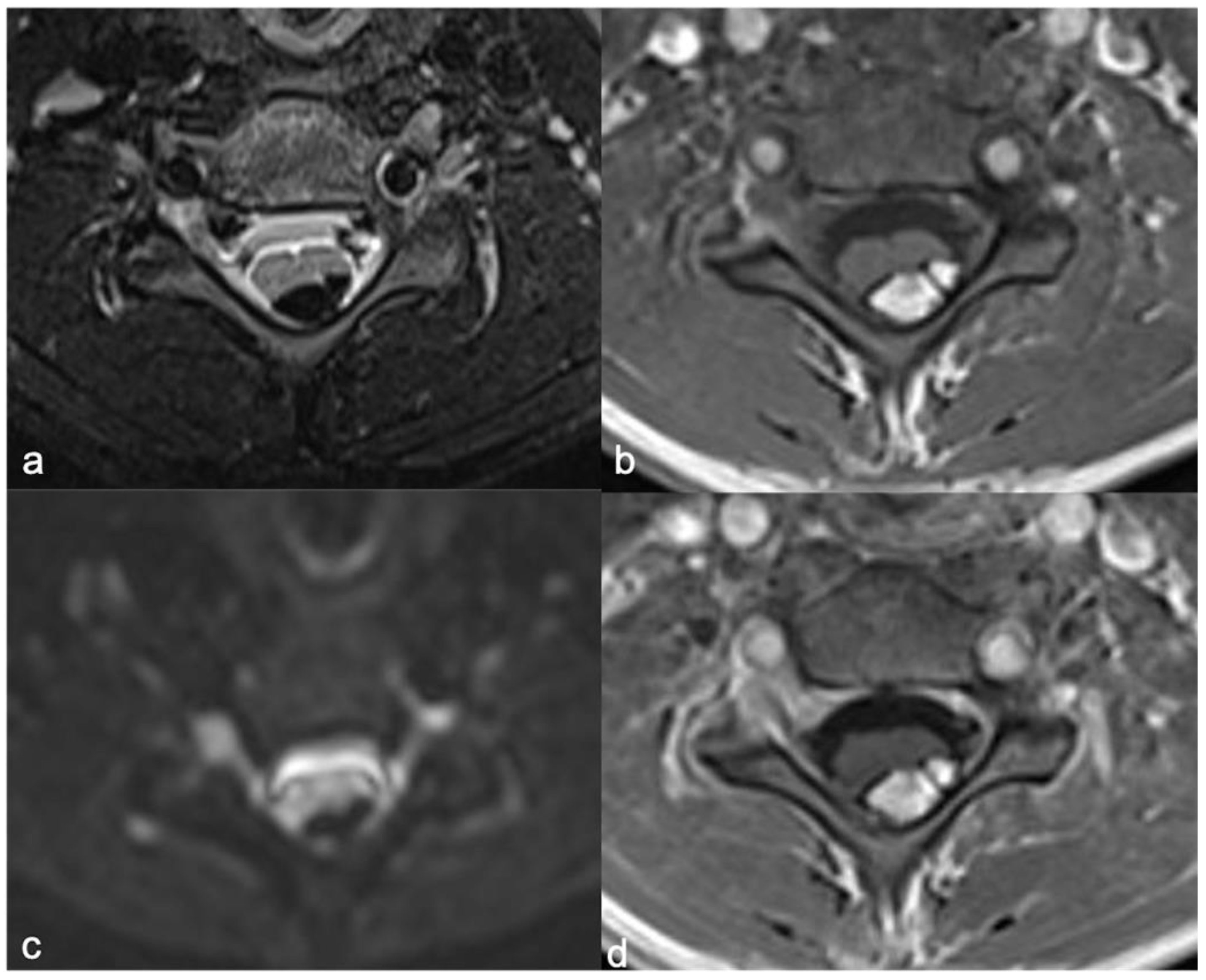
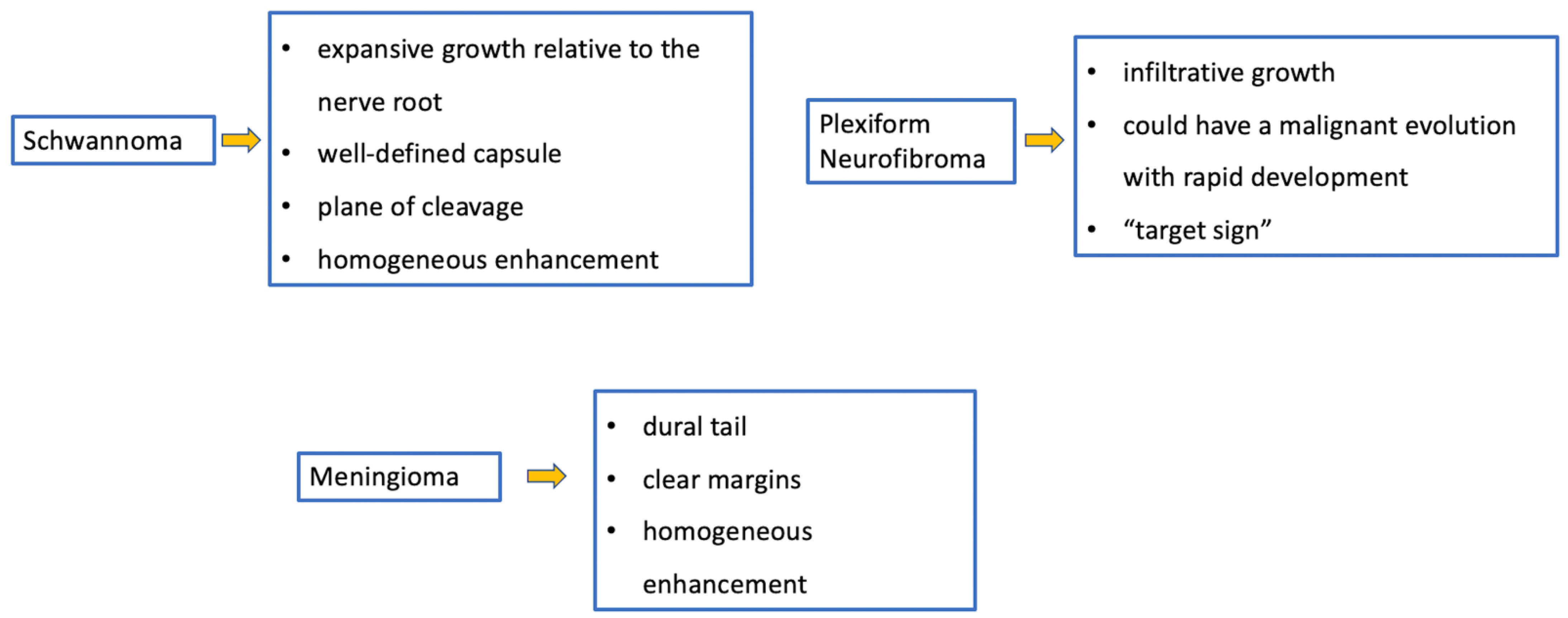
5. Meningiomas and Tumors of the Paraspinal Nerves
6. Embryonal Tumors
6.1. Atypical Teratoid/Rhabdoid Tumor
6.2. Embryonal Tumor with Multilayered Rosettes
7. Genetic Syndromes
8. Imaging Technique and Differential Diagnoses
9. Targeted Therapies
10. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Koeller, K.K.; Rosenblum, R.S.; Morrison, A.L. Neoplasms of the spinal cord and filum terminale: Radiologic-Pathologic correlation. RadioGraphics 2000, 20, 1721–1749. [Google Scholar] [CrossRef] [PubMed]
- Wilson, P.E.; Oleszek, J.L.; Clayton, G.H. Pediatric spinal cord tumors and masses. J. Spinal Cord Med. 2007, 30, S15–S20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, A.; Gandolfo, C.; Morana, G.; Tortori-Donati, P. Tumors of the Spine in children. Neuroimaging Clin. N. Am. 2007, 17, 17–35. [Google Scholar] [CrossRef] [PubMed]
- Huisman, T.A.G.M. Pediatric tumors of the spine. Cancer Imaging 2009, 9, S45–S48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rifkinson-Mann, S.; Wisoff, J.H.; Epstein, F. The association of hydrocephalus with intramedullary spinal cord tumors: A series of 25 patients. Neurosurgery 1990, 27, 749–754. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.B.; Soderlund, K.A.; Rushing, E.J.; Smirniotopolous, J.G. Radiologic-Pathologic correlation of pediatric and adolescent spinal neoplasms: Part 1, intramedullary spinal neoplasms. Am. J. Roentgenol. 2012, 198, 34–43. [Google Scholar] [CrossRef]
- Tortori-Donati, P.; Rossi, A. Pediatric Neuroradiology; Springer: Berlin/Heidelberg, Germany, 2005; ISBN 978-3-540-41077-5. [Google Scholar]
- Seo, H.S.; Kim, J.-H.; Lee, D.H.; Lee, Y.H.; Suh, S.-I.; Kim, S.Y.; Na, D.G. Nonenhancing intramedullary astrocytomas and other MR imaging features: A retrospective study and systematic review. Am. J. Neuroradiol. 2010, 31, 498–503. [Google Scholar] [CrossRef] [Green Version]
- Cacchione, A.; Mastronuzzi, A.; Cefalo, M.G.; Colafati, G.S.; Diomedi-Camassei, F.; Rizzi, M.; De Benedictis, A.; Carai, A. Pediatric spinal glioblastoma of the Conus Medullaris: A Case Report of Long Survival. Chin. J. Cancer 2016, 35, 44. [Google Scholar] [CrossRef] [Green Version]
- Pollack, I.F. Intramedullary spinal cord astrocytomas in children. Pediatr. Blood Cancer 2004, 43, 617–618. [Google Scholar] [CrossRef]
- Mori, K.; Imai, S.; Shimizu, J.; Taga, T.; Ishida, M.; Matsusue, Y. Spinal glioblastoma multiforme of the Conus Medullaris with Holocordal and intracranial spread in a child: A case report and review of the literature. Spine J. 2012, 12, e1–e6. [Google Scholar] [CrossRef]
- Grob, S.T.; Nobre, L.; Campbell, K.R.; Davies, K.D.; Ryall, S.; Aisner, D.L.; Hoffman, L.; Zahedi, S.; Morin, A.; Crespo, M.; et al. Clinical and Molecular characterization of a multi-institutional cohort of pediatric spinal cord low-grade gliomas. Neurooncol. Adv. 2020, 2, vdaa103. [Google Scholar] [CrossRef]
- Konovalov, N.A.; Asyutin, D.S.; Shayhaev, E.G.; Kaprovoy, S.V.; Timonin, S.Y. Molecular Biomarkers of brain and spinal cord astrocytomas. Acta Nat. 2019, 11, 17–27. [Google Scholar] [CrossRef]
- Hawkins, C.; Walker, E.; Mohamed, N.; Zhang, C.; Jacob, K.; Shirinian, M.; Alon, N.; Kahn, D.; Fried, I.; Scheinemann, K.; et al. BRAF-KIAA1549 Fusion Predicts Better Clinical Outcome in Pediatric Low-Grade Astrocytoma. Clin. Cancer Res. 2011, 17, 4790–4798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, A.; Jakacki, R.I.; Onar-Thomas, A.; Wu, S.; Nicolaides, T.; Young Poussaint, T.; Fangusaro, J.; Phillips, J.; Perry, A.; Turner, D.; et al. A phase I trial of the MEK inhibitor selumetinib (AZD6244) in pediatric Patients with recurrent or refractory low-grade glioma: A Pediatric brain tumor consortium (PBTC) study. Neuro-Oncol. 2017, 19, 1135–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindler, G.; Capper, D.; Meyer, J.; Janzarik, W.; Omran, H.; Herold-Mende, C.; Schmieder, K.; Wesseling, P.; Mawrin, C.; Hasselblatt, M.; et al. Analysis of BRAF V600E mutation in 1320 Nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol. 2011, 121, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Perwein, T.; Benesch, M.; Kandels, D.; Pietsch, T.; Schmidt, R.; Quehenberger, F.; Bison, B.; Warmuth-Metz, M.; Timmermann, B.; Krauss, J.; et al. High frequency of disease progression in pediatric spinal cord low-grade glioma (lgG): Management Strategies and results from the German LGG Study Group. Neuro-Oncol. 2020, 23, 1148–1162. [Google Scholar] [CrossRef]
- Okuda, T.; Hata, N.; Suzuki, S.O.; Yoshimoto, K.; Arimura, K.; Amemiya, T.; Akagi, Y.; Kuga, D.; Oba, U.; Koga, Y.; et al. Pediatric ganglioglioma with an H3 K27M mutation arising from the cervical spinal cord. Neuropathology 2018, 38, 422–427. [Google Scholar] [CrossRef]
- Chai, R.-C.; Zhang, Y.-W.; Liu, Y.-Q.; Chang, Y.-Z.; Pang, B.; Jiang, T.; Jia, W.-Q.; Wang, Y.-Z. The molecular characteristics of spinal cord gliomas with or without H3 K27M mutation. Acta Neuropathol. Commun. 2020, 8, 40. [Google Scholar] [CrossRef]
- Hamburger, C.; Büttner, A.; Weis, S. Ganglioglioma of the spinal cord: Report of two rare cases and review of the literature. Neurosurgery 1997, 41, 1410–1415. [Google Scholar] [CrossRef]
- Ryall, S.; Tabori, U.; Hawkins, C. Pediatric Low-grade glioma in the era of molecular diagnostics. Acta Neuropathol. Commun. 2020, 8, 30. [Google Scholar] [CrossRef]
- Houten, J.K.; Weiner, H.L. Pediatric Intramedullary spinal cord tumors: Special considerations. J. Neurooncol. 2000, 47, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Patel, U.; Pinto, R.S.; Miller, D.C.; Handler, M.S.; Rorke, L.B.; Epstein, F.J.; Kricheff, I.I. MR of Spinal cord ganglioglioma. Am. J. Neuroradiol. 1998, 19, 879–887. [Google Scholar] [PubMed]
- Lang, F.F.; Epstein, F.J.; Ransohoff, J.; Allen, J.C.; Wisoff, J.; Abbott, I.R.; Miller, D.C. Central nervous system gangliogliomas. Part 2: Clinical Outcome. J. Neurosurg. 1993, 79, 867–873. [Google Scholar] [CrossRef] [PubMed]
- Deora, H.; Sumitra, S.; Nandeesh, B.N.; Bhaskara Rao, M.; Arivazhagan, A. Spinal Intramedullary ganglioglioma in children: An unusual location of a common pediatric tumor. Pediatr. Neurosurg. 2019, 54, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Gardiman, M.P.; Fassan, M.; Orvieto, E.; D’Avella, D.; Denaro, L.; Calderone, M.; Severino, M.; Scarsello, G.; Viscardi, E.; Perilongo, G. Diffuse Leptomeningeal glioneuronal tumors: A new entity? Brain Pathol. 2010, 20, 361–366. [Google Scholar] [CrossRef]
- Chiang, J.C.H.; Harreld, J.H.; Orr, B.A.; Sharma, S.; Ismail, A.; Segura, A.D.; Ellison, D.W. Low-Grade spinal glioneuronal tumors with BRAF Gene fusion and 1p deletion but without leptomeningeal dissemination. Acta Neuropathol. 2017, 134, 159–162. [Google Scholar] [CrossRef]
- Deng, M.Y.; Sill, M.; Chiang, J.; Schittenhelm, J.; Ebinger, M.; Schuhmann, M.U.; Monoranu, C.-M.; Milde, T.; Wittmann, A.; Hartmann, C.; et al. Molecularly defined diffuse leptomeningeal glioneuronal tumor (DLGNT) Comprises two subgroups with distinct clinical and genetic features. Acta Neuropathol. 2018, 136, 239–253. [Google Scholar] [CrossRef] [Green Version]
- Peer, S.; Murumkar, V.; Kulanthaivelu, K.; Prasad, C.; Rao, S.; Santosh, V. Diffuse Leptomeningeal glioneuronal tumor with high-grade features masquerading as tubercular meningitis—A case report. Egypt. J. Radiol. Nucl. Med. 2021, 52, 146. [Google Scholar] [CrossRef]
- Nemoto, Y.; Inoue, Y.; Tashiro, T.; Mochizuki, K.; Oda, J.; Kogame, S.; Katsuyama, J.; Hakuba, A.; Onoyama, Y. Intramedullary Spinal cord tumors: Significance of Associated hemorrhage at MR imaging. Radiology 1992, 182, 793–796. [Google Scholar] [CrossRef]
- Ghasemi, D.R.; Sill, M.; Okonechnikov, K.; Korshunov, A.; Yip, S.; Schutz, P.W.; Scheie, D.; Kruse, A.; Harter, P.N.; Kastelan, M.; et al. MYCN amplification drives an aggressive form of spinal ependymoma. Acta Neuropathol. 2019, 138, 1075–1089. [Google Scholar] [CrossRef] [Green Version]
- Swanson, A.A.; Raghunathan, A.; Jenkins, R.B.; Messing-Jünger, M.; Pietsch, T.; Clarke, M.J.; Kaufmann, T.J.; Giannini, C. Spinal Cord ependymomas with MYCN amplification show aggressive clinical behavior. J. Neuropathol. Exp. Neurol. 2019, 78, 791–797. [Google Scholar] [CrossRef]
- Wippold, F.J.; Smirniotopoulos, J.G.; Moran, C.J.; Suojanen, J.N.; Vollmer, D.G. MR imaging of myxopapillary ependymoma: Findings and value to determine extent of tumor and its relation to intraspinal structures. Am. J. Roentgenol. 1995, 165, 1263–1267. [Google Scholar] [CrossRef] [PubMed]
- Benesch, M.; Weber-Mzell, D.; Gerber, N.U.; von Hoff, K.; Deinlein, F.; Krauss, J.; Warmuth-Metz, M.; Kortmann, R.-D.; Pietsch, T.; Driever, P.H.; et al. Ependymoma of the spinal cord in children and adolescents: A Retrospective series from the HIT database. J. Neurosurg. Pediatr. 2010, 6, 137–144. [Google Scholar] [CrossRef]
- Gläsker, S.; Vergauwen, E.; Koch, C.A.; Kutikov, A.; Vortmeyer, A.O. Von Hippel-Lindau Disease: Current challenges and future prospects. OncoTargets Ther. 2020, 13, 5669–5690. [Google Scholar] [CrossRef] [PubMed]
- Huvos, A.G.; Rosen, G.; Dabska, M.; Marcove, R.C. Mesenchymal chondrosarcoma. A clinicopathologic analysis of 35 patients with emphasis on treatment. Cancer 1983, 51, 1230–1237. [Google Scholar] [CrossRef]
- Andersson, C.; Österlundh, G.; Enlund, F.; Kindblom, L.-G.; Hansson, M. Primary spinal intradural mesenchymal chondrosarcoma with detection of fusion gene HEY1-NCOA2: A paediatric case report and review of the literature. Oncol. Lett. 2014, 8, 1608–1612. [Google Scholar] [CrossRef] [PubMed]
- Di Giannatale, A.; Colletti, M.; Russo, I.; Ferruzzi, V.; Anna, V.A.D.; Cozza, R.; Colafati, G.S.; Messina, R.; Mastronuzzi, A.; De Vito, R.; et al. Intraspinal Mesenchymal chondrosarcoma: Report of a Pediatric case and literature review. Tumori 2017, 103, S66–S72. [Google Scholar] [CrossRef]
- Wang, L.; Motoi, T.; Khanin, R.; Olshen, A.; Mertens, F.; Bridge, J.; Dal Cin, P.; Antonescu, C.R.; Singer, S.; Hameed, M.; et al. Identification of a novel, recurrent HEY1-NCOA2 Fusion in mesenchymal chondrosarcoma based on a genome-wide screen of exon-level expression data. Genes Chromosomes Cancer 2012, 51, 127–139. [Google Scholar] [CrossRef] [Green Version]
- Bishop, M.W.; Somerville, J.M.; Bahrami, A.; Kaste, S.C.; Interiano, R.B.; Wu, J.; Mao, S.; Boop, F.A.; Williams, R.F.; Pappo, A.S.; et al. Mesenchymal chondrosarcoma in children and young adults: A single institution retrospective review. Sarcoma 2015, 2015, 608279. [Google Scholar] [CrossRef] [Green Version]
- Pathmanaban, O.N.; Sadler, K.V.; Kamaly-Asl, I.D.; King, A.T.; Rutherford, S.A.; Hammerbeck-Ward, C.; McCabe, M.G.; Kilday, J.-P.; Beetz, C.; Poplawski, N.K.; et al. Association of genetic predisposition with solitary schwannoma or meningioma in children and young adults. JAMA Neurol. 2017, 74, 1123–1129. [Google Scholar] [CrossRef]
- Soderlund, K.A.; Smith, A.B.; Rushing, E.J.; Smirniotopolous, J.G. Radiologic-Pathologic correlation of pediatric and adolescent spinal neoplasms: Part 2, Intradural extramedullary spinal neoplasms. Am. J. Roentgenol. 2012, 198, 44–51. [Google Scholar] [CrossRef]
- Van Noesel, M.M.; Orbach, D.; Brennan, B.; Kelsey, A.; Zanetti, I.; de Salvo, G.L.; Gaze, M.N.; Craigie, R.J.; McHugh, K.; Francotte, N.; et al. Outcome and prognostic factors in pediatric malignant peripheral nerve sheath tumors: An analysis of the European Pediatric Soft Tissue Sarcoma Group (EpSSG) NRSTS-2005 prospective study. Pediatr. Blood Cancer 2019, 66, e27833. [Google Scholar] [CrossRef] [Green Version]
- Balogun, J.A.; Halliday, W.; Bouffet, E.; Kulkarni, A.V. Spinal Clear Cell Meningioma in a 3-Year-Old: A Case Report. Pediatric Neurosurg. 2013, 49, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Alameda, F.; Lloreta, J.; Ferrer, M.D.; Corominas, J.M.; Galitó, E.; Serrano, S. Clear cell meningioma of the lumbo-sacral spine with chordoid features. Ultrastruct. Pathol. 1999, 23, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Chang, K.-H.; Choe, G.; Chi, J.G.; Chung, C.-K.; Kim, I.H.; Han, M.H.; Park, S.-W.; Shin, S.J.; Koh, Y.H. MR Imaging features of clear-cell meningioma with diffuse leptomeningeal seeding. Am. J. Neuroradiol. 2000, 21, 130–132. [Google Scholar] [PubMed]
- Yu, K.B.; Lim, M.K.; Kim, H.J.; Suh, C.H.; Park, H.C.; Kim, E.Y.; Han, H.S. Clear-Cell meningioma: CT and MR Imaging findings in two cases involving the spinal canal and cerebellopontine angle. Korean J. Radiol. 2002, 3, 125–129. [Google Scholar] [CrossRef] [Green Version]
- Clear Cell Meningioma. A clinicopathologic study of a potentially aggressive variant of meningioma. Am. J. Surg. Pathol. 1995, 19, 493–505. [Google Scholar] [CrossRef]
- Wu, L.; Yang, C.; Liu, T.; Fang, J.; Yang, J.; Xu, Y. Clinical Features and long-term outcomes of pediatric spinal meningiomas. J. Neurooncol. 2017, 133, 347–355. [Google Scholar] [CrossRef]
- Smith, M.J.; O’Sullivan, J.; Bhaskar, S.S.; Hadfield, K.D.; Poke, G.; Caird, J.; Sharif, S.; Eccles, D.; Fitzpatrick, D.; Rawluk, D.; et al. Loss-of-Function mutations in SMARCE1 Cause an inherited disorder of multiple spinal meningiomas. Nat. Genet. 2013, 45, 295–298. [Google Scholar] [CrossRef]
- Biswas, A.; Kashyap, L.; Kakkar, A.; Sarkar, C.; Julka, P.K. Atypical Teratoid/rhabdoid tumors: Challenges and search for solutions. Cancer Manag. Res. 2016, 8, 115–125. [Google Scholar] [CrossRef] [Green Version]
- Rickert, C.H.; Paulus, W. Epidemiology of central nervous system tumors in childhood and adolescence based on the new WHO classification. Childs Nerv. Syst. 2001, 17, 503–511. [Google Scholar] [CrossRef]
- Woehrer, A.; Slavc, I.; Waldhoer, T.; Heinzl, H.; Zielonke, N.; Czech, T.; Benesch, M.; Hainfellner, J.A.; Haberler, C. Austrian Brain Tumor Registry incidence of atypical teratoid/rhabdoid tumors in children: A population-based study by the Austrian Brain Tumor Registry, 1996–2006. Cancer 2010, 116, 5725–5732. [Google Scholar] [CrossRef] [PubMed]
- Frühwald, M.C.; Biegel, J.A.; Bourdeaut, F.; Roberts, C.W.M.; Chi, S.N. Atypical Teratoid/rhabdoid tumors—Current concepts, advances in biology, and potential future therapies. Neuro-Oncol. 2016, 18, 764–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johann, P.D.; Erkek, S.; Zapatka, M.; Kerl, K.; Buchhalter, I.; Hovestadt, V.; Jones, D.T.W.; Sturm, D.; Hermann, C.; Segura Wang, M.; et al. Atypical Teratoid/rhabdoid tumors are comprised of three epigenetic subgroups with distinct enhancer landscapes. Cancer Cell 2016, 29, 379–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benesch, M.; Nemes, K.; Neumayer, P.; Hasselblatt, M.; Timmermann, B.; Bison, B.; Ebetsberger-Dachs, G.; Bourdeaut, F.; Dufour, C.; Biassoni, V.; et al. Spinal cord atypical teratoid/rhabdoid tumors in children: Clinical, genetic, and outcome characteristics in a Representative European Cohort. Pediatr. Blood Cancer 2020, 67, e28022. [Google Scholar] [CrossRef]
- Biegel, J.A.; Tan, L.; Zhang, F.; Wainwright, L.; Russo, P.; Rorke, L.B. Alterations of the HSNF5/INI1 Gene in central nervous system atypical teratoid/rhabdoid tumors and renal and extrarenal rhabdoid tumors. Clin. Cancer Res. 2002, 8, 3461–3467. [Google Scholar] [PubMed]
- Chamberlain, M.C.; Tredway, T.L. Adult primary intradural spinal cord tumors: A review. Curr. Neurol. Neurosci. Rep. 2011, 11, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Ginn, K.F.; Gajjar, A. Atypical Teratoid rhabdoid tumor: Current therapy and future directions. Front. Oncol. 2012, 2, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, J.M.; Abraham, M.; Nandeesh, B.N.; Nair, S.N. Pediatric Suprasellar atypical teratoid rhabdoid tumor arising from the third ventricle: A Rare tumor at a very rare location. Asian J. Neurosurg. 2018, 13, 873–876. [Google Scholar] [CrossRef] [PubMed]
- Korshunov, A.; Sturm, D.; Ryzhova, M.; Hovestadt, V.; Gessi, M.; Jones, D.T.W.; Remke, M.; Northcott, P.; Perry, A.; Picard, D.; et al. Embryonal tumor with abundant neuropil and true rosettes (ETANTR), ependymoblastoma, and medulloepithelioma share molecular similarity and comprise a single clinicopathological entity. Acta Neuropathol. 2014, 128, 279–289. [Google Scholar] [CrossRef] [Green Version]
- Spence, T.; Sin-Chan, P.; Picard, D.; Barszczyk, M.; Hoss, K.; Lu, M.; Kim, S.-K.; Ra, Y.-S.; Nakamura, H.; Fangusaro, J.; et al. CNS-PNETs with C19MC amplification and/or LIN28 Expression comprise a distinct histogenetic diagnostic and therapeutic entity. Acta Neuropathol. 2014, 128, 291–303. [Google Scholar] [CrossRef] [Green Version]
- Mayr, L.; Gojo, J.; Peyrl, A.; Azizi, A.A.; Stepien, N.M.; Pletschko, T.; Czech, T.; Dorfer, C.; Lambo, S.; Dieckmann, K.; et al. Potential importance of early focal radiotherapy following gross total resection for long-term survival in children with embryonal tumors with multilayered rosettes. Front. Oncol. 2020, 10, 584681. [Google Scholar] [CrossRef]
- Eberhart, C.G.; Brat, D.J.; Cohen, K.J.; Burger, P.C. Pediatric neuroblastic brain tumors containing abundant neuropil and true rosettes. Pediatr. Dev. Pathol. 2000, 3, 346–352. [Google Scholar] [CrossRef]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO Classification of Tumours of the Central Nervous System. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [Green Version]
- Lambo, S.; von Hoff, K.; Korshunov, A.; Pfister, S.M.; Kool, M. ETMR: A Tumor entity in its infancy. Acta Neuropathol. 2020, 140, 249–266. [Google Scholar] [CrossRef]
- Nowak, J.; Seidel, C.; Berg, F.; Pietsch, T.; Friedrich, C.; von Hoff, K.; Rutkowski, S.; Warmuth-Metz, M. MRI characteristics of ependymoblastoma: Results from 22 centrally reviewed cases. Am. J. Neuroradiol. 2014, 35, 1996–2001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dangouloff-Ros, V.; Tauziède-Espariat, A.; Roux, C.-J.; Levy, R.; Grévent, D.; Brunelle, F.; Gareton, A.; Puget, S.; Beccaria, K.; Blauwblomme, T.; et al. CT and multimodal MR Imaging features of embryonal tumors with multilayered rosettes in children. Am. J. Neuroradiol. 2019, 40, 32–736. [Google Scholar] [CrossRef]
- Vijapura, C.; Saad Aldin, E.; Capizzano, A.A.; Policeni, B.; Sato, Y.; Moritani, T. Genetic syndromes associated with central nervous system tumors. RadioGraphics 2016, 37, 258–280. [Google Scholar] [CrossRef] [PubMed]
- Hoa, M.; Slattery, W.H. Neurofibromatosis 2. Otolaryngol. Clin. N. Am. 2012, 45, 315–332. [Google Scholar] [CrossRef]
- Legius, E.; Messiaen, L.; Wolkenstein, P.; Pancza, P.; Avery, R.A.; Berman, Y.; Blakeley, J.; Babovic-Vuksanovic, D.; Cunha, K.S.; Ferner, R.; et al. Revised Diagnostic criteria for neurofibromatosis Type 1 and legius syndrome: An International consensus recommendation. Genet. Med. 2021, 23, 1506–1513. [Google Scholar] [CrossRef]
- Gutmann, D.H.; Rasmussen, S.A.; Wolkenstein, P.; MacCollin, M.M.; Guha, A.; Inskip, P.D.; North, K.N.; Poyhonen, M.; Birch, P.H.; Friedman, J.M. Gliomas presenting after age 10 in Individuals with neurofibromatosis Type 1 (NF1). Neurology 2002, 59, 759–761. [Google Scholar] [CrossRef]
- Rodriguez, F.J.; Perry, A.; Gutmann, D.H.; O’Neill, B.P.; Leonard, J.; Bryant, S.; Giannini, C. Gliomas in neurofibromatosis type 1: A clinicopathologic study of 100 patients. J. Neuropathol. Exp. Neurol. 2008, 67, 240–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakkar, S.D.; Feigen, U.; Mautner, V.F. Spinal Tumours in Neurofibromatosis type 1: An MRI Study of frequency, multiplicity and variety. Neuroradiology 1999, 41, 625–629. [Google Scholar] [CrossRef] [PubMed]
- Ruggieri, M.; Polizzi, A.; Spalice, A.; Salpietro, V.; Caltabiano, R.; D’Orazi, V.; Pavone, P.; Pirrone, C.; Magro, G.; Platania, N.; et al. The natural history of spinal neurofibromatosis: A Critical review of clinical and genetic features. Clin. Genet. 2015, 87, 401–410. [Google Scholar] [CrossRef]
- Ducatman, B.S.; Scheithauer, B.W.; Piepgras, D.G.; Reiman, H.M.; Ilstrup, D.M. Malignant Peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer 1986, 57, 2006–2021. [Google Scholar] [CrossRef]
- Shofty, B.; Barzilai, O.; Khashan, M.; Lidar, Z.; Constantini, S. Spinal manifestations of neurofibromatosis type 1. Childs Nerv. Syst. 2020, 36, 2401–2408. [Google Scholar] [CrossRef] [PubMed]
- Goertz, O.; Langer, S.; Uthoff, D.; Ring, A.; Stricker, I.; Tannapfel, A.; Steinau, H.-U. Diagnosis, treatment and survival of 65 patients with malignant peripheral nerve sheath tumors. Anticancer Res. 2014, 34, 777–783. [Google Scholar]
- Uhlmann, E.J.; Plotkin, S.R. Neurofibromatoses. Adv. Exp. Med. Biol. 2012, 724, 266–277. [Google Scholar] [CrossRef]
- Evans, D.G.R. Neurofibromatosis type 2 (NF2): A Clinical and molecular review. Orphanet J. Rare Dis. 2009, 4, 16. [Google Scholar] [CrossRef] [Green Version]
- Mautner, V.F.; Tatagiba, M.; Lindenau, M.; Fünsterer, C.; Pulst, S.M.; Baser, M.E.; Kluwe, L.; Zanella, F.E. Spinal Tumors in patients with neurofibromatosis type 2: MR imaging study of frequency, multiplicity, and variety. Am. J. Roentgenol. 1995, 165, 951–955. [Google Scholar] [CrossRef]
- Coy, S.; Rashid, R.; Stemmer-Rachamimov, A.; Santagata, S. An update on the CNS manifestations of neurofibromatosis type 2. Acta Neuropathol. 2020, 139, 643–665. [Google Scholar] [CrossRef] [Green Version]
- Baser, M.E.; Friedman, J.M.; Aeschliman, D.; Joe, H.; Wallace, A.J.; Ramsden, R.T.; Evans, D.G.R. Predictors of the risk of mortality in neurofibromatosis 2. Am. J. Hum. Genet. 2002, 71, 715–723. [Google Scholar] [CrossRef] [Green Version]
- Halliday, D.; Emmanouil, B.; Pretorius, P.; MacKeith, S.; Painter, S.; Tomkins, H.; Evans, D.G.; Parry, A. Genetic severity score predicts clinical phenotype in NF2. J. Med. Genet. 2017, 54, 657–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maher, E.R.; Iselius, L.; Yates, J.R.; Littler, M.; Benjamin, C.; Harris, R.; Sampson, J.; Williams, A.; Ferguson-Smith, M.A.; Morton, N. Von Hippel-Lindau Disease: A genetic study. J. Med. Genet. 1991, 28, 443–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, H.P.; Wiestler, O.D. Clustering of features of von Hippel-Lindau Syndrome: Evidence for a complex genetic locus. Lancet 1991, 337, 1052–1054. [Google Scholar] [CrossRef]
- Grasparil, A.D.; Gottumukkala, R.V.; Greer, M.-L.C.; Gee, M.S. Whole-Body MRI Surveillance of cancer predisposition syndromes: Current Best practice guidelines for use, performance, and interpretation. Am. J. Roentgenol. 2020, 215, 1002–1011. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Martinetti, C.; Morana, G.; Severino, M.; Tortora, D. Diagnostic approach to pediatric spine disorders. Magn. Reson. Imaging Clin. N. Am. 2016, 24, 621–644. [Google Scholar] [CrossRef] [PubMed]
- Vargas, M.I.; Delattre, B.M.A.; Boto, J.; Gariani, J.; Dhouib, A.; Fitsiori, A.; Dietemann, J.L. Advanced Magnetic Resonance Imaging (MRI) Techniques of the Spine and Spinal Cord in Children and Adults. Insights Imaging 2018, 9, 549–557. [Google Scholar] [CrossRef] [Green Version]
- Ishizaka, K.; Kudo, K.; Fujima, N.; Zaitsu, Y.; Yazu, R.; Tha, K.K.; Terae, S.; Haacke, E.M.; Sasaki, M.; Shirato, H. Detection of normal spinal veins by using susceptibility-weighted imaging. J. Magn. Reson. Imaging 2010, 31, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Fujima, N.; Kudo, K.; Terae, S.; Hida, K.; Ishizaka, K.; Zaitsu, Y.; Asano, T.; Yoshida, D.; Tha, K.K.; Haacke, E.M.; et al. Spinal arteriovenous malformation: Evaluation of change in venous oxygenation with susceptibility-weighted MR Imaging after treatment. Radiology 2010, 254, 891–899. [Google Scholar] [CrossRef]
- Fornasa, F. Diffusion-Weighted magnetic resonance imaging: What makes water run fast or slow? J. Clin. Imaging Sci. 2011, 1, 27. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Tian, W.; Chen, H.; LoStracco, T.A.; Zhang, J.; Li, M.Y.; Germin, B.; Wang, H.Z. Advanced neuroimaging in the evaluation of spinal cord tumors and tumor mimics: Diffusion tensor and perfusion-weighted imaging. Semin. Ultrasound CT MRI 2017, 38, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.B.; Poplawski, M.M.; Pawha, P.S.; Naidich, T.P.; Tanenbaum, L.N. Diffusion-Weighted MRI “Claw Sign” improves differentiation of infectious from degenerative modic type 1 signal changes of the spine. Am. J. Neuroradiol. 2014, 35, 1647–1652. [Google Scholar] [CrossRef] [Green Version]
- Mazura, J.C.; Karimi, S.; Pauliah, M.; Banihashemi, M.A.; Gobin, Y.P.; Bilsky, M.H.; Patsalides, A. Dynamic contrast-enhanced magnetic resonance perfusion compared with digital subtraction angiography for the evaluation of extradural spinal metastases: A pilot study. Spine 2014, 39, E950–E954. [Google Scholar] [CrossRef] [PubMed]
- Hock, A.; Henning, A.; Boesiger, P.; Kollias, S.S. 1H-MR Spectroscopy in the Human spinal cord. Am. J. Neuroradiol. 2013, 34, 1682–1689. [Google Scholar] [CrossRef] [Green Version]
- Holly, L.T.; Ellingson, B.M.; Salamon, N. Metabolic Imaging using proton magnetic spectroscopy as a predictor of outcome after surgery for cervical spondylotic myelopathy. Clin. Spine Surg. 2017, 30, E615–E619. [Google Scholar] [CrossRef]
- Rossi, A. Pediatric spinal infection and inflammation. Neuroimaging Clin. N. Am. 2015, 25, 173–191. [Google Scholar] [CrossRef]
- Indrajit, I.; Ganesan, S. Magnetic REsonance Imaging in Intracranial tuberculosis. Med. J. Armed Forces India 2001, 57, 292–297. [Google Scholar] [CrossRef] [Green Version]
- D’Amico, A.; Mazio, F.; Ugga, L.; Cuocolo, R.; Cirillo, M.; Santoro, C.; Perrotta, S.; Melis, D.; Brunetti, A. Medullary Unidentified bright objects in neurofibromatosis type 1: A Case series. BMC Pediatrics 2018, 18, 91. [Google Scholar] [CrossRef] [Green Version]
- Klonou, A.; Piperi, C.; Gargalionis, A.N.; Papavassiliou, A.G. Molecular Basis of pediatric brain tumors. Neuromol. Med. 2017, 19, 256–270. [Google Scholar] [CrossRef]
- Del Bufalo, F.; Carai, A.; Figà-Talamanca, L.; Pettorini, B.; Mallucci, C.; Giangaspero, F.; Antonelli, M.; Badiali, M.; Moi, L.; Bianco, G.; et al. Response of recurrent BRAFV600E mutated ganglioglioma to vemurafenib as single agent. J. Transl. Med. 2014, 12, 356. [Google Scholar] [CrossRef]
- Hargrave, D.R.; Bouffet, E.; Tabori, U.; Broniscer, A.; Cohen, K.J.; Hansford, J.R.; Geoerger, B.; Hingorani, P.; Dunkel, I.J.; Russo, M.W.; et al. Efficacy and safety of dabrafenib in pediatric patients with BRAF V600 mutation—Positive relapsed or refractory low-grade glioma: Results from a phase I/IIa study. Clin. Cancer Res. 2019, 25, 7303–7311. [Google Scholar] [CrossRef] [PubMed]
- Lassaletta, A.; Zapotocky, M.; Bouffet, E.; Hawkins, C.; Tabori, U. An integrative molecular and genomic analysis of pediatric hemispheric low-grade gliomas: An update. Childs Nerv. Syst. 2016, 32, 1789–1797. [Google Scholar] [CrossRef] [PubMed]
- Cacchione, A.; Lodi, M.; Carai, A.; Miele, E.; Tartaglia, M.; Megaro, G.; Del Baldo, G.; Alessi, I.; Colafati, G.S.; Carboni, A.; et al. Upfront Treatment with MTOR Inhibitor Everolimus in Pediatric Low-Grade Gliomas: A Single-Center Experience. Int. J. Cancer 2020, 148, 2522–2534. [Google Scholar] [CrossRef] [PubMed]
- Tiong, I.S.; Wei, A.H. New Drugs Creating New Challenges in Acute Myeloid Leukemia. Genes Chromosomes Cancer 2019, 58, 903–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agnihotri, S.; Jalali, S.; Wilson, M.R.; Danesh, A.; Li, M.; Klironomos, G.; Krieger, J.R.; Mansouri, A.; Khan, O.; Mamatjan, Y.; et al. The genomic landscape of schwannoma. Nat. Genet. 2016, 48, 1339–1348. [Google Scholar] [CrossRef] [PubMed]






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumors | Location * | Molecolar/Genetics |
|---|---|---|
| LGG | Cervico and thoracic tract | KIAA1549–BRAF fusion or BRAFV600E mutation; NF1 mutation |
| HGG | / | H3K27M |
| DL-GNT | Leptomeningeal dissemination | KIAA1549–BRAF, NTRK1/2/3, or TRIM33:RAF1 fusion |
| Ependymomas | Cervical/lumbo-sacral tract | RELA-/YAP1- fusion; nMyc amplification NF2 mutation |
| Hemangioblastomas | Variable epicenter | VHL mutation |
| Mesenchymal chondrosarcomas | Thoracic tract | HEY1/NCOA2 fusion |
| Meningiomas | / | NF2 mutation; SMARCE1, SMARCB1, or SUFU mutation |
| Schwannomas | Nerve sheaths | NF2 mutation; LZTR1 or SMARCB1 mutation |
| Plexiform neurofibromas | Nerve sheaths | NF1 mutation |
| Atypical teratoid/rhabdoid tumor | / | SMARCB1 mutation |
| Embryonal tumor with multilayered rosettes | / | C19MC amplification |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marrazzo, A.; Cacchione, A.; Rossi, S.; Carboni, A.; Gandolfo, C.; Carai, A.; Mastronuzzi, A.; Colafati, G.S. Intradural Pediatric Spinal Tumors: An Overview from Imaging to Novel Molecular Findings. Diagnostics 2021, 11, 1710. https://doi.org/10.3390/diagnostics11091710
Marrazzo A, Cacchione A, Rossi S, Carboni A, Gandolfo C, Carai A, Mastronuzzi A, Colafati GS. Intradural Pediatric Spinal Tumors: An Overview from Imaging to Novel Molecular Findings. Diagnostics. 2021; 11(9):1710. https://doi.org/10.3390/diagnostics11091710
Chicago/Turabian StyleMarrazzo, Antonio, Antonella Cacchione, Sabrina Rossi, Alessia Carboni, Carlo Gandolfo, Andrea Carai, Angela Mastronuzzi, and Giovanna Stefania Colafati. 2021. "Intradural Pediatric Spinal Tumors: An Overview from Imaging to Novel Molecular Findings" Diagnostics 11, no. 9: 1710. https://doi.org/10.3390/diagnostics11091710