
A Male Subject with Congenital Adrenal Hyperplasia due to 21-Hydroxylase Deficiency Which Was Diagnosed at 31 Years Old due to Infertility
 ,
,
Abstract
:1. Introduction
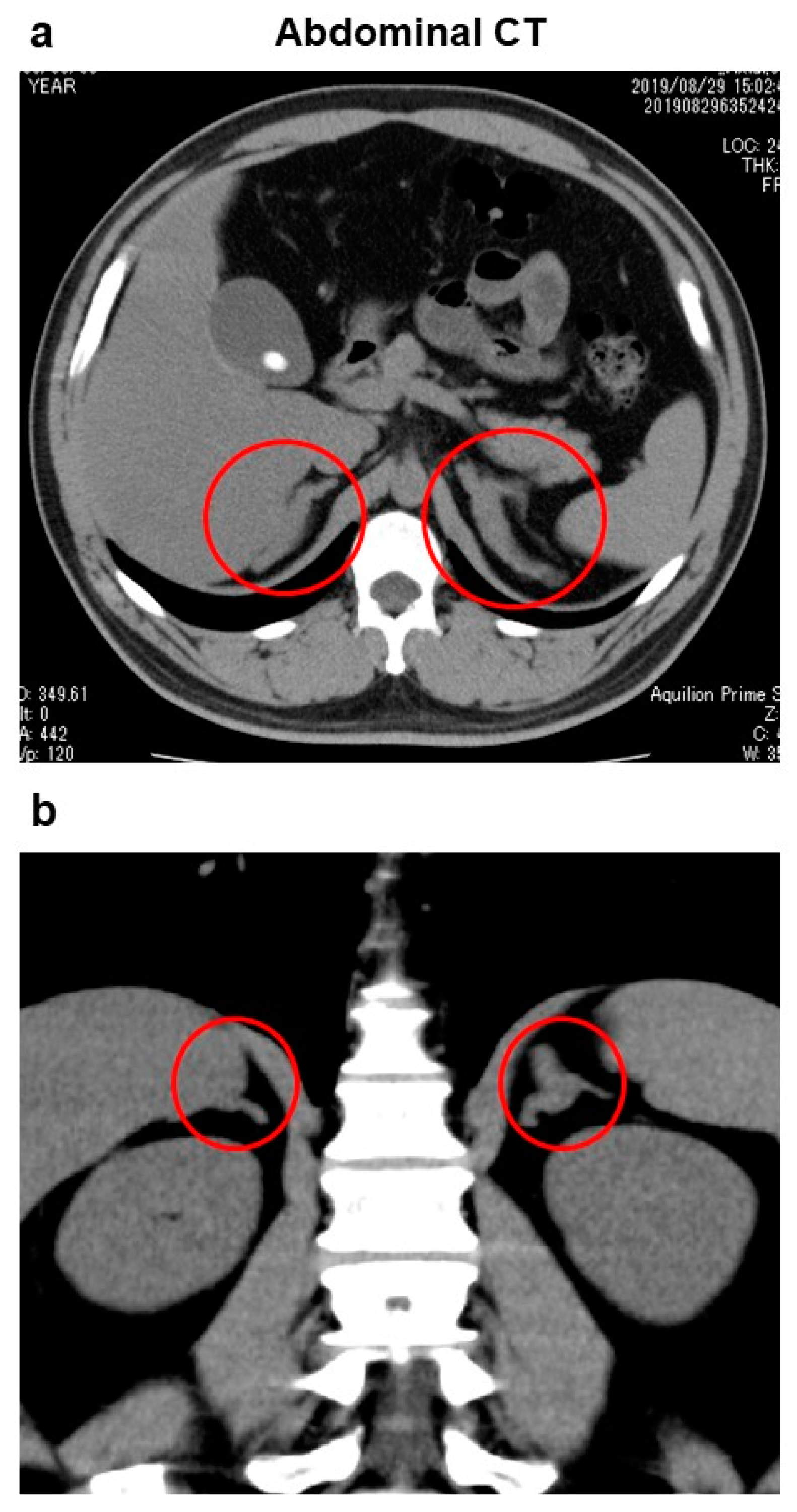
2. Case Presentation
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Speiser, P.W.; White, P.C. Congenital Adrenal Hyperplasia. New Engl. J. Med. 2003, 349, 776–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merke, D.P.; Auchus, R.J. Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency. N. Engl. J. Med. 2020, 383, 1248–1261. [Google Scholar] [CrossRef]
- Finkielstain, G.P.; Vieites, A.; Bergadá, I.; Rey, R.A. Disorders of Sex Development of Adrenal Origin. Front. Endocrinol. 2021, 12, 770782. [Google Scholar] [CrossRef] [PubMed]
- Nordenström, A.; Lajic, S.; Falhammar, H. Clinical outcomes in 21-hydroxylase deficiency. Curr. Opin. Endocrinol. Diabetes 2021, 28, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Claahsen-van der Grinten, H.L.; Speiser, P.W.; Ahmed, S.F.; Arlt, W.; Auchus, R.J.; Falhammar, H.; E Flück, C.; Guasti, L.; Huebner, A.; Kortmann, B.B.M.; et al. Congenital Adrenal Hyperplasia—Current Insights in Pathophysiology, Diagnostics, and Management. Endocr. Rev. 2021, 43, 91–159. [Google Scholar] [CrossRef]
- Morikawa, S.; Nakamura, A.; Fujikura, K.; Fukushi, M.; Hotsubo, T.; Miyata, J.; Ishizu, K.; Tajima, T. Results from 28 Years of Newborn Screening for Congenital Adrenal Hyperplasia in Sapporo. Clin. Pediatr. Endocrinol. 2014, 23, 35–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, T.; Kashimada, K.; Amano, N.; Takasawa, K.; Nakamura-Utsunomiya, A.; Yatsuga, S.; Mukai, T.; Ida, S.; Isobe, M.; Fukushi, M.; et al. Clinical guidelines for the diagnosis and treatment of 21-hydroxylase deficiency (2021 revision). Clin. Pediatr. Endocrinol. 2022, 31, 116–143. [Google Scholar] [CrossRef]
- El-Maouche, D.; Arlt, W.; Merke, D.P. Congenital adrenal hyperplasia. Lancet 2017, 390, 2194–2210. [Google Scholar] [CrossRef] [PubMed]
- Jha, S.; Turcu, A.F. Nonclassic Congenital Adrenal Hyperplasia: What do endocrinologists need to know? Endocrinol. Metab. Clin. N. Am. 2021, 50, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Simeoli, C.; de Angelis, C.; Veneri, A.D.; Menafra, D.; Di Paola, N.; Pivonello, C.; Di Somma, C.; Valerio, P.; Melis, D.; Alviggi, C.; et al. Severe impact of late diagnosis of congenital adrenal hyperplasia on gender identity, sexual orientation and function: Case report and review of the literature. Front. Genet. 2022, 13, 902844. [Google Scholar] [CrossRef] [PubMed]
- Ben Simon, A.; Brener, A.; Segev-Becker, A.; Yackobovitch-Gavan, M.; Uretzky, A.; Davidov, A.S.; Alaev, A.; Oren, A.; Eyal, O.; Weintrob, N.; et al. Body composition in children and adolescents with non-classic congenital adrenal hyperplasia and the risk for components of metabolic syndrome: An observational study. Front. Endocrinol. 2022, 13, 22752. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Zhang, Y.; Yu, Y.; Zhang, L.; Ullah, K.; Ji, M.; Jin, B.; Shu, J. Getting pregnant with congenital adrenal hyperplasia: Assisted reproduction and pregnancy complications. A systematic review and meta-analysis. Front. Endocrinol. 2022, 13, 982953. [Google Scholar] [CrossRef] [PubMed]
- Charmandari, E.; Matthews, D.R.; Johnston, A.; Brook, C.G.D.; Hindmarsh, P.C. Serum Cortisol and 17-Hydroxyprogesterone Interrelation in Classic 21-Hydroxylase Deficiency: Is Current Replacement Therapy Satisfactory? J. Clin. Endocrinol. Metab. 2001, 86, 4679–4685. [Google Scholar] [CrossRef] [PubMed]
- Auchus, R.J.; Arlt, W. Approach to the Patient: The Adult with Congenital Adrenal Hyperplasia. J. Clin. Endocrinol. Metab. 2013, 98, 2645–2655. [Google Scholar] [CrossRef]
- Bachelot, A.; Grouthier, V.; Courtillot, C.; Dulon, J.; Touraine, P. MANAGEMENT OF ENDOCRINE DISEASE: Congenital adrenal hyperplasia due to 21-hydroxylase deficiency: Update on the management of adult patients and prenatal treatment. Eur. J. Endocrinol. 2017, 176, R167–R181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auchus, R.J.; Courtillot, C.; Dobs, A.; El-Maouche, D.; Falhammar, H.; Lacroix, A.; Farrar, M.; O’Donoghue, C.; Anatchkova, M.; Cutts, K.; et al. Treatment patterns and unmet needs in adults with classic congenital adrenal hyperplasia: A modified Delphi consensus study. Front. Endocrinol. 2022, 13, 5963. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Peripheral Blood | Blood Biochemistry | Endocrine Markers | |||
|---|---|---|---|---|---|
| Red blood cells | 571 × 104/μL | Total protein | 7.4 g/dL | TSH | 3.62 μIU/mL |
| Hemoglobin | 17.0 g/dL | Albumin | 4.3 g/dL | FT3 | 3.47 pg/mL |
| Hematocrit | 49.1% | Total bilirubin | 0.5 mg/dL | FT4 | 1.18 ng/mL |
| White blood cells | 6210/μL | AST | 54 U/L | ACTH | 172.2 pg/mL |
| Neutrophils | 53.9% | ALT | 123 U/L | Cortisol | 6.9 μg/dL |
| Lymphocytes | 35.3% | γ-GTP | 66 U/L | DHEA-S | 738 mg/dL |
| Monocytes | 7.4% | LDH | 189 U/L | GH | 0.12 ng/mL |
| Eosinophils | 2.4% | ALP | 245 U/L | IGF-1 | 194 ng/mL |
| Basophils | 1.0% | Creatinine | 0.81 mg/dL | Prolactin | 13.5 ng/mL |
| Platelets | 21.6 × 104/μL | BUN | 20 mg/dL | LH | <0.10 mIU/L |
| Electrolytes | UA | 6.2 mg/dL | FSH | 0.22 mIU/mL | |
| Sodium | 137 mmol/L | CRP | 0.15 mg/dL | Free testosterone | 29.7 pg/mL |
| Potassium | 4.0 mmol/L | Plasma glucose | 109 mg/dL | Estradiol | 63.1 pg/mL |
| Chloride | 100 mmol/L | HbA1c | 8.1% | Progesterone | 8.15 ng/mL |
| Calcium | 9.7 mg/dL | LDL-C | 87 mg/dL | PRA | 16.5 ng/mL/hr |
| Phosphorus | 4.8 mg/dL | HDL-C | 28 mg/dL | Aldosterone | 659 pg/mL |
| Magnesium | 2.2 mg/dL | Triglyceride | 448 mg/dL | Adrenaline | 16 pg/mL |
| Noradrenaline | 194 pg/mL | ||||
| Pre | 15 Min | 30 Min | 60 Min | 120 Min | 180 Min | |
|---|---|---|---|---|---|---|
| ACTH (pg/mL) | 69.2 | 271.6 | 422.2 | 202.0 | 139.0 | 210.3 |
| Cortisol (μg/dL) | 8.8 | 8.9 | 8.7 | 8.1 | 8.9 | 8.9 |
| LH (mIU/mL) | <0.1 | 0.43 | 0.55 | 0.46 | 0.42 | |
| FSH (mIU/mL) | <0.1 | 0.31 | 0.38 | 0.57 | 0.79 | |
| GH (ng/mL) | 0.95 | 1.84 | 1.78 | 3.17 | 1.51 | 1.17 |
| TSH (mIU/mL) | 3.71 | 52.00 | 55.10 | 25.69 | 9.91 | 4.29 |
| Prolactin (ng/mL) | 8.2 | 88.3 | 65.7 | 25.3 | 19.0 |
| Urine Collection Test | Urine Steroid Fraction Analysis | ||||
|---|---|---|---|---|---|
| Parameter | Value | Reference Range | Parameter | Value | Reference Range |
| Cortisol | 33.9 μg/day | Pregnanetriol | 10.1 mg/gCre | 0.0–0.3 | |
| Aldosterone | 43 μg/day | 11OH-androstenedione | 22.5 mg/gCre | 0.00–0.35 | |
| Pregnanediol | 1.78 mg/day | 0.16–0.79 | |||
| Pregnanetriol | 36.12 mg/day | 0.13–1.60 | Other analysis | ||
| 17-KGS | 0.67 mg/day | 6.00–18.40 | Parameter | Value | Reference range |
| (17-KS fraction) | 17aOH-progesterone | 52.4 ng/mL | 0.0–0.6 | ||
| Androsterone | 7.18 mg/day | 1.10–4.20 | (Direct method) | ||
| Etiocholanolone | 1.82 mg/day | 0.55–2.60 | 17aOH-progesterone | 42.7 ng/mL | 0.00–0.35 |
| DHEA | 4.42 mg/day | 0.12–5.20 | (Extraction method) | ||
| 11-ketoandrosterone | 1.27 mg/day | 0.00–0.12 | |||
| 11-ketoetiocholanolone | 3.00 mg/day | 0.04–0.65 | |||
| 11OH-androsterone | 28.51 mg/day | 0.40–2.30 | |||
| 11OH-etiocholanolone | 0.06 mg/day | 0.03–0.65 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaneto, H.; Isobe, H.; Sanada, J.; Tatsumi, F.; Kimura, T.; Shimoda, M.; Nakanishi, S.; Kaku, K.; Mune, T. A Male Subject with Congenital Adrenal Hyperplasia due to 21-Hydroxylase Deficiency Which Was Diagnosed at 31 Years Old due to Infertility. Diagnostics 2023, 13, 505. https://doi.org/10.3390/diagnostics13030505
Kaneto H, Isobe H, Sanada J, Tatsumi F, Kimura T, Shimoda M, Nakanishi S, Kaku K, Mune T. A Male Subject with Congenital Adrenal Hyperplasia due to 21-Hydroxylase Deficiency Which Was Diagnosed at 31 Years Old due to Infertility. Diagnostics. 2023; 13(3):505. https://doi.org/10.3390/diagnostics13030505
Chicago/Turabian StyleKaneto, Hideaki, Hayato Isobe, Junpei Sanada, Fuminori Tatsumi, Tomohiko Kimura, Masashi Shimoda, Shuhei Nakanishi, Kohei Kaku, and Tomoatsu Mune. 2023. "A Male Subject with Congenital Adrenal Hyperplasia due to 21-Hydroxylase Deficiency Which Was Diagnosed at 31 Years Old due to Infertility" Diagnostics 13, no. 3: 505. https://doi.org/10.3390/diagnostics13030505